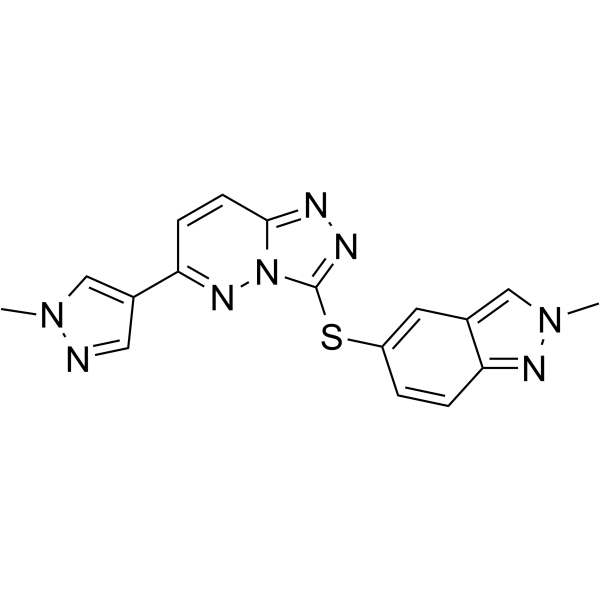
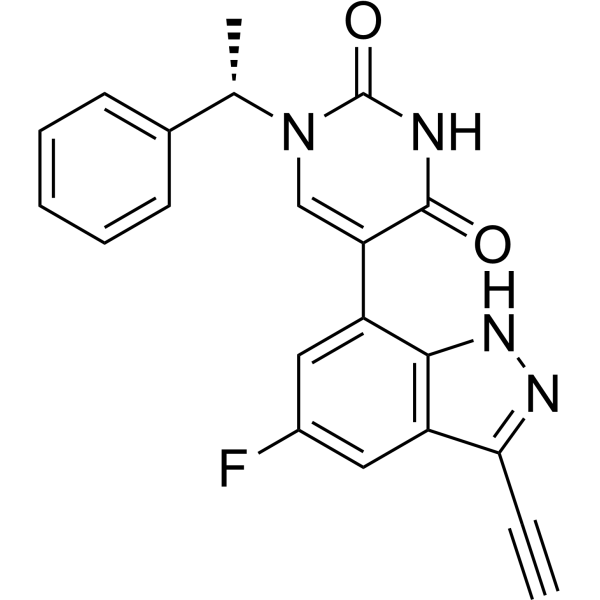
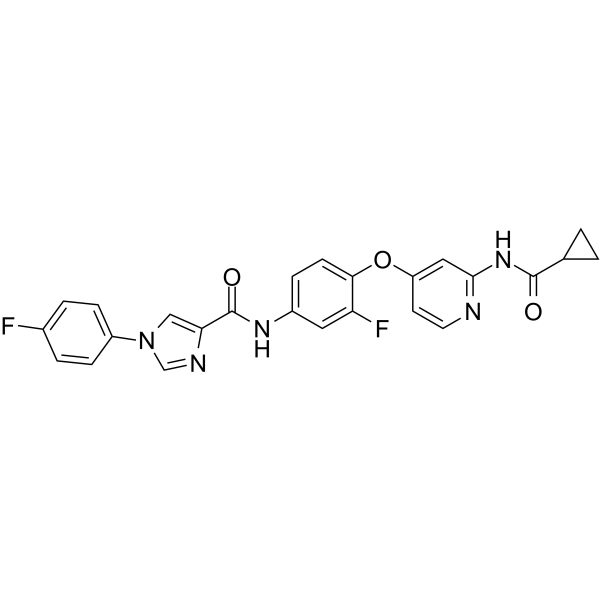
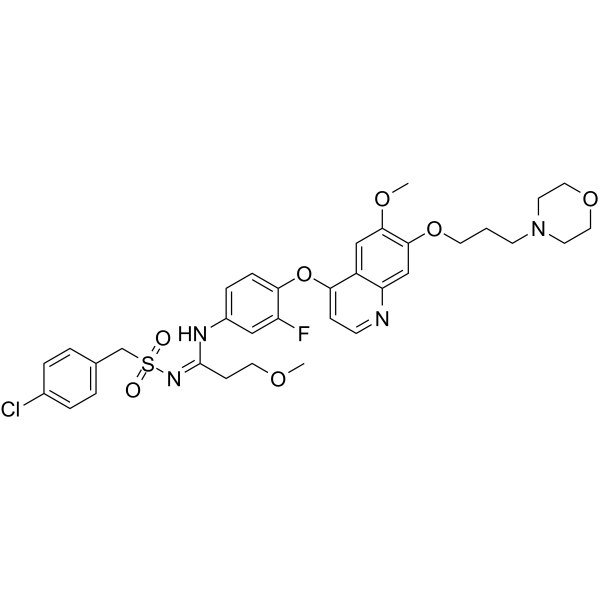
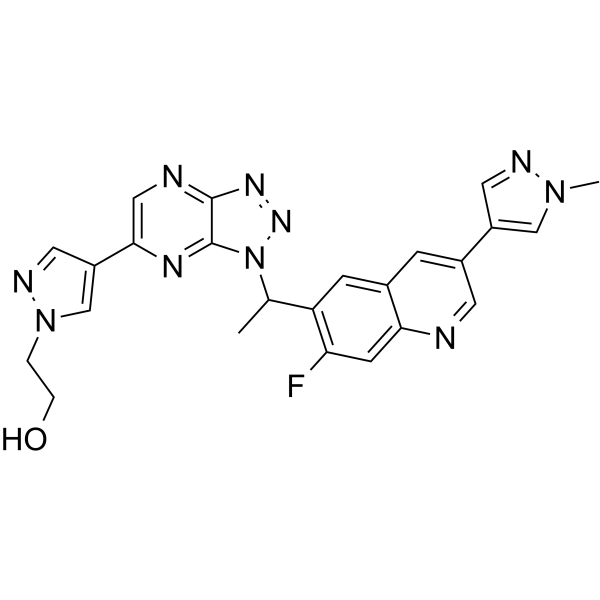
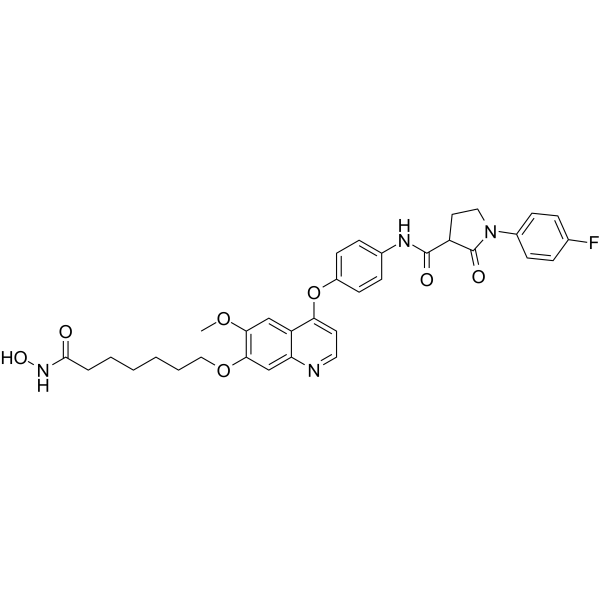
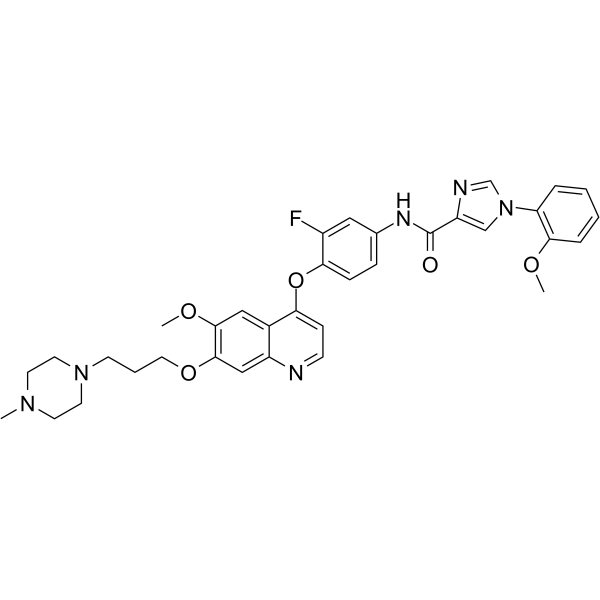
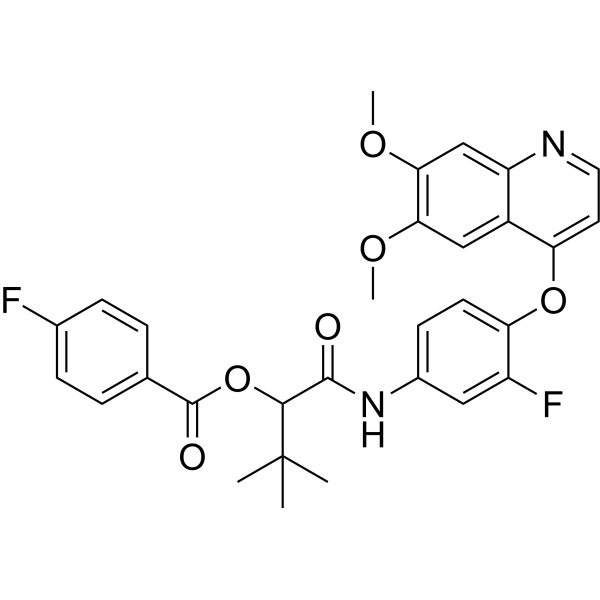
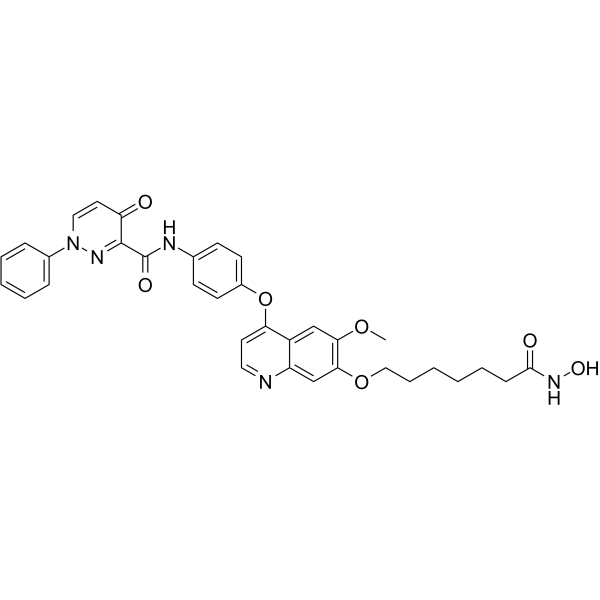
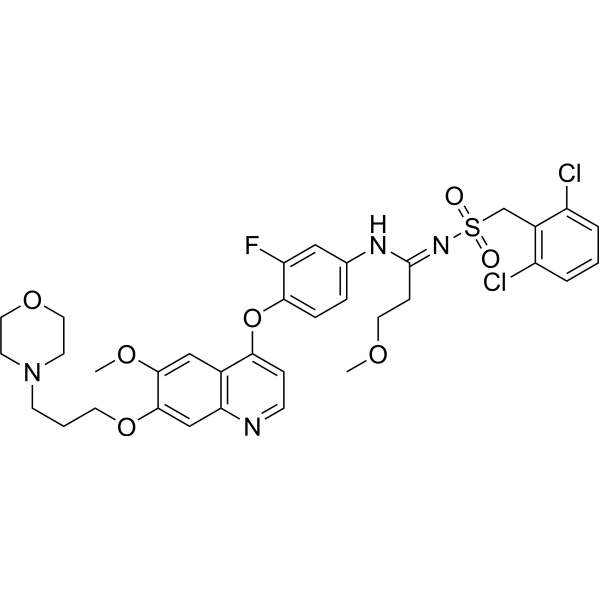
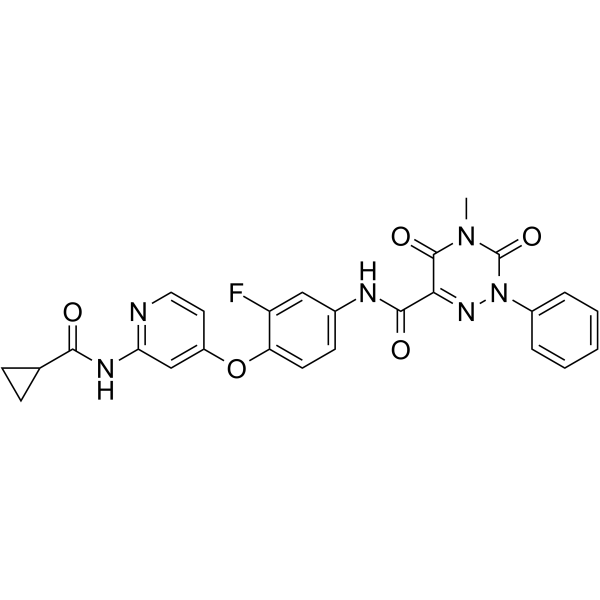
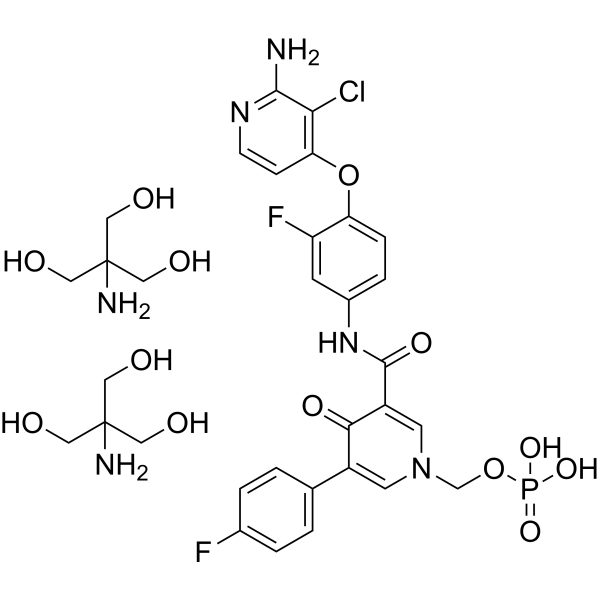
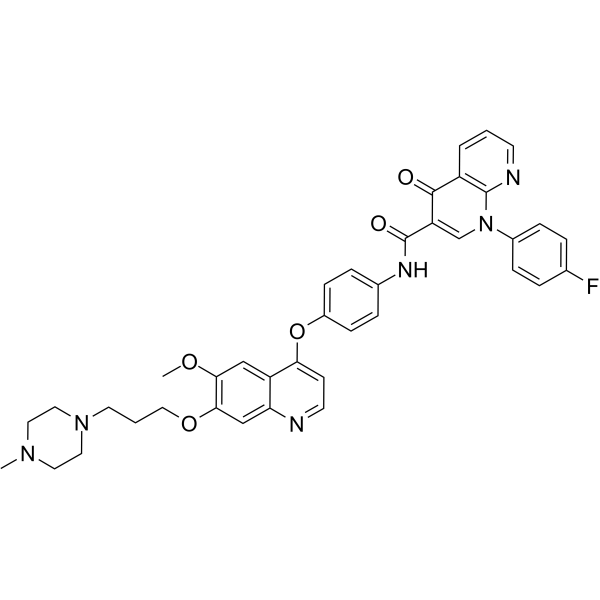
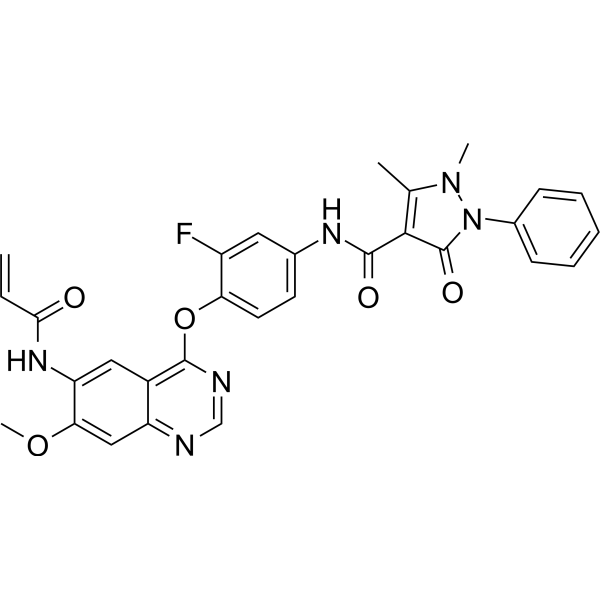
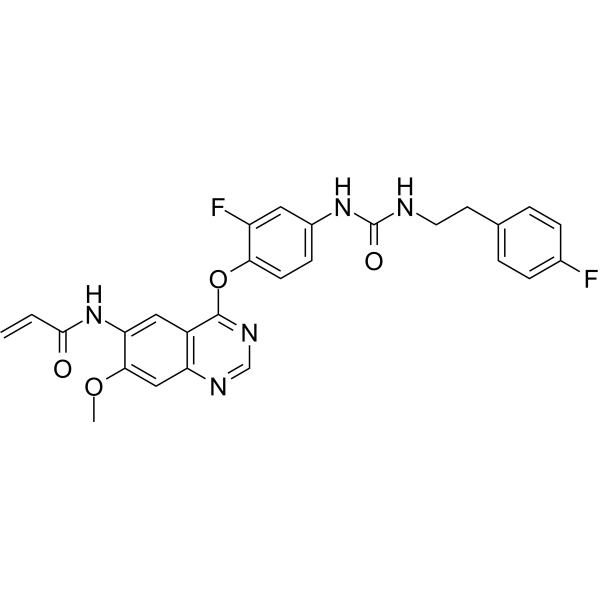
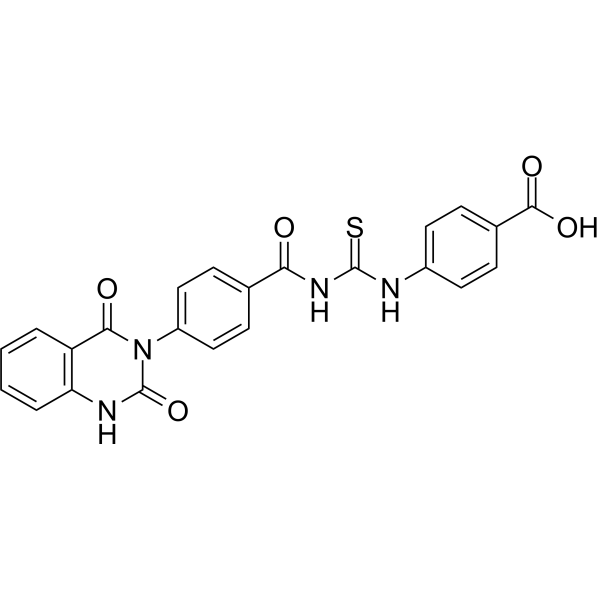
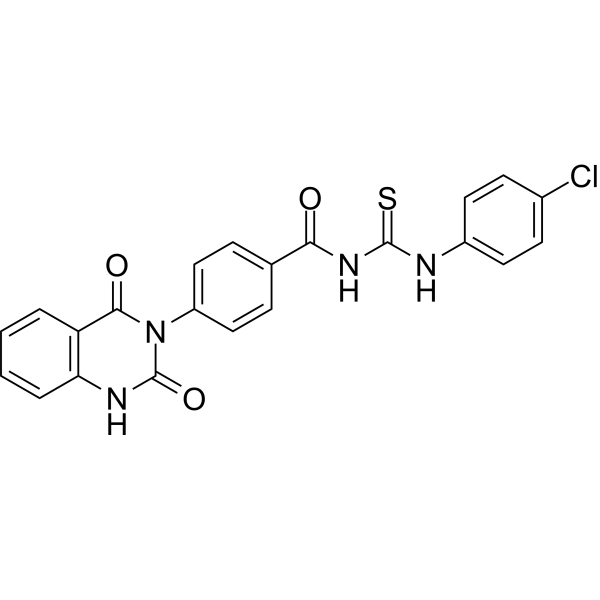
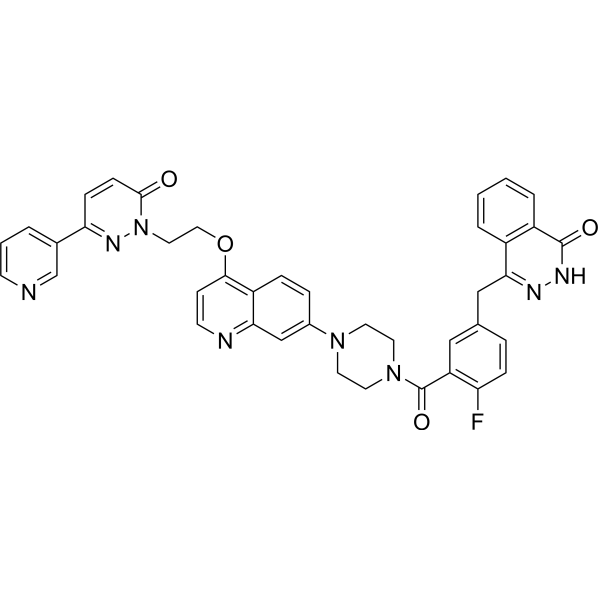
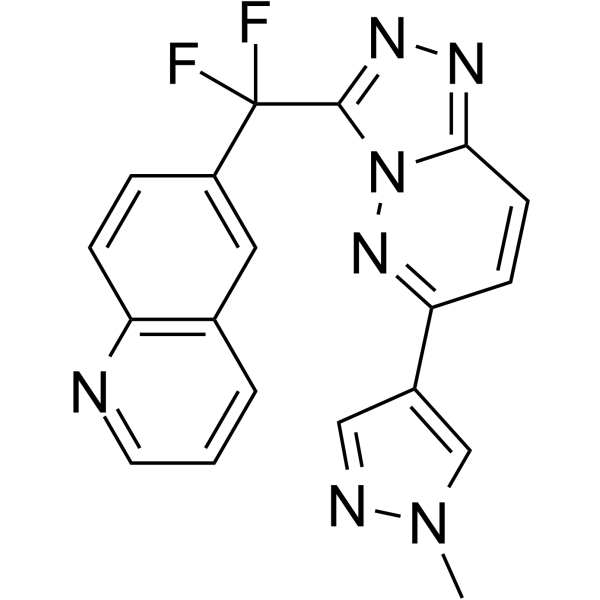
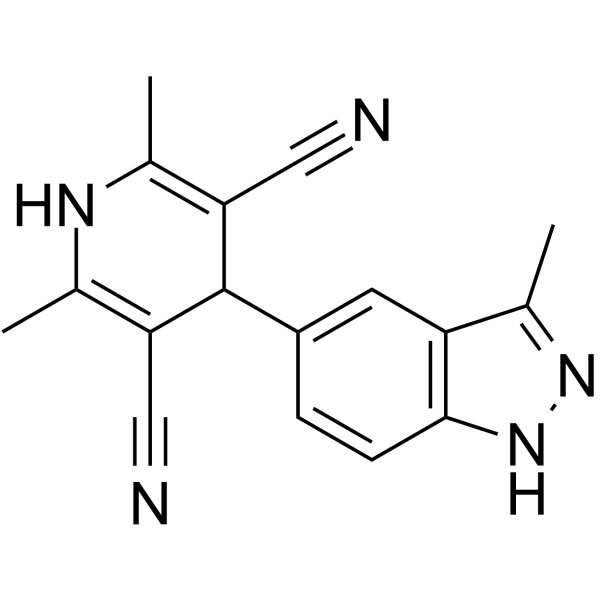
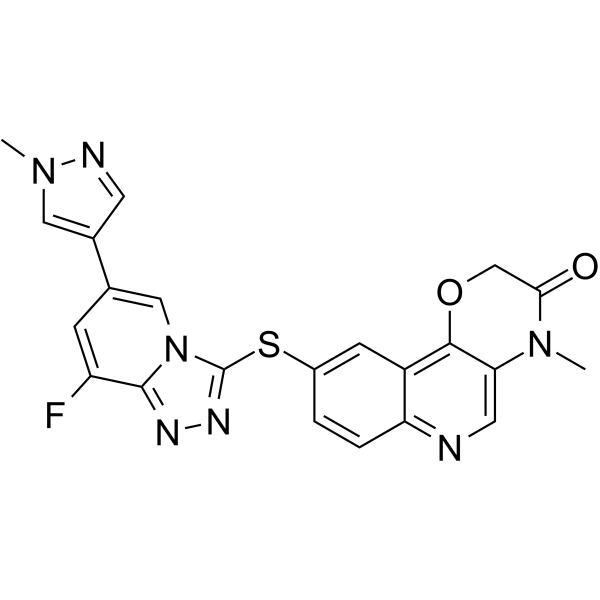
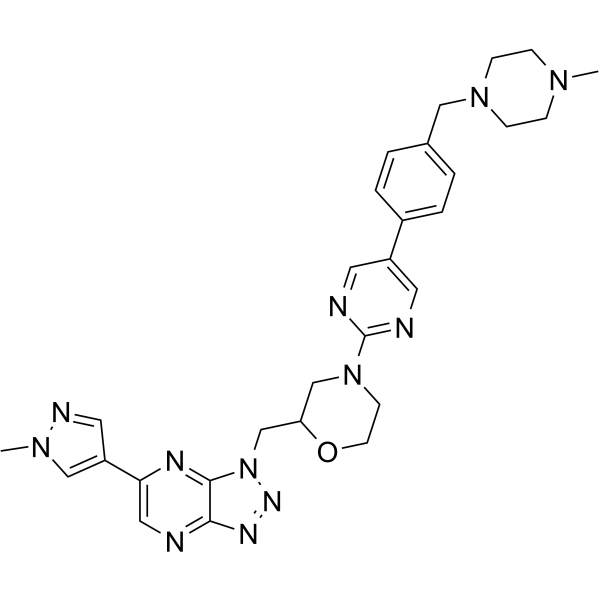
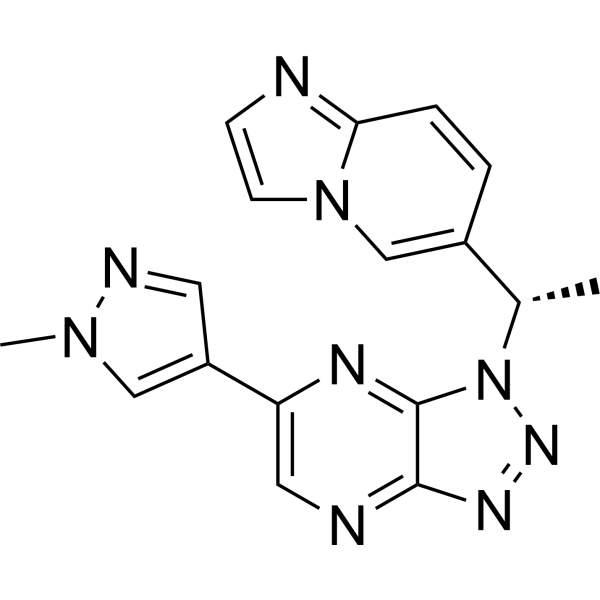
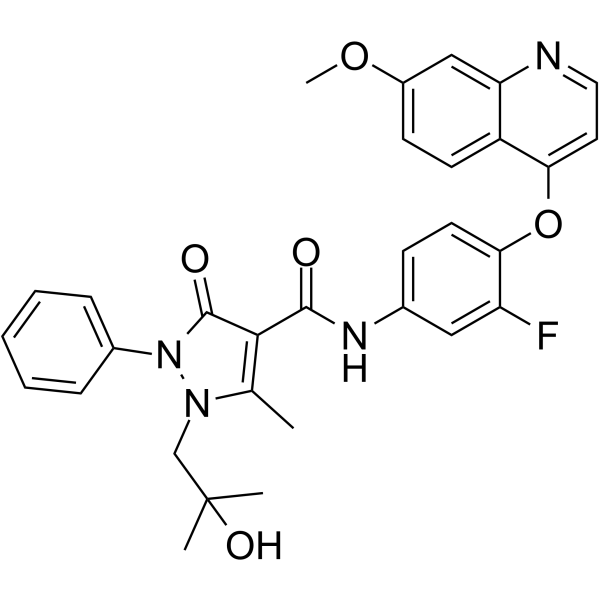
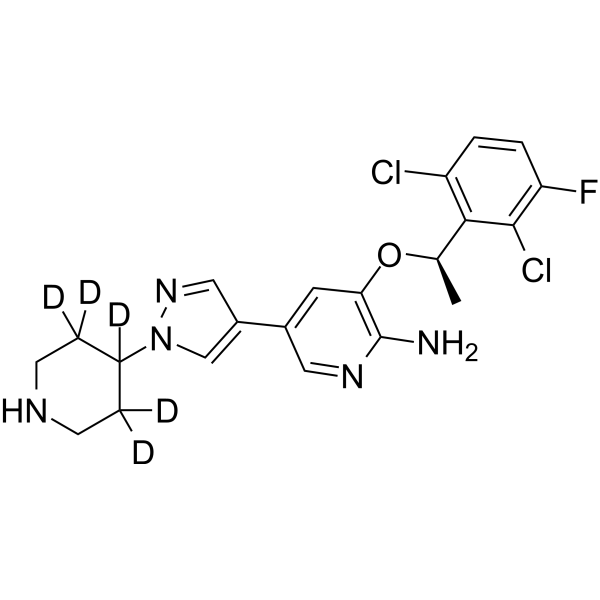
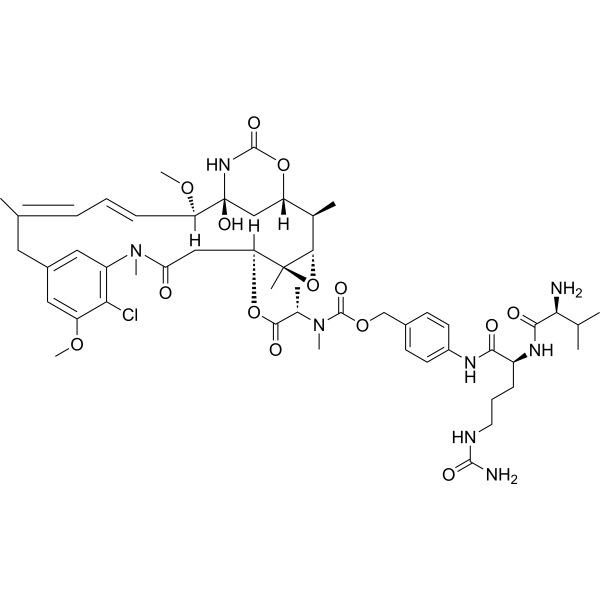
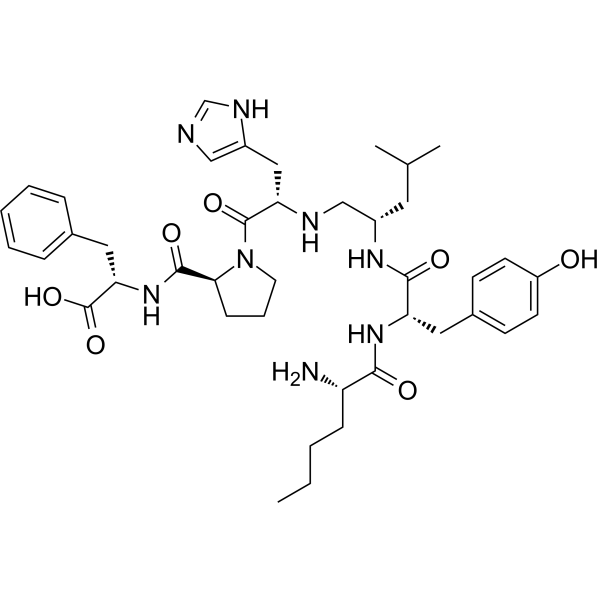
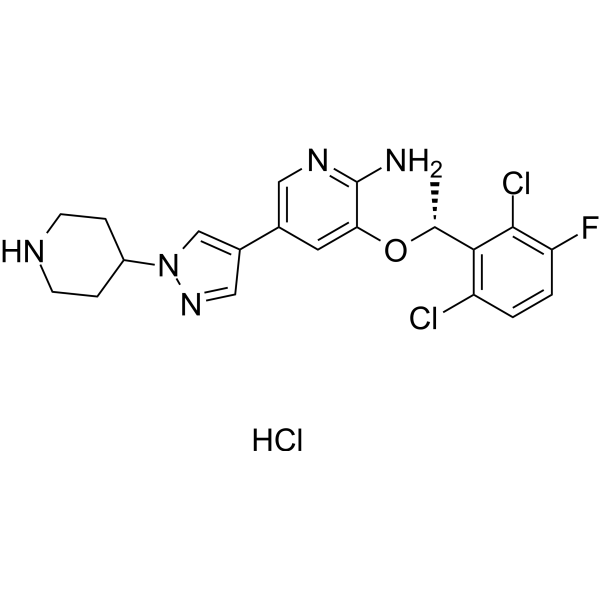
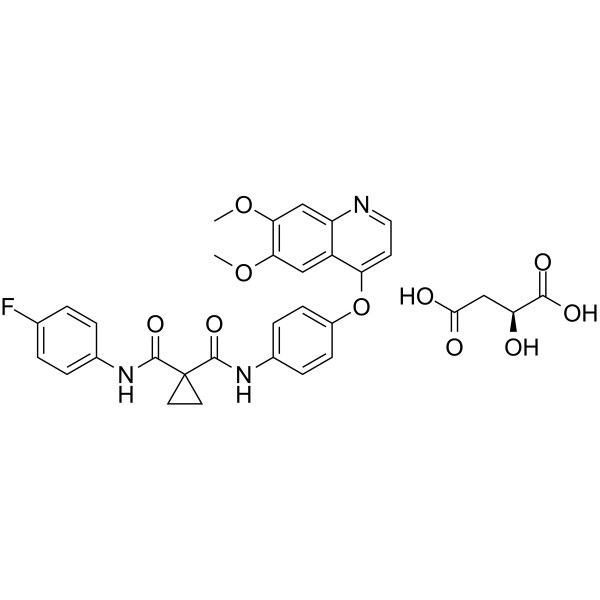
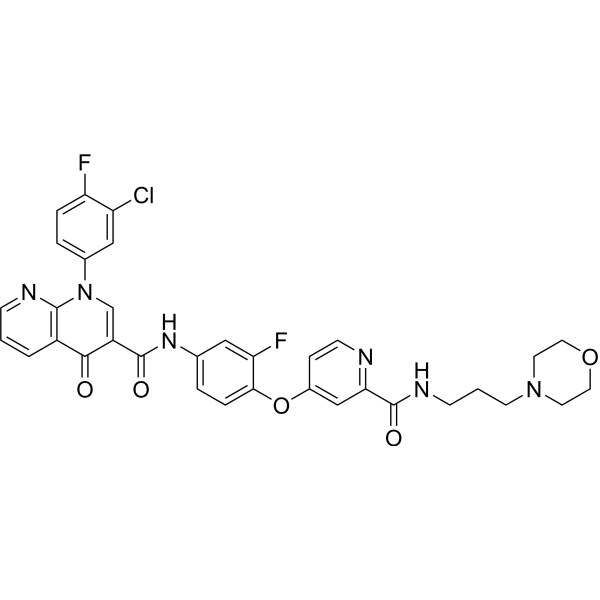
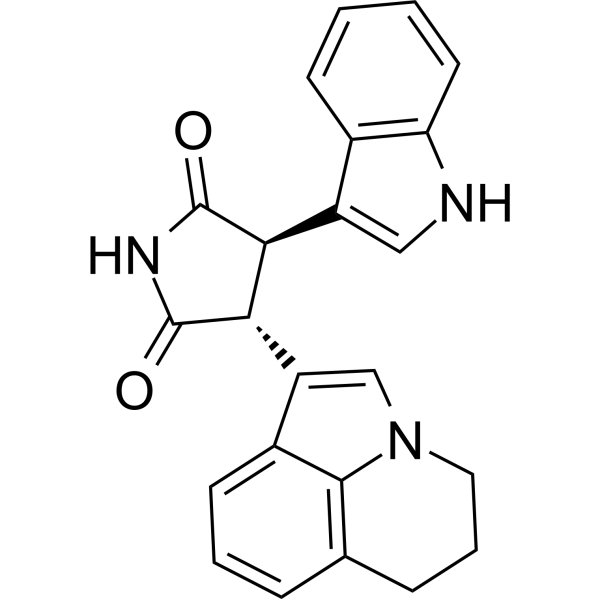
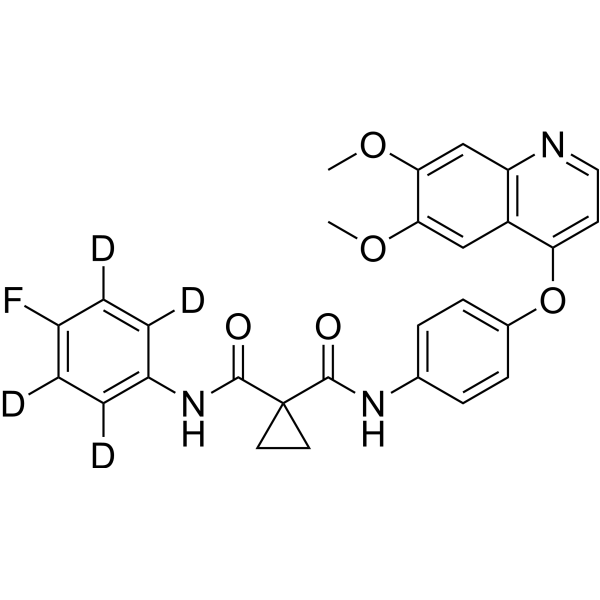
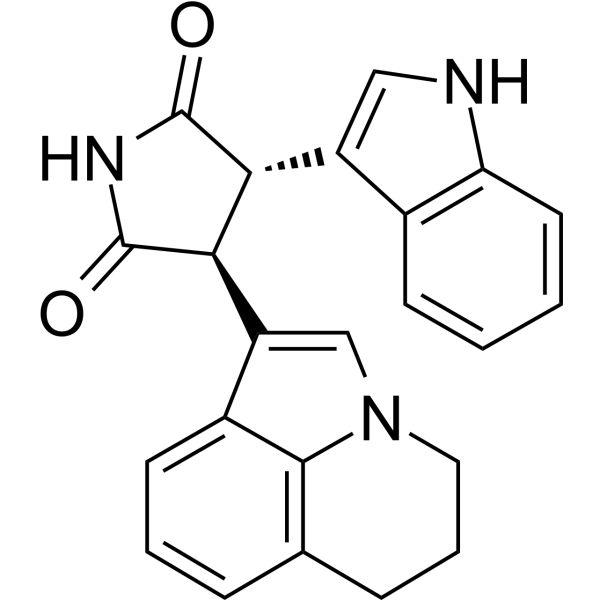
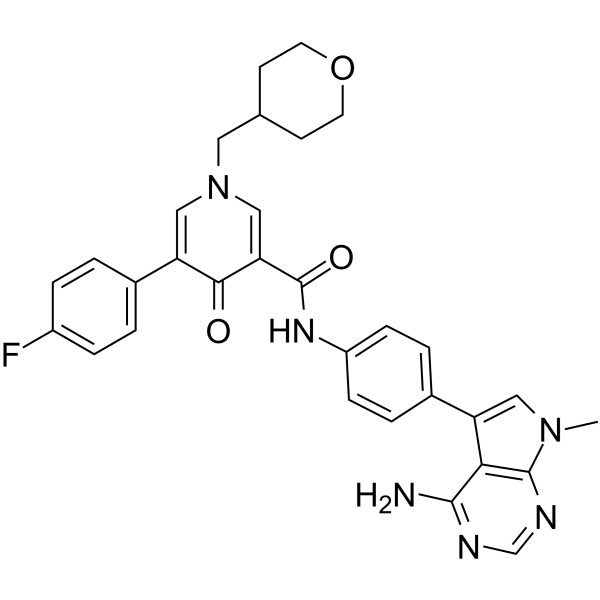
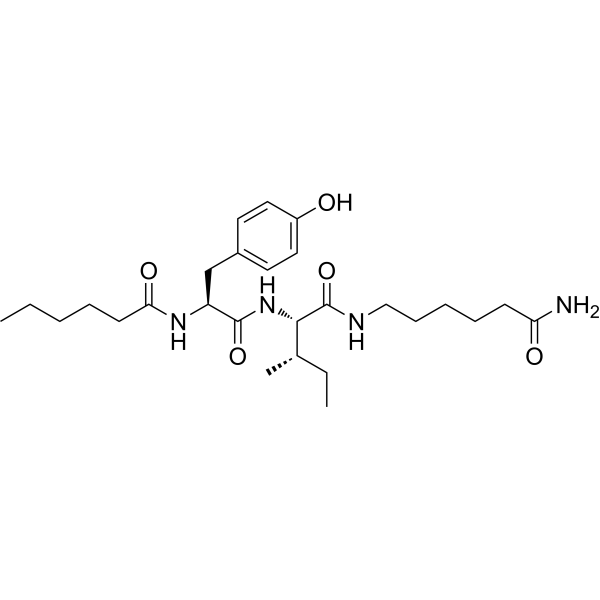
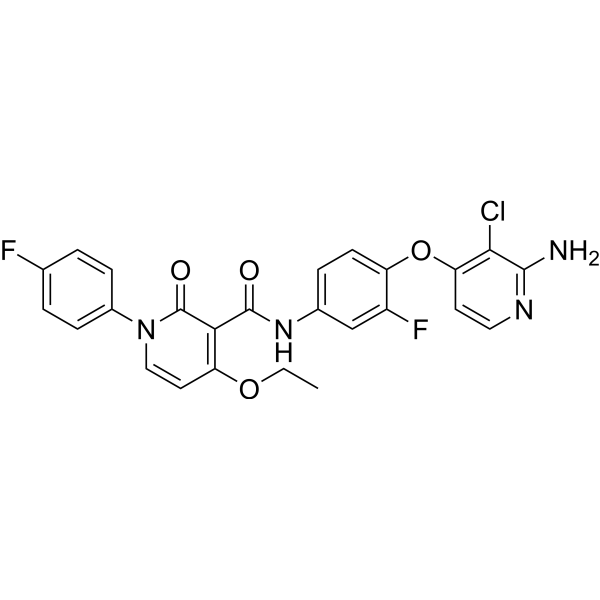
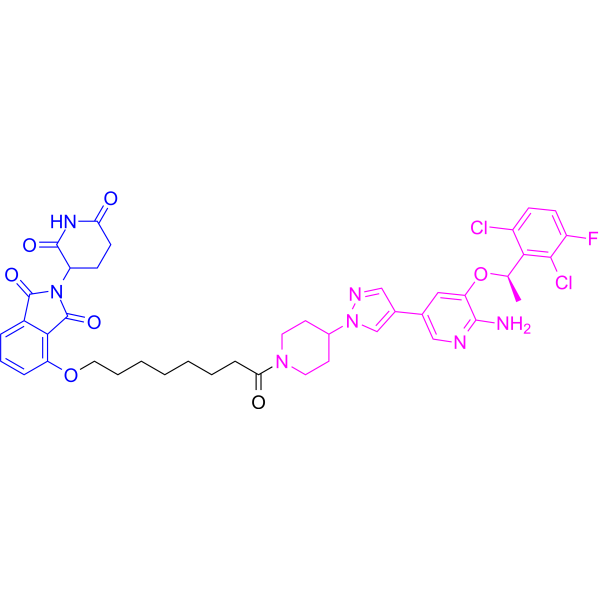
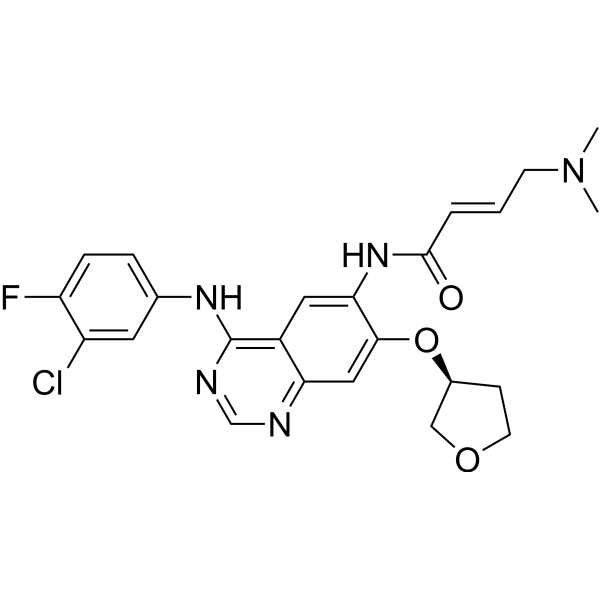
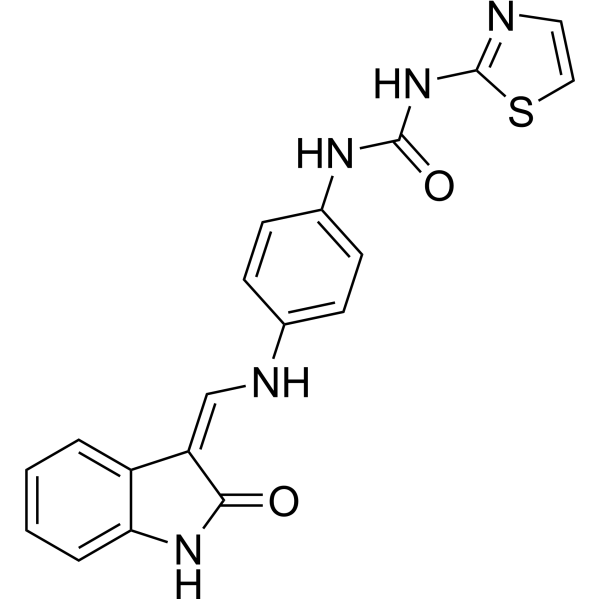
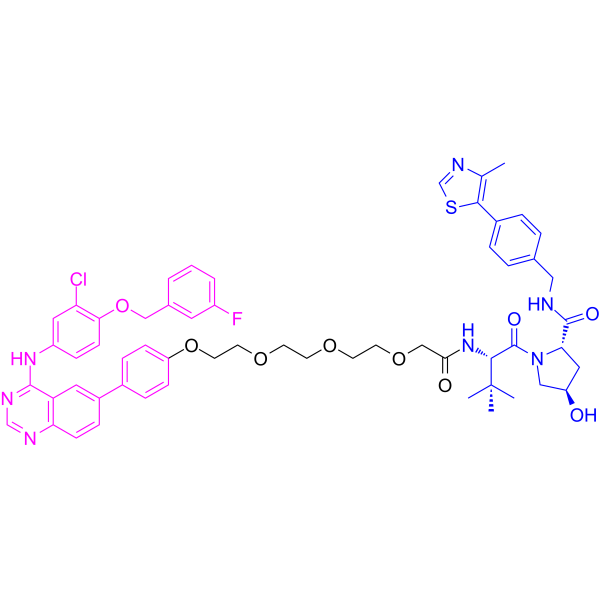
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
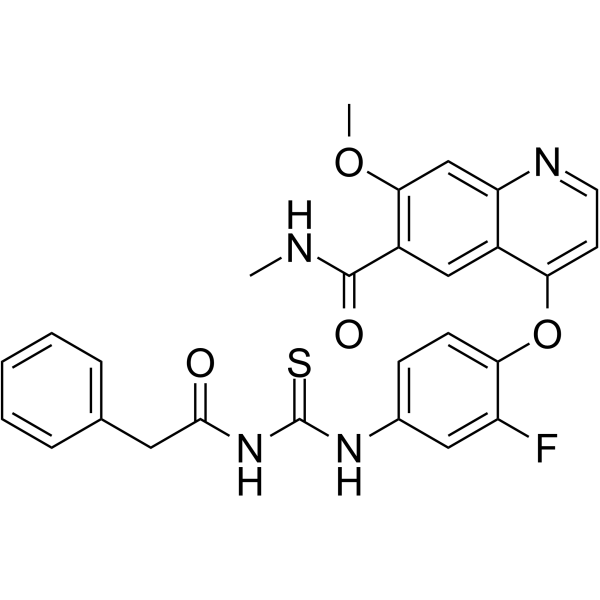
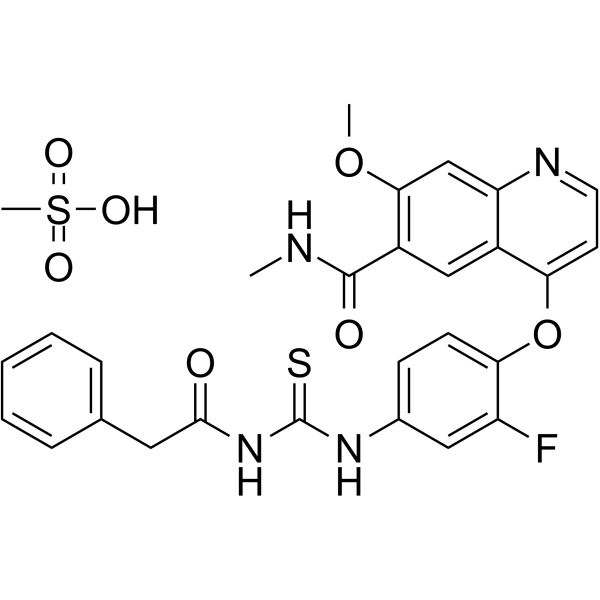
c-Met inhibitor 1 is an inhibitor of the c-Met receptor signaling pathway useful for the treatment of cancer including gastric, glioblastoma, and pancreatic cancer.
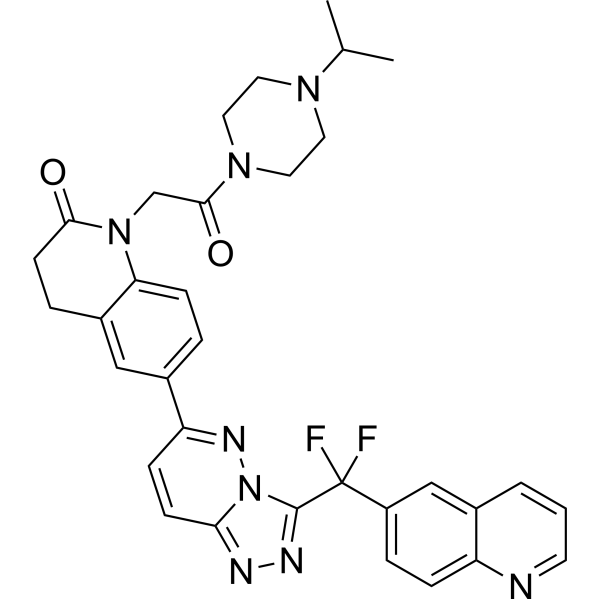
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
c-Met-IN-9, a 4-phenoxypyridine derivative, is a c-Met kinas inhibitor with an IC50 of 12 nM. c-Met-IN-9 induces cells apoptosis, and has antitumor activities .
c-Met-IN-14 (compound 26af) is a selective inhibitor of c-Met kinase from N-sulfonylamidine-based derivatives, with an IC50 value of 2.89 nM. c-Met-IN-14 shows anticancer activity by blocking phosphorylation of c-Met, and arrests cell cycle at G2/M phase. c-Met-IN-14 induces apoptosis of A549 cells in a dose-dependent manner .
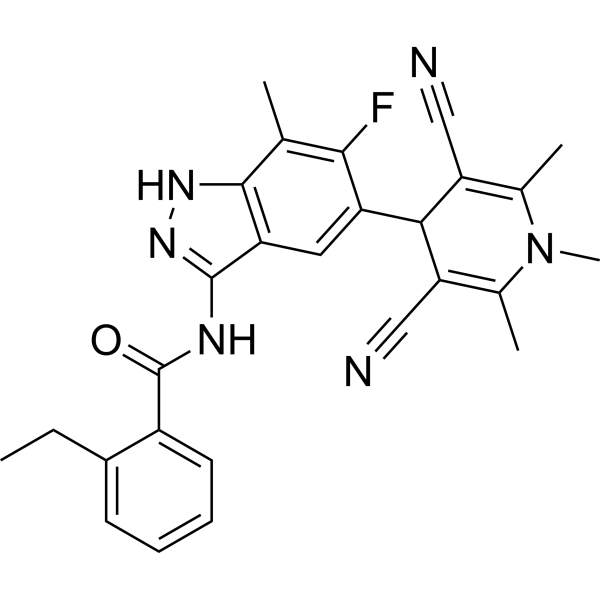
c-Met-IN-22 (compound 51am) is an orally active inhibitor against c-Met with an IC50 value of 2.54 nM. c-Met-IN-22 has antiproliferative and antitumor activities. c-Met-IN-22 induces cell apoptosis .
c-Met-IN-17 is a potent c-Met kinase inhibitor with an IC50 of 0.031 μM. c-Met-IN-17 can be used in anticancer research. . c-Met-IN-17 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
c-Met-IN-12 (compound 4r) is an orally active, potent and selective type II c-Met kinase inhibitor, with an IC50 of 10.6 nM. c-Met-IN-12 displays high inhibitory effects (inhibition rate > 80% in 1 μM) against AXL, Mer and TYRO3 kinases. c-Met-IN-12 can be used a scaffold for further kinase selectivity enhancement. c-Met-IN-12 shows antitumor efficacy .
c-Met-IN-23 (Compound 12g) is a c-Met inhibitor (IC50 = 0.052 μM for c-Met). c-Met-IN-23 also inhibits MDR1 and MRP1/2 pumps in the cancerous HepG2 and BxPC3 cells. c-Met-IN-23 is an anticancer agent .
c-Met/HDAC-IN-3 (Compound 15f) is a dual c-Met and HDAC inhibitor with IC50 values of 12.50 nM and 26.97 nM against c-Met and HDAC1, respectively. c-Met/HDAC-IN-3 induces apoptosis and cause cell cycle arrest in G2/M phase .
c-Met-IN-13 is a potent c-Met inhibitor with an IC50 value of 2.43 nM. c-Met-IN-13 shows excellent cytotoxicity for cancer cells. c-Met-IN-13 shows antiproliferative activity in a concentration- and time- dependent manner. c-Met-IN-13 has the potential for the research of cancer .
c-Met/HDAC-IN-2 is a highly potent c-Met and HDAC dual inhibitor with IC50s of 18.49 nM and 5.40 nM for HDAC1 and c-Met, respectively. c-Met/HDAC-IN-2 has antiproliferative activities against certain cancer cell lines. c-Met/HDAC-IN-2 can cause G2/M-phase arrest and induce apoptosis in HCT-116. c-Met/HDAC-IN-2 can be used for researching anti-cancer resistance .
c-Met-IN-10 (compound 26a) is a highly potent c-Met kinase inhibitor with an IC50 value of 16 nM. c-Met-IN-10 has inhibitory activity against cancer cells A549, H460 and HT-29 with IC50s of 0.56 ~ 1.59 μM. c-Met-IN-10 suppresses the colony formation on HT-29 cells, induces HT-29 and A549 cells apoptosis, and inhibits A549 cells motility. c-Met-IN-10 can be used for researching anticancer .
SCR-1481B1 (Metatinib anhydrous) is a potent compound that has activity against cancers dependent upon Met activation and also has activity against cancers as a VEGFR inhibitor.
EGFR/c-Met-IN-1 (compound TS-41) is a dual-target inhibitor of EGFR/c-Met. The IC50 for inhibiting EGFR L858R and c-Met is 68.1 nM and 0.26 nM respectively. . EGFR/c-Met-IN-1 induces apoptosis and cell cycle arrest in A549-P cells, downregulating the phosphorylation of EGFR, c-Met, and downstream AKT. EGFR/c-Met-IN-1 inhibits tumor growth in vitro and in vivo .
EGFR/ C-Met-in-2 (Compound H-22) is a dual inhibitor of EGFR/c-Met. EGFR/c-Met-IN-2 inhibits cell proliferation by arresting G2/M phase. EGFR/c-Met-IN-2 has antitumor activity .
VEGFR-2/c-Met-IN-2 (compound 3e) is a VEGFR-2/c-Met inhibitor, with IC50 values of 83 and 48 nM, respectively. VEGFR-2/c-Met-IN-2 exhibits cytotoxic activity against HCT-116 cell line (IC50: 3.403 µM) .
PARP1/c-Met-IN-1 (Compound 16) is a selective dual inhibitor for PARP1 and c-Met, with IC50s of 3.3 and 32.2 nM, respectively. PARP1/c-Met-IN-1 induces cell apoptosis and cell cycle arrest in G2/M phase in MDA-MB-231 cells. PARP1/c-Met-IN-1 exhibits antitumor activity in mice .
JNJ-38877605 is an orally active ATP-competitive inhibitor of c-Met with an IC50 of 4 nM, 600-fold selective for c-Met than 200 other tyrosine and serine-threonine kinases . JNJ-38877605 inhibits c-Met phosphorylation and regulates lipid accumulation. JNJ-38877605 can be used for tumor and metabolic disease reseach .
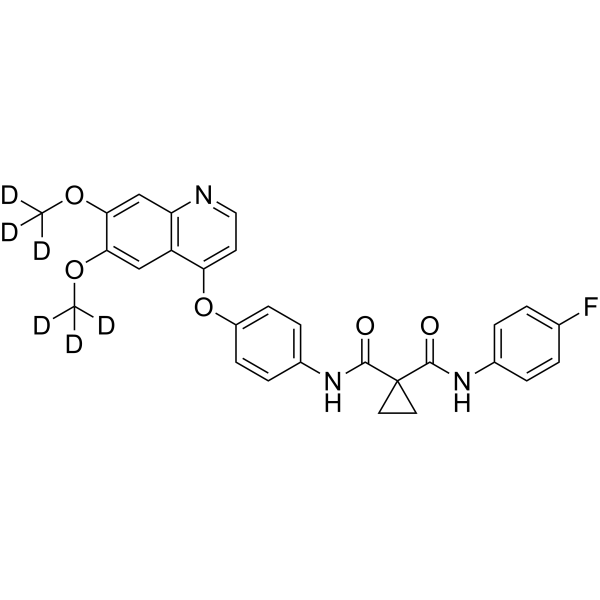
Dalmelitinib is an orally active selective c-Met kinase inhibitor (IC50: 2.9 nM) that binds to the ATP-binding region of c-Met. Dalmelitinib induces the phosphorylation of MET, partially or completely inhibits the phosphorylation of AKT and ERK. Dalmelitinib potently inhibits cancer cell (c-Met oncogene amplification) proliferation, and is used for the research of cancers like human non-small cell lung cancer (NSCLC) .
ABN401 is a highly potent and selective ATP-competitive c-MET inhibitor with an IC50 value of 10 nM. ABN401 has cytotoxic activity against MET-addicted cancer cells. ABN401 can inhibit c-MET phosphorylation in tumor tissues. ABN401 can be used for researching anticancer .
Savolitinib (AZD-6094) is a potent, highly selective, and orally bioavailable c-Met inhibitor with IC50 s of 5 nM and 3 nM for c-Met and p-Met, respectively. Savolitinib (AZD-6094) selectively binds to and inhibits the activation of c-Met in an ATP-competitive manner, and disrupts c-Met signal transduction pathways. Antineoplastic activity .
SYN1143 is a potent, selective and orally active dual inhibitor of c-Met/RON, with IC50s of 4 and 9 nM, respectively. SYN1143 has weak inhibitory activity on Lck, Tie2, Src, and BTK with IC50s ranging from 160 to 710 nM. SYN1143 can be used for the research of cancers that RON and c-Met are activated .
SJF-8240 (PROTAC 7) is a proteolysis targeting chimera (PROTAC) degrader. SJF-8240 induces polyubiquitination of c-Met and inhibits the proliferation of GTL16 cells (IC50=66.7 nM) .
Zgwatinib (SOMG-833) is a potent, selective, and ATP-competitive c-MET inhibitor, with an IC50 of 0.93 nM against c-MET, over 10,000-fold more potent compared with 19 tyrosine kinases (including c-MET family members and highly homologous kinases). Zgwatinib potently inhibits c-MET-driven cell proliferation. Zgwatinib as a potential candidate agent for c-MET-driven human cancers research .
Glumetinib (SCC244) is a highly selective, orally bioavailable, ATP-competitive c-Met inhibitor with an IC50 of 0.42 nM. Glumetinib has greater than 2400-fold selectivity for c-Met over those 312 kinases evaluated, including the c-Met family member RON and highly homologous kinases Axl, Mer, TyrO3. Antitumor activity .
Antitumor agent-45 (Compound 21) could induce and stimulate A549 cells apoptosis in G0/G1 and G2/M phase. Antitumor agent-45 (Compound 21) inhibits c-Met expression to regulate the growth of tumor cells .
Tepotinib (EMD-1214063) is a potent and highly selective c-Met inhibitor with an IC50 of 4 nM, >200-fold selective for c-Met than IRAK4, TrkA, Axl, IRAK1, and Mer. Antitumor effects .
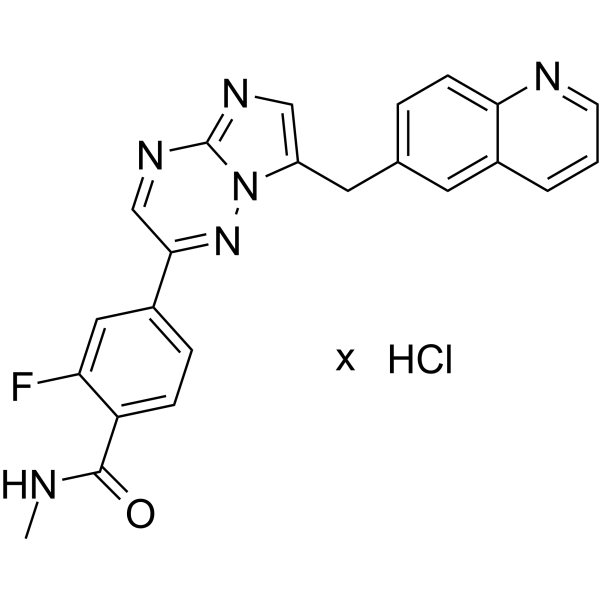
Capmatinib (INC280; INCB28060) is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) hydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib hydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib hydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib hydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
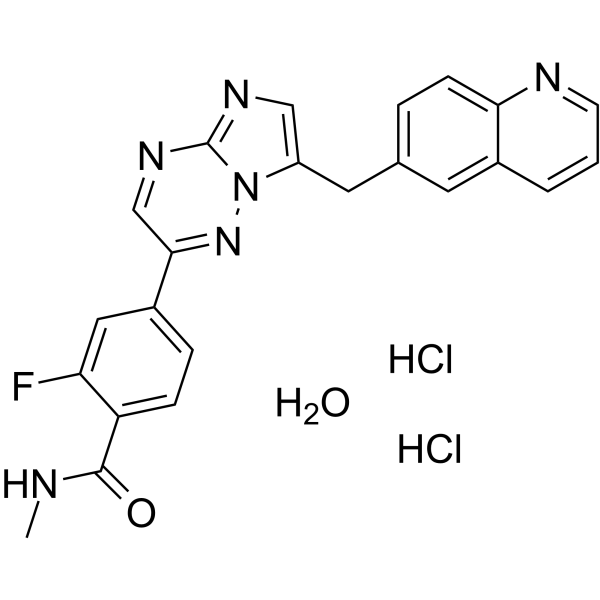
Capmatinib (INC280; INCB28060) dihydrochloride hydrate is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride hydrate can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride hydrate potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride hydrate is largely metabolized by CYP3A4 and aldehyde oxidase .
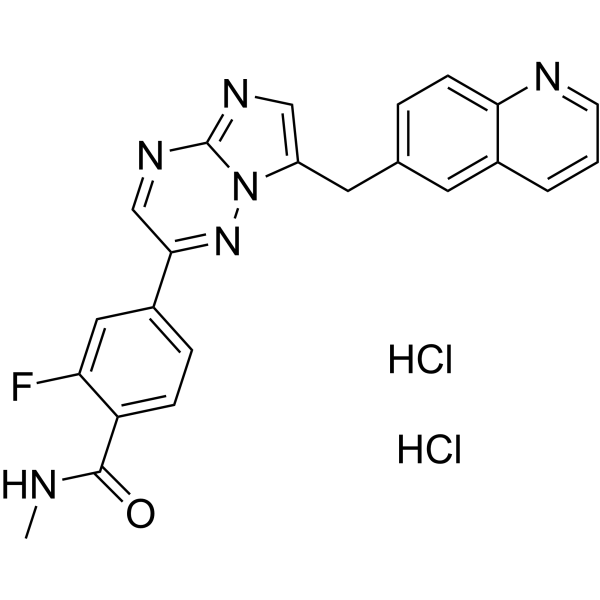
Capmatinib (INC280; INCB28060) dihydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
PF-04217903 is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
PF-04217903 mesylate is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 mesylate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
PF-04217903 phenolsulfonate is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 phenolsulfonate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
Arylsulfonamide 64B (HIF inhibitor 64B) is an inhibitor of the hypoxia-induced factor (HIF). Arylsulfonamide 64B inhibits hypoxia/HIF-induced expression of c-Met and CXCR4 and reduces primary tumor growth and metastasis of uveal melanoma mouse model .
Ficlatuzumab is a monoclonal antibody (McAb) targeting human hepatocyte growth factor (HGF). Ficlatuzumab blocks the activation of the HGF/c-Met signaling pathway, and inhibits c-Met receptor-mediated cancer cell proliferation, migration, and invasion .
Poloppin is a potent, cell penetrant inhibitor of the mitotic Polo-like kinase (PLK) (IC50=26.9 μM) and prevents the protein-protein interaction via the Polo-box domain (PBD) (Kd= 29.5 μM). Poloppin selectively kills cells expressing mutant KRAS, enhancing death in mitosis. Poloppin is used for the study of KRAS-mutant cancers as single agents, or in combination with c-MET inhibitors .
Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
Crizotinib (PF-02341066) acetate is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib acetate inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib acetate is also a ROS1 inhibitor. Crizotinib acetate has effective tumor growth inhibition .
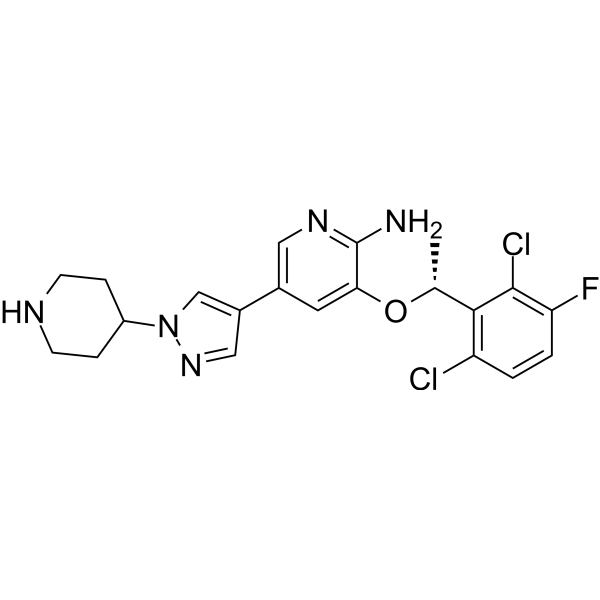
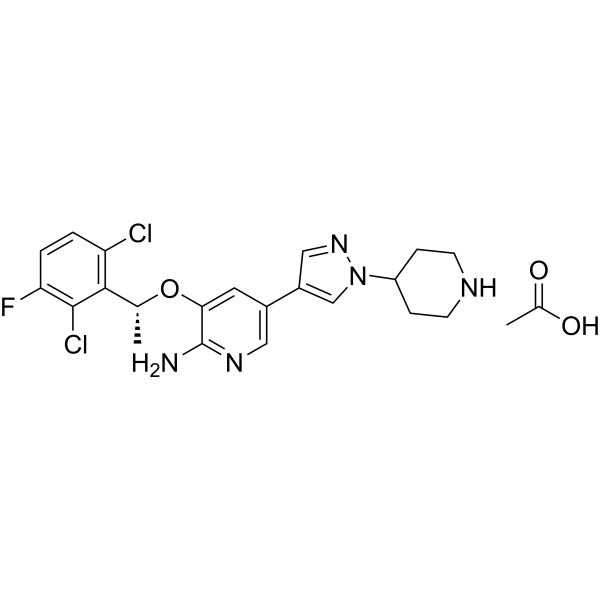
ABBV-303 is a multi-specific NK cell antibody targeting c-Met designed based on TriNKET technology for natural killer (NK) cell redirection therapy. The ABBV-303 antibody consists of three functional arms, a single-chain variable fragment (scFV) targeting c-Met, a Fab arm targeting NKG2D, and an Fc fragment targeting NK cell CD16a .
PHA-665752 is a selective, ATP-competitive, and active-site inhibitor of the catalytic activity of c-Met kinase (Ki=4 nM; IC50=9 nM). PHA-665752 exhibits >50-fold selectivity for c-Met compared with a panel of diverse tyrosine and serine-threonine kinases. PHA-665752 induces apoptosis and cell cycle arrest, and exhibits cytoreductive antitumor activity .
AMG-208 is an orally active c-Met/RON dual selective inhibitor with an IC50 of 9 nM for c-Met. AMG-208 is a CYP3A4 inhibitor with an IC50 of 32 μM. AMG-208 has anti-cancer activity .
(Rac)-Tivantinib is the isomer of Tivantinib (HY-50686), and can be used as an experimental control. Tivantinib is a highly selective c-Met tyrosine kinase inhibitor with a Ki of 355 nM.
Meleagrin is a roquefortine C-derived alkaloid produced by fungi of the genus Penicillium and has antimicrobial and anti-proliferative activities. Meleagrin is a class of FabI inhibitor. Meleagrin is a lead c-Met inhibitory entity useful for the control of c-Met-dependent metastatic and invasive breast malignancies .
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
Val-Cit-amide-Ph-Maytansine is an antibody and bispecific antigen-binding mol. that bind hepatocyte growth factor receptor c-Met (MET) or antibody-drug conjugates (ADCs) .
Norleual, an angiotensin (Ang) IV analog, is a hepatocyte growth factor (HGF)/c-Met inhibitor with an IC50 of 3 pM. Norleual is an AT4 receptor antagonist and exhibits potent antiangiogenic activities .
Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
EGFR-IN-8 is a dual EGFR and c-Met inhibitor, compound 48. EGFR-IN-8 can be a promising candidate for further development to target EGFR TKI-resistant NSCLC .
LAH-1 is a c-Met inhibitor with oral activity and membrane permeability with an IC50 of 49 nM. LAH-1 has anticancer activity and can induce apoptosis, migration, and invasion .
Norleual TFA, an angiotensin (Ang) IV analog, is a hepatocyte growth factor (HGF)/c-Met inhibitor with an IC50 of 3 pM. Norleual TFA is an AT4 receptor antagonist and exhibits potent antiangiogenic activities .
Ningetinib Tosylate is a potent, orally bioavailable small molecule tyrosine kinase inhibitor (TKI) with IC50s of 6.7, 1.9 and <1.0 nM for c-Met, VEGFR2 and Axl, respectively.
Ningetinib is a potent, orally bioavailable small molecule tyrosine kinase inhibitor (TKI) with IC50s of 6.7, 1.9 and <1.0 nM for c-Met, VEGFR2 and Axl, respectively.
Val-Cit-amide-Cbz-N(Me)-Maytansine is an antibody and bispecific antigen-binding mol. that bind hepatocyte growth factor receptor c-Met (MET) or antibody-drug conjugates (ADCs) .
MK-8033 is an orally active ATP competitive c-Met/Ron dual inhibitor (IC50s: 1 nM (c-Met),7 nM (Ron)), with preferential binding to the activated kinase conformation. MK-8033 can be used in the research of cancers, such as breast and bladder cancers, non-small cell lung cancers (NSCLCs) .
MK-8033 hydrochloride is an orally active ATP competitive c-Met/Ron dual inhibitor (IC50s: 1 nM (c-Met),7 nM (Ron)), with preferential binding to the activated kinase conformation. MK-8033 hydrochloride can be used in the research of cancers, such as breast and bladder cancers, non-small cell lung cancers (NSCLCs) .
Vabametkib is a potent inhibitor of hepatocyte growth factor receptor (HGFR). Vabametkib inhibits Hs746T cells proliferation and inhibits c-Met with an IC50 value of 7 nM. Vabametkib can be used as an antineoplastic agent .
MGCD-265 analog is a potent and oral active inhibitor of c-Met and VEGFR2 tyrosine kinases, with IC50s of 29 nM and 10 nM, respectively. MGCD-265 analog has significant antitumor activity .
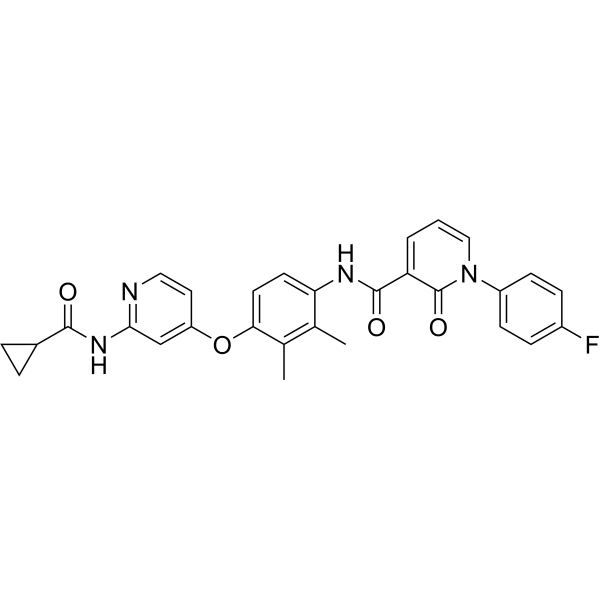
D6808 is a highly selective and potent c‑Met inhibitor with an IC50 value of 2.9 nM. D6808 induces cell apoptosis and cell cycle arrest. D6808 can be used for the research of NSCLC and gastric cancers .
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
PF-07265807 is a potent TAM and c-Met kinase inhibitor with IC50 values of 6.1 nM, 13.2 nM and 21.6 nM for AXL, MER and TYRO3, respectively. PF-07265807 can be used for researching anticancer .
BPI-9016M is a potent, orally active, and selective dual c-Met and AXL tyrosine kinases inhibitor. BPI-9016M suppresses tumor cell growth, migration and invasion of lung adenocarcinoma .
Bozitinib (PLB-1001) is a highly selective c-MET kinase inhibitor with blood-brain barrier permeability. Bozitinib (PLB-1001) is a ATP-competitive small-molecule inhibitor, binds to the conventional ATP-binding pocket of the tyrosine kinase superfamily .
AC-386 is a highly potent c-Met inhibitor with IC50 value of 7.42 nM. AC-386 has antiproliferative activities against certain cancer cell lines. AC-386 can be used for researching anti-cancer resistance .
Cabozantinib S-malate (XL184 S-malate) is a potent multiple receptor tyrosine kinases inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
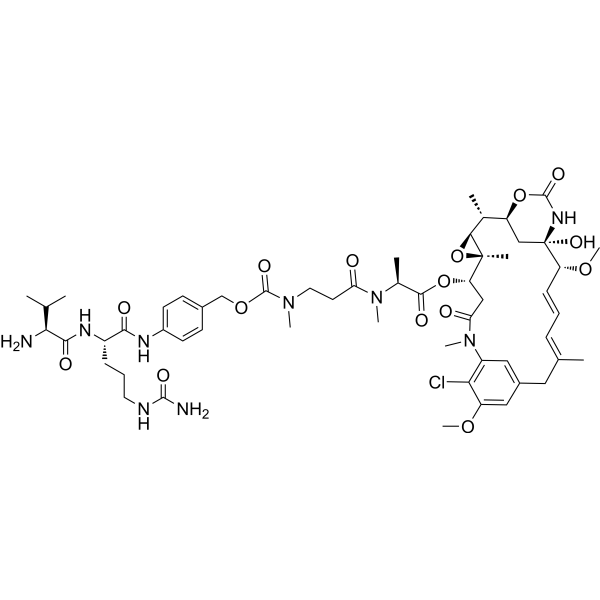
Telisotuzumab vedotin (ABBV-399) is an antibody-drug conjugate (ADCs) targeting c-Met. Telisotuzumab vedotin consists of Monomethyl Auristatin E (MMAE), Telisotuzumab antibody and a cleavable mc-val-cit-PABC type linker. Telisotuzumab vedotin can be used in cancer research .
Antitumor agent-111 (compound 46) is an c-Met kinase inhibitor (IC50=46 nM) with antitumor and antiproliferative activity. Antitumor agent-111 arrests cell cycle at G0/G1 phase, and induces apoptosis .
(rel)-Tivantinib is a potent and highly selective inhibitor of the receptor tyrosine kinase c-MET. (rel)-Tivantinib has two novel targets, GSK3α and GSK3β, which play an important role in the cellular mechanism of non-small cell lung cancer (NSCLC) .
Cabozantinib-d4 is deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
Cabozantinib-d6 is the deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively[1][2][3].
RIPK3-IN-1 is a RIPK3 type II DFG-out inhibitor with an IC50 of 9.1 nM. RIPK3-IN-1 inhibits RIPK1 and RIPK2 with IC50s of 5.5 and >10 μM. RIPK3-IN-1 is also a c-Met kinase inhibitor with an IC50 of 1.1 μM .
Amuvatinib (MP470) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib (MP470) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
Amuvatinib hydrochloride (MP470 hydrochloride) is an orally bioavailable multi-targeted tyrosine kinase inhibitor with potent activity against mutant c-Kit, PDGFRα, Flt3, c-Met and c-Ret. Amuvatinib hydrochloride (MP470 hydrochloride) is also a DNA repair suppressor through suppression of DNA repair protein RAD51, thereby disrupting DNA damage repair . Antineoplastic activity .
(3S,4S)-Tivantinib is a potent and highly selective inhibitor of the receptor tyrosine kinase c-MET. (3S,4S)-Tivantinib has two novel targets, GSK3α and GSK3β, which play an important role in the cellular mechanism of non-small cell lung cancer (NSCLC) .
Axl-IN-8 (NO.1) is a potent AXL inhibitor, with an IC50 of <1 nM. Axl-IN-8 also inhibits c-MET, with an IC50 of 1-10 nM. Axl-IN-8 shows anti-proliferative activity against BaF3/TEL-AXL, MKN45, and EBC-1 cells, with IC50 values of <10, 226.6 and 120.3 nM, respectively .
Dihexa, an oligopeptide drug, is an orally active and blood-brain barrier-permeable angiotensin IV analog. Dihexa binds to hepatocyte growth factor (HGF) with high affinity (Kd=65 pM) and potentiates its activity at its receptor, c-Met. Dihexa exhibits excellent antidementia activity and improves cognitive function in animal models. Dihexa may have therapeutic potential as a treatment Alzheimer’s disease .
BMS 777607 (BMS 817378) is a Met-related inhibitor for c-Met, Axl, Ron and Tyro3 with IC50s of 3.9 nM, 1.1 nM, 1.8 nM and 4.3 nM, respectively, and 40-fold more selective for Met-related targets than Lck, VEGFR-2, and TrkA/B, with more than 500-fold greater selectivity versus all other receptor and non receptor kinases .
TAK-285 is a potent, selective, ATP-competitive and orally active HER2 and EGFR(HER1) inhibitor with IC50 of 17 nM and 23 nM, respectively. TAK-285 is >10-fold selectivity for HER1/2 than HER4, and less potent to MEK1/5, c-Met, Aurora B, Lck, CSK etc. TAK-285 has effective antitumor activity . TAK-285 can cross the blood-brain barrier (BBB) .
Merestinib (LY2801653) is a potent, orally bioavailable c-Met inhibitor (Ki=2 nM) with anti-tumor activities. Merestinib (LY2801653) also has potent activity against MST1R (IC50=11 nM), FLT3 (IC50=7 nM), AXL (IC50=2 nM), MERTK (IC50=10 nM), TEK (IC50=63 nM), ROS1, DDR1/2 (IC50=0.1/7 nM) and MKNK1/2 (IC50=7 nM) .
MAPK-IN-2 (compound 3h) is a potent MAPK inhibitor with antineoplastic activity. MAPK-IN-2 inhibits cancer cell proliferation among serval cancer cell lines, and suppresses MAPK pathway with potant efficacy (EGFR WTIC50=281 nM, c-METIC50=205 nM, B-RAF WTIC50=112 nM, and CDK4/6 IC50=95 and 184 nM, respectively). MAPK-IN-2 even shows a remarkable potency against mutated EGFR and B-RAF (EGFR T790MIC50=69 nM and B-RAF V600EIC50=83 nM) .
Pamufetinib (TAS-115) is a potent VEGFR and hepatocyte growth factor receptor (c-Met/HGFR)-targeted kinase inhibitor with IC50s of 30 and 32 nM for rVEGFR2 and rMET, respectively.
Pamufetinib (TAS-115) mesylate is a potent VEGFRand hepatocyte growth factor receptor (c-Met/HGFR)-targeted kinase inhibitor, with IC50s of 30 and 32 nM for rVEGFR2 and rMET, respectively.
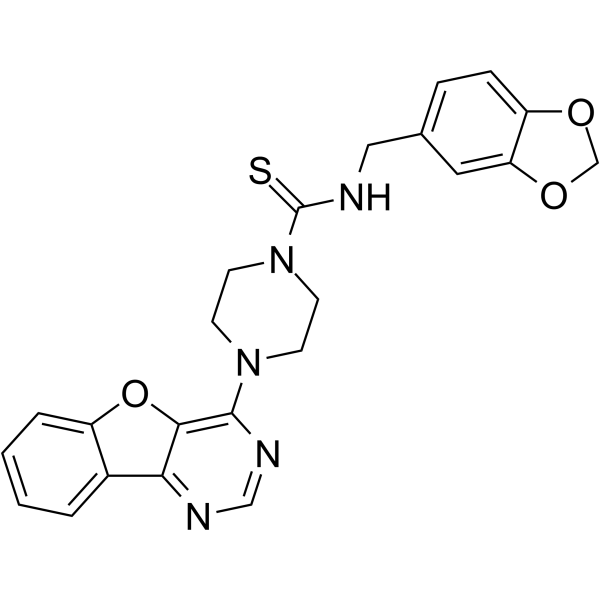
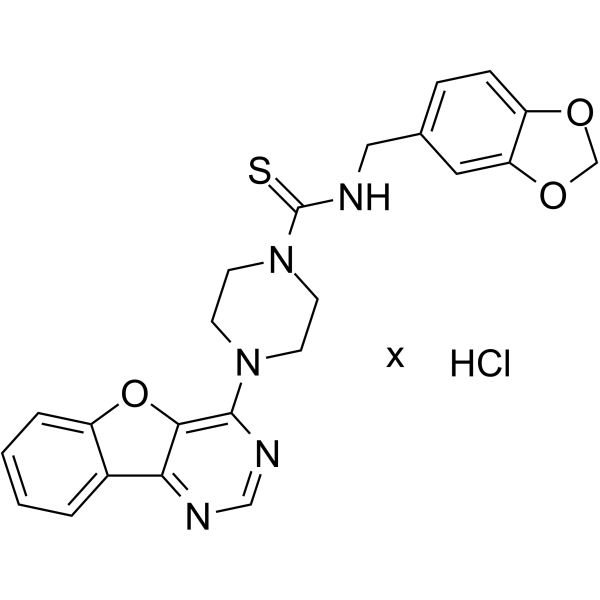
1D228 is a c-Met/TRK inhibitor with antitumor activity. 1D228 inhibits cyclin D1 to induce G0/G1 arrest and inhibit cancer cell proliferation and migration. 1D228 can be used in the study of gastric, liver and vascular tumors .
OSI-296 is a potent, oral and selective inhibitor of cMET and RON kinases. OSI-296 shows in vivo efficacy in MKN45 tumor xenografts models and well tolerated .
PRO-6E is an oral active PROTAC based on Cereblon ligand, and induces the degradation of MET with maximum degradation of 81.9% at 1 μM in MKN-45 cells. PRO-6E inhibits tumor growth in vivo and in vitro. PRO-6E induces cell apoptosis and induces cell arrest (Sturcture Note:(Blue: Cereblon ligand (HY-103596), Black: linker;Pink: ALK/c-Met inhibitor Crizotinib (HY-50878)) .
(E/Z)-Afatinib ((E/Z)-BIBW 2992) is the mixture of (E)-Afatinib and (Z)-Afatinib. Afatinib (HY-10261) is an irreversible inhibitor of EGFR, by irreversibly binding to their ATP binding site to block activation of EGFR, HER2, HER4, and EGFRvIII. Afatinib used in co-administration with Temozolomide (HY-17364), potently targeting to EGFRvIII-cMet signaling in glioblastoma cells .
Multi-kinase-IN-5 (compound 15c) is a promising multi-kinase inhibitory agent. Multi-kinase-IN-5 inhibits a panel of protein kinases (RET, KIT, cMet, VEGFR1,2, FGFR1, PDGFR and BRAF), showing % inhibition of 74%, 31%, 62%, 40%, 73%, 74%, 59%, and 69%, respectively, and IC50 of 1.287, 0.117 and 1.185 μM against FGFR1, VEGFR, and RET kinases, respectively .
SJF-1521 is a selective EGFR PROTAC degrader. SJF-1521 contains the EGFR inhibitor lapatinib (HY-50898). SJF-1521 can induce EGFR degradation in OVCAR8 cells .
BAY-3827 is a potent and selective AMPK inhibitor with IC50 values of 1.4 nM at low (10 µM ATP concentration) and 15 nM at high (2 mM ATP concentration). BAY-3827 shows over 500-fold selectivity for most of the 331 kinases. BAY-3827 prevents phosphorylation of acetyl-CoA carboxylase 1 and shows strongest anti-proliferative activity in androgen-dependent prostate cancer cell lines .
Protein tyrosine kinases (PTKs) are key signaling molecules and important drug targets. Two classes of PTKs are present in cells: the transmembrane receptor PTKs (RTKs) and the nonreceptor PTKs. The RTK family includes the receptors for insulin and for many growth factors, such as EGFR, FGFR, PDGFR, VEGFR, and NGFR. RTKs are transmembrane glycoproteins that are activated by the binding of their ligands, and they transduce the extracellular signal to the cytoplasm by phosphorylating tyrosine residues on the receptors themselves (autophosphorylation) and on downstream signaling proteins. Their principal functions of PTKs involve the regulation of multicellular aspects of the organism. Cell to cell signals concerning growth, differentiation, adhesion, motility, and death are frequently transmitted through tyrosine kinases. In humans, tyrosine kinases have been demonstrated to play significant roles in the development of many disease states, including diabetes and cancers.
MCE designs a unique collection of 1064 compounds that act as a useful tool for PTKs-related drug screening and disease research.
Liver cancer is one of the leading malignancies which occupies the second position in cancer deaths worldwide, becoming serious threat to human health. Hepatocellular carcinoma (HCC), also known as hepatoma is the most common type accounting for approximately 90% of all liver cancers.
Current evidence indicates that during hepatocarcinogenesis, two main pathogenic mechanisms prevail: (1) cirrhosis associated with hepatic regeneration after tissue damage caused by hepatitis infection, toxins or metabolic influences, and (2) mutations occurring in single or multiple oncogenes or tumor suppressor genes. Both mechanisms have been linked with alterations in several important cellular signaling pathways. These include the RAF/MEK/ERK pathway, PI3K/AKT/mTOR pathway, WNT/b-catenin pathway, insulin-like growth factor pathway, c-MET/HGFR pathway , etc.
MCE offers a unique collection of 1828 compounds with identified and potential anti-liver cancer activity. MCE anti-liver cancer compound library is a useful tool for anti-liver cancer drugs screening and other related research.
Norleual TFA, an angiotensin (Ang) IV analog, is a hepatocyte growth factor (HGF)/c-Met inhibitor with an IC50 of 3 pM. Norleual TFA is an AT4 receptor antagonist and exhibits potent antiangiogenic activities .
Norleual, an angiotensin (Ang) IV analog, is a hepatocyte growth factor (HGF)/c-Met inhibitor with an IC50 of 3 pM. Norleual is an AT4 receptor antagonist and exhibits potent antiangiogenic activities .
Telisotuzumab (ABT-700) is a human recombinant bivalent antibody, a therapeutic antibody against the hepatocyte growth factor receptor (MET) that binds c-Met with high affinity and inhibits c-Met signaling. Telisotuzumab has antitumor activity .
Ficlatuzumab is a monoclonal antibody (McAb) targeting human hepatocyte growth factor (HGF). Ficlatuzumab blocks the activation of the HGF/c-Met signaling pathway, and inhibits c-Met receptor-mediated cancer cell proliferation, migration, and invasion .
ABBV-303 is a multi-specific NK cell antibody targeting c-Met designed based on TriNKET technology for natural killer (NK) cell redirection therapy. The ABBV-303 antibody consists of three functional arms, a single-chain variable fragment (scFV) targeting c-Met, a Fab arm targeting NKG2D, and an Fc fragment targeting NK cell CD16a .
Meleagrin is a roquefortine C-derived alkaloid produced by fungi of the genus Penicillium and has antimicrobial and anti-proliferative activities. Meleagrin is a class of FabI inhibitor. Meleagrin is a lead c-Met inhibitory entity useful for the control of c-Met-dependent metastatic and invasive breast malignancies .
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, His-Avi) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-Avi, C-His labeled tag. The total length of HGFR Protein, Human (HEK293, His-Avi) is 908 a.a., with molecular weight of 45-60 kDa & 80-100 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. FITC-Labeled HGFR Protein, Human (HEK293, His) is the recombinant human-derived FITC-Labeled HGFR protein, expressed by HEK293 , with His labeled tag. The total length of FITC-Labeled HGFR Protein, Human (HEK293, His) is 908 a.a., with molecular weight of 30-40 kDa & 90-100 kDa & 150 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (Biotinylated, HEK293, His-Avi) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-Avi, C-6*His labeled tag. The total length of HGFR Protein, Human (Biotinylated, HEK293, His-Avi) is 908 a.a., with molecular weight of 75-95 & 40-50 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, His) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-6*His labeled tag. The total length of HGFR Protein, Human (HEK293, His) is 908 a.a., with molecular weight of 130 & 80-95 & 40-50 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (HEK293, Fc) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-hFc labeled tag. The total length of HGFR Protein, Human (HEK293, Fc) is 908 a.a., with molecular weight of 170 & 100-130 & 45-50 kDa, respectively.
The HGFR protein is a receptor tyrosine kinase that transduces signals by binding to hepatocyte growth factor/HGF ligand. It regulates proliferation, scattering, morphogenesis, and cell survival. HGFR Protein, Human (495a.a, HEK293, His) is the recombinant human-derived HGFR protein, expressed by HEK293 , with C-6*His labeled tag. The total length of HGFR Protein, Human (495a.a, HEK293, His) is 495 a.a., with molecular weight of 32 & 35-57 kDa, respectively.
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
Cabozantinib-d6 is the deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively[1][2][3].
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
Cabozantinib-d4 is deuterium labeled Cabozantinib. Cabozantinib is a potent multiple receptor tyrosine kinases (RTKs) inhibitor that inhibits VEGFR2, c-Met, Kit, Axl and Flt3 with IC50s of 0.035, 1.3, 4.6, 7 and 11.3 nM, respectively.
Met (C-Met) Antibody is a non-conjugated and Rabbit origined monoclonal antibody about 156 kDa, targeting to Met (C-Met). It can be used for WB,ICC/IF,IHC-P,FC assays with tag free, in the background of Human.
Met; Hepatocyte growth factor receptor; HGF receptor; HGF/SF receptor; Proto-oncogene c-Met; Scatter factor receptor; SF receptor; Tyrosine-protein kinase Met
WB, IP
Human
Phospho-c-Met (Tyr1349) Antibody is a non-conjugated and Rabbit origined monoclonal antibody about 156 kDa, targeting to Phospho-c-Met (Tyr1349). It can be used for WB,IP assays with tag free, in the background of Human.
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
c-Met-IN-17 is a potent c-Met kinase inhibitor with an IC50 of 0.031 μM. c-Met-IN-17 can be used in anticancer research. . c-Met-IN-17 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Inquiry Online
Your information is safe with us. * Required Fields.