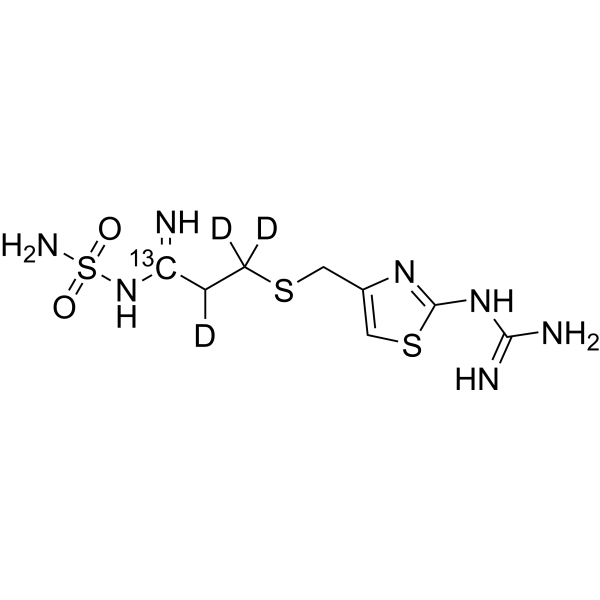Search Result
Results for "
competitive inhibition
" in MedChemExpress (MCE) Product Catalog:
11
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-N0598
-
|
20(S)-Ginsenoside F1
|
Cytochrome P450
Endogenous Metabolite
|
Cancer
|
|
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
|
-
-
- HY-101578
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
2614W94 is a selective, reversible inhibitor of monoamine oxidase-A with a competitive mechanism of inhibition and IC50 of 5 nM and Ki of 1.6 nM with serotonin as substrate.
|
-
-
- HY-161507
-
|
|
Carbonic Anhydrase
Cholinesterase (ChE)
|
Metabolic Disease
|
|
hCAI/II-IN-8 (Compound 8) is a hydrazide derivative based on 4-hydroxybenzaldehyde. hCAI/II-IN-8 primarily targets human carbonic anhydrase isomerase I (hCA I) and II (hCA II) for inhibition (IC50 = 21.35 ± 0.39 nM (hCA I); 7.12 ± 0.12 nM (hCA II)). hCAI/II-IN-8 inhibits AChE and BChE as well(IC50 = 46.27 ±0.75 nM (AChE); 43.38 ± 0.83 nM (BChE)). . .
|
-

-
- HY-N12427
-
|
|
Tyrosinase
|
Cancer
|
|
Litchinol B (compound 2) is a non-competitive tyrosinase inhibitor with an inhibition constant of 5.70 μM .
|
-
-
- HY-130199
-
|
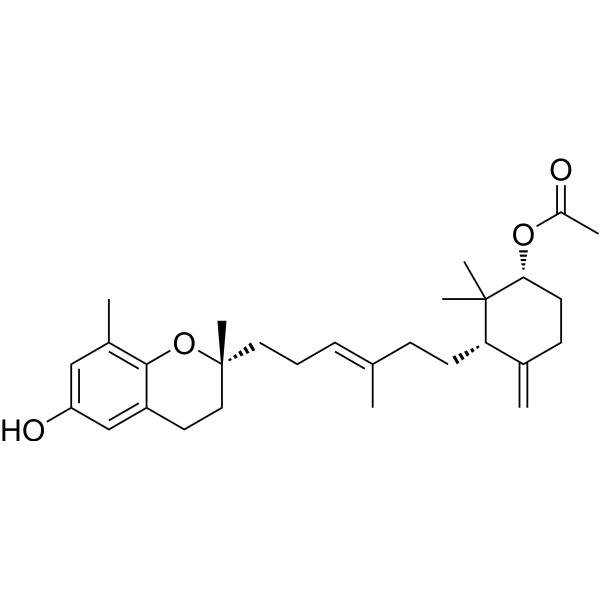
Parellic acid
|
Others
|
Metabolic Disease
|
|
Psoromic acid is a potent and selective RabGGTase (Rab geranylgeranyl transferase) inhibitor with an IC50 value of 1.3 µM. Psoromic acid is an antioxidative agent. Psoromic acid exhibits a competitive type of HMGR inhibition and mixed type of ACE (angiotensin converting enzyme) inhibition .
|
-
-
- HY-B0377
-
-
-
- HY-N3394
-
|
|
Others
|
Infection
|
|
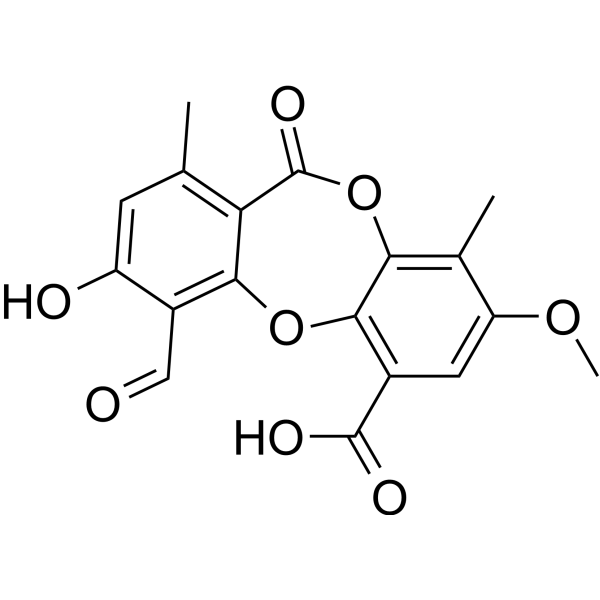
Lecanoric acid is a histidine-decarboxylase inhibitor isolated from fungus. The inhibition by lecanoric acid is competitive with histidineand noncompetitive with pyridoxal phosphate. Lecanoric acid did not inhibit aromatic amino acid decarboxylase .
|
-
-
- HY-15663
-
|
|
PAK
|
Cancer
|
|
IPA-3 is a selective non-ATP competitive PAK1 inhibitor with IC50 of 2.5 μM, and shows no inhibition to group II PAKs (PAKs 4-6).
|
-
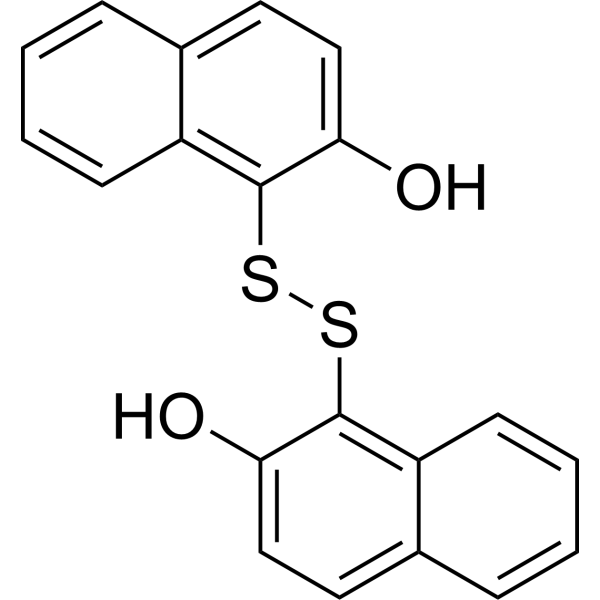
-
- HY-N7389B
-
|
|
Endogenous Metabolite
|
Metabolic Disease
|
|
GDP-α-D-mannose disodium is the donor substrate for mannosyltransferases and the precursor of GDP-β-L-fucose. GDP-α-D-mannose disodium gives a competitive inhibition with respect to GTP (Ki 14.7 μM) and an uncompetitive inhibition with respect to mannose-1-P (Ki 115 μM) .
|
-
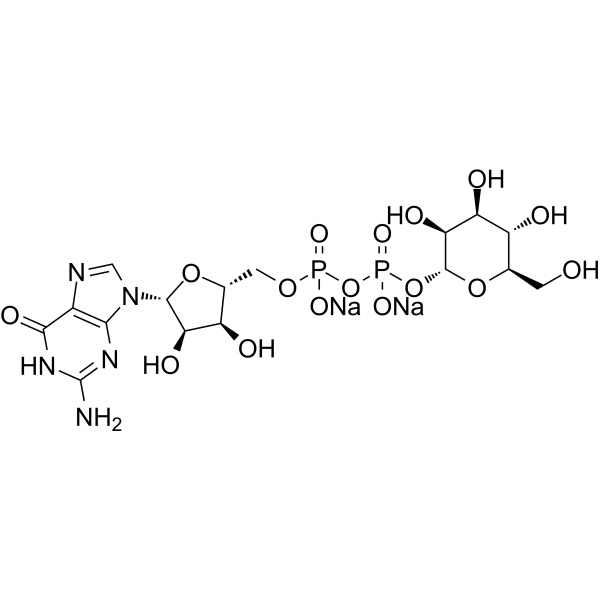
-
- HY-N7389A
-
|
|
Endogenous Metabolite
|
Metabolic Disease
|
|
GDP-α-D-mannose disodium is the donor substrate for mannosyltransferases and the precursor of GDP-β-L-fucose. GDP-α-D-mannose disodium gives a competitive inhibition with respect to GTP (Ki 14.7 μM) and an uncompetitive inhibition with respect to mannose-1-P (Ki 115 μM) .
|
-
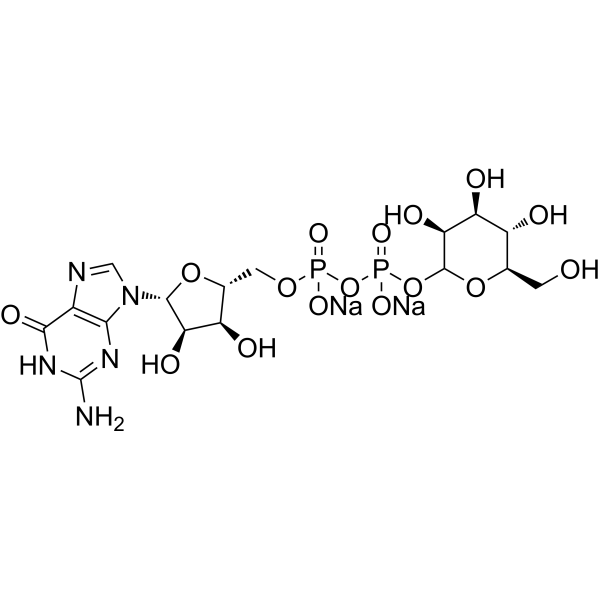
-
- HY-N3136
-
|
|
Histamine Receptor
|
Others
|
|
Onitin is a natural product, that can be isolated from Onychium siliculosum. Onitin is also a non-competitive antagonist of histamine. Onitin shows activity in blocking the peristaltic reflex of the guinea-pig ileum, in inhibition of the responses of guinea-pig ileum to histamine and of inhibition of the responses of guinea-pig tracheal muscle to histamine .
|
-
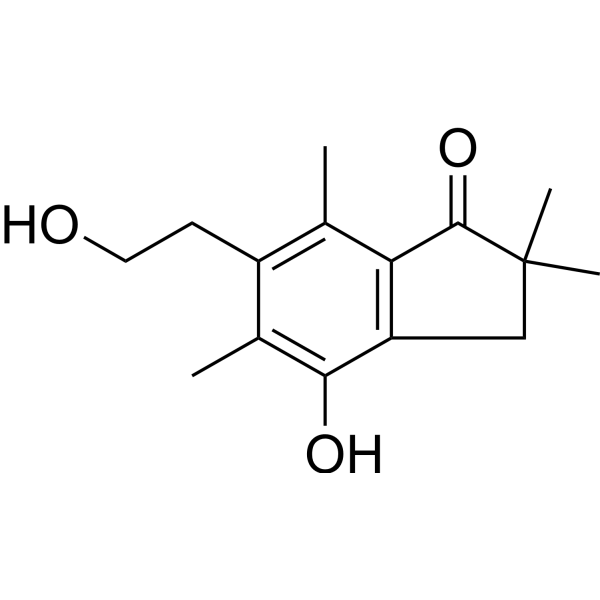
-
- HY-13007
-
|
PF-03758309
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
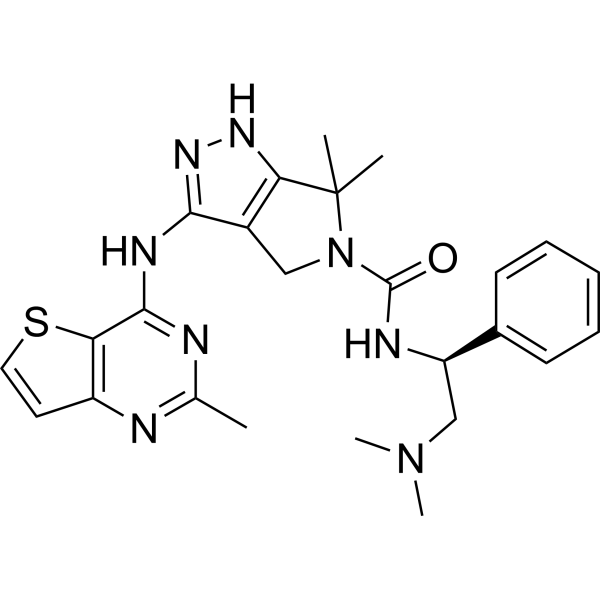
-
- HY-13007B
-
|
PF-03758309 dihydrochloride
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) dihydrochloride is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 dihydrochloride has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
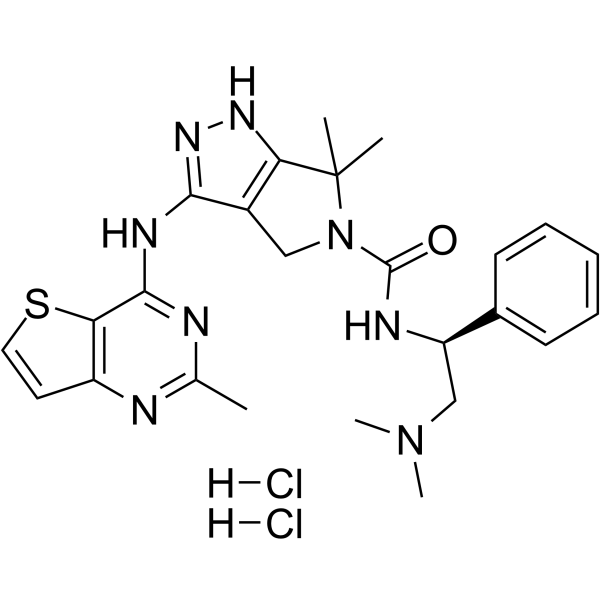
-
- HY-13007A
-
|
PF-03758309 hydrochloride
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) hydrochloride is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 hydrochloride has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
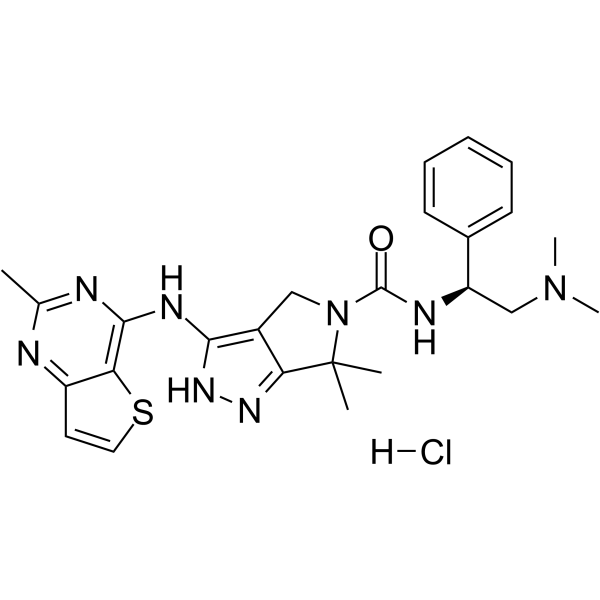
-
- HY-17000S
-
-
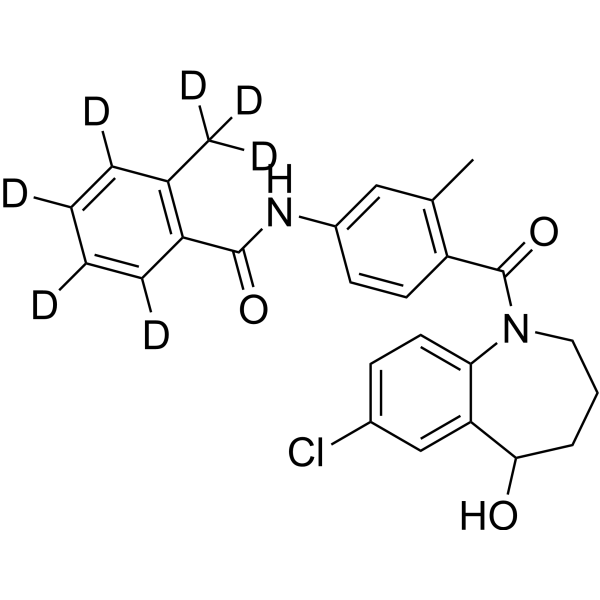
-
- HY-111173
-
|
|
Dipeptidyl Peptidase
|
Others
|
|
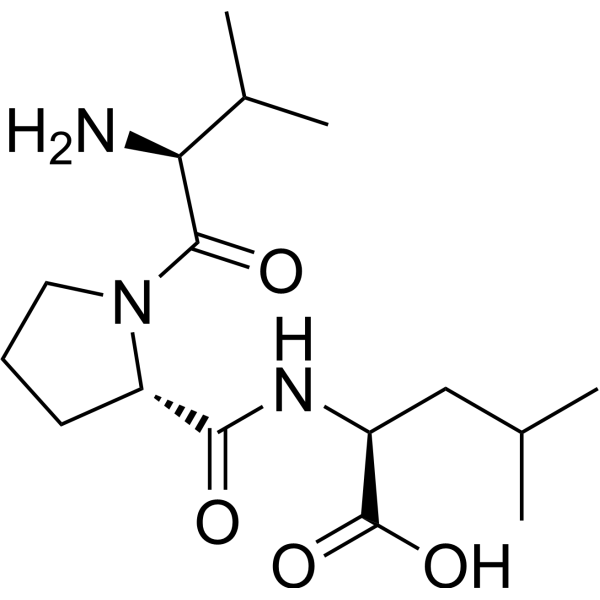
Diprotin B is a dipeptidyl aminopeptidase IV (DPP IV) inhibitor. The apparent competitive inhibition of DPP-IV by the diprotins is a kinetic artifact, derived from the substrate-like nature of tripeptides containing a penultimate proline residue .
|
-
-
- HY-117769
-
|
|
Fatty Acid Synthase (FASN)
|
Metabolic Disease
|
|
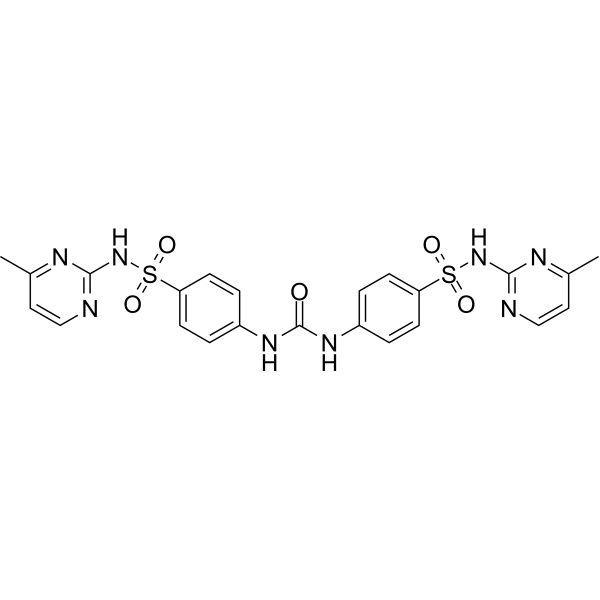
GSK837149A is a selective inhibitor of human Fatty Acid Synthase (FASN) targeting the KR domain. GSK837149A has reversible inhibition effect on FASN and selectivity for type I FASN (Ki=30 nM). GSK837149A is also a competitive inhibitor of NADPH and a non-competitive inhibitor of acetoacetyl-CoA. GSK837149A can be used for the research of obesity and breast cancer .
|
-
-
- HY-15687A
-
|
|
ROCK
|
Metabolic Disease
|
|
SAR407899 is a selective, potent and ATP-competitive ROCK inhibitor, with an IC50 of 135 nM for ROCK-2, and Kis of 36 nM and 41 nM for human and rat ROCK-2, respectively. SAR407899 shows stable inhibition of migrasome formation.
|
-
-
- HY-B0690
-
-
-
- HY-145701
-
|
|
MEK
|
Cancer
|
|
MEK1/2-IN-2 is a potent ATP-competitive MEK1/2 inhibitor and shows equipotent inhibition of WT MEK1/2 and a panel of MEK1/2 mutant cell lines .
|
-
-
- HY-B0377S
-
-
-
- HY-146142
-
|
|
Cholinesterase (ChE)
Amyloid-β
|
Neurological Disease
|
|
AChE/BuChE-IN-2 (Compound 5f) is an orally active AChE and BuChE inhibitor with IC50 values of 0.72 μM and 0.16 μM, respectively. AChE/BuChE-IN-2 shows a non-competitive inhibition with AChE and shows potent self-induced β-amyloid (Aβ) aggregation inhibition with an IC50 of 62.52 μM. AChE/BuChE-IN-2 can cross the BBB .
|
-
-
- HY-10195B
-
|
LY333531 hydrochloride
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
-
- HY-10195
-
|
LY333531
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
-
- HY-161421
-
|
|
Glucosidase
|
Metabolic Disease
|
α-Glucosidase-IN-61 (Compd 1j), a competitive α-glucosidase inhibitor, demonstrates excellent inhibition with an IC50 of 0.73 μM. α-Glucosidase-IN-61 (Compd 1j) is used for the research of type 2 diabetes mellitus .
|
-
-
- HY-157220
-
|
|
Vasopressin Receptor
|
Cardiovascular Disease
|
|
Tolvaptan phosphate ester sodium, a prodrug of Tolvaptan (HY-17000), can be used in the study of cardiac edema. Tolvaptan is a selective, competitive and orally active vasopressin receptor 2 (V2R) antagonist with an IC50 of 1.28 μM for the inhibition of arginine vasopressin (AVP)-induced platelet aggregation .
|
-
-
- HY-17000
-
|
OPC-41061
|
Vasopressin Receptor
Autophagy
|
Cardiovascular Disease
Endocrinology
Cancer
|
|
Tolvaptan is a selective, competitive and orally active vasopressin receptor 2 (V2R) antagonist with an IC50 of 1.28 μM for the inhibition of arginine vasopressin (AVP)-induced platelet aggregation. Tolvaptan induces cell apoposis and affects cell cycle. Tolvaptan can be used for the research of hyponatremia .
|
-
-
- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-
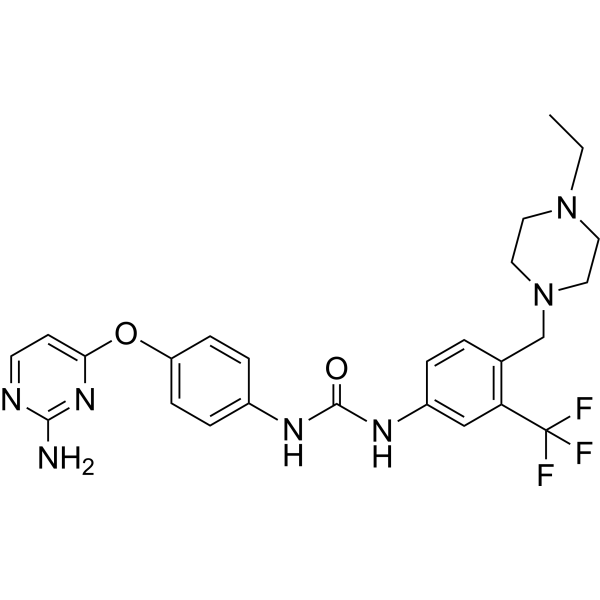
-
- HY-13965
-
|
ML161
|
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Parmodulin 2 (ML161) is an allosteric inhibitor of protease-activated receptor 1 (PAR1) with an IC50 of 0.26 μM . Parmodulin 2 is a potent and non-competitive inhibitor of SFLLRN-induced P-selectin expression leading to inhibition of platelet aggregation in vitro and platelet thrombus formation in vivo .
|
-
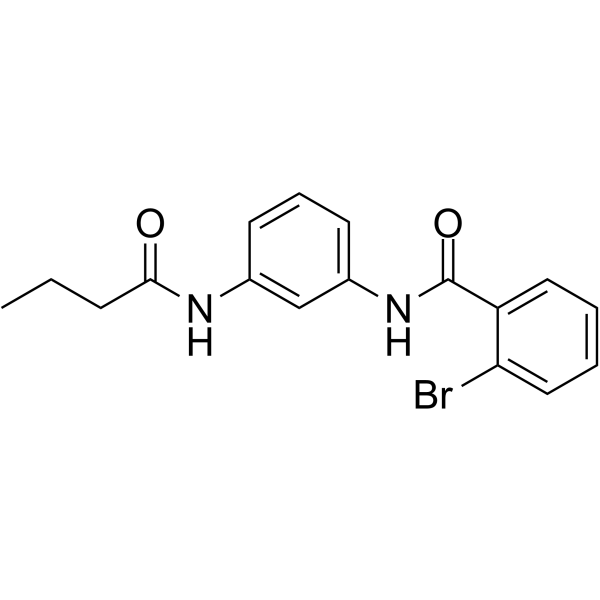
-
- HY-P1115
-
|
|
Akt
|
Others
|
|
AKTide-2T is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-

-
- HY-137506
-
-
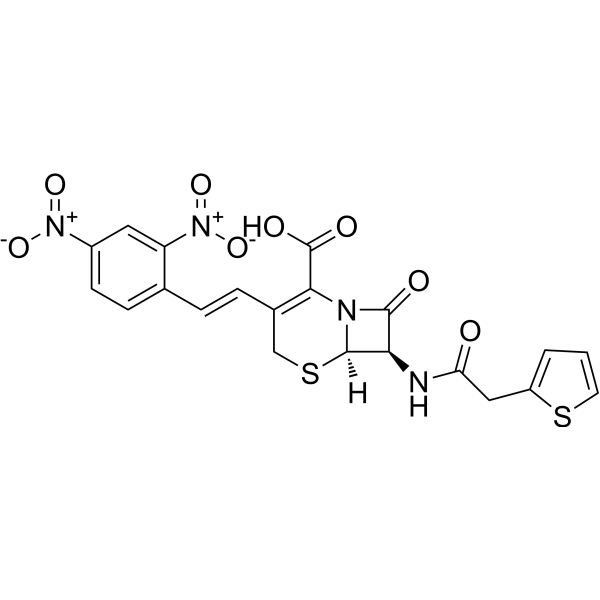
-
- HY-108913
-
|
|
Beta-lactamase
Antibiotic
|
Infection
|
|
Nitrocefin is a chromogenic β-lactamase substrate that undergoes a distinctive color change from yellow to red as the amide bond in the β-lactam ring is hydrolyzed by β-lactamase. Nitrocefin is used in competitive inhibition studies in developmental work on β-lactamase-resistant antibiotics .
|
-
-
- HY-157068
-
|
|
Ferroptosis
|
Cancer
|
|
icFSP1 is a potent ferroptosis suppressor protein-1 (FSP1) inhibitor. icFSP1 does not competitively inhibit FSP1 enzyme activity, but instead triggers subcellular relocalization of FSP1 from the membrane and FSP1 condensation before ferroptosis induction, in synergism with GPX4 inhibition .
|
-
-
- HY-135111
-
|
|
Drug Metabolite
|
Infection
Metabolic Disease
Cancer
|
|
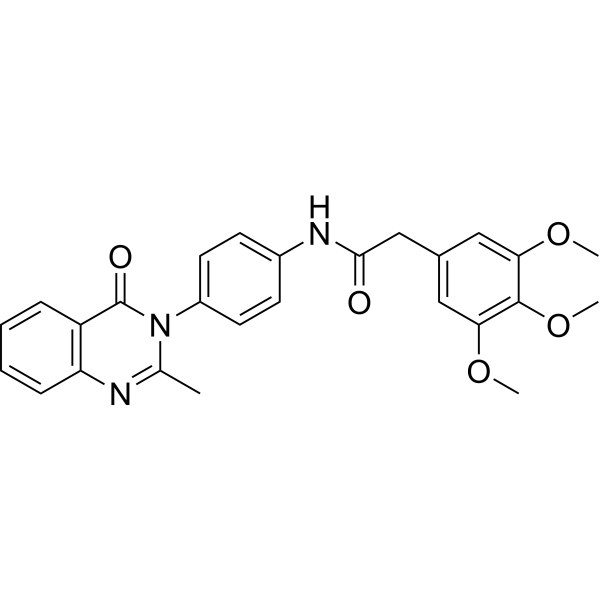
4-Desmethoxy Omeprazole is the active metabolite of Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .
|
-
-
- HY-P1115A
-
|
|
Akt
|
Others
|
|
AKTide-2T TFA is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T TFA mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-
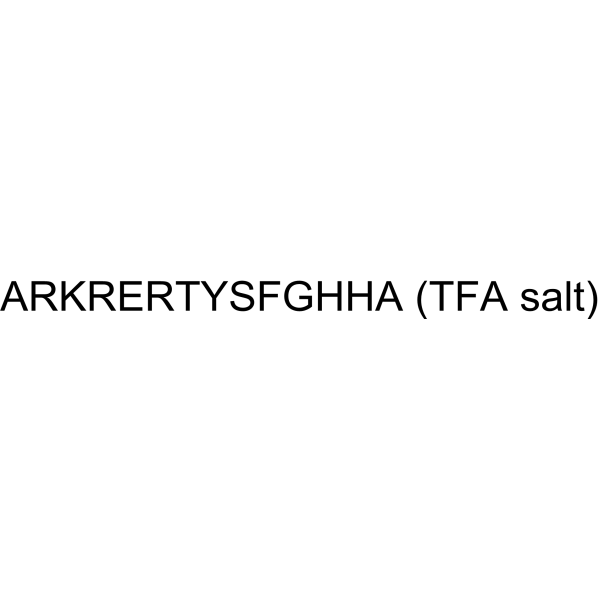
-
- HY-163174
-
|
|
Others
|
Cardiovascular Disease
Neurological Disease
Metabolic Disease
Cancer
|
|
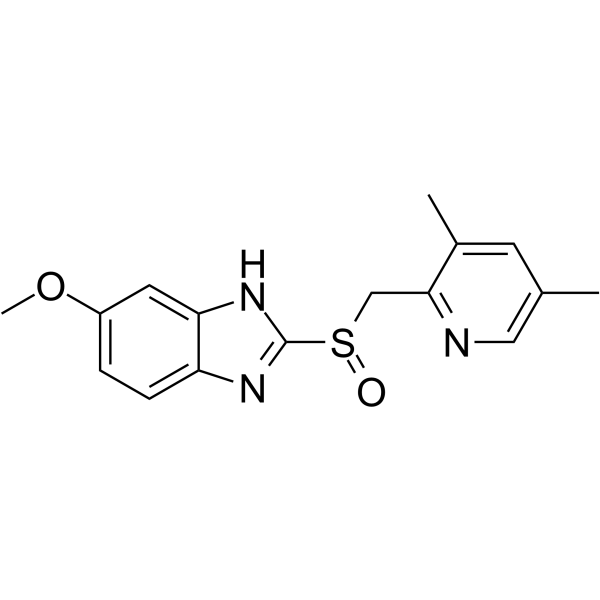
II399 is a potent, selective NNMT bisubstrate inhibitor containing an unconventional SAM mimic, with a Ki of 5.9 nM. II399 exhibits an explicit pattern of competitive inhibition for NAM. II399 occupies both the substrate and cofactor binding pockets. II399 has the potential for the research of cancers, metabolic, cardiovascular, and neurodegenerative diseases .
|
-
-
- HY-B0113S
-
|
H 16868-d3
|
Proton Pump
Bacterial
Autophagy
|
Infection
Metabolic Disease
Cancer
|
|
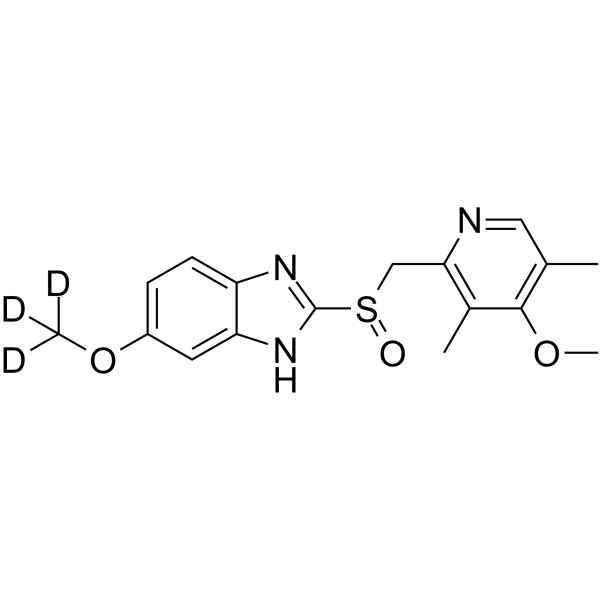
Omeprazole-d3 is deuterium labeled Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].
|
-
-
- HY-12481
-
|
|
PI3K
Autophagy
|
Cancer
|
|
SAR405 is a first-in-class, selective, and ATP-competitive PI3K class III (PIK3C3) isoform Vps34 inhibitor (IC50=1.2 nM; Kd=1.5 nM). SAR405 inhibits autophagy induced either by starvation or by mTOR inhibition. Anticancer activity .
|
-
-
- HY-10195BS
-
|
|
Isotope-Labeled Compounds
PKC
|
Metabolic Disease
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM[1][2].
|
-
-
- HY-112179
-
|
|
Others
|
Inflammation/Immunology
|
|
GSK180 is a selective, competitive, and potent inhibitor of kynurenine-3-monooxygenase (KMO), a key enzyme of tryptophan metabolism (IC50, ~6 nM), but shows negligible activity against other enzymes on the tryptophan pathway. GSK180 rapidly changes levels of kynurenine pathway metabolites, and acts as a useful tool to probe the therapeutic potential of KMO inhibition .
|
-
-
- HY-145702
-
|
|
MEK
ERK
|
Cancer
|
|
MAP855 is a highly potent, selective, ATP-competitive and orally active MEK1/2 kinase inhibitor (MEK1 ERK2 cascade IC50=3 nM, pERK EC50=5 nM). MAP855 shows equipotent inhibition of wild-type and mutant MEK1/2 .
|
-
-
- HY-114858
-
|
|
Casein Kinase
|
Cancer
|
|
Epiblastin A is an ATP competitive casein kinase 1 (CK1) inhibitor with IC50s of 8.9, 0.5, and 4.7 µM for CK1α, CK1δ, and CK1 ɛ, respectively. Epiblastin A induces reprogramming of epiblast stem cells into embryonic stem cells by inhibition of CK1 .
|
-
-
- HY-107727
-
|
|
Neuropeptide Y Receptor
|
Neurological Disease
|
|
BMS-193885 (L-Lactic acid) is a potent, selective, and brain-penetrant neuropeptide Y1 receptor antagonist. BMS-193885 has a Ki value of 3.3 nM for the neuropeptide Y1 receptor, competitively acts on the neuropeptide Y binding site, and can reduce food intake and body weight through central Y1 inhibition .
|
-
-
- HY-15814
-
|
|
Bcr-Abl
PDGFR
c-Kit
Src
JAK
Apoptosis
|
Cancer
|
|
HG-7-85-01 is a type II ATP competitive inhibitor of wild-type and gatekeeper mutations forms of Bcr-Abl, PDGFRα, Kit, and Src kinases. HG-7-85-01 inhibits T315I mutant Bcr-Abl kinase, KDR and RET with IC50s of 3 nM, 20 nM and 30 nM, and is only weak or no inhibition of other kinases (IC50>2 μM). HG-7-85-01 inhibits the cell proliferation, which is mediated by the induction of apoptosis, and inhibition of cell-cycle progression .
|
-
-
- HY-12037A
-
|
ON-01910
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib (ON-01910) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3 kinase/Akt pathway, promots the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
-
- HY-B0113
-
|
H 16868
|
Proton Pump
Autophagy
Bacterial
Phospholipase
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
-
- HY-100414
-
|
BYK61359
|
Proton Pump
|
Metabolic Disease
|
|
Soraprazan (BYK61359) is a selective, reversible K-competitive inhibitor of the H,K-ATPase (Ki=6.4 nM), with an IC50 of 0.19 μM in gastric glands. Soraprazan binds to the H,K-ATPase with a Kd of 28.27 nM. Soraprazan shows immediate inhibition of acid secretion and is more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases .
|
-
-
- HY-B0113A
-
|
H 16868 sodium
|
Proton Pump
Autophagy
Bacterial
Phospholipase
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole sodium (H 16868 sodium), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole sodium shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole sodium also inhibits growth of Gram-positive and Gram-negative bacteria . Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
-
- HY-18953
-
|
|
mTOR
|
Cancer
|
|
mTOR inhibitor-23 (compound DHM25) is a selective, competitive, irreversible and covalent inhibitor of mTOR. mTOR inhibitor-23 has the mechanism of inhibition occurs mainly through its capacity to covalently interact with a nucleophilic amino acid inside the ATP pocket. mTOR inhibitor-23 exerts potent antitumor activity against triple-negative breast tumor cell lines .
|
-

-
- HY-108913R
-
|
|
Beta-lactamase
Antibiotic
|
Infection
|
|
Nitrocefin (Standard) is the analytical standard of Nitrocefin. This product is intended for research and analytical applications. Nitrocefin is a chromogenic β-lactamase substrate that undergoes a distinctive color change from yellow to red as the amide bond in the β-lactam ring is hydrolyzed by β-lactamase. Nitrocefin is used in competitive inhibition studies in developmental work on β-lactamase-resistant antibiotics .
|
-

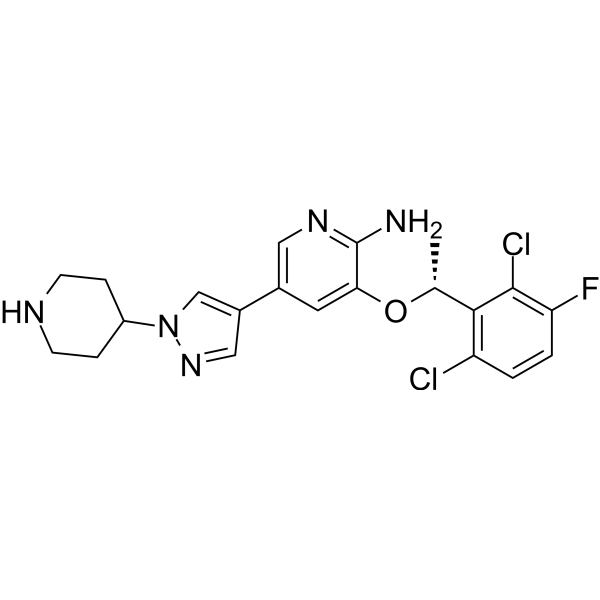
- HY-50878
-
Crizotinib
Maximum Cited Publications
62 Publications Verification
PF-02341066
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
- HY-50878B
-
|
PF-02341066 acetate
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
c-Met/HGFR
|
Cancer
|
|
Crizotinib (PF-02341066) acetate is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib acetate inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib acetate is also a ROS1 inhibitor. Crizotinib acetate has effective tumor growth inhibition .
|
-
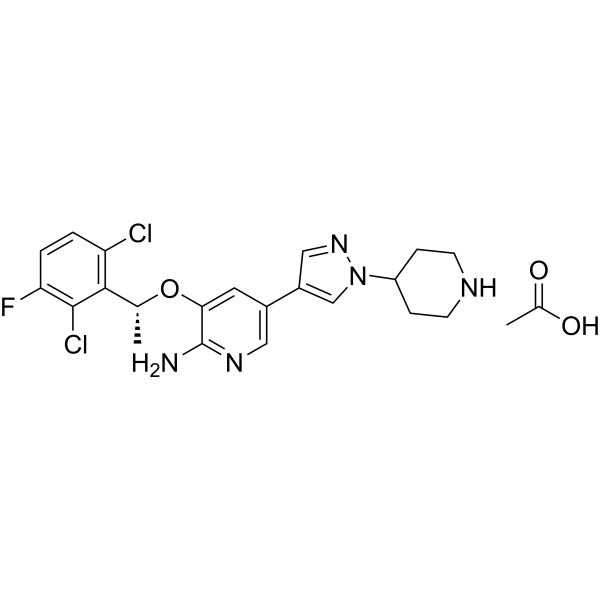
- HY-12037
-
|
ON-01910 sodium
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib sodium (ON-01910 sodium) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3K/Akt pathway, promotes the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib sodium is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
- HY-162480
-
|
|
Glucosidase
Phosphatase
|
Endocrinology
|
PTP1B-IN-27 (Compound 7i) is an inhibitor of protein tyrosine phosphatase 1-B (PTP‐1B)(IC50=8.2 µM). PTP1B-IN-27 also inhibits α-Glucosidase (IC50=120 µM) and shows competitive inhibition (Ki=118 µM) .
|
-
- HY-114923
-
|
|
DNA-PK
PI3K
|
Cancer
|
|
SU-11752 is an inhibitor for DNA-dependent protein kinase (DNA-PK) with an IC50 of 0.13 μM. SU-11752 inhibits PI3K p110γ kinase with IC50 of 1.1 μM. SU-11752 binds competitively for ATP-site in DNA-PK, results in inhibition of intracellular DNA double-strand break repair and increases the sensitivity of cells to radiotherapy .
|
-

- HY-120214
-
|
|
Syk
RET
|
Inflammation/Immunology
|
|
TAS05567 is a potent, highly selective, ATP-competitive and orally active Syk inhibitor with an IC50 of 0.37 nM. In a panel of 192 kinases, TAS05567 only shows >70% inhibition of Syk and 4 other kinases (FLT3, JAK2, KDR and RET with IC50s of 10 nM, 4.8 nM, 600 nM and 29 nM, respectively). TAS05567 can be used for humoral immune-mediated inflammatory conditions such as autoimmune and allergic diseases .
|
-
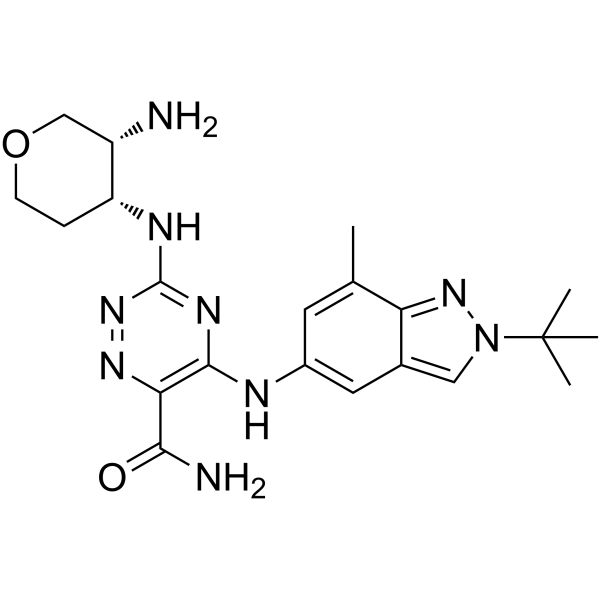
- HY-N6796
-
|
|
Farnesyl Transferase
Ras
Apoptosis
Phospholipase
|
Infection
Cancer
|
|
Manumycin A is an antibiotic. Manumycin A acts as a selective, competitive inhibitor of protein farnesyltransferase (FTase) with respect to farnesylpyrophosphate (Ki =1.2 μM), and as a noncompetitive inhibitor with respect to the Ras protein. Manumycin A induces apoptosis and exerts antitumor activity . Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling . Manumycin A is a nSMase inhibitor (EC50=0.25 μM) .
|
-
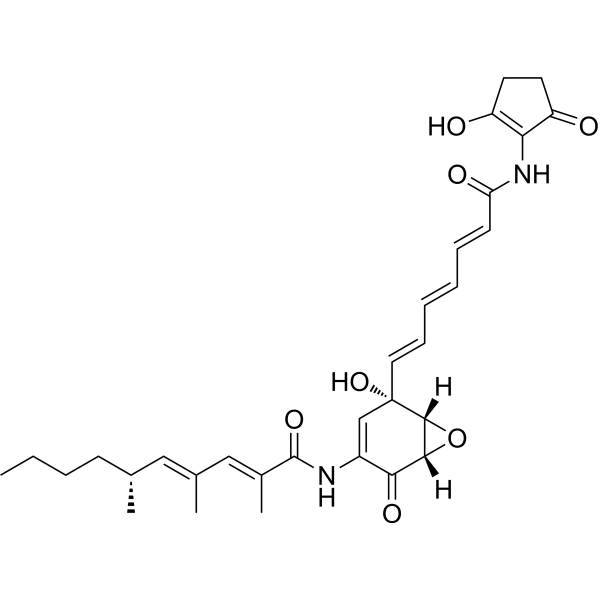
- HY-115570
-
|
GW108X
|
Kinesin
ULK
Autophagy
|
Cancer
|
|
GW406108X is a specific Kif15 (Kinesin-12) inhibitor with an IC50 of 0.82 uM in ATPase assays. GW406108X, a potent autophagy inhibitor, shows ATP competitive inhibition against ULK1 with a pIC50 of 6.37 (427 nM). GW406108X inhibits ULK1 kinase activity and blocks autophagic flux, without affecting the upstream signaling kinases mTORC1 and AMPK .
|
-
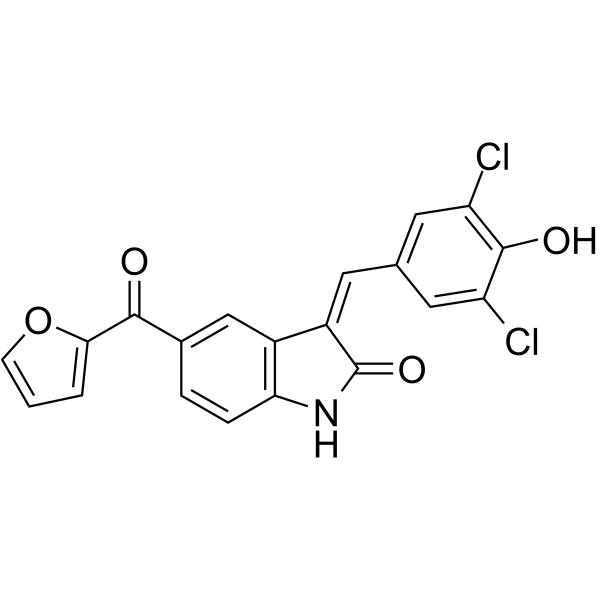
- HY-15186
-
|
GDC-0068; RG7440
|
Organoid
Akt
Apoptosis
|
Cancer
|
|
Ipatasertib (GDC-0068) is an orally active, highly selective and ATP-competitive pan-Akt inhibitor with IC50 values of 5, 18, 8 nM for Akt1/2/3, respectively. Ipatasertib synchronously activates FoxO3a and NF-κB through inhibition of Akt leading to p53-independent activation of PUMA. Ipatasertib also induces apoptosis in cancer cells and inhibits tumor growth in xenograft mouse models .
|
-
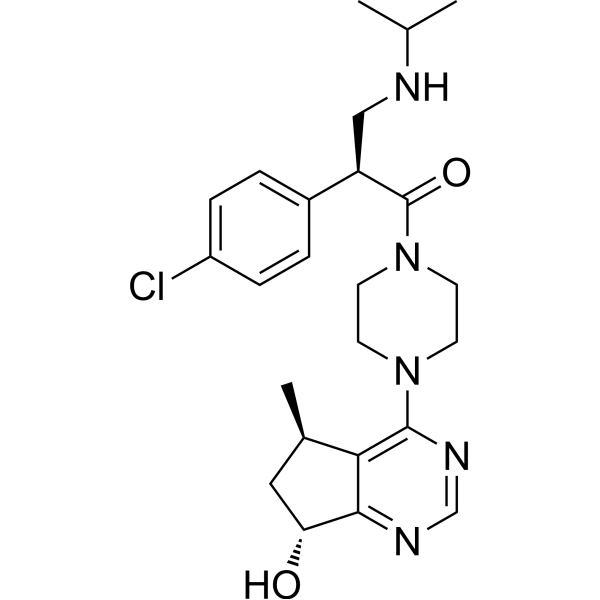
- HY-50878S
-
|
PF-02341066-d5
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
- HY-131339
-
|
|
Aurora Kinase
|
Cancer
|
|
SP-96 is a highly potent, selective and non-ATP-competitive Aurora B (IC50=0.316 nM) inhibitor and shows >2000 fold selectivity against FLT3 and KIT. SP-96 shows selective growth inhibition in NCI60 screening, incluing MDA-MD-468 (GI50=107 nM). SP-96 can be used for the research of triple negative breast cancer (TNBC) .
|
-
- HY-144636
-
|
|
Atg4
Cathepsin
Phospholipase
Autophagy
|
Cancer
|
|
Atg4B-IN-2 is a potent competitive Atg4B inhibitor with Ki value of 3.1 μM, also possesses declining PLA2 inhibitory potency, IC50s of 11 μM and 3.5 μM for Atg4B and PLA2, respectively. Atg4B-IN-2 enhances the anticancer activity of anti-castration-resistant prostate cancer agents via autophagy inhibition .
|
-
- HY-162485
-
|
|
Dengue virus
Virus Protease
|
Infection
|
|
DV-B-120 is a competitive inhibitor for dengue virus (DENV) with IC50s of 5.35, 7.39, 10.49 and 8.58 μM, for DENV-1, DENV-2, DENV-3 and DENV-4, respectively, by inhibiting NS2B-NS3 protease. DV-B-120 exhibits antiviral activity through inhibition of DENV replication .
|
-

- HY-B0113R
-
|
|
Proton Pump
Autophagy
Bacterial
Phospholipase
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole (Standard) is the analytical standard of Omeprazole. This product is intended for research and analytical applications. Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-

- HY-103430
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 hydrobromide is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for the research of parkinson’s disease and nicotine craving alleviation .
|
-
- HY-103430A
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for research of parkinson’s disease and nicotine craving alleviation .
|
-
- HY-50878A
-
|
PF-02341066 hydrochloride
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
- HY-N1584
-
|
RU-19110
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
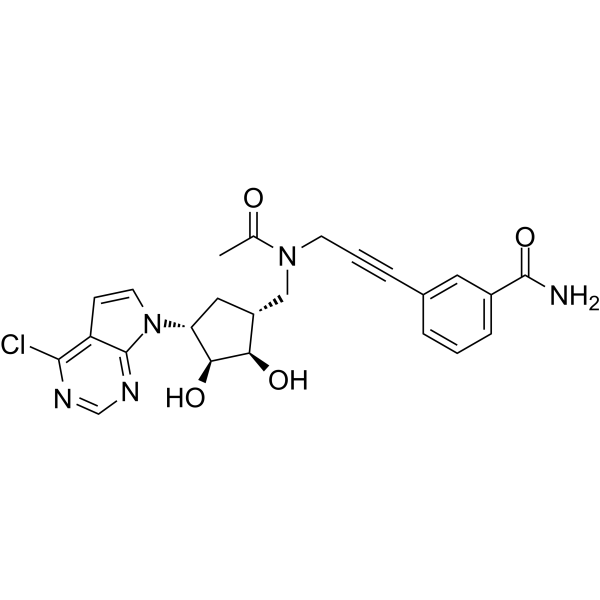
Halofuginone (RU-19110), a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
- HY-B0113S1
-
|
H 16868-d3-1
|
Proton Pump
Autophagy
Bacterial
Phospholipase
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole-d3-1 is the deuterium labeled Omeprazole. Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor)[3].
|
-
- HY-N6948S
-
|
|
Isotope-Labeled Compounds
|
Inflammation/Immunology
|
|
Linalyl acetate-d6 is deuterated labeled Omeprazole (HY-B0113). Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
- HY-B0113S4
-
|
H 16868-d3 sodium
|
Autophagy
Phospholipase
Proton Pump
Bacterial
Isotope-Labeled Compounds
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole-d3 sodium is deuterated labeled Omeprazole (HY-B0113). Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
- HY-50878R
-
|
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib (Standard) is the analytical standard of Crizotinib. This product is intended for research and analytical applications. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-

- HY-B0113S3
-
|
H 16868-13C,d3
|
Proton Pump
Autophagy
Bacterial
Phospholipase
|
Infection
Metabolic Disease
Cancer
|
|
Omeprazole- 13C,d3 is a 13C-labeled and deuterium labeled Omeprazole. Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor)[3].
|
-
- HY-N1584A
-
|
RU-19110 hydrobromide
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
- HY-P5272
-
|
|
Bacterial
|
Inflammation/Immunology
|
|
Histatin-3 TFA, a 32 amino acid peptide, possesses powerful antimicrobial properties. Histatin-3 TFA behaves as a substrate for proprotein convertase 1 (PC1), being cleaved by this endoprotease primarily at a site carboxy terminal to the single Arg25 residue (HRGYR decrease SN). Histatin-3 TFA is a moderately potent, reversible and competitive inhibitor of the furin-mediated cleavage of the pentapeptide pGlu-Arg-Thr-Lys-Arg-MCA fluorogenic substrate, with an estimated inhibition constant Ki of 1.98 μM .
|
-

- HY-N1584B
-
|
RU-19110 hydrochloride
|
Calcium Channel
DNA/RNA Synthesis
Parasite
Sodium Channel
TGF-beta/Smad
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM. Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity. Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
- HY-17422S1
-
|
Aciclovir-d4; Acycloguanosine-d4
|
Isotope-Labeled Compounds
HSV
Bacterial
Apoptosis
Antibiotic
|
Infection
Cancer
|
|
Acyclovir-d4 is the deuterium labeled Acyclovir. Acyclovir (Aciclovir) is a guanosine analogue and an orally active antiviral agent. Acyclovir inhibits HSV-1 (IC50 of 0.85 μM), HSV-2 (IC50 of 0.86 μM) and varicella-zoster virus. Acyclovir can be phosphorylated by viral thymidine kinase (TK), and Acyclovir triphosphate interferes with viral DNA polymerization through competitive inhibition with guanosine triphosphate and obligatory chain termination[1][2][3]. Acyclovir prevents bacterial infections during induction therapy for acute leukaemia[4].
|
-
- HY-110137A
-
|
DB75; NSC 305831
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine (DB75) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine is also an antiparasite agent .
|
-
- HY-110137
-
|
DB75 dihydrochloride; NSC 305831 dihydrochloride
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine dihydrochloride (DB75 dihydrochloride) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine dihydrochloride is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine dihydrochloride is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine dihydrochloride is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine dihydrochloride is also an antiparasite agent .
|
-
- HY-120826
-
|
|
Monoamine Oxidase
Adenosine Receptor
|
Neurological Disease
|
|
A2AAR/hMAO-B-IN-1 (compoudn 17) is a non-xanthine dual-target inhibitor targeting the A2A adenosine receptor (A2AAR) (IC50: 34.9 nM) andMAO-B (Ki: 39.5 nM, human). A2AAR/hMAO-B-IN-1 inhibits A2AAR-induced cAMP accumulation and exhibits competitive, reversible inhibition of MAO-B. A2AAR/hMAO-B-IN-1 can be used in the study of neurodegenerative diseases such as Parkinson's disease (PD) .
|
-
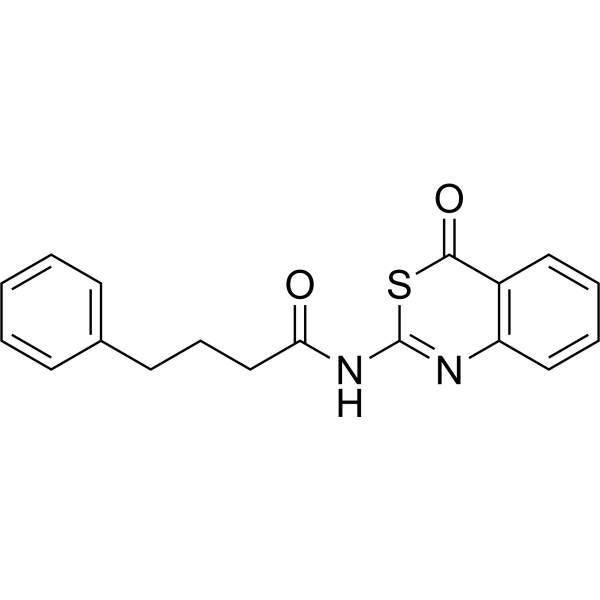
- HY-50878AS
-
|
PF-02341066-d9 hydrochloride
|
c-Met/HGFR
Autophagy
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Isotope-Labeled Compounds
|
Cancer
|
|
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
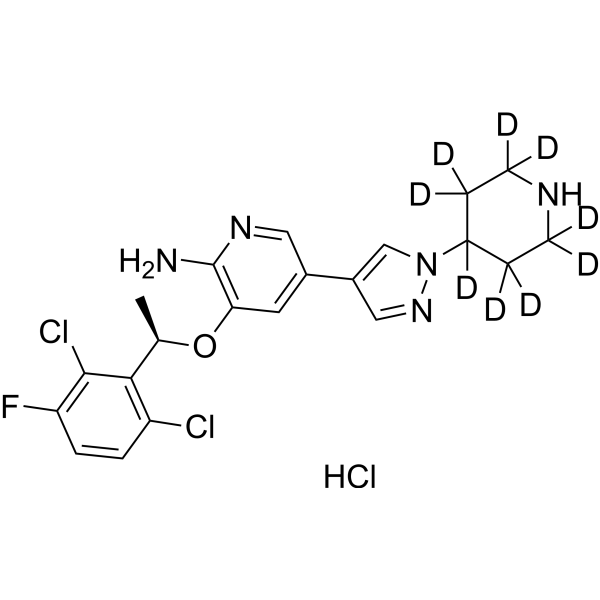
- HY-N1584AR
-
|
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (hydrobromide) (Standard) is the analytical standard of Halofuginone (hydrobromide). This product is intended for research and analytical applications. Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-

- HY-121998
-
|
|
Aurora Kinase
|
Others
|
|
Binucleine 2 is an isoform-specific and ATP-competitive inhibitor of Drosophila Aurora B kinase (Ki=0.36 μM), a kinase involved in cell division. It is specific for Drosophila Aurora B kinase, inhibiting it in a dose-dependent manner, with minimal inhibition of human or X. laevis Aurora B kinases at concentrations up to 100 μM. Binucleine 2 induces mitotic and cytokinesis defects in Drosophila Kc167 cells. It prevents Drosophila S2 cells from assembling a contractile ring during cell division when used at a concentration of 40 μM but does not affect ring ingression, suggesting that Aurora B kinase activity is not required for that step.
|
-

- HY-P0299A
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) TFA is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) TFA can readily crosse the blood-brain barrier .
|
-
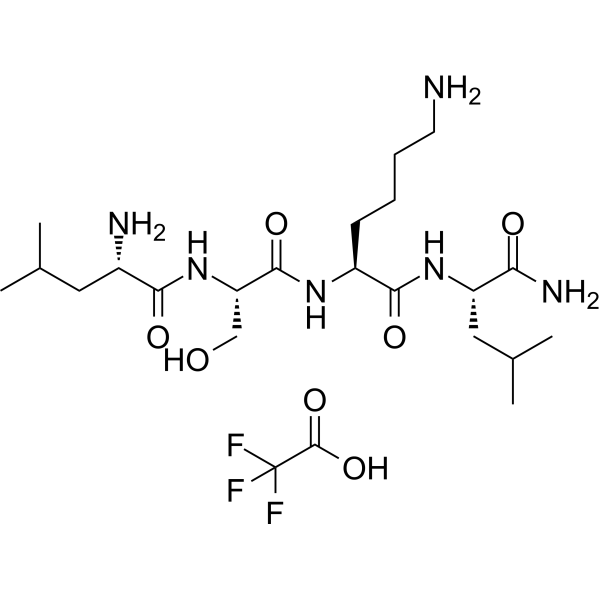
- HY-P0299
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) can readily crosse the blood-brain barrier .
|
-
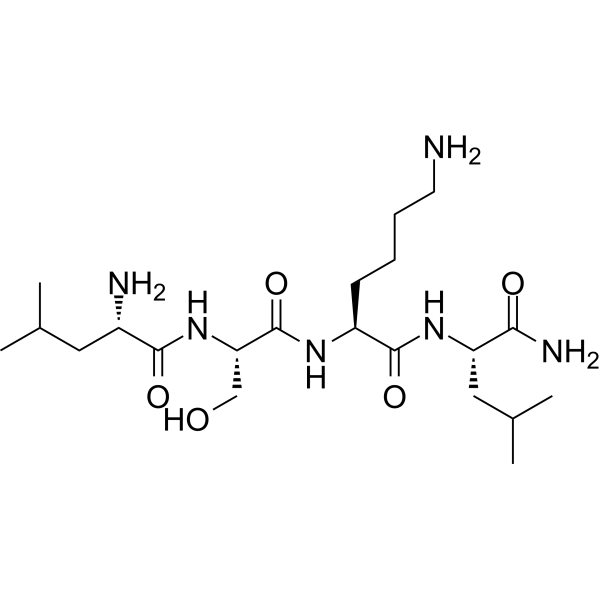
- HY-161449
-
|
|
11β-HSD
|
Metabolic Disease
|
|
JTT-654 is an orally active, potent and selective11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor. The IC50 of JTT-654 for 11β-HSD1 is 4.65, 0.97, and 0.74 nM in human, rat, and mouse recombinant enzymes, respectively. JTT-654 showed competitive inhibition against human recombinant enzyme. The IC50 value for human 11β-HSD2 is > 30 μM (human 11β-HSD2 is responsible for the reverse reaction against human 11β-HSD1). JTT-654 ameliorates insulin resistance and non-obese type 2 diabetes by inhibiting adipose tissue and liver 11β-HSD1 .
|
-

- HY-A0139
-
|
NSC 108165; Navan; Navane
|
Others
|
Others
|
|
Thiothixene is a typical antipsychotic. It selectively binds to dopamine D2 over D1, D3, and D4 receptors (Kis=0.417, 338, 186.2, and 363.1 nM, respectively). Thiothixene also binds to various serotonin (5-HT), histamine H1, α1- and α2-adrenergic, muscarinic acetylcholine, and sigma receptors (Kis=15-5,754 nM) as well as the dopamine, norepinephrine, and serotonin transporters (Kis=3.16-30 μM). In vivo, thiothixene reduces spontaneous and amphetamine-induced locomotor activity in rats. It enhances latent inhibition, as measured by a decreased lick latency in response to light and foot shock stimuli, which is a measure of selective attention in rats.3 Thiothixene also increases competitive behavior in submissive mice, indicating antidepressant-like behavior.
|
-

| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P5272
-
|
|
Bacterial
|
Inflammation/Immunology
|
|
Histatin-3 TFA, a 32 amino acid peptide, possesses powerful antimicrobial properties. Histatin-3 TFA behaves as a substrate for proprotein convertase 1 (PC1), being cleaved by this endoprotease primarily at a site carboxy terminal to the single Arg25 residue (HRGYR decrease SN). Histatin-3 TFA is a moderately potent, reversible and competitive inhibitor of the furin-mediated cleavage of the pentapeptide pGlu-Arg-Thr-Lys-Arg-MCA fluorogenic substrate, with an estimated inhibition constant Ki of 1.98 μM .
|
-
- HY-W142169
-
|
Formyl-L-histidine
|
Peptides
|
Others
|
|
N-Formyl-L-histidine shows binding affinity to histidyl-tRNA synthetase with a Ki value of 4.6 μM. N-Formyl-L-histidine shows a competitive inhibition against L-histidine ammonia-lyase, inhibits urocanic acid formation from L-histidine with a Ki value of 4.26 mM .
|
-
- HY-111173
-
|
|
Dipeptidyl Peptidase
|
Others
|
|
Diprotin B is a dipeptidyl aminopeptidase IV (DPP IV) inhibitor. The apparent competitive inhibition of DPP-IV by the diprotins is a kinetic artifact, derived from the substrate-like nature of tripeptides containing a penultimate proline residue .
|
-
- HY-P10346
-
|
Smooth-Muscle Myosin Light-Chain Kinase (796-815)
|
Peptides
|
Cardiovascular Disease
Inflammation/Immunology
|
|
smMLCK peptide is a specific inhibitor of smooth muscle myosin light chain kinase (smMLCK). The smMLCK peptide mimics the substrate and competitively inhibits the binding of the actual substrate to the enzyme, thereby inhibiting the kinase activity. This inhibition prevents the phosphorylation of the myosin light chain, thus inhibiting muscle contraction .
|
-
- HY-P1115A
-
|
|
Akt
|
Others
|
|
AKTide-2T TFA is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T TFA mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-
- HY-P0299A
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) TFA is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) TFA can readily crosse the blood-brain barrier .
|
-
- HY-P0299
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) can readily crosse the blood-brain barrier .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
| Cat. No. |
Product Name |
Chemical Structure |
-
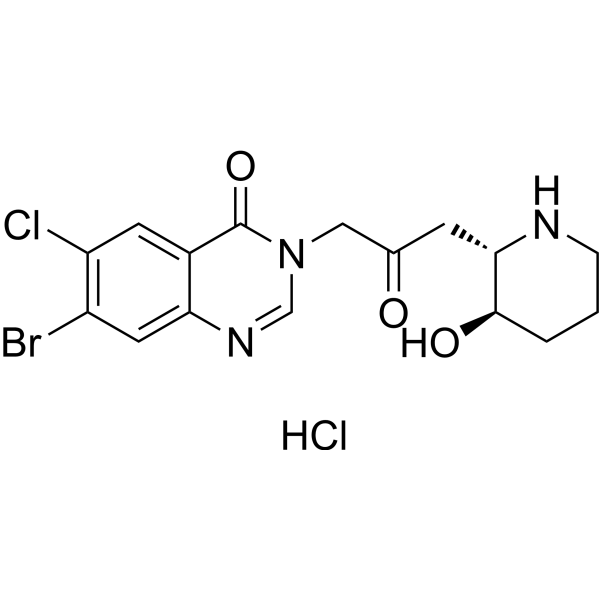
- HY-17000S
-
|
|
|
Tolvaptan-d7 is the deuterium labeled Tolvaptan. Tolvaptan is a selective, competitive arginine vasopressin receptor 2 antagonist with an IC50 of 1.28μM for the inhibition of AVP-induced platelet aggregation[1][2].
|
-
-
- HY-B0113S
-
|
|
|
Omeprazole-d3 is deuterium labeled Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].
|
-
-
- HY-50878S
-
|
|
|
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
-
- HY-B0377S
-
|
|
|
Famotidine- 13C,d3 is the 13C- and deuterium labeled Famotidine. Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion.
|
-
-
- HY-10195BS
-
|
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM[1][2].
|
-
-
- HY-B0113S1
-
|
|
|
Omeprazole-d3-1 is the deuterium labeled Omeprazole. Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor)[3].
|
-
-
- HY-N6948S
-
|
|
|
Linalyl acetate-d6 is deuterated labeled Omeprazole (HY-B0113). Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
-
- HY-B0113S4
-
|
|
|
Omeprazole-d3 sodium is deuterated labeled Omeprazole (HY-B0113). Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-
-
- HY-B0113S3
-
|
|
|
Omeprazole- 13C,d3 is a 13C-labeled and deuterium labeled Omeprazole. Omeprazole (H 16868), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM[1]. Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria[2].Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor)[3].
|
-
-
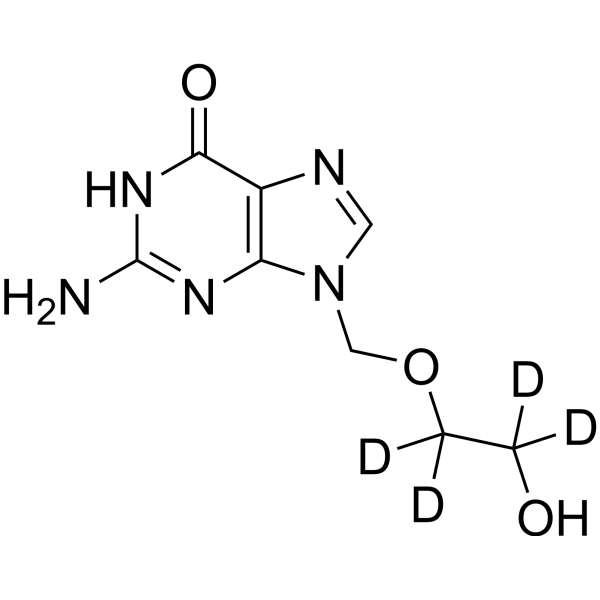
- HY-17422S1
-
|
|
|
Acyclovir-d4 is the deuterium labeled Acyclovir. Acyclovir (Aciclovir) is a guanosine analogue and an orally active antiviral agent. Acyclovir inhibits HSV-1 (IC50 of 0.85 μM), HSV-2 (IC50 of 0.86 μM) and varicella-zoster virus. Acyclovir can be phosphorylated by viral thymidine kinase (TK), and Acyclovir triphosphate interferes with viral DNA polymerization through competitive inhibition with guanosine triphosphate and obligatory chain termination[1][2][3]. Acyclovir prevents bacterial infections during induction therapy for acute leukaemia[4].
|
-
-
- HY-50878AS
-
|
|
|
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent: