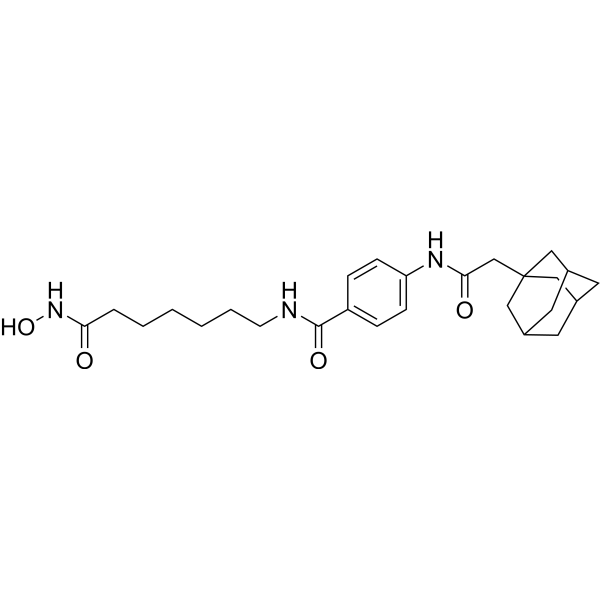
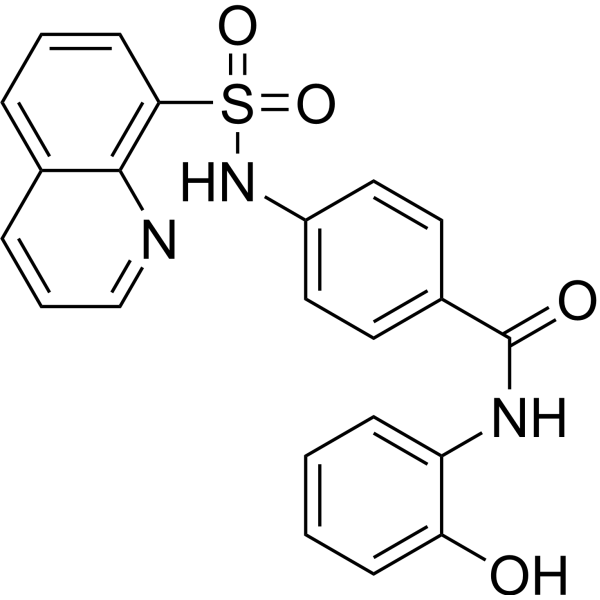
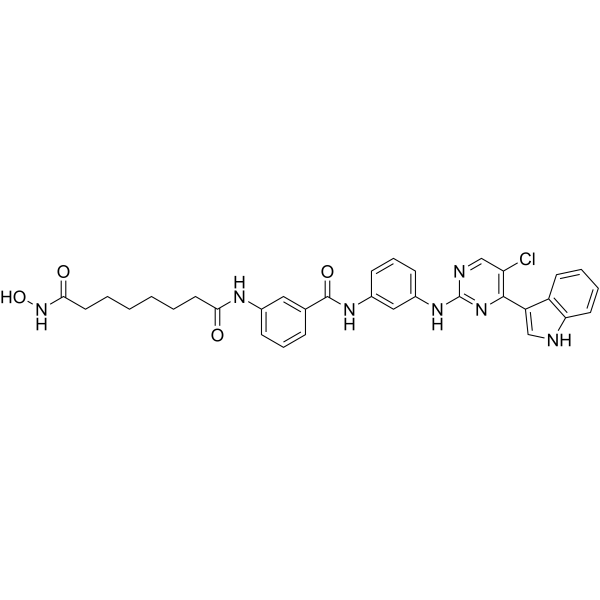
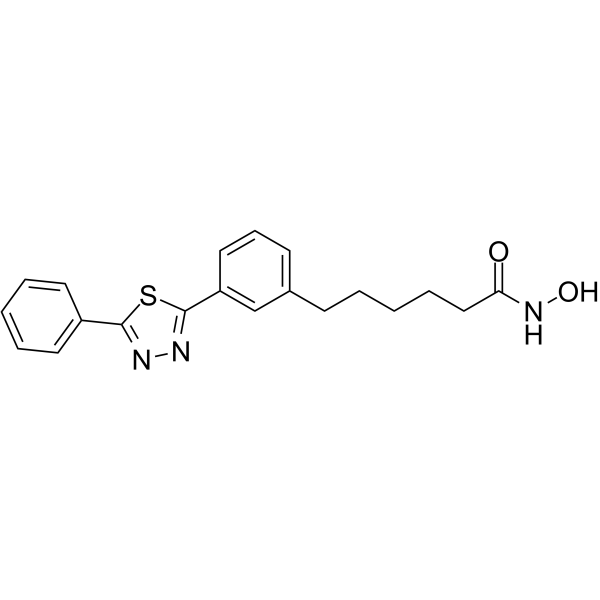
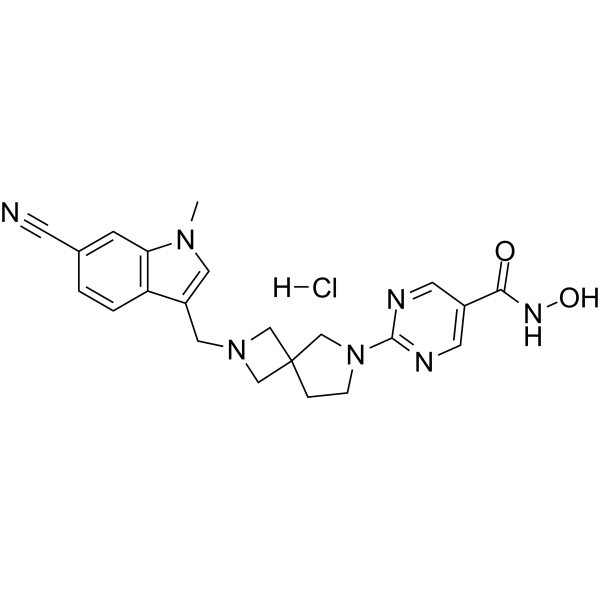
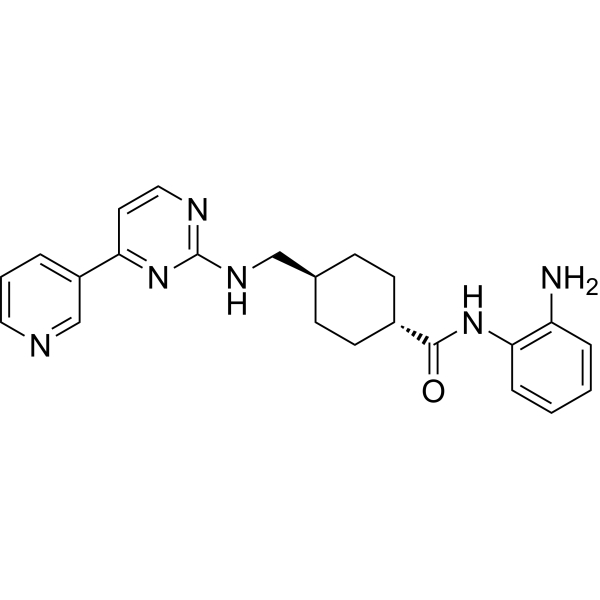
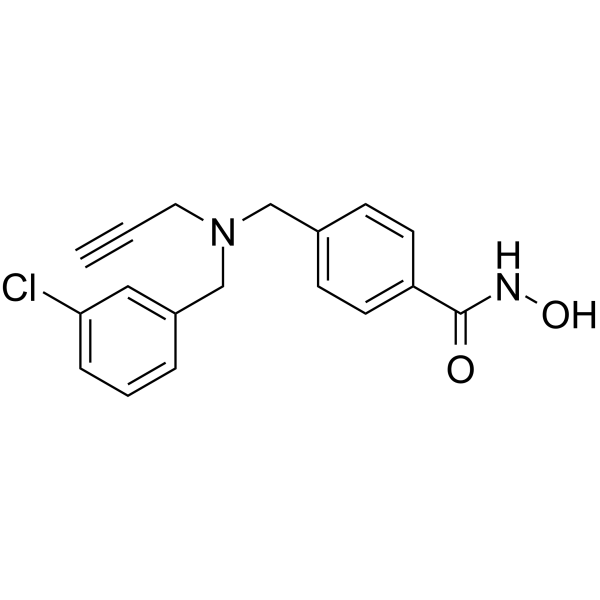
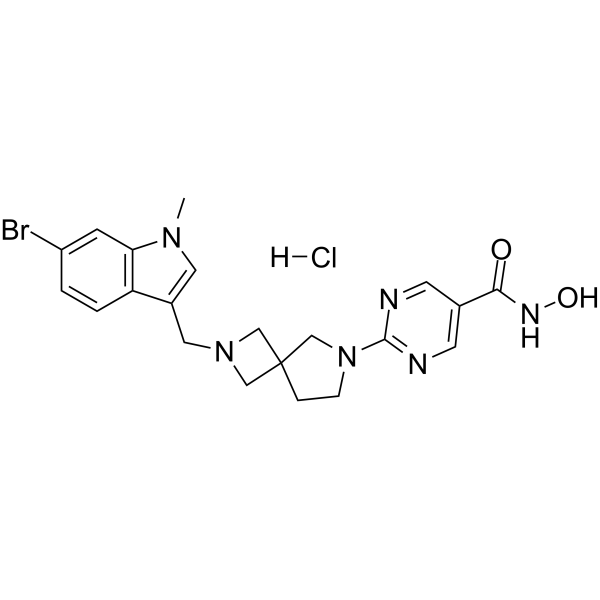
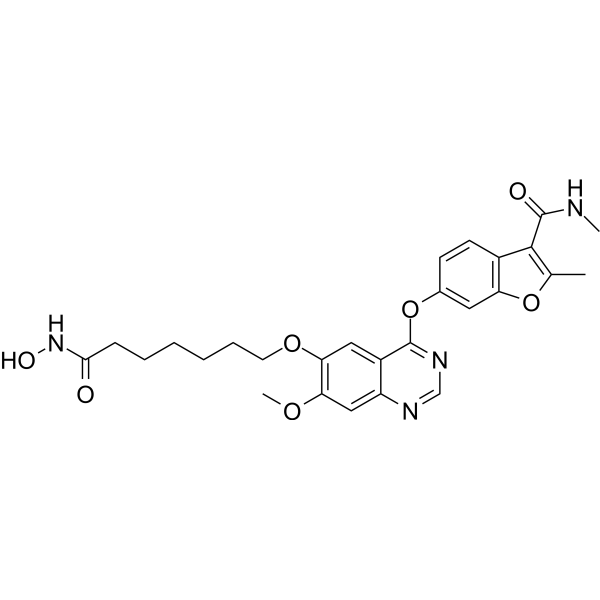
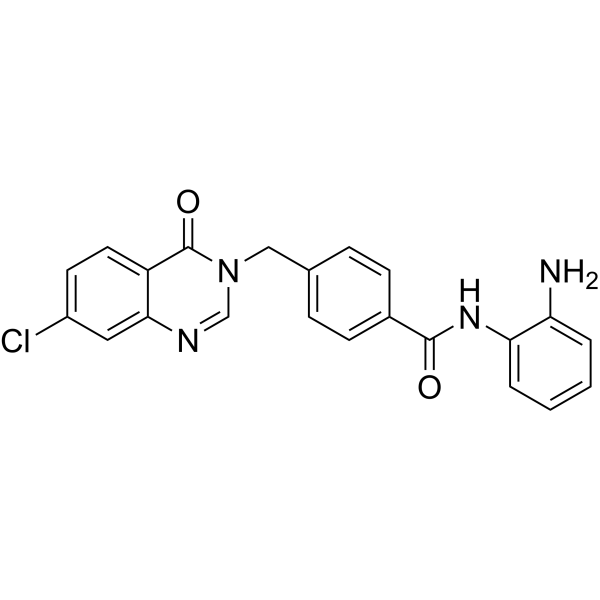
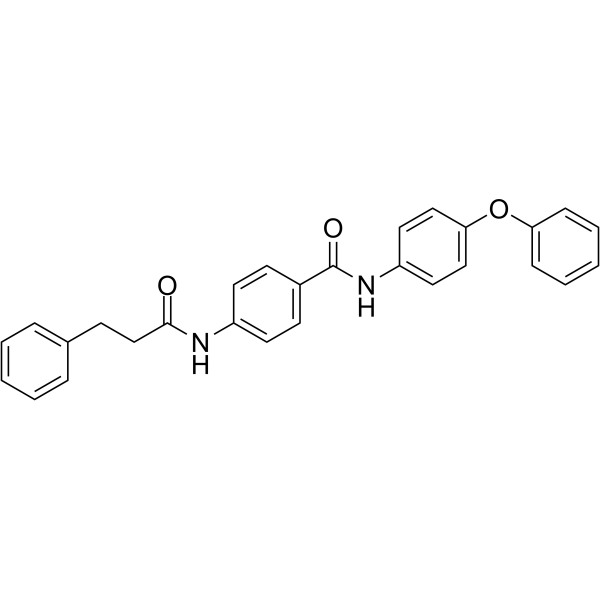
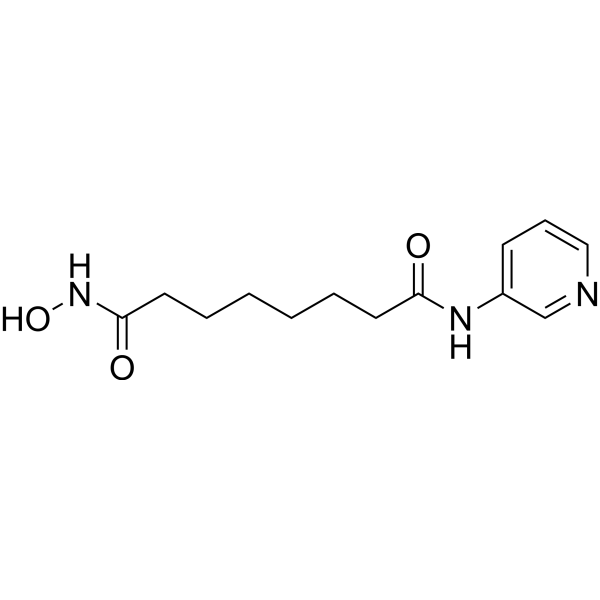
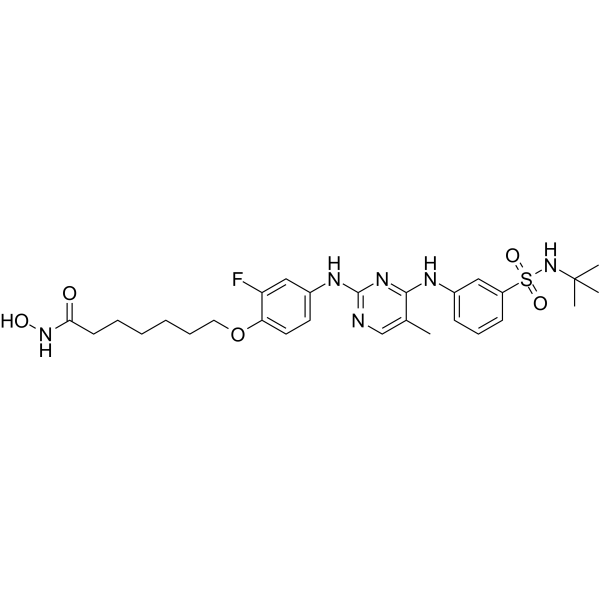
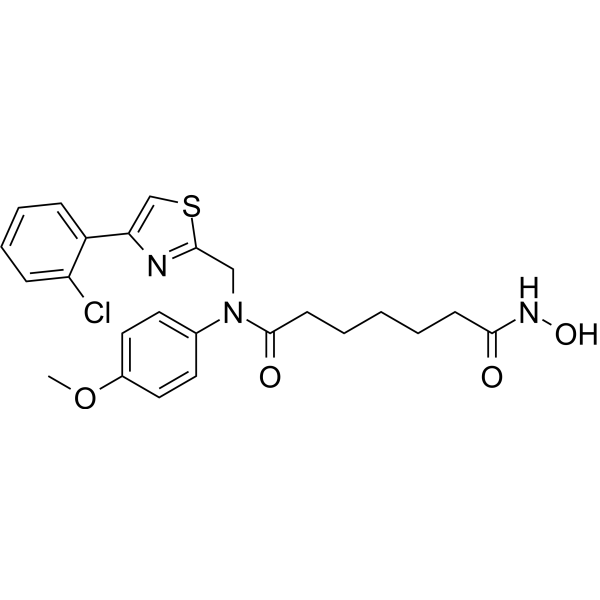
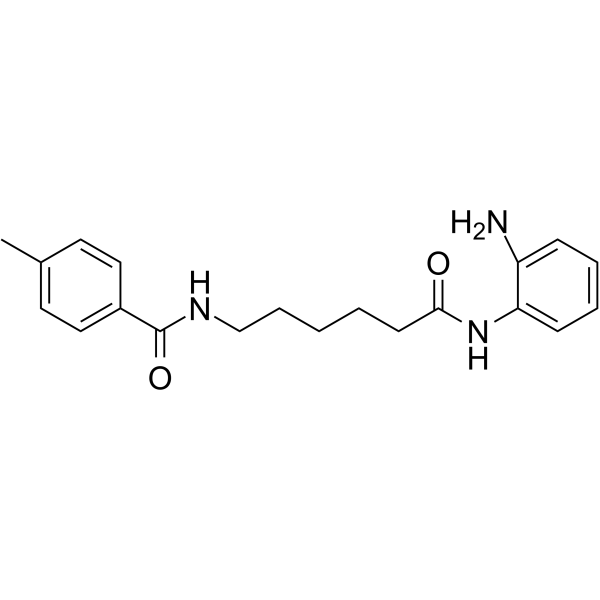
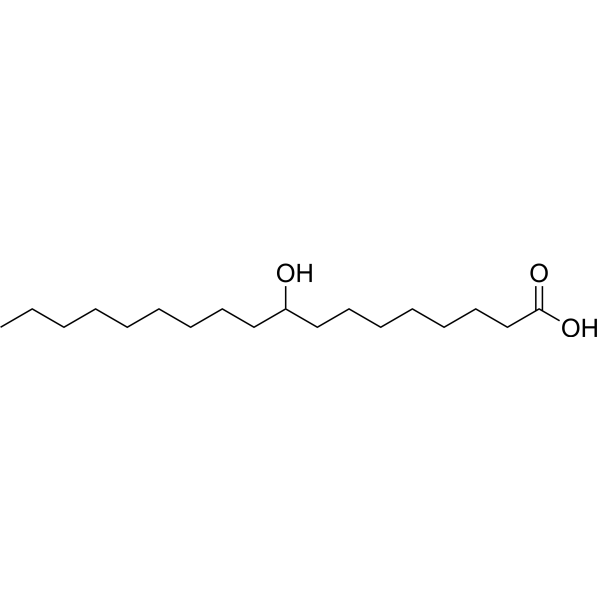
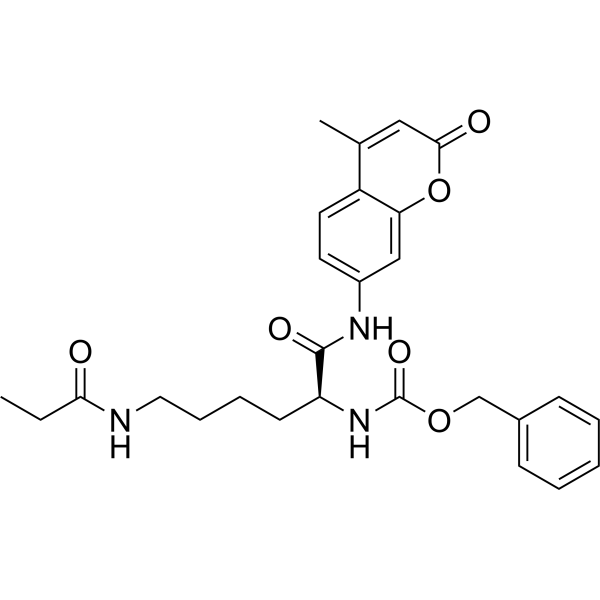
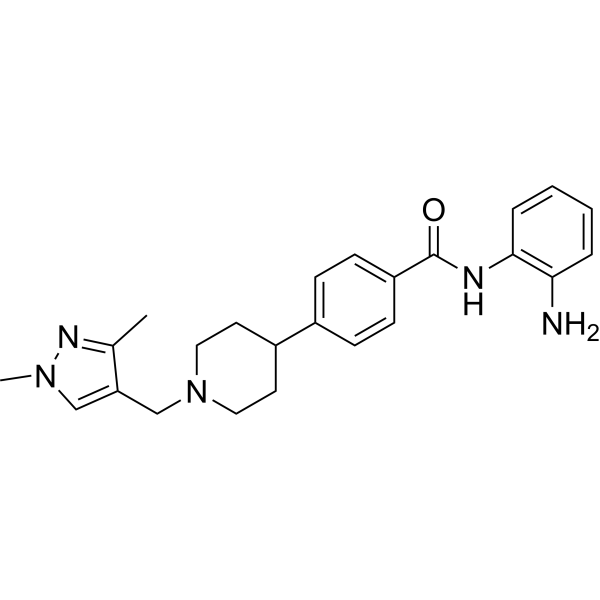
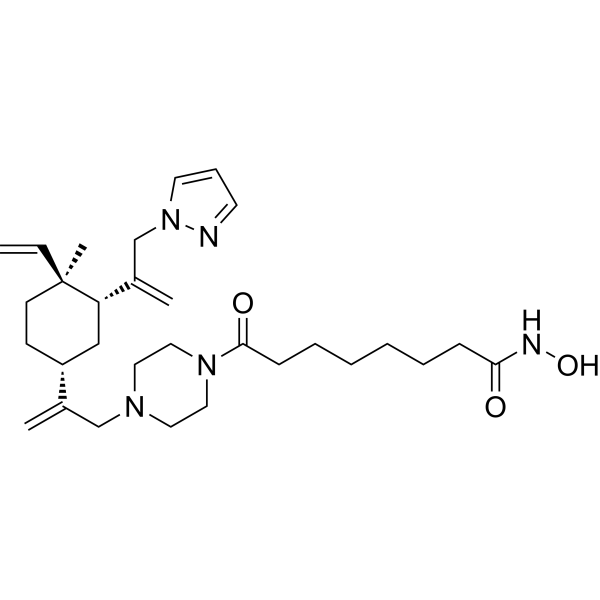
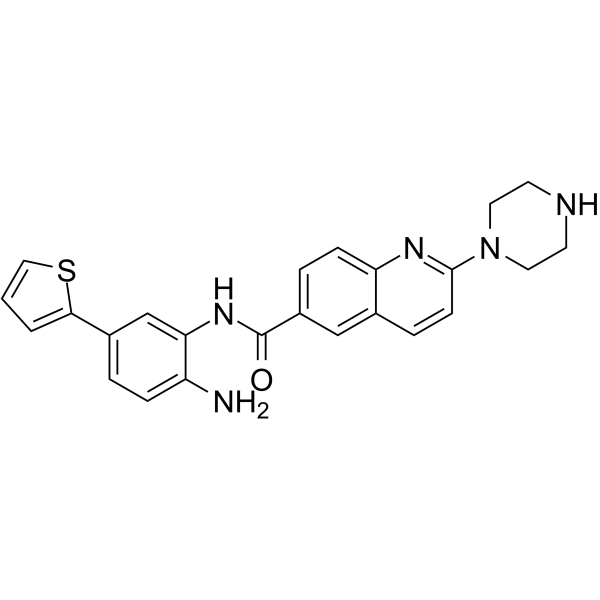
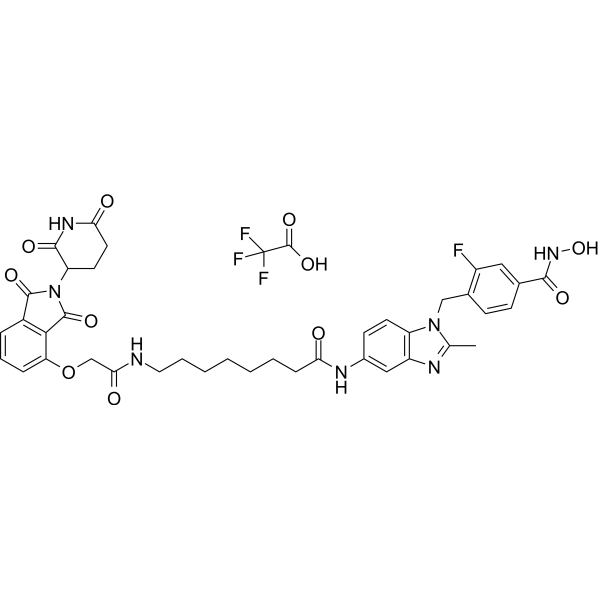
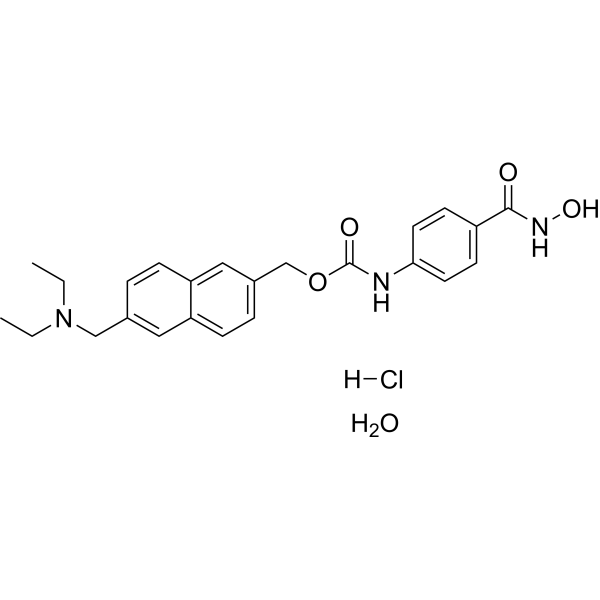
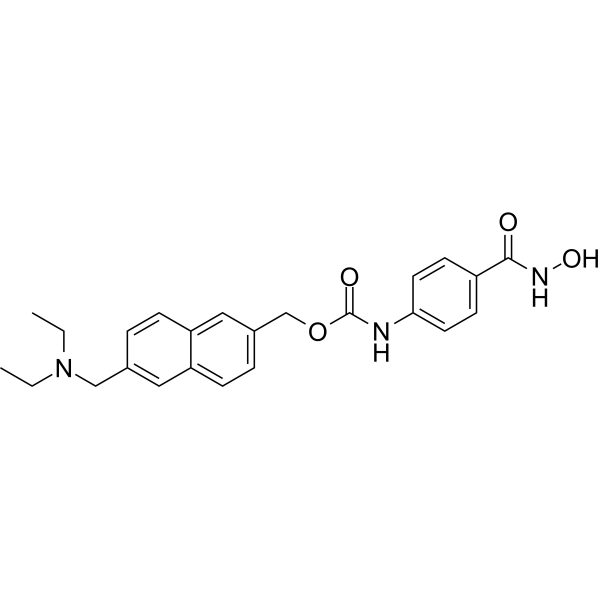
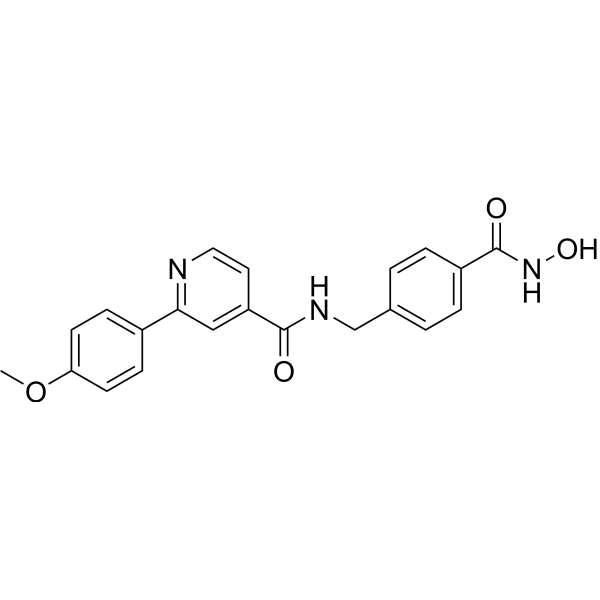
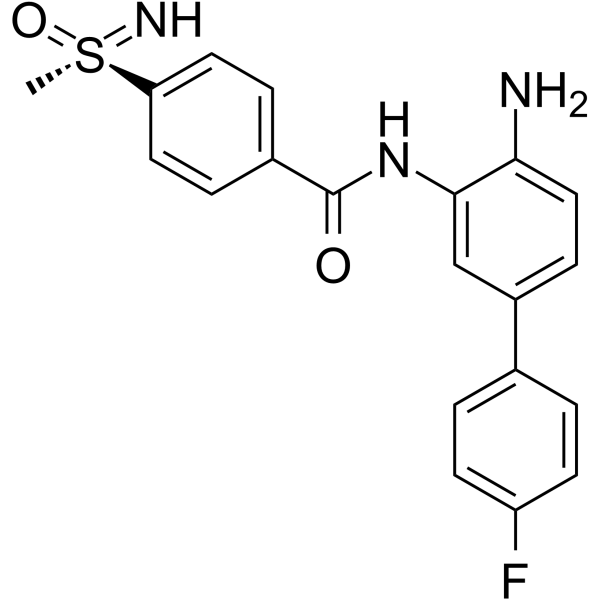
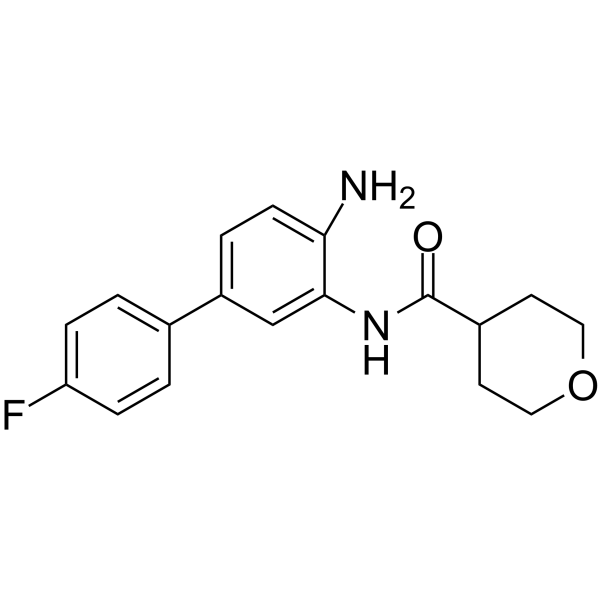
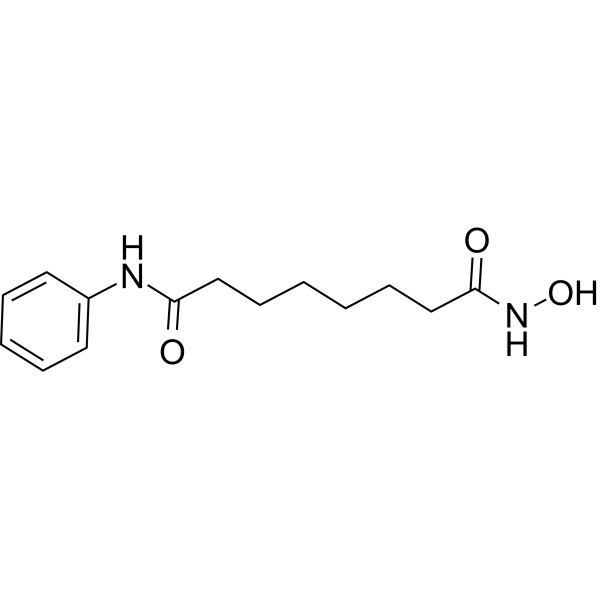
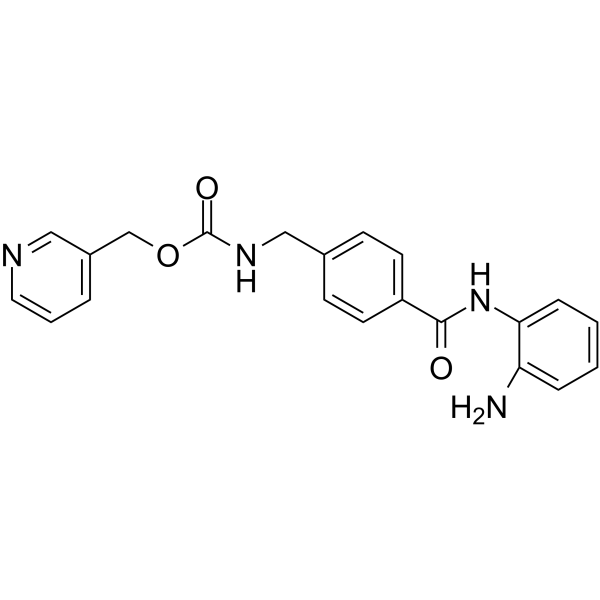
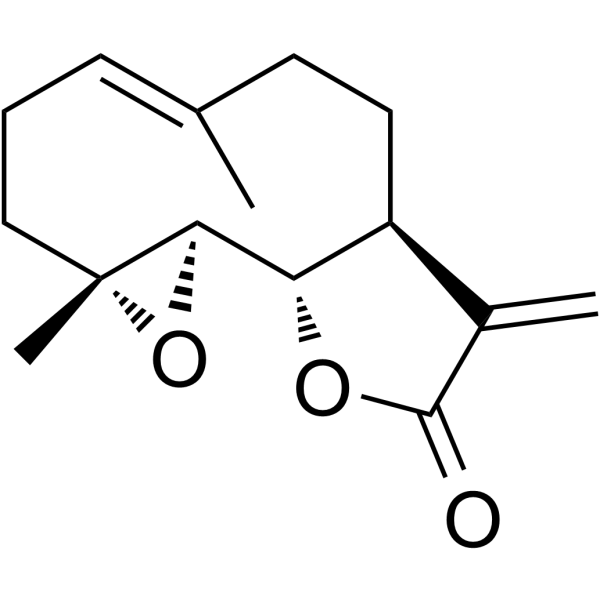
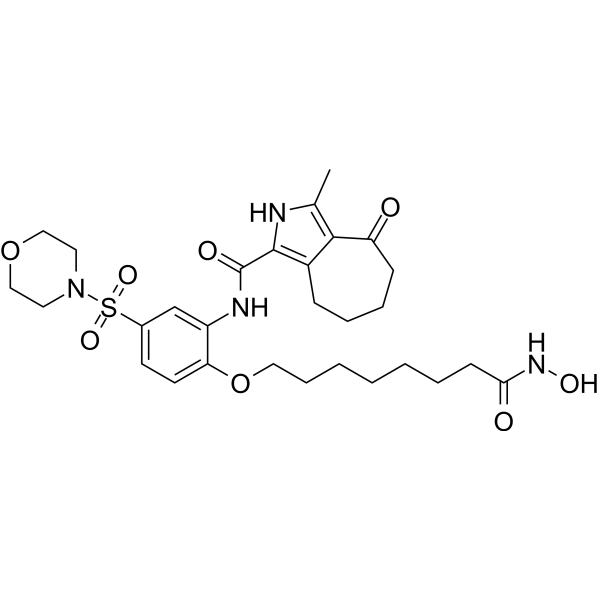
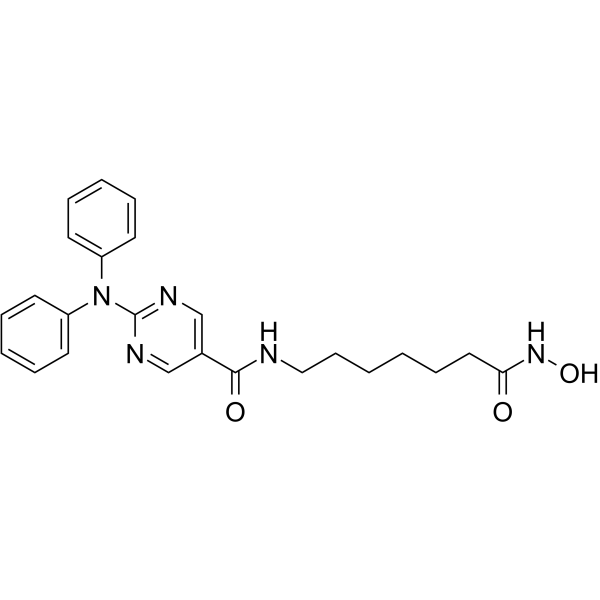
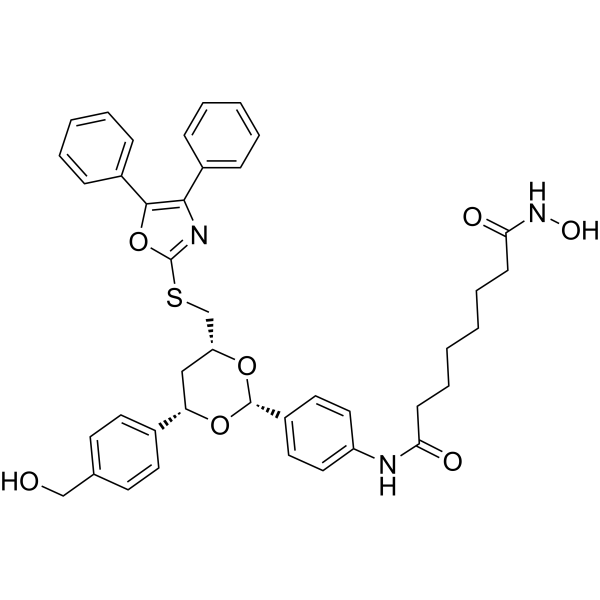
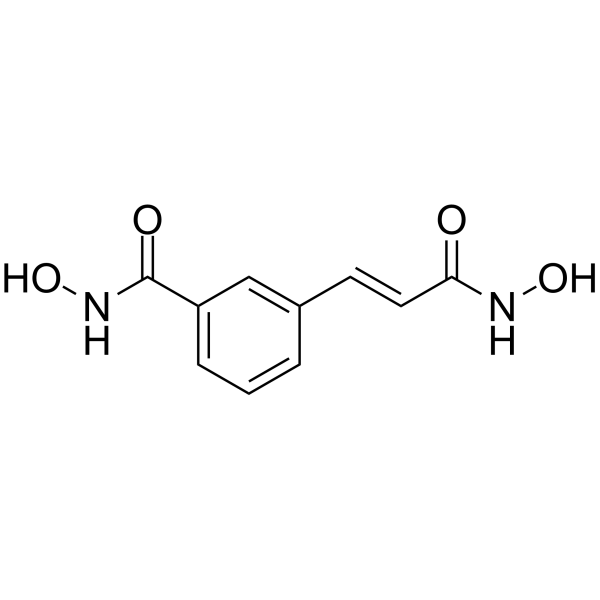
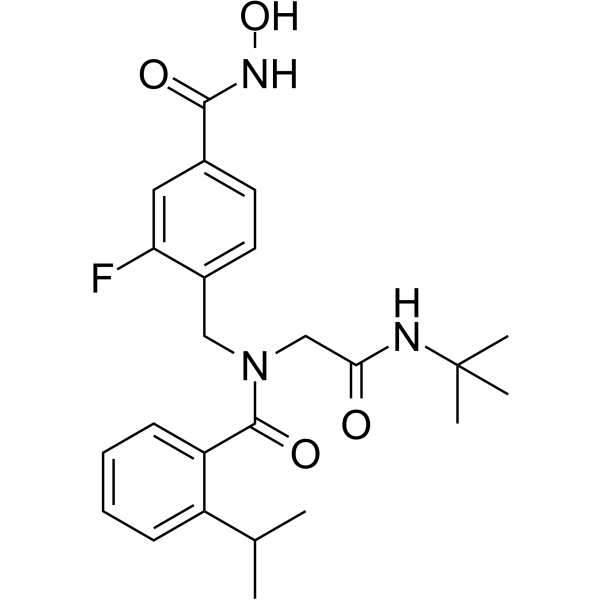
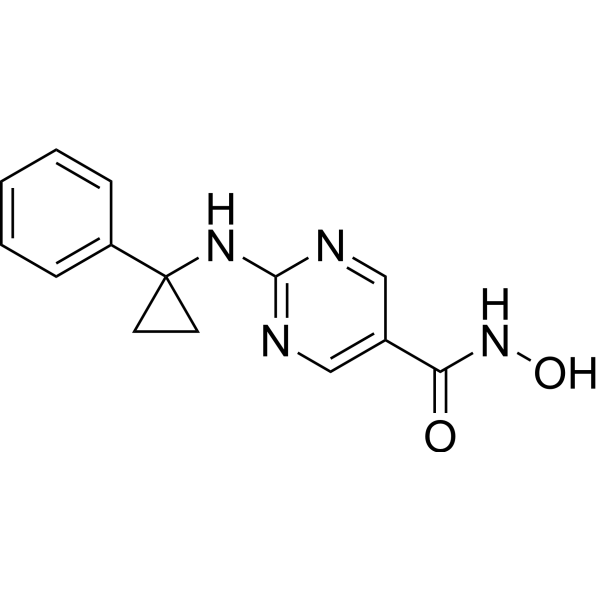
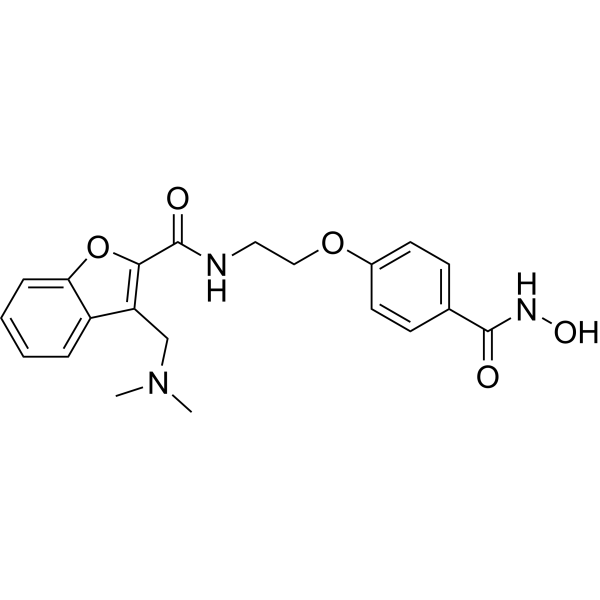
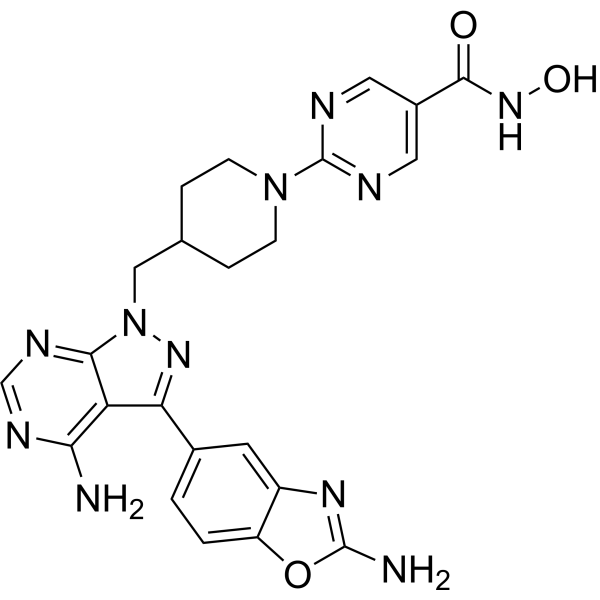
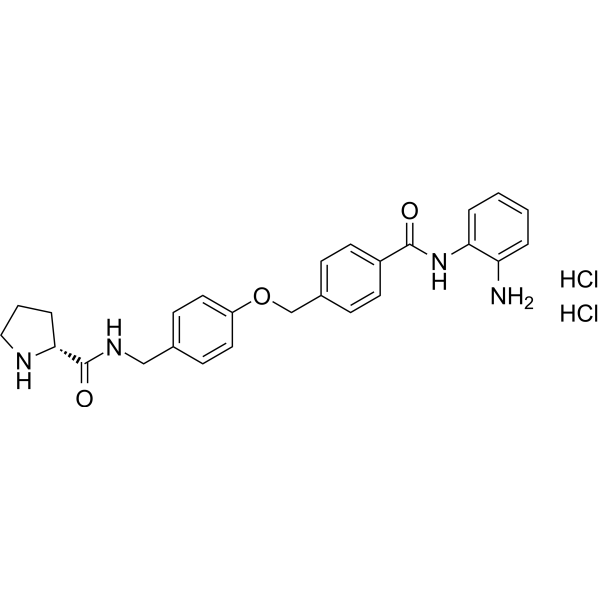
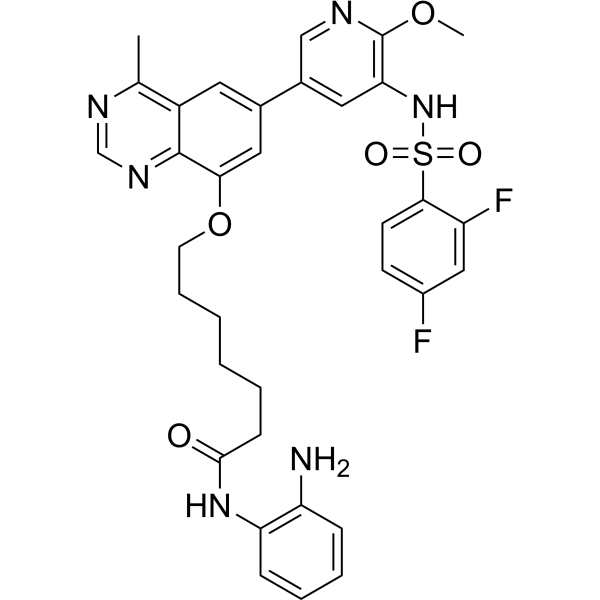
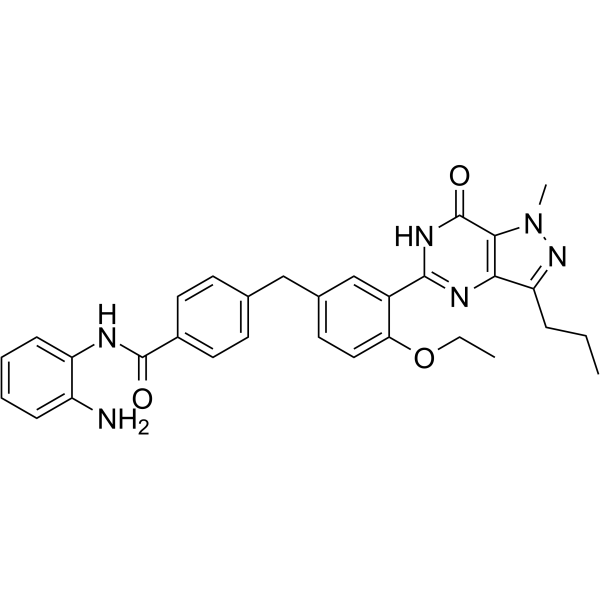
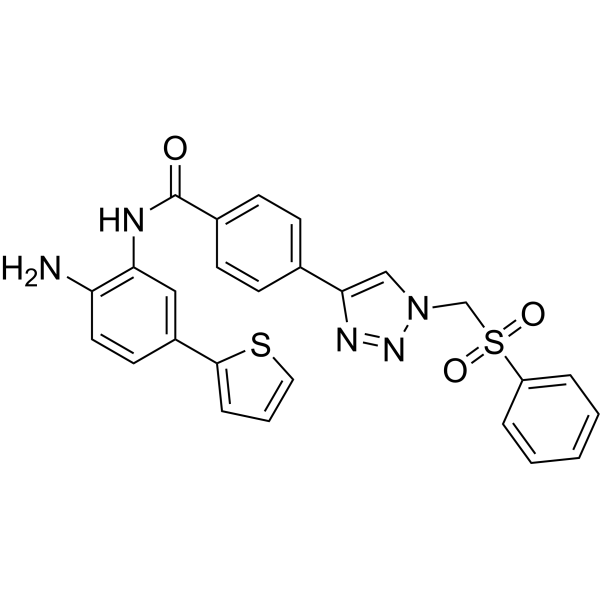
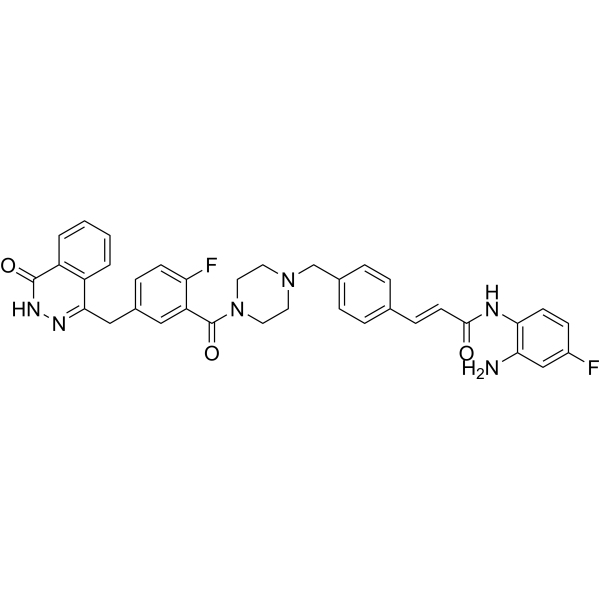
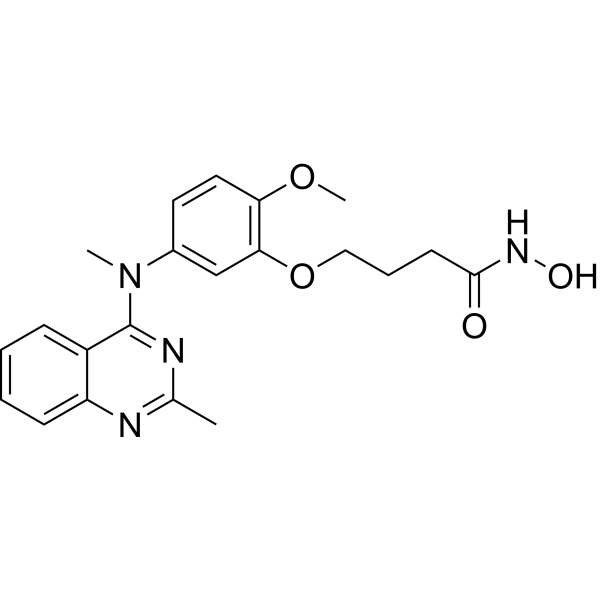
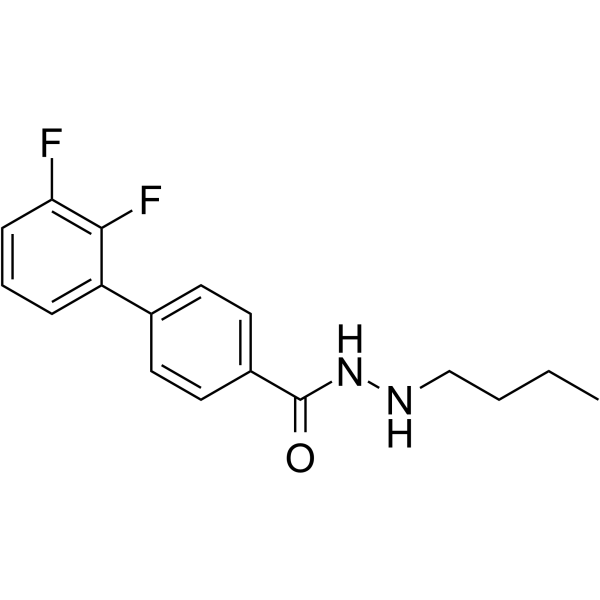
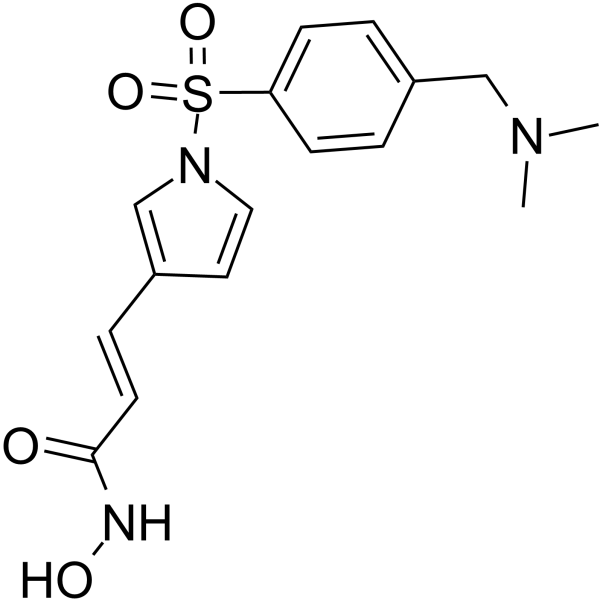
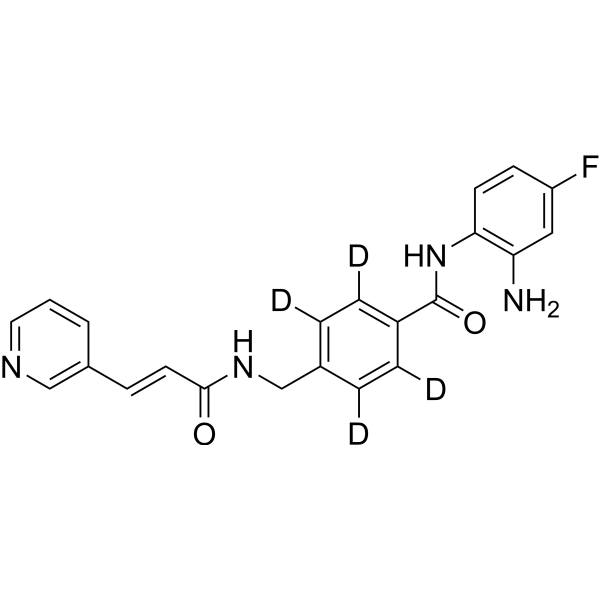
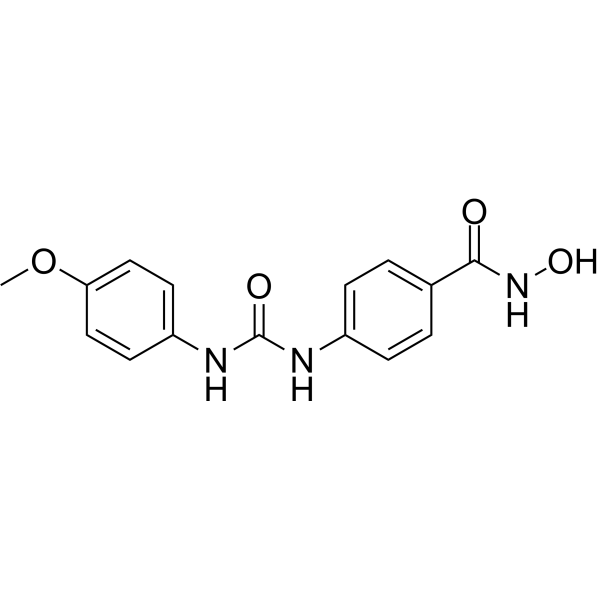
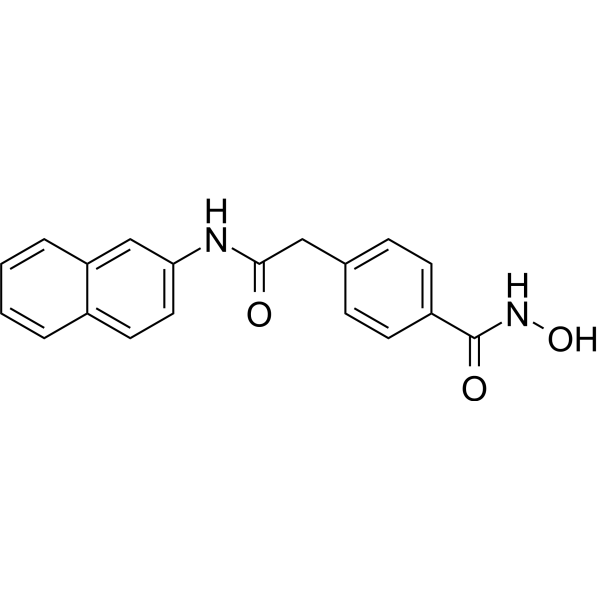
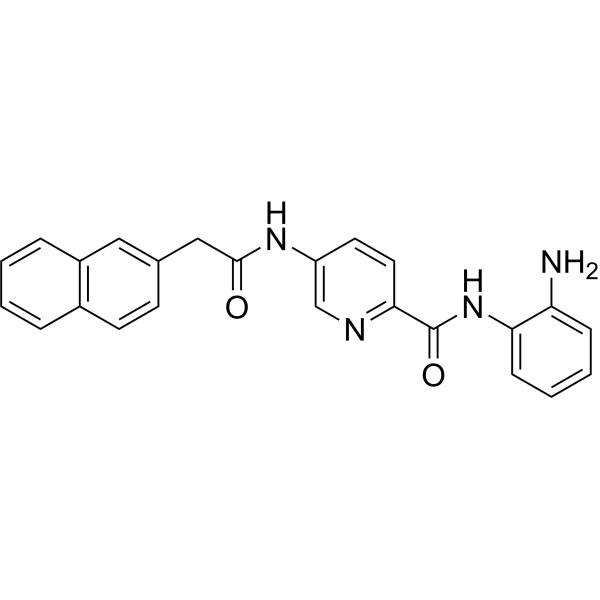
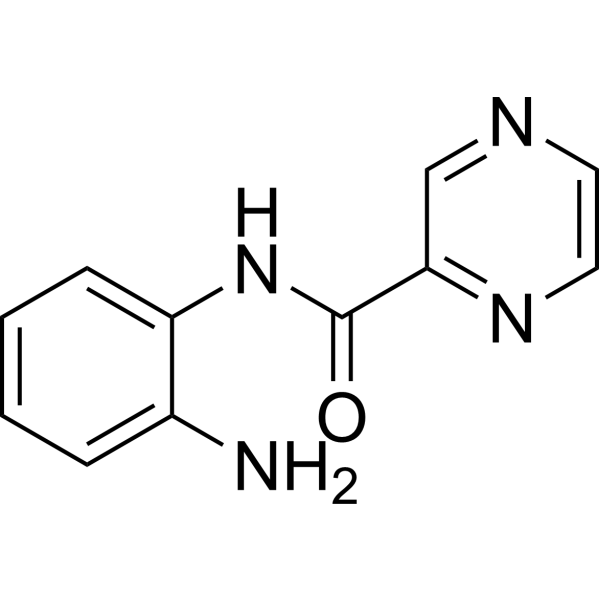
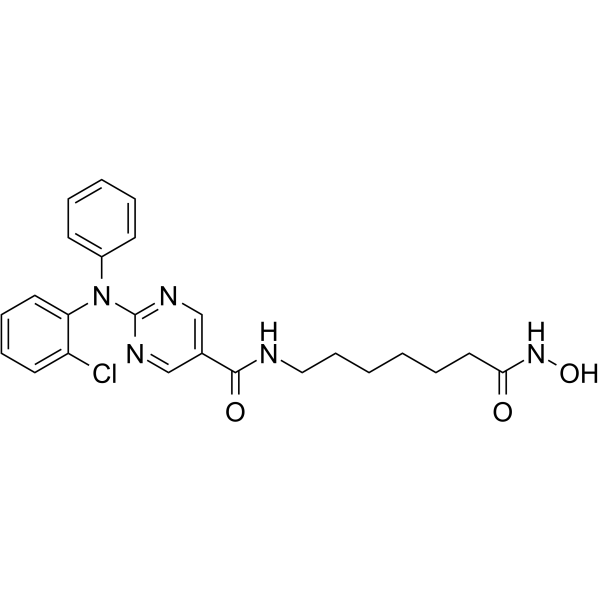
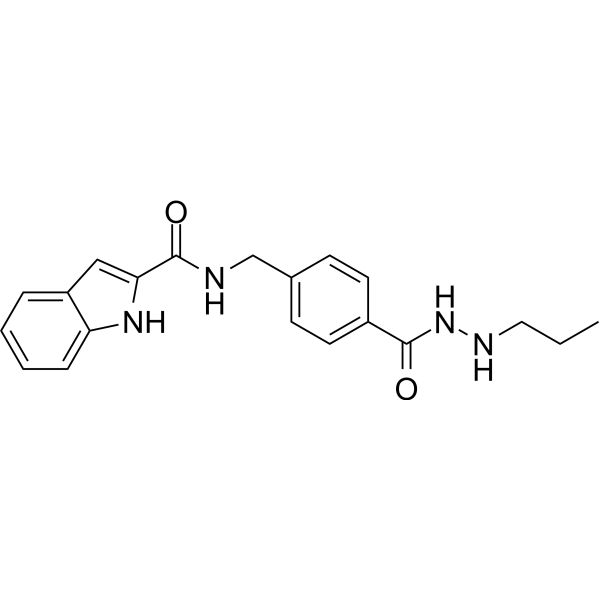
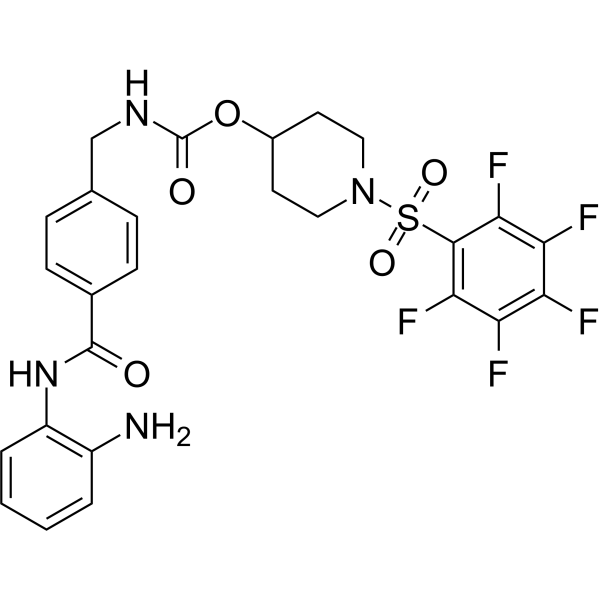
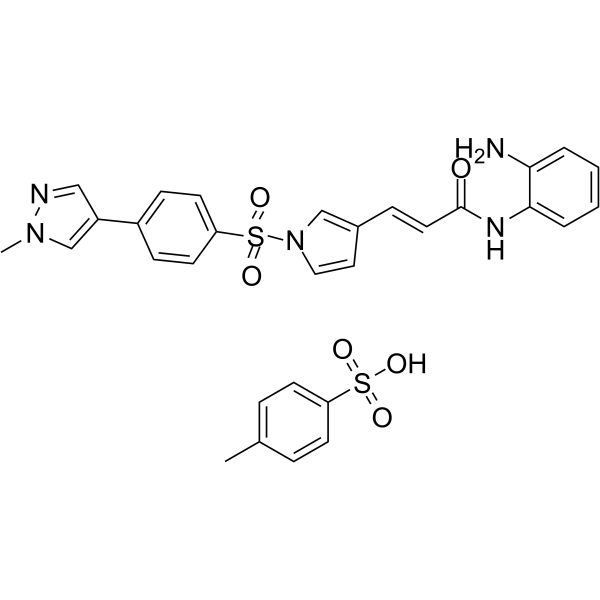
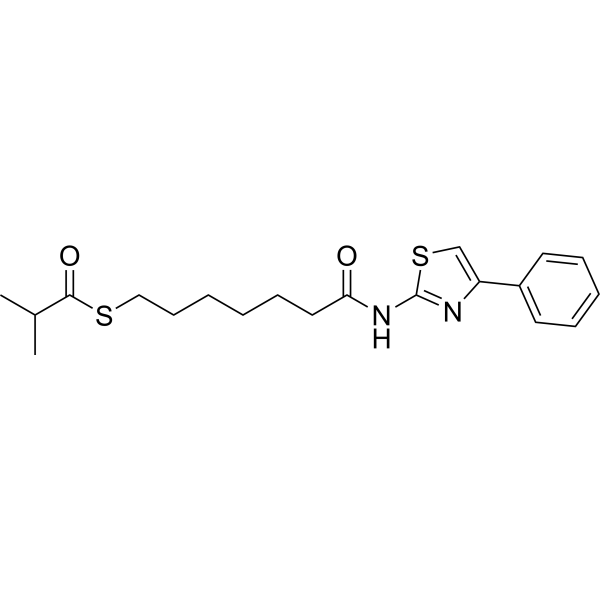
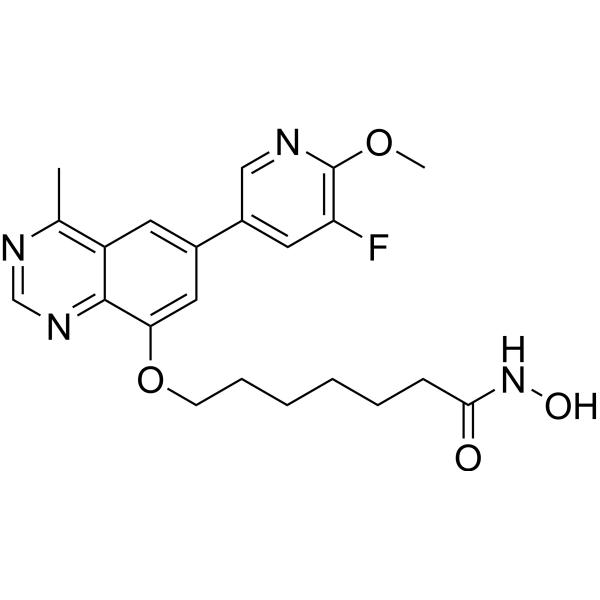
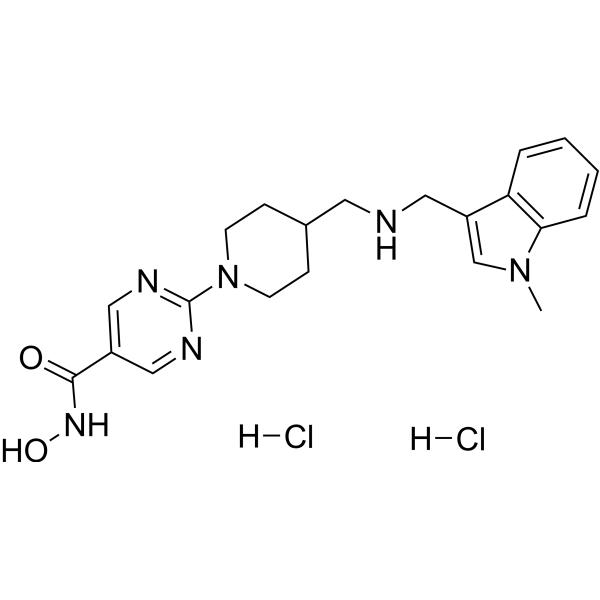
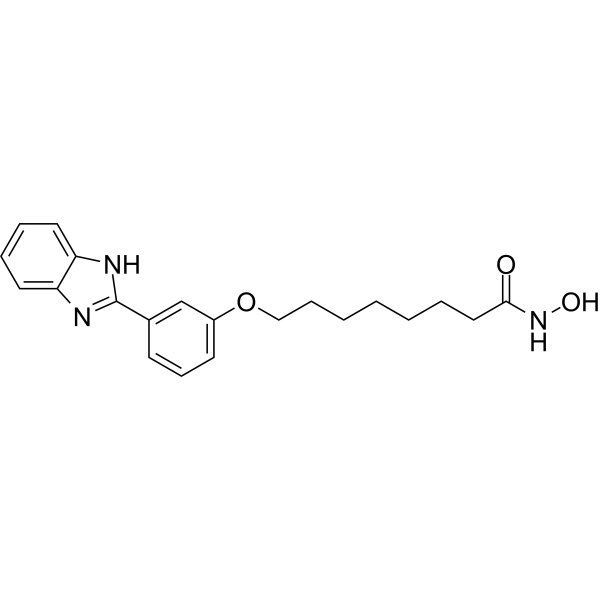
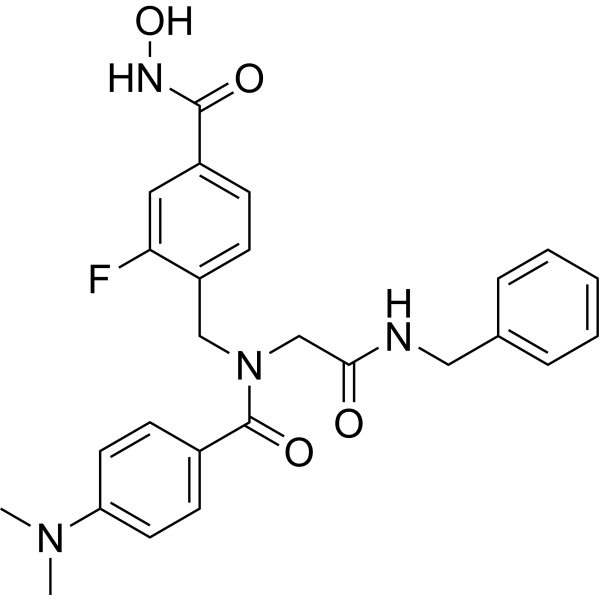
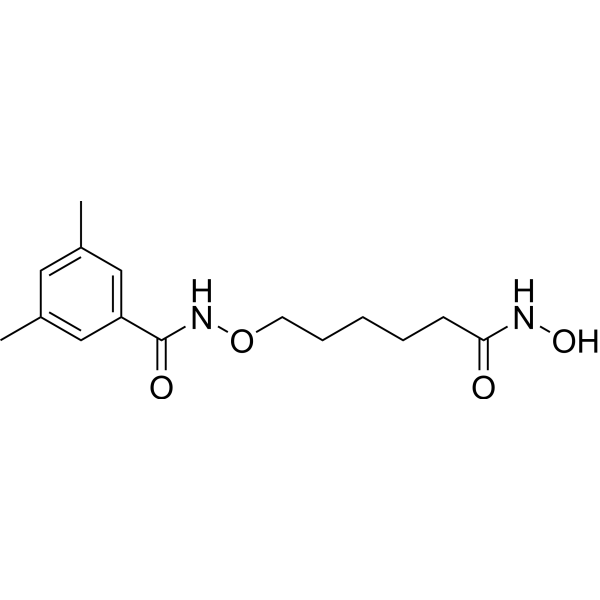
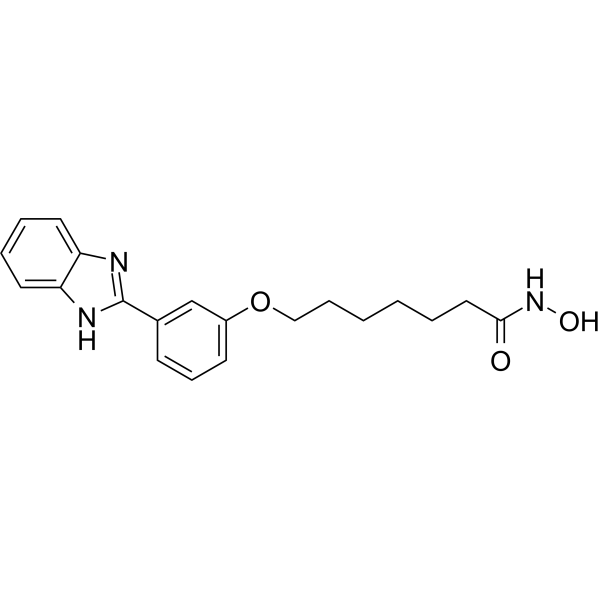
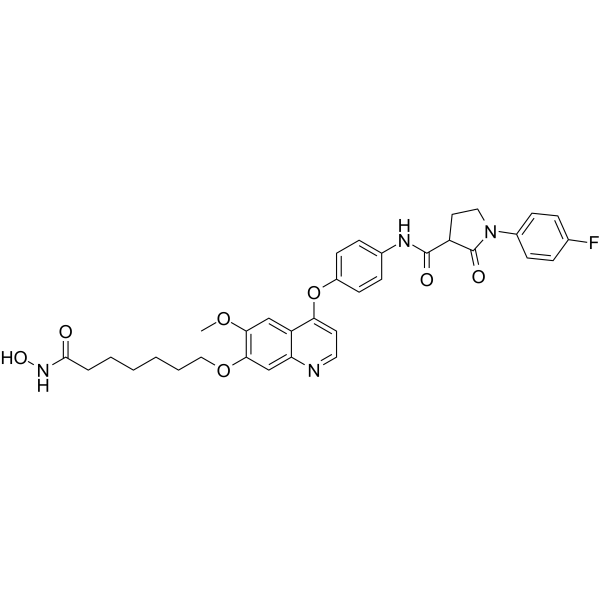
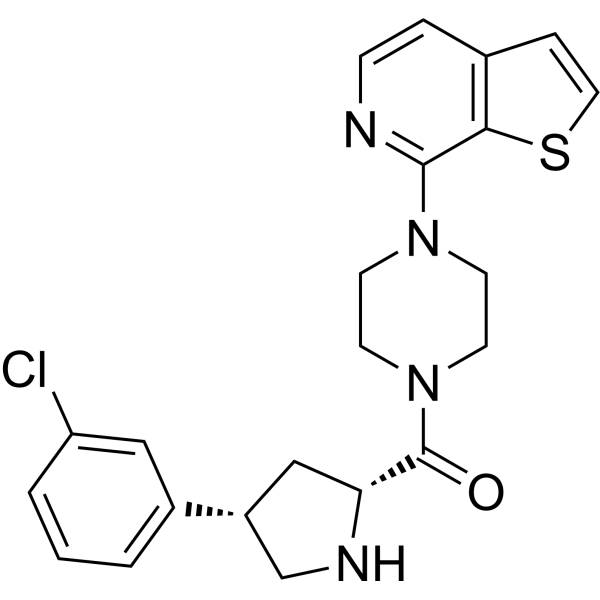
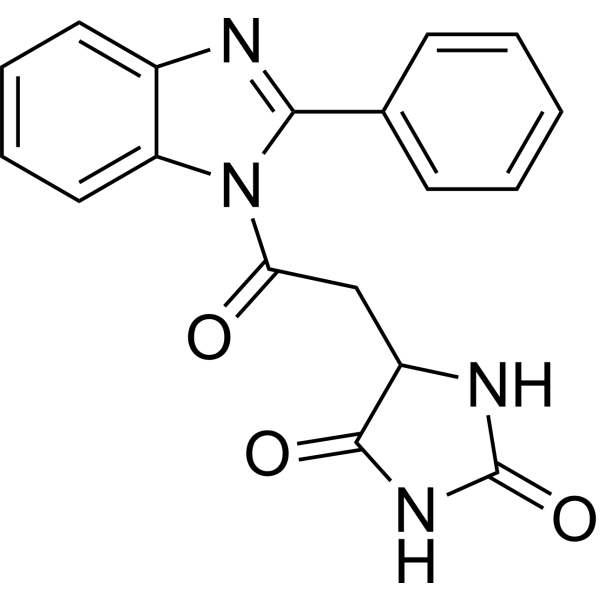
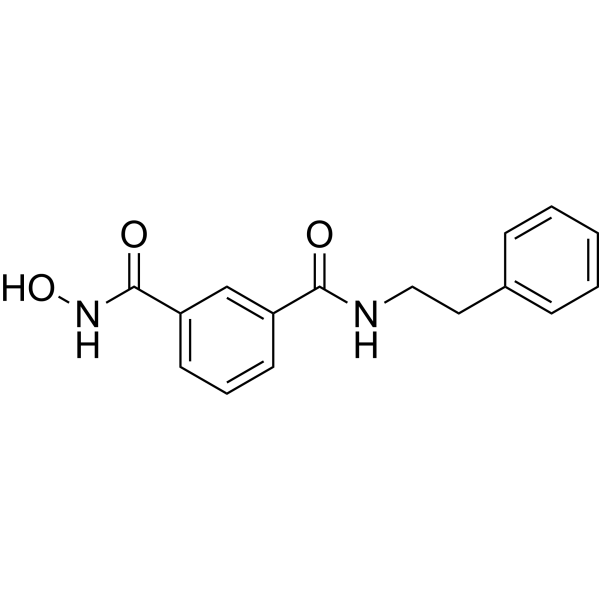
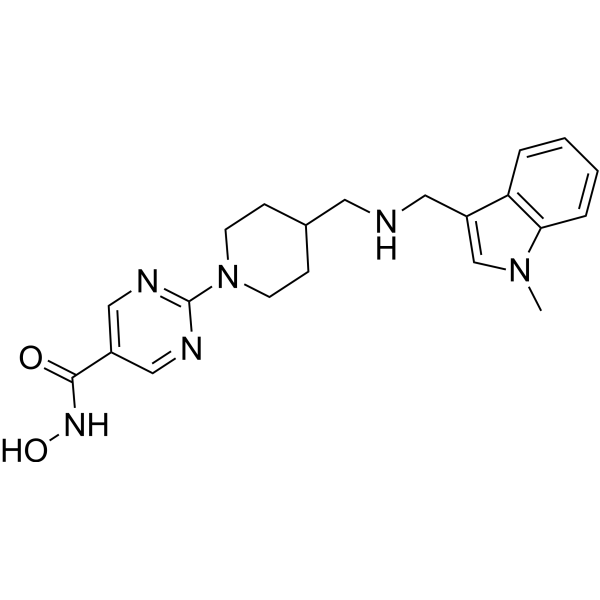
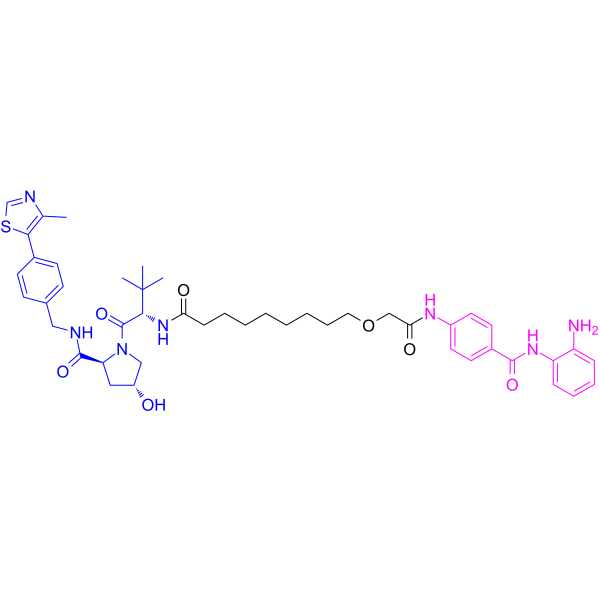
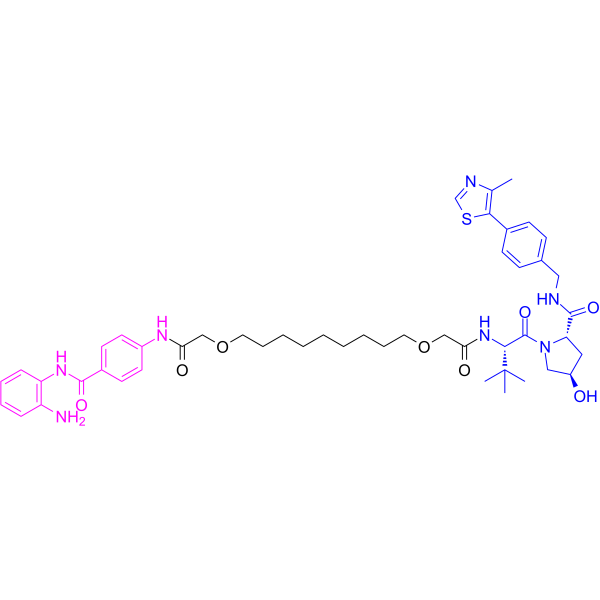
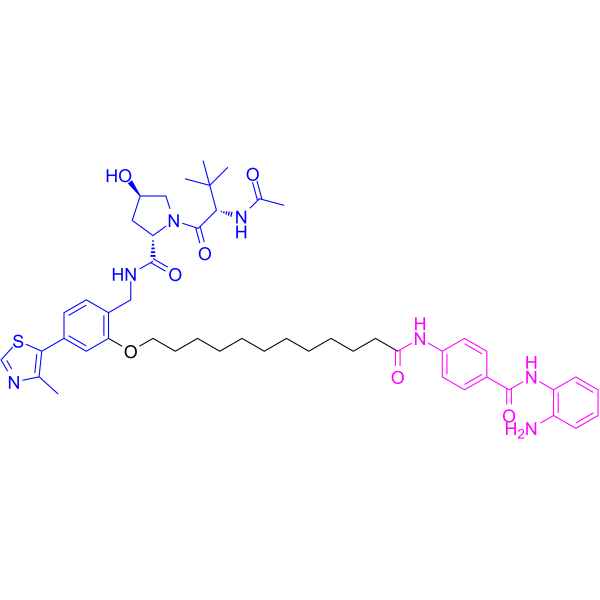
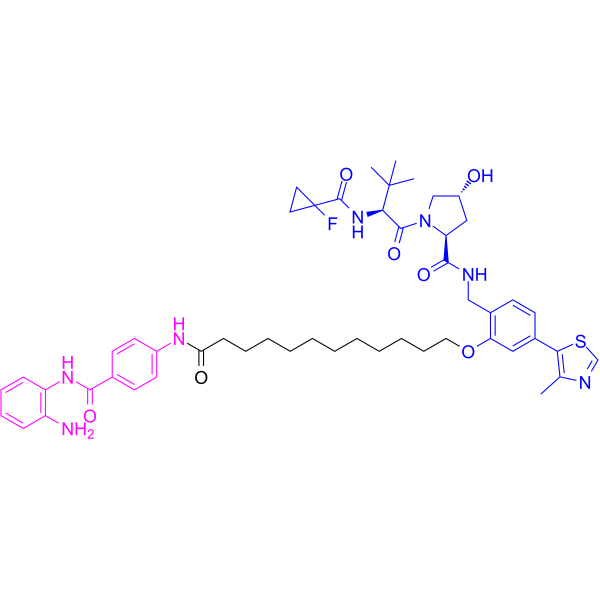
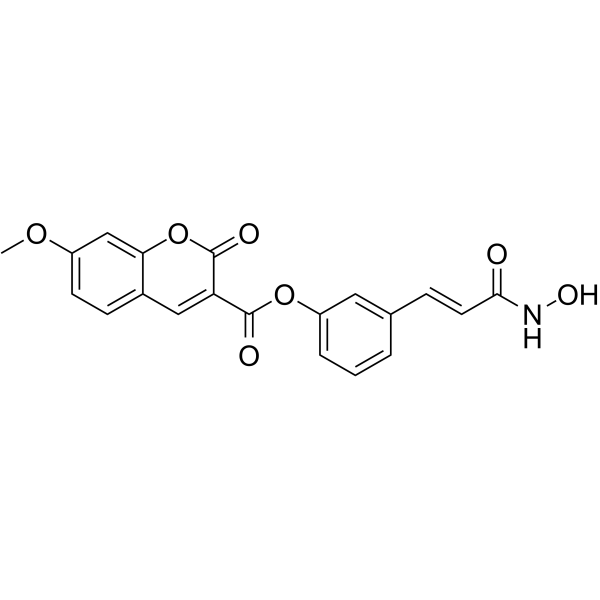
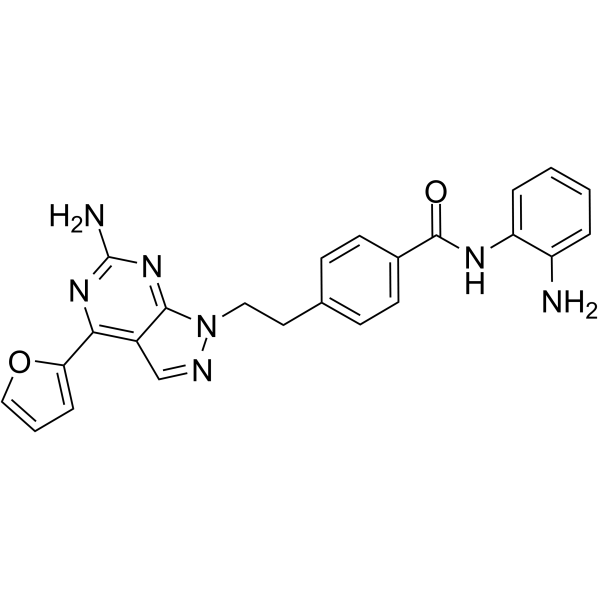
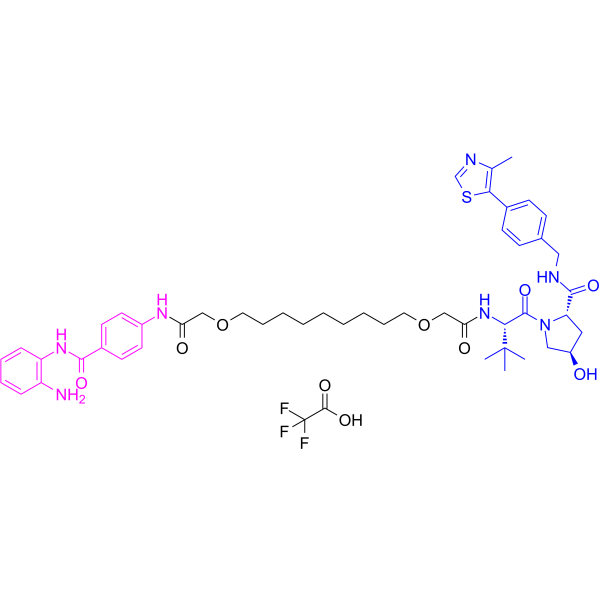
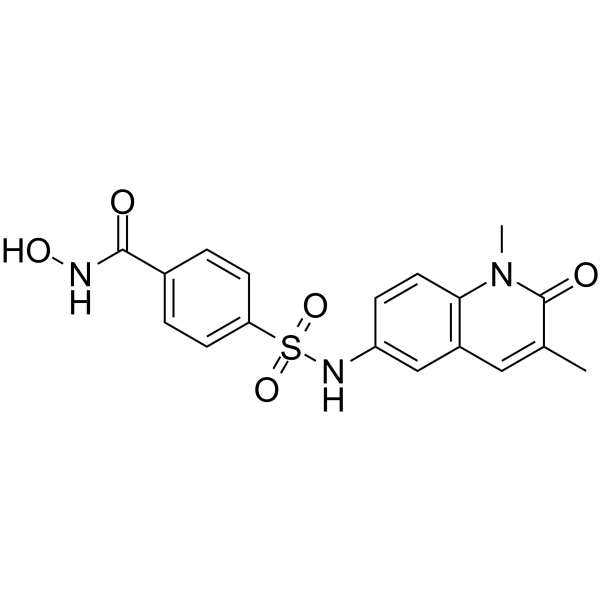
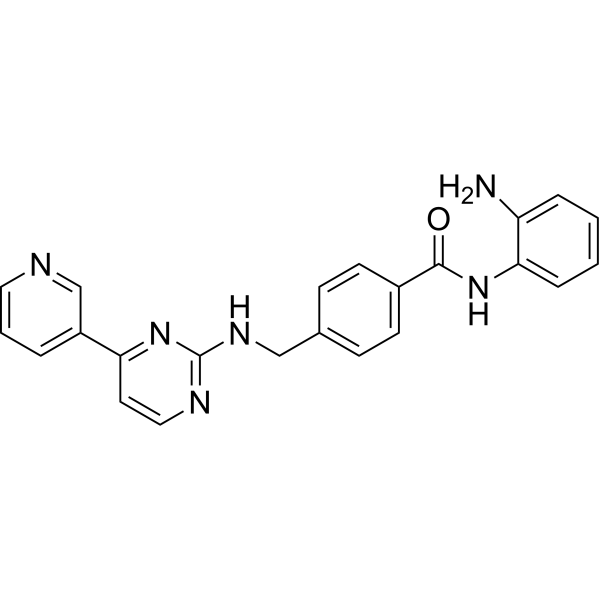
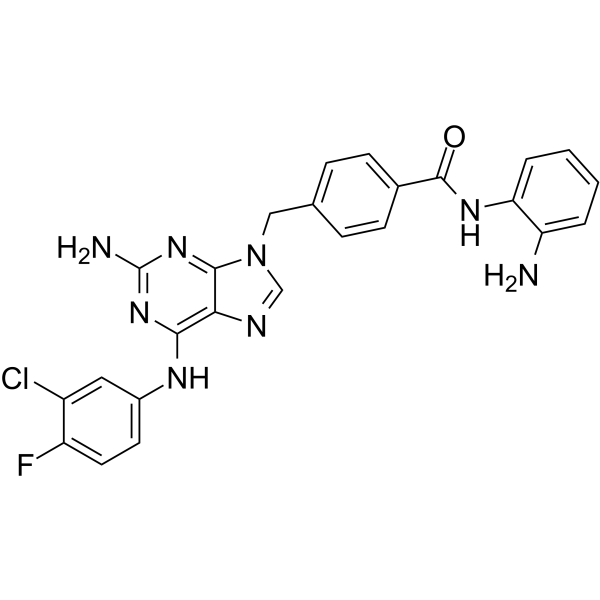
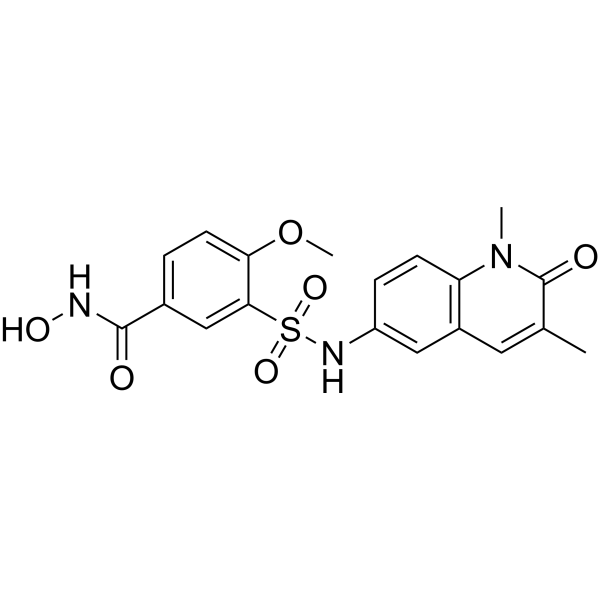
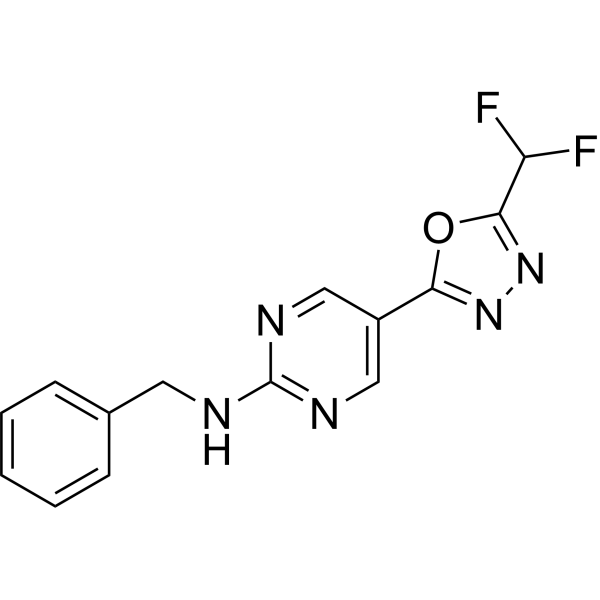
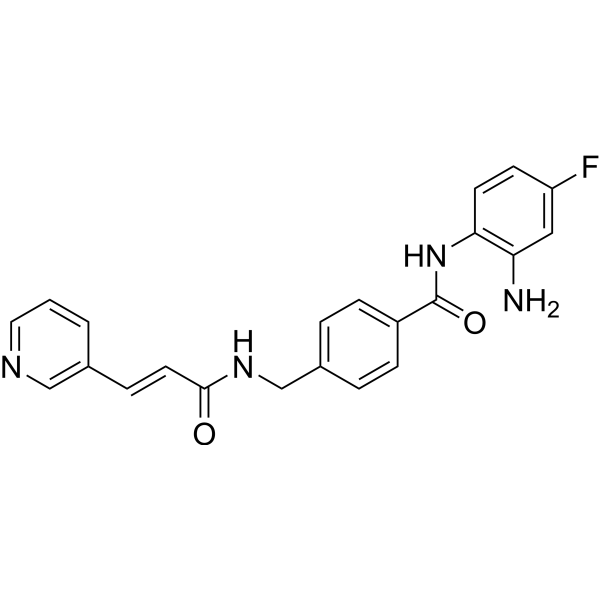
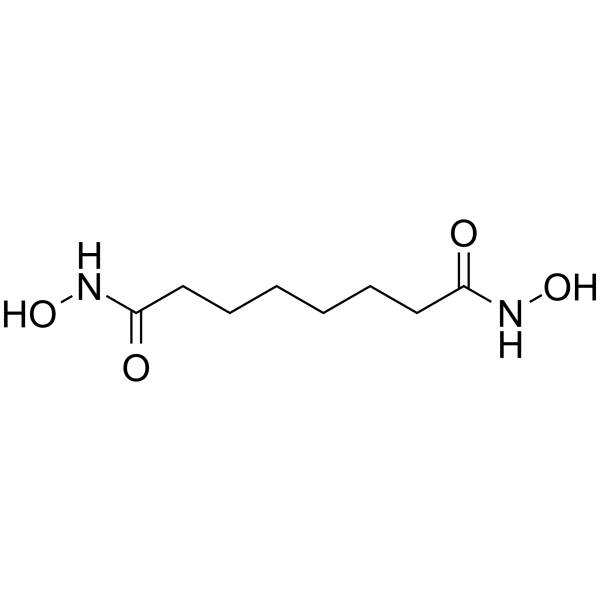
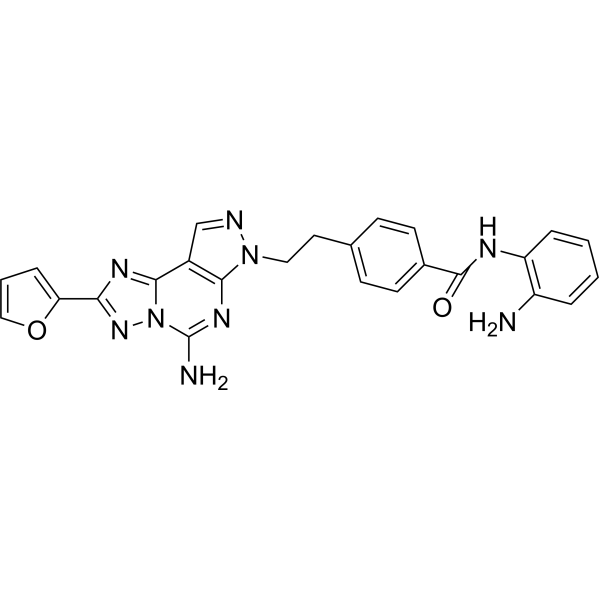
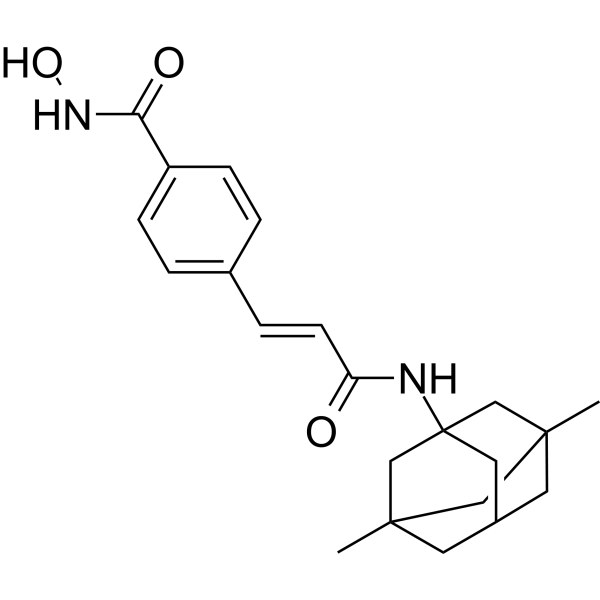
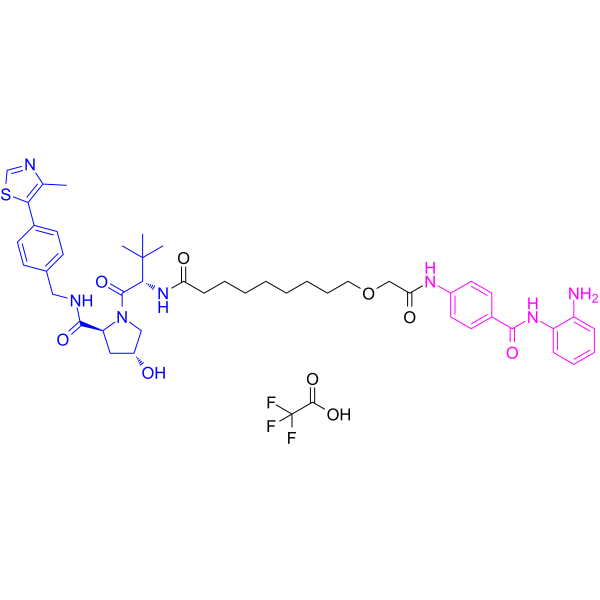
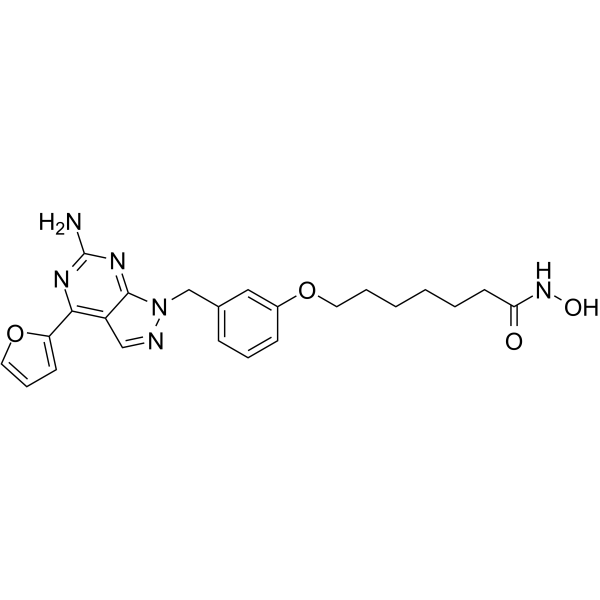
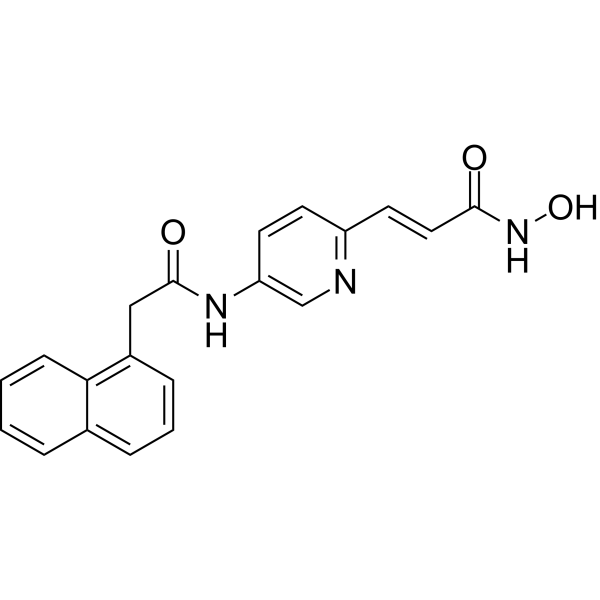
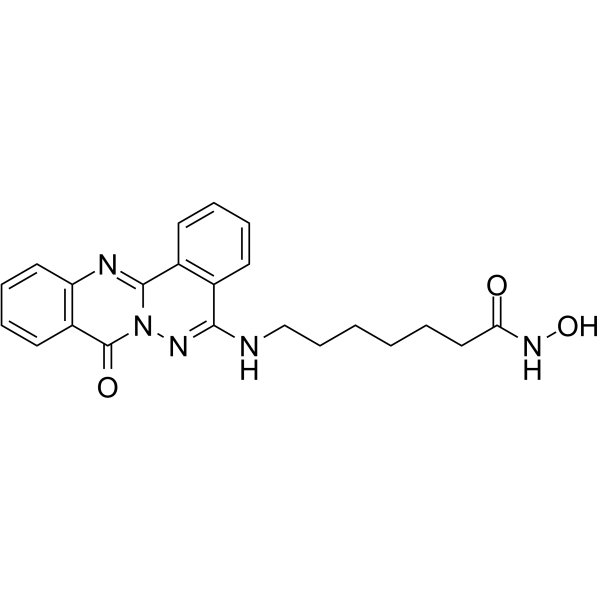
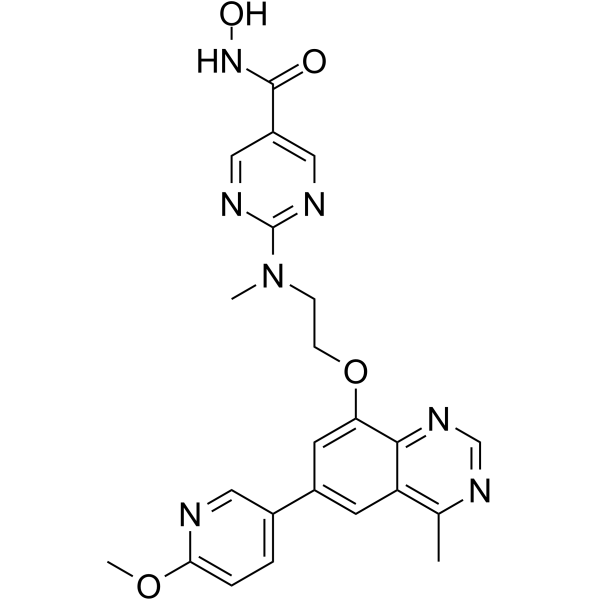
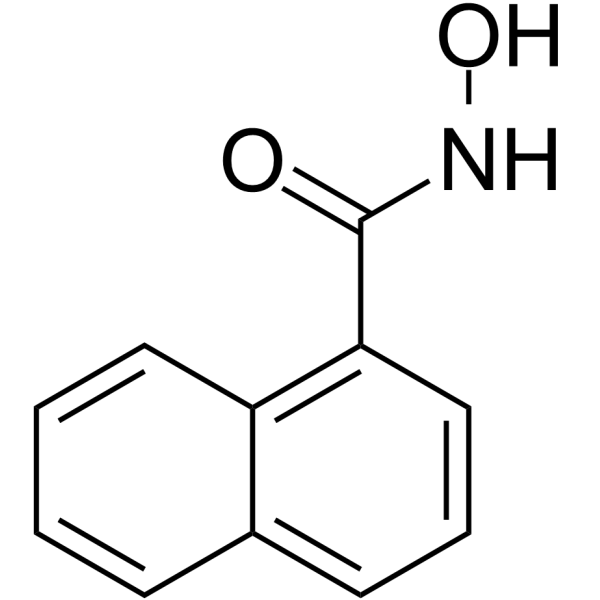
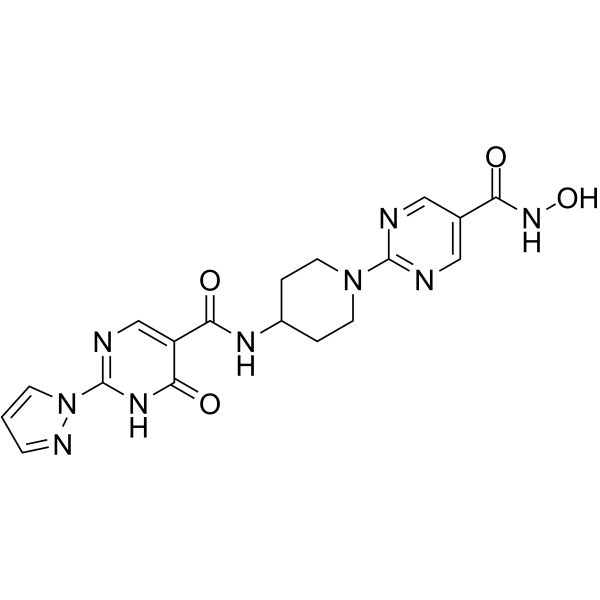
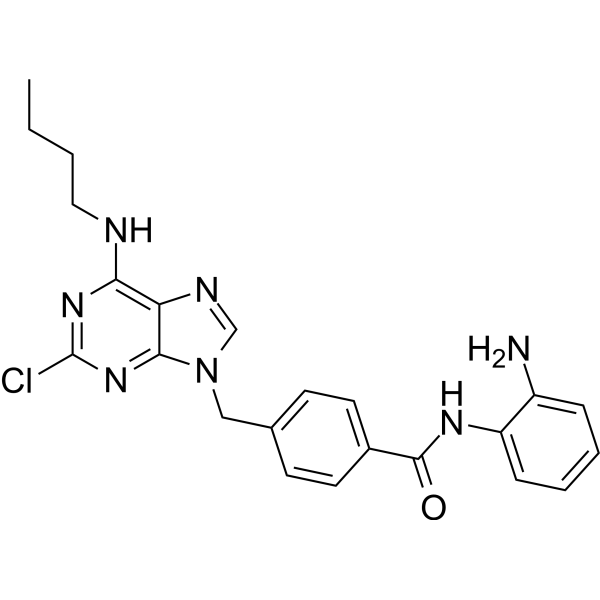
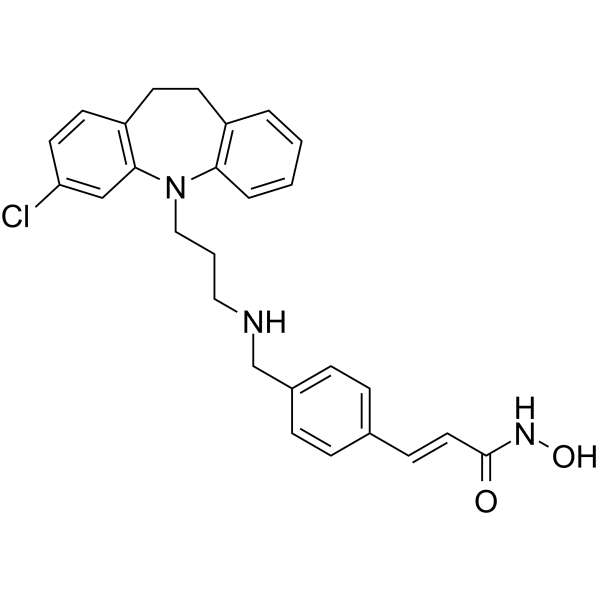
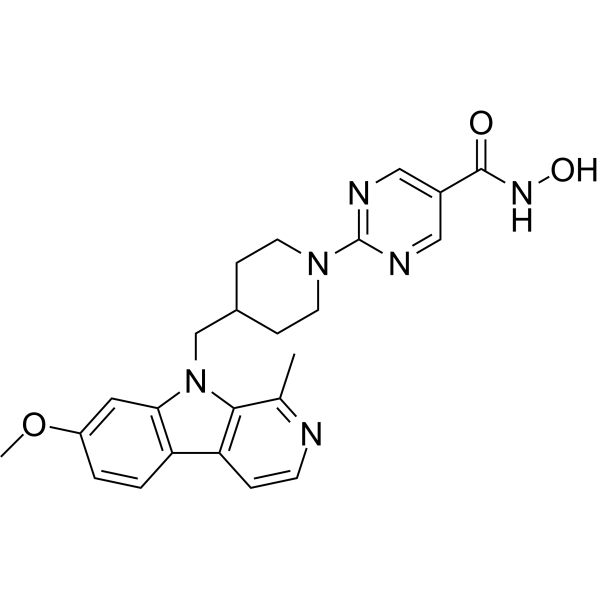
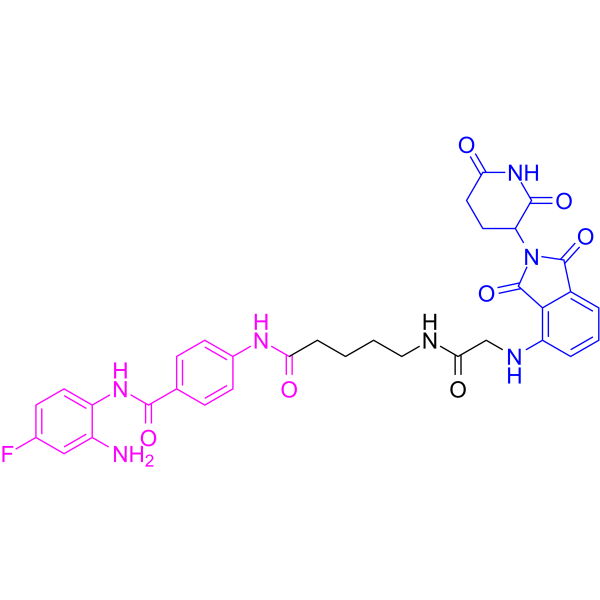
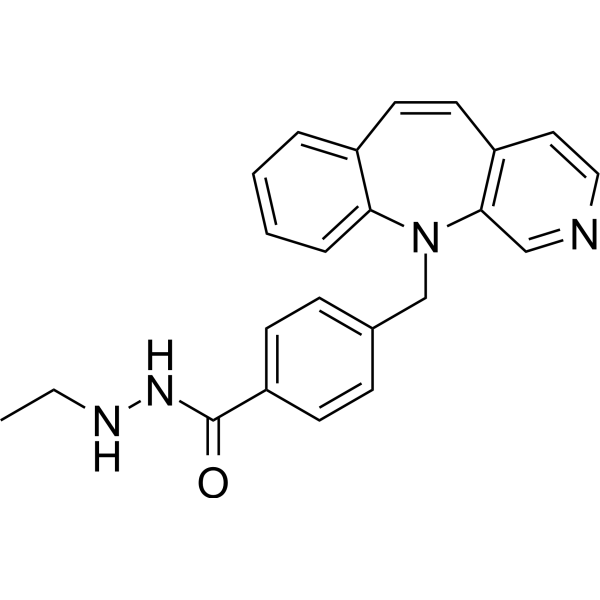
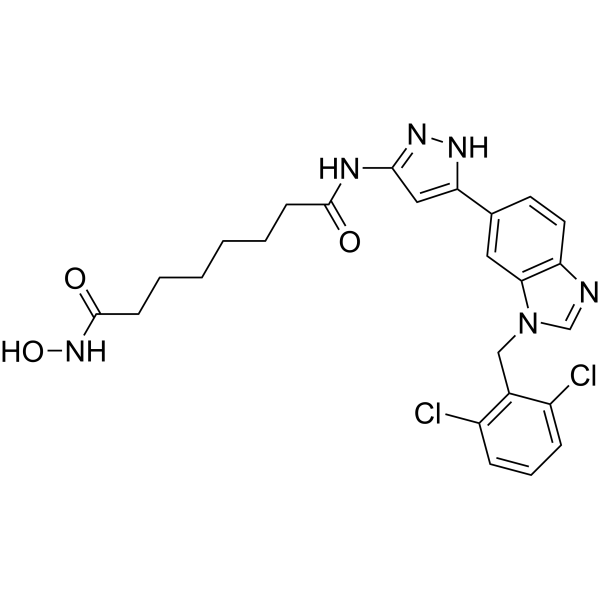
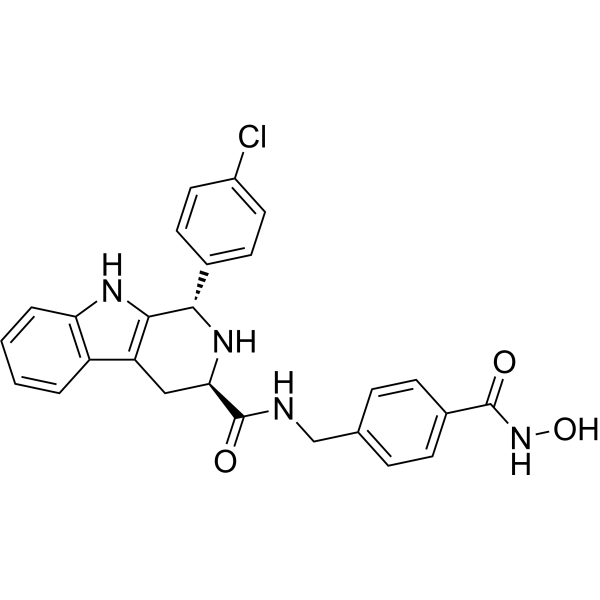
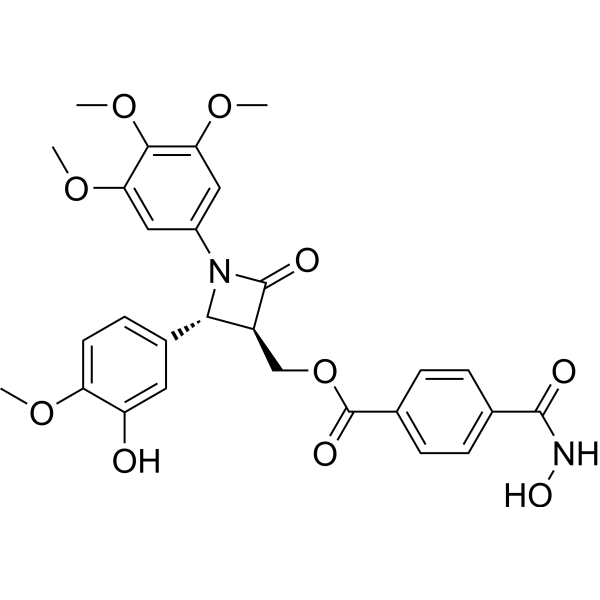
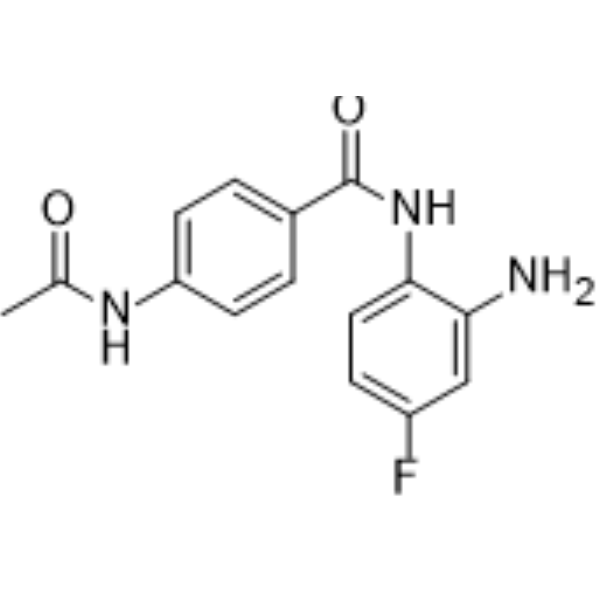
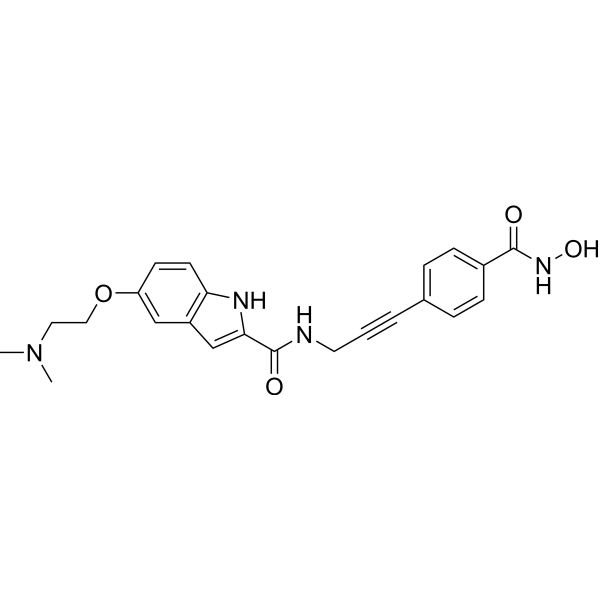
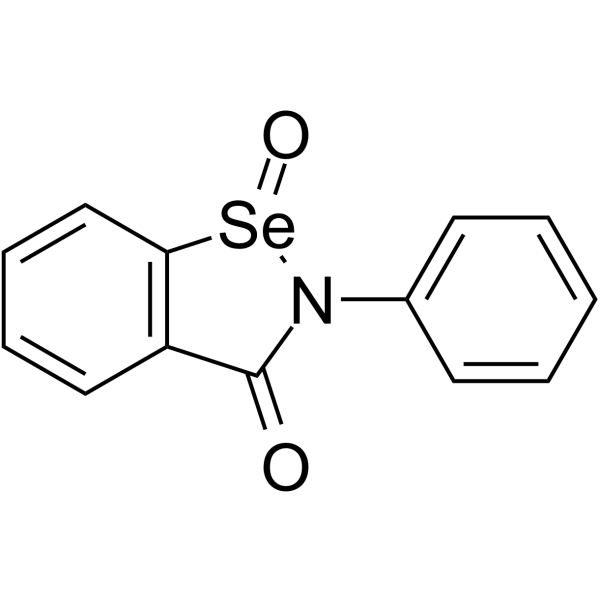
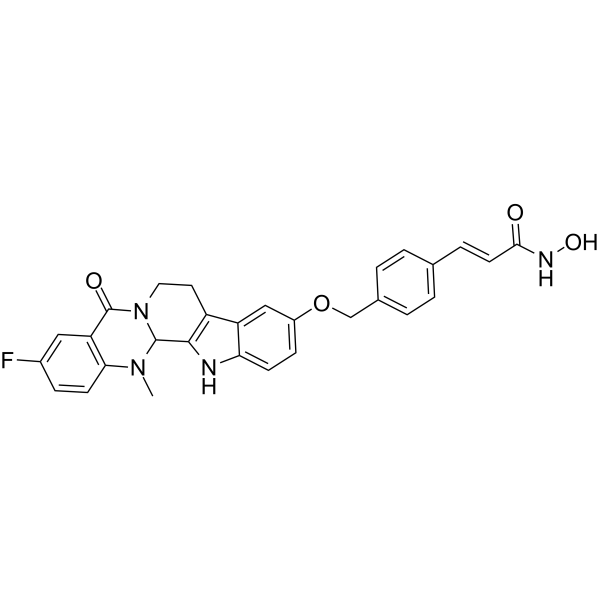
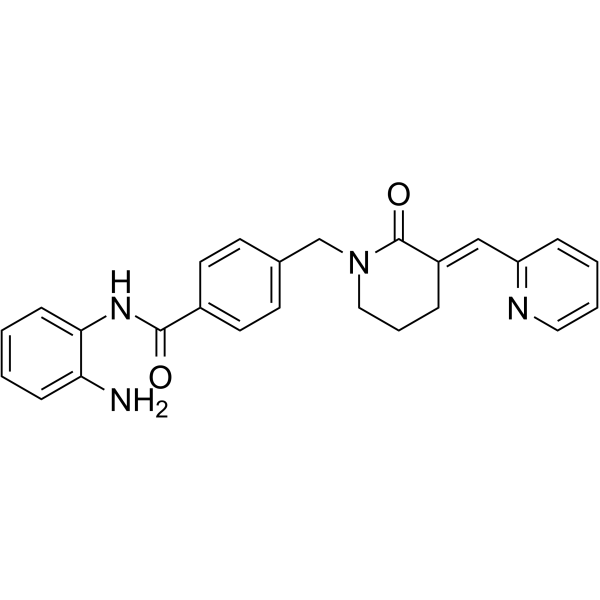
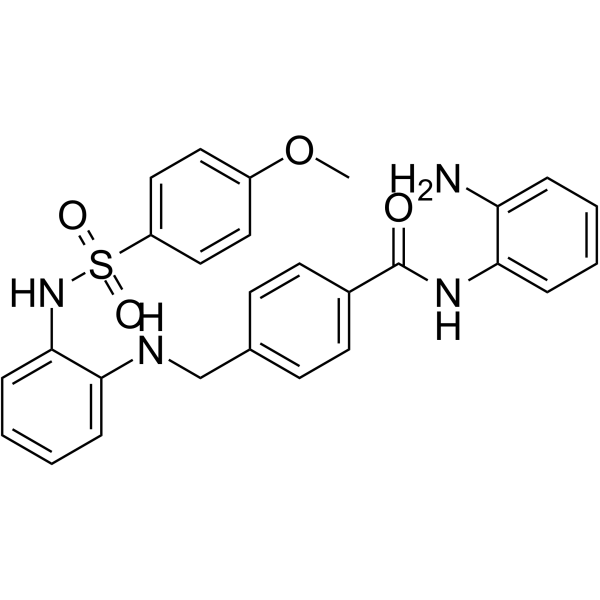
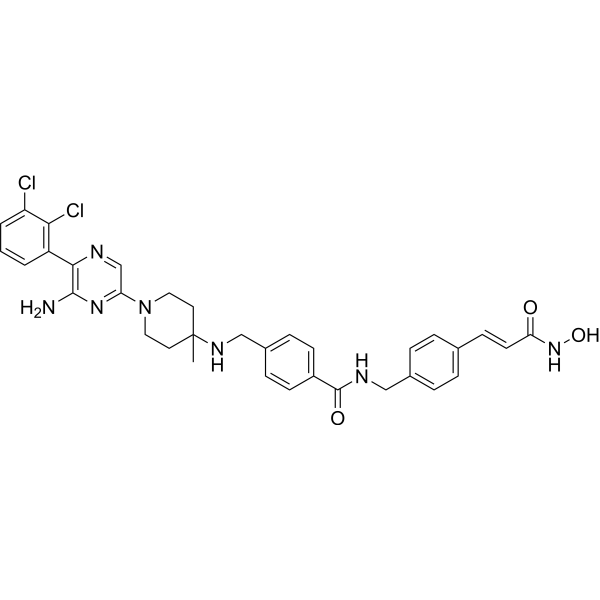
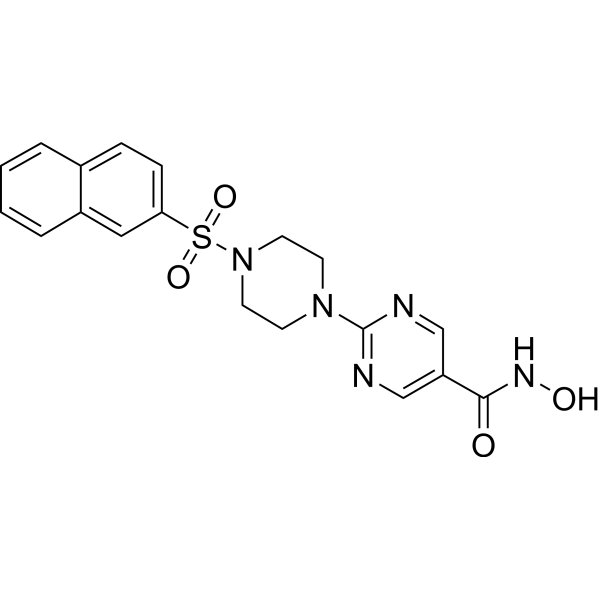
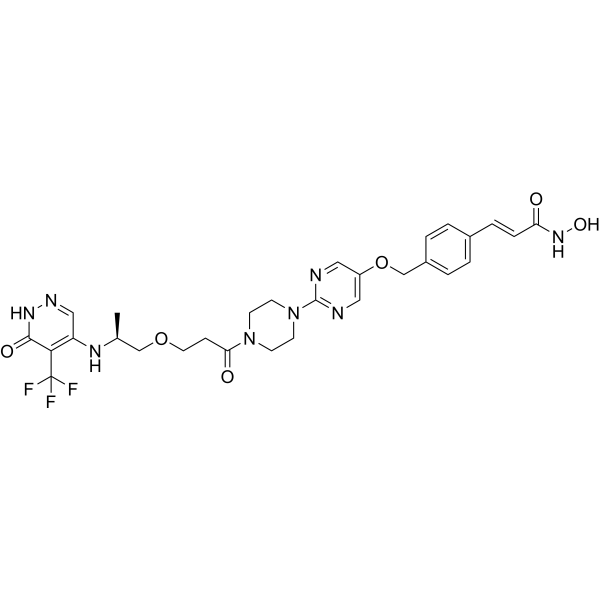
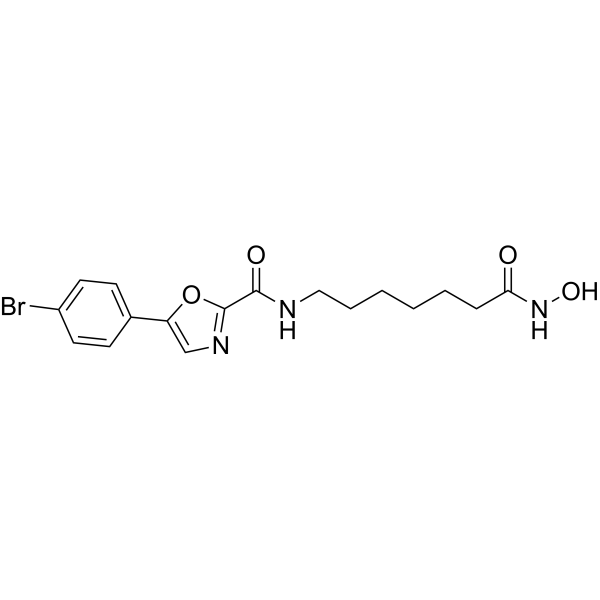
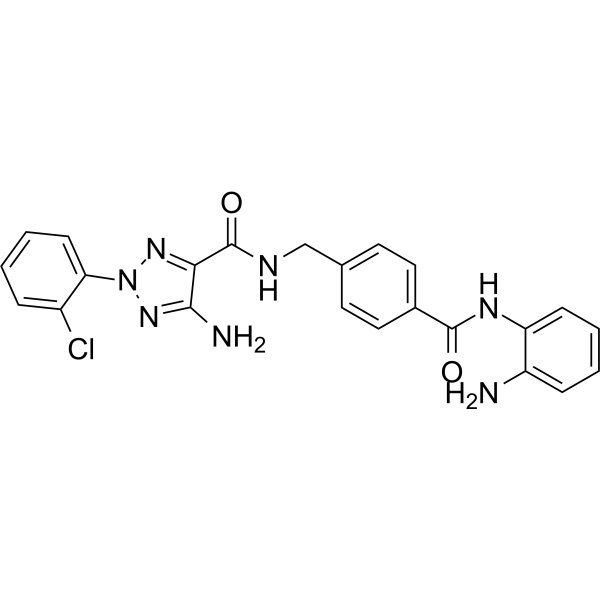
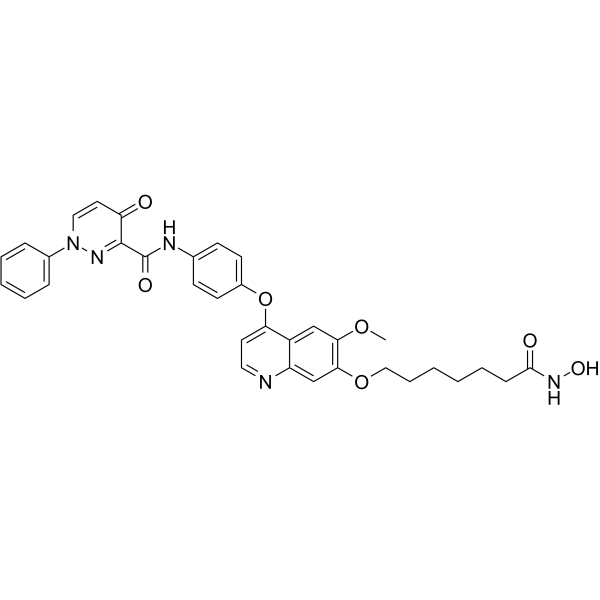
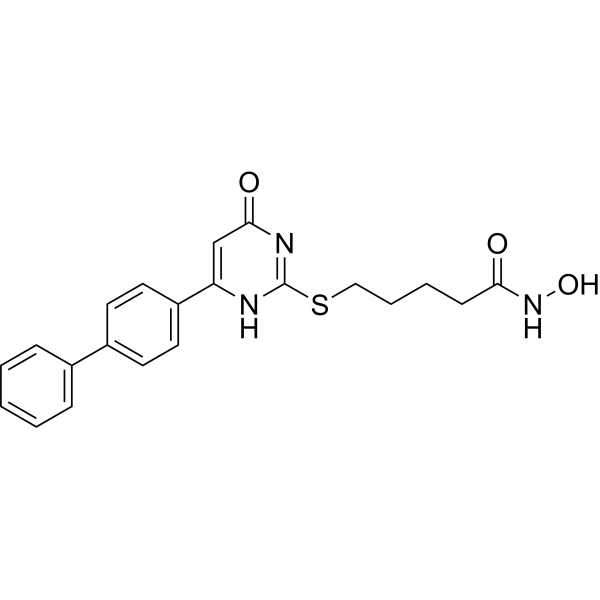
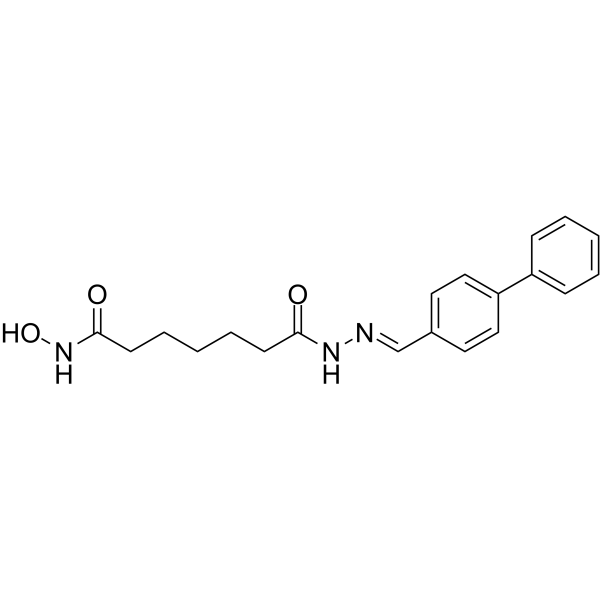
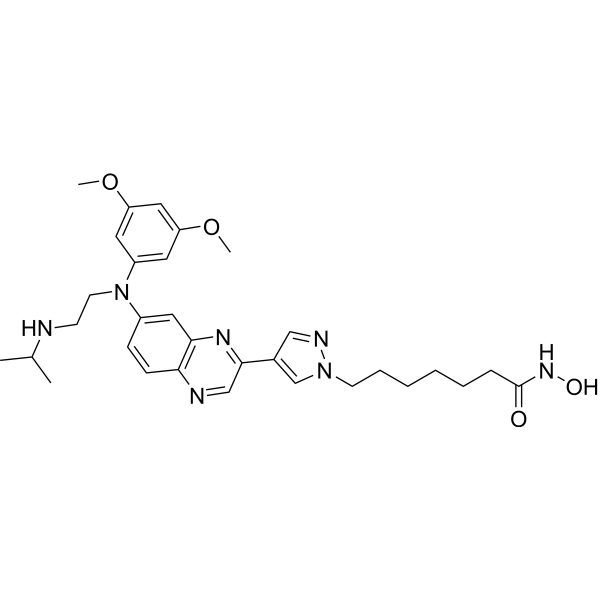
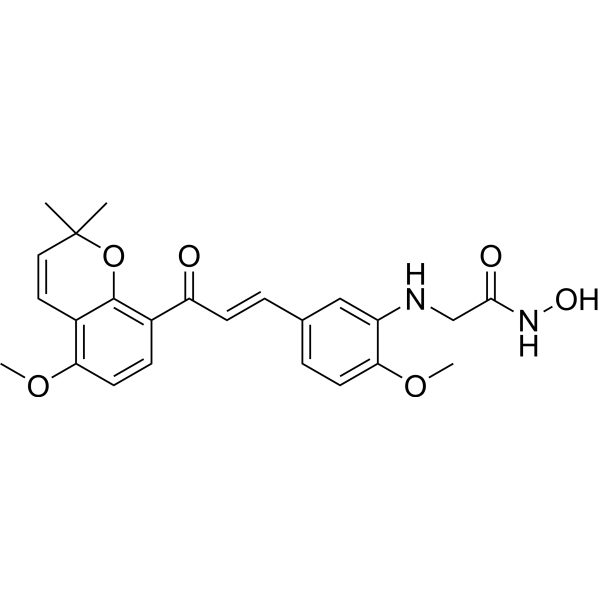
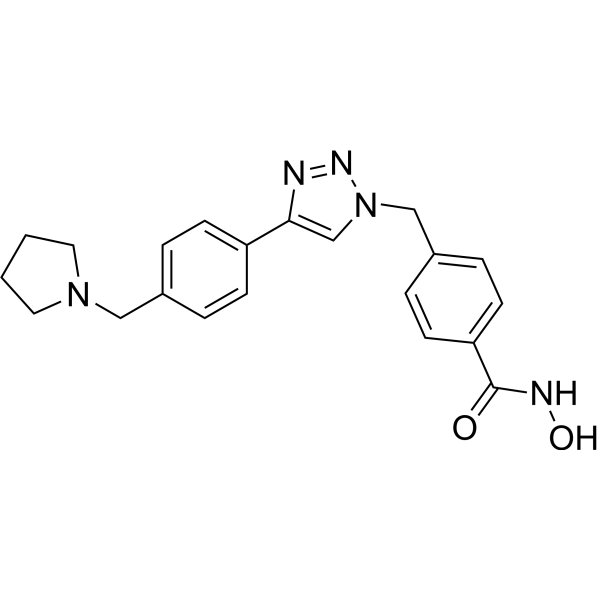
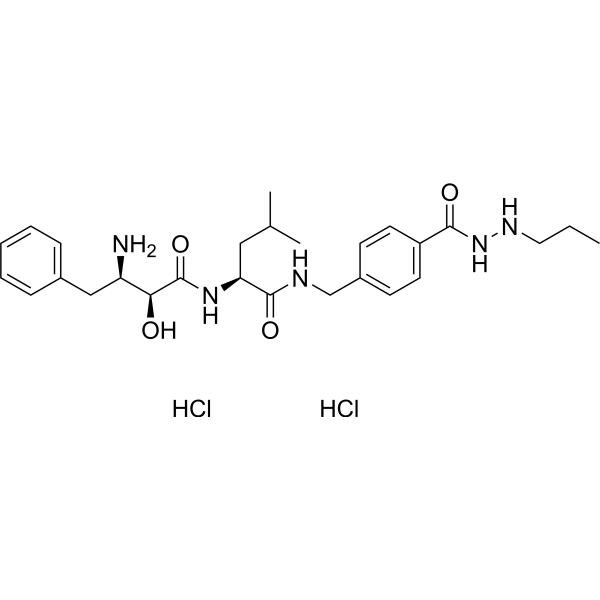
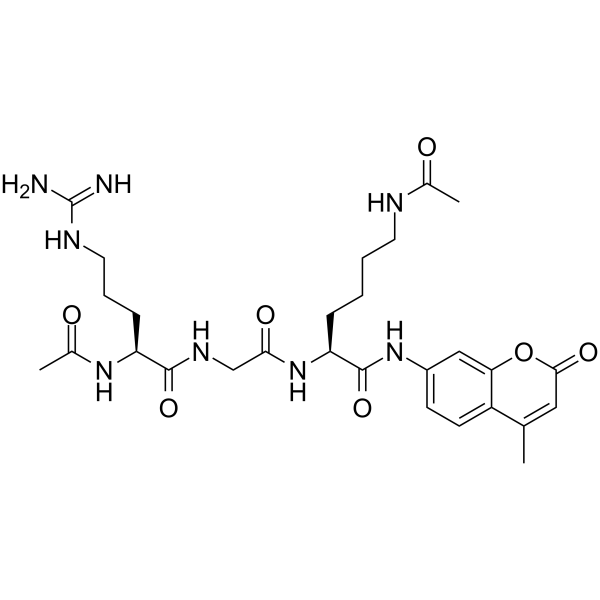
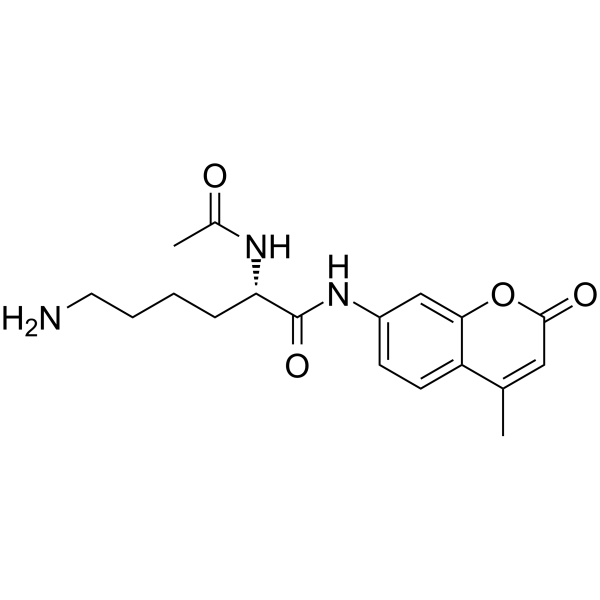
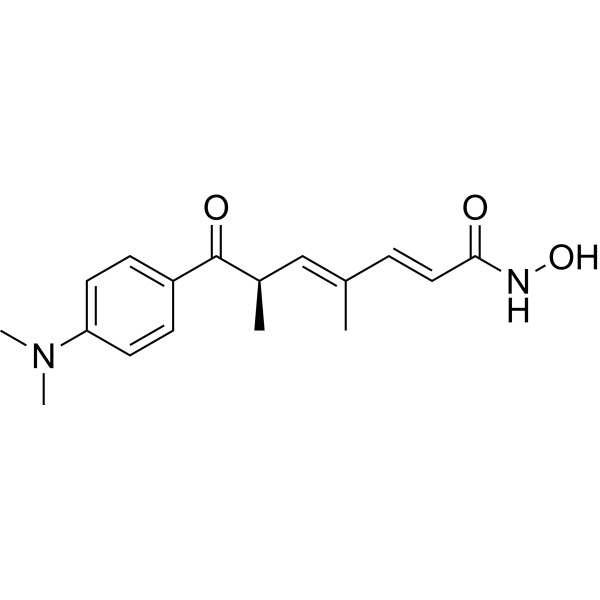
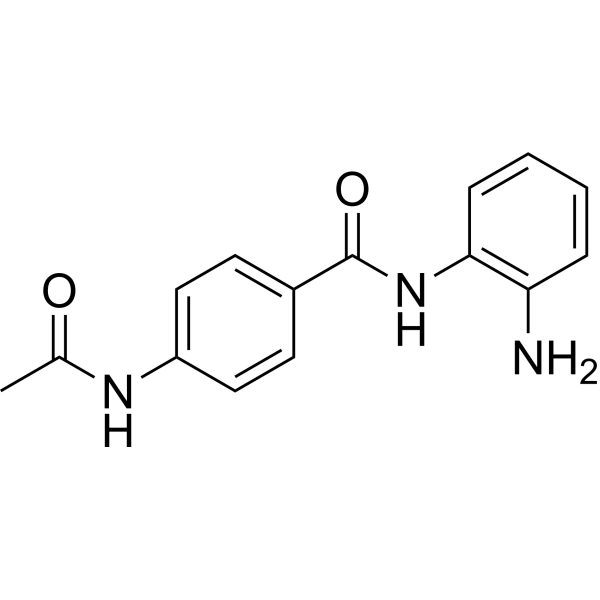
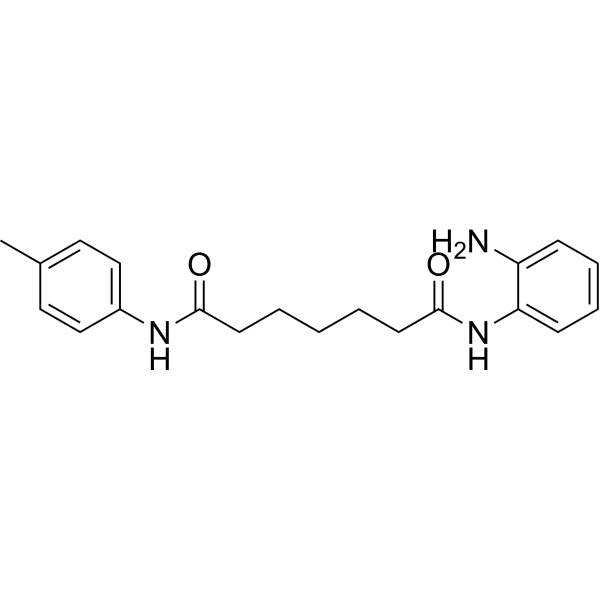
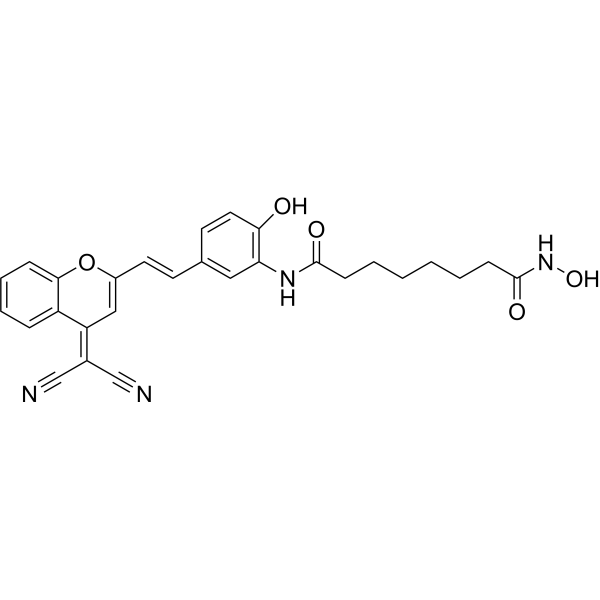
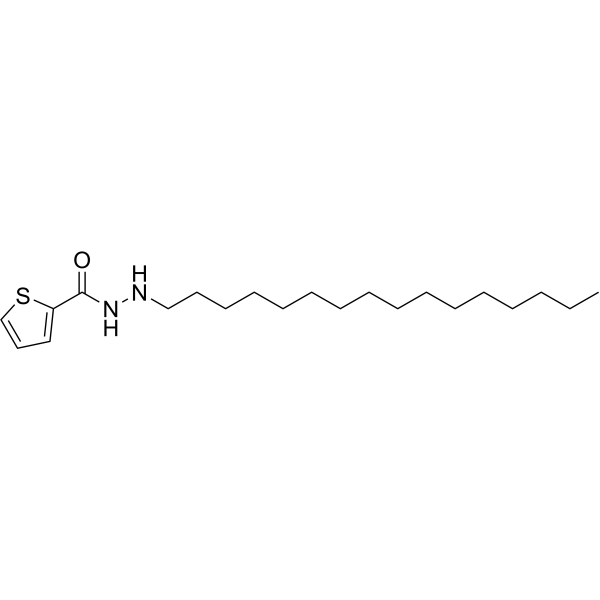
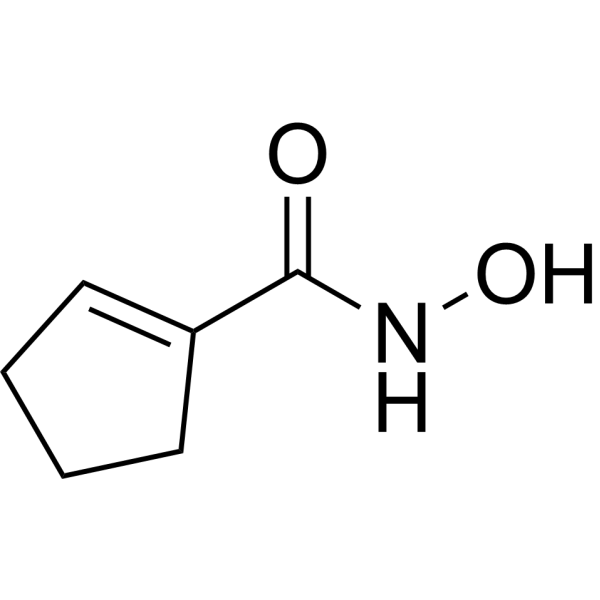
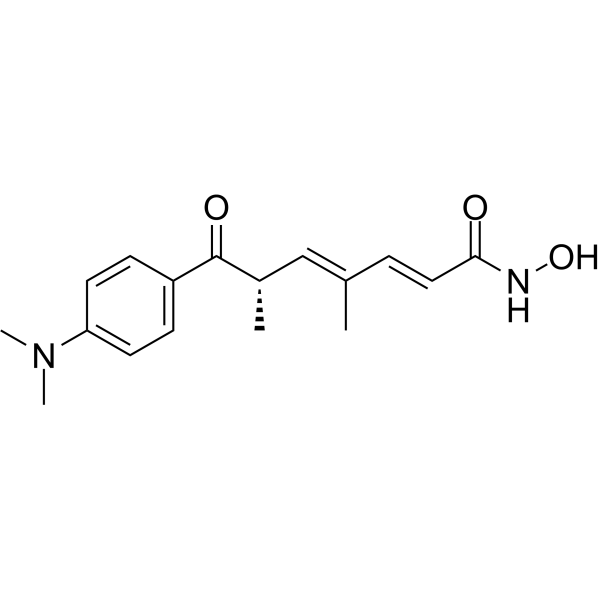
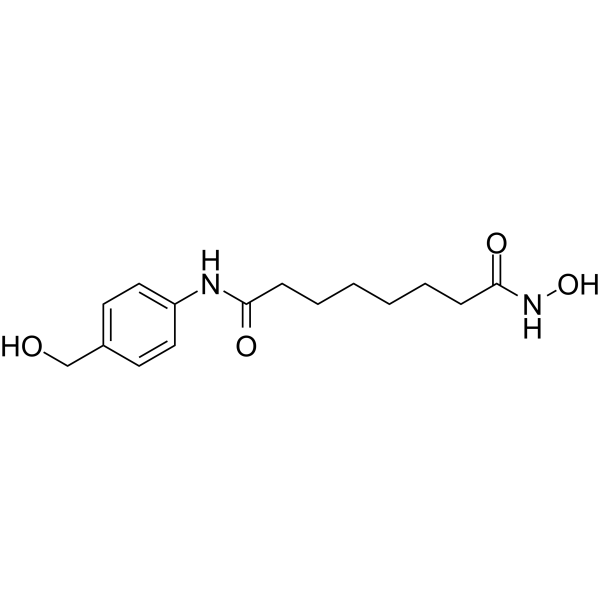
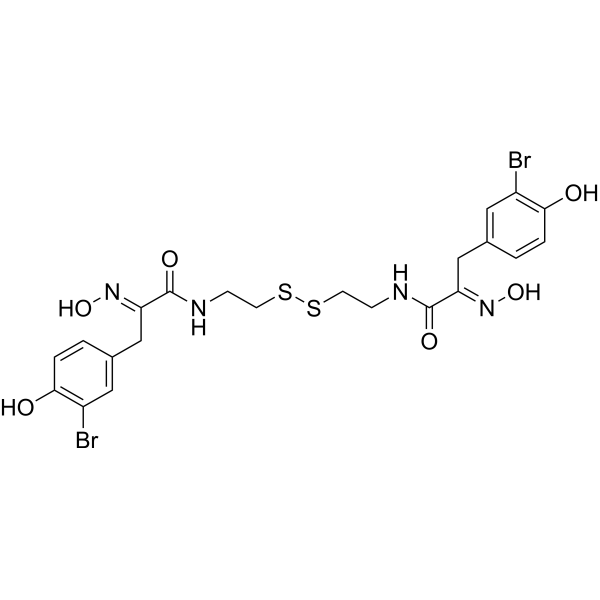
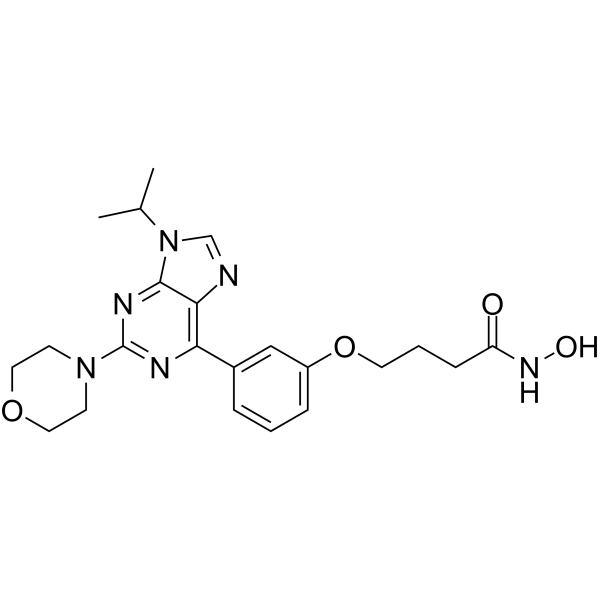
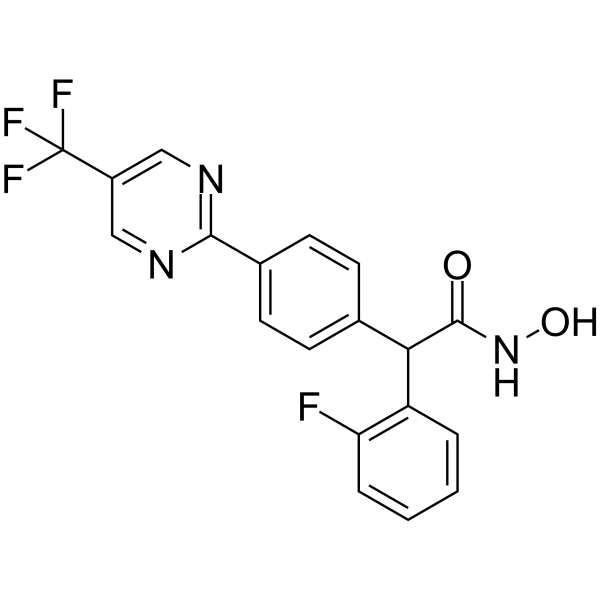
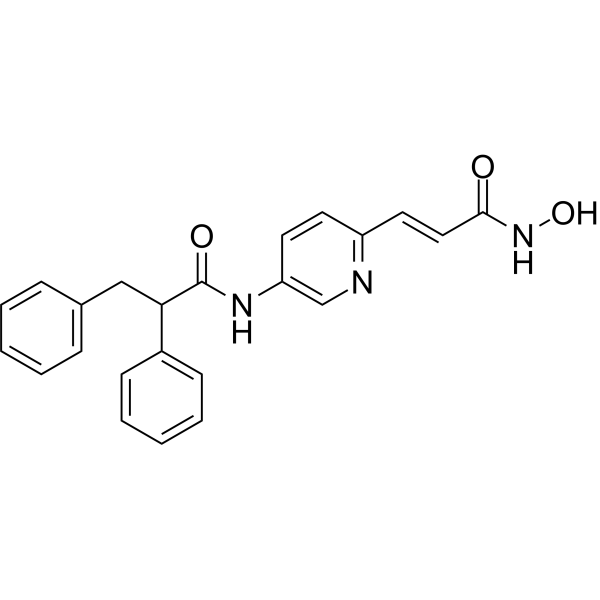
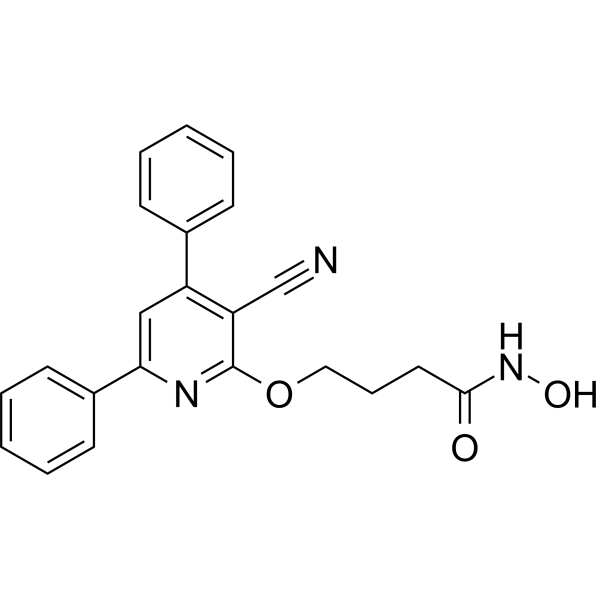
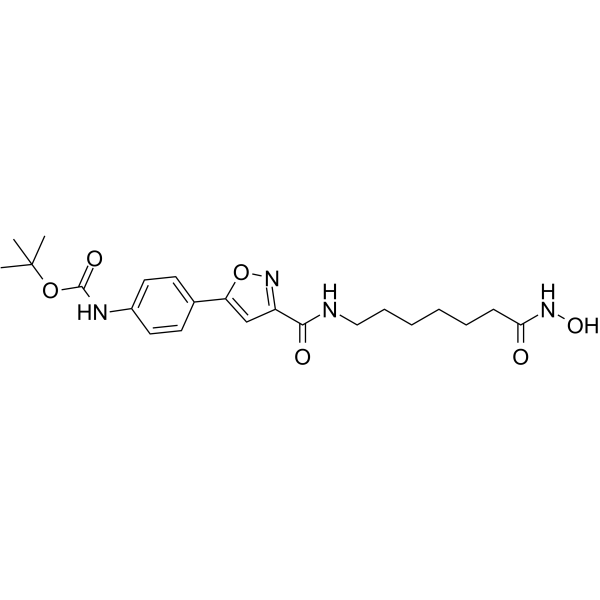From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
HDAC1 Human Pre-designed siRNA Set A contains three designed siRNAs for HDAC1 gene (Human), as well as a negative control, a positive control, and a FAM-labeled negative control.
Hdac1 Mouse Pre-designed siRNA Set A contains three designed siRNAs for Hdac1 gene (Mouse), as well as a negative control, a positive control, and a FAM-labeled negative control.
Hdac1 Rat Pre-designed siRNA Set A contains three designed siRNAs for Hdac1 gene (Rat), as well as a negative control, a positive control, and a FAM-labeled negative control.
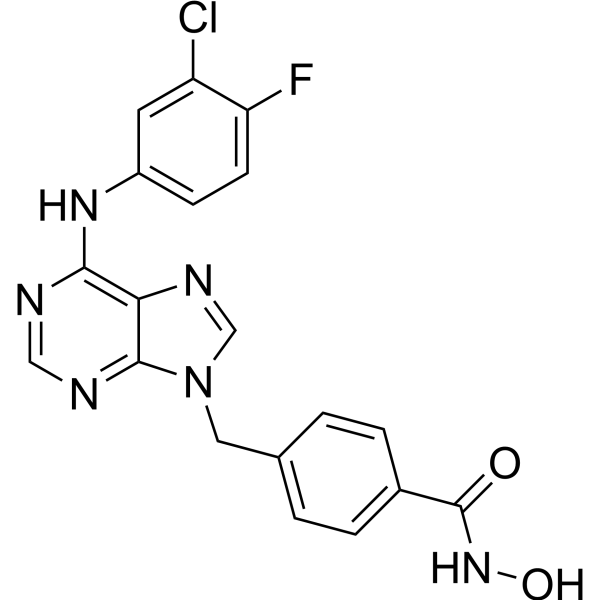
HDAC1/CDK7-IN-1 (compound 8e) is a dual CDK7 and HDAC1 inhibitor with IC50s of 893 nM and 248 nM, respectively. HDAC1/CDK7-IN-1 inhibits the growth cells of MDA-MB-231, MCF-7, A549, and HCT-116 cancer cells. HDAC1/CDK7-IN-1 induces cell cycle arrest and apoptosis in HCT-116 cells, as well as hindered the migration of HCT-116 cells [1].
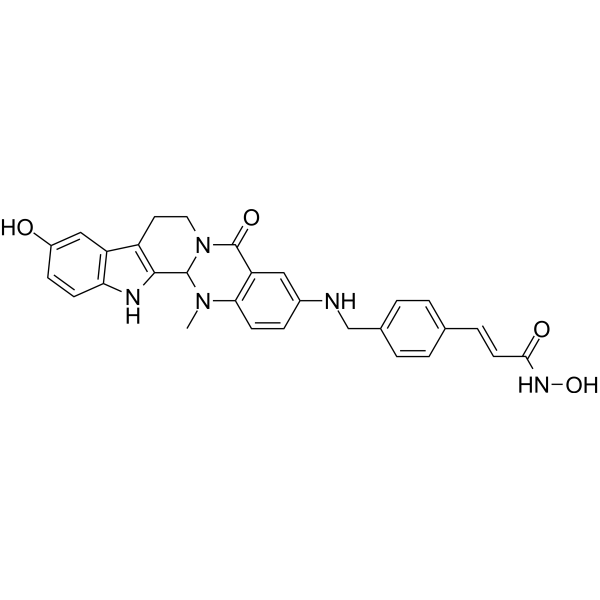
HDAC1-IN-5 is a potent HDAC1 inhibitor with IC50 values of 15 nM and 20 nM for HDAC1 and HDAC6, respectively. HDAC1-IN-5 can enhance the acetylation of histone H3 and α-tubulin, as well as promote the activation of caspase 3 in cancer cells, thereby inducing apoptosis. HDAC1-IN-5 induces chromatin damage by binding with DNA. HDAC1-IN-5 has strong inhibitory activity against tumor growth in xenograft mice [1].
HDAC1-IN-3 is a potent Pf HDAC1 inhibitor. HDAC1-IN-3 shows antimalarial activity in wild-type and multidrug-resistant parasite strains. HDAC1-IN-3 shows a significant in vivo killing effect against all life cycles of parasites [1].
HDAC1-IN-6 (compound 1) is an inhibitor of HDAC1 and 11, with an IC50 of 1.9 μM and 1.6 μM, respectively. HDAC1-IN-6 induces differentiation in AML cells [1].
HDAC1/MAO-B-IN-1 is a potent, selective and cross the blood-brain barrier HDAC1/MAO-B inhibitor with IC50 values of 21.4 nM and 99.0 nM for HDAC1 and MAO-B, respectively. HDAC1/MAO-B-IN-1 has the potential for the research of Alzheimer’s disease [1].
HDAC1/6-IN-1 (compound D7) is a potent multitarget inhibitor of GLP, HDAC6 and HDAC1, with IC50 values of 1.3, 13, and 89 nM, respectively. HDAC1/6-IN-1 can inhibit the methylation and deacetylation of H3K9 on protein level. HDAC1/6-IN-1 induces cancer cell apoptosis, G0/G1 cell cycle arrest, and blocks migration and invasion [1].
HDAC1/2 and CDK2-IN-1 (compound 14d) is a potent HDAC1, HDAC2 and CDK2 dual inhibitor, with IC50 values of 70.7, 23.1 and 0.80 μM, respectively. HDAC1/2 and CDK2-IN-1 can block the cell cycle and induce apoptosis. HDAC1/2 and CDK2-IN-1 exhibits desirable in vivo antitumor activity [1].
VEGFR2/HDAC1-IN-1 (compound 13) is a potent VEGFR-2/HDAC dual inhibitor, with IC50s of 57.83 nM and 9.82 nM, respectively. VEGFR2/HDAC1-IN-1 arrests the cell cycle at the S and G2 phases, and induces apoptosis in HeLa cells. VEGFR2/HDAC1-IN-1 exhibits anti-angiogenic effect [1].
Chlopynostat (Compound 6c) is a HDAC1 inhibitor with a IC50 value of 67 nM. Chlopynostat reverses STAT4/p66Shc defects by inhibiting HDAC1-induced < b>Apoptosis [1].
SB-429201 is a potent and selective HDAC1(IC50~1.5 μM). SB-429201 displays at least a 20-fold preference for HDAC1 inhibition over HDAC3 and HDAC8 [1] .
Pyroxamide is a potent inhibitor of histone deacetylase 1(HDAC1) with an ID50 of 100 nM. Pyroxamide can induce apoptosis and cell cycle arrest in leukemia [1].
HR488B is an efficient HDAC1 inhibitor. HR488B specifically suppressed the growth of CRC cells by inducing cell cycle G0/G1 arrest and apoptosis. HR488B causes mitochondrial dysfunction, reactive oxygen species (ROS) generation, and DNA damage accumulation [1].
RG2833 is a brain-penetrant HDAC inhibitor with IC50s of 60 nM and 50 nM for HDAC1 and HDAC3, respectively. The Ki values for HDAC1 and HDAC3 are 32 and 5 nM, respectively [1].
Zabadinostat (CXD101) is a potent, selective and orally active class I HDAC inhibitor with IC50s of 63 nM, 570 nM and 550 nM for HDAC1, HDAC2 and HDAC3, respectively. Zabadinostat has no activity against HDAC class II. Zabadinostat has antitumor activity [1] .
HDAC-IN-67 (compound 27f) is an HDAC inhibitor against HDAC1 and HDAC6, with IC50 values of 22 nM and 8 nM, respectively. HDAC-IN-67 inhibits cell proliferation and induces cell apoptosis. HDAC-IN-67 exhibits antitumor activity [1].
ACY-957 is an orally active and selective inhibitor of HDAC1 and HDAC2, with IC50s of 7 nM, 18 nM, and 1300 nM against HDAC1/2/3, respectively, and shows no inhibition on HDAC4/5/6/7/8/9 [1].
HDAC6 degrader-3 is a potent and selective HDAC6 degrader via ternary complex formation and the ubiquitin-proteasome pathway with a DC50 value of 19.4 nM. HDAC6 degrader-3 has IC50s of 4.54 nM and 0.647 μM for HDAC6 and HDAC1, respectively. HDAC6 degrader-3 causes strong hyperacetylation of α-tubulin [1].
Givinostat hydrochloride monohydrate (ITF-2357 hydrochloride monohydrate) is a HDAC inhibitor with an IC50 of 198 and 157 nM for HDAC1 and HDAC3, respectively.
HDAC-IN-57 is an orally active inhibitor of histone deacetylases (HDAC), with IC50s of 2.07 nM, 4.71 nM, 2.4 nM and 107 nM for HDAC1, HDAC2, HDAC6, HDAC8, respectively. HDAC-IN-57 can inhibits LSD1, with IC50 of 1.34 μΜ. HDAC-IN-57 induces apoptosis, and has anti-tumor activity [1].
Antitumor agent-123 (Copmound 4d) effectively inhibits multiple kinase targets with anti-cancer effects, including JAK2, JAK3, HDAC1 and HDAC6, with IC50 values of 34.6 and 2.6 μM for JAK2 and JAK3, respectively. Antitumor agent-123 exhibits moderate activity in solid tumor models [1].
BRD-6929 is a potent, selective brain-penetrant inhibitor of class I histone deacetylase HDAC1 and HDAC2 inhibitor with IC50 of 1 nM and 8 nM, respectively. BRD-6929 shows high-affinity to HDAC1 and HDAC2 with Ki of 0.2 and 1.5 nM, respectively. BRD-6929 can be used for mood-related behavioral model research .
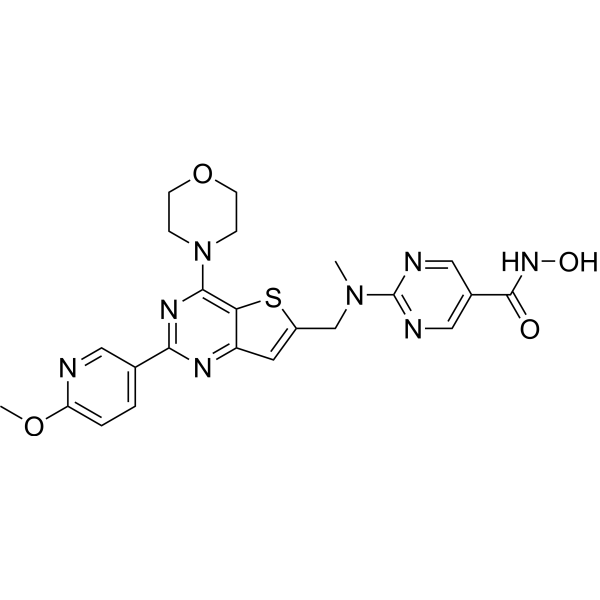
TNG260 is a CoREST-selective deacetylase (CoreDAC) inhibitor. TNG260 inhibits HDAC1 with 10-fold selectivity over HDAC3. TNG260 leads to HDAC1 inhibition, reverses anti-PD1 resistance driven by loss of STK11. TNG260 decreases intratumoral infiltration of neutrophils. TNG260 exhibits immune-mediated cell killing [1].
Vorinostat (SAHA) is a potent and orally active pan-inhibitor of HDAC1, HDAC2 and HDAC3 (Class I), HDAC6 and HDAC7 (Class II) and HDAC11 (Class IV), with ID50 values of 10 nM and 20 nM for HDAC1 and HDAC3, respectively. Vorinostat induces cell apoptosis[1] . Vorinostat is also an effective inhibitor of human papillomaviruse (HPV)-18 DNA amplification .
cis-BG47 is an cis-isomer of BG47, BG47 is a prototypical histone deacetylases HDAC1 and HDAC2 selective, optoepigenetic probe. BG47 can bind to and competitively inhibits the deacetylase activity of HDAC targets upon a light-induced trans-to-cis isomerization, and increases Histone Methyltransferase H3K9 acetylation. cis-BG47 can be used for neurological disease research [1].
Corin is a dual inhibitor of histone lysine specific demethylase (LSD1) and histone deacetylase (HDAC), with a Ki(inact) of 110 nM for LSD1 and an IC50 of 147 nM for HDAC1.
(S)-TNG260 is an isomer of TNG260 (HY-153358). TNG260 is a CoREST selective deacetylase (CoreDAC) inhibitor. TNG260 inhibits HDAC1 with 10-fold selectivity over HDAC3. TNG260 causes HDAC1 inhibition and reverses anti-PD1 resistance driven by STK11 deletion. TNG260 reduces intratumoral infiltration of neutrophils. TNG260 exhibits immune-mediated cell killing.
Vorinostat-d5 (SAHA-d5) is the deuterium labeled Vorinostat. Vorinostat (SAHA) is a potent and orally active pan-inhibitor of HDAC1, HDAC2 and HDAC3 (Class I), HDAC7 (Class II) and HDAC11 (Class IV), with ID50 values of 10 nM and 20 nM for HDAC1 and HDAC3, respectively. Vorinostat induces cell apoptosis[1] . Vorinostat is also an effective inhibitor of human papillomaviruse (HPV)-18 DNA amplification .
GK444 (Compound 15a) is a HDAC1/2 inhibitor (IC50: 100 and 92 nM for HDAC1/2 respectively). GK444 inhibits Caco-2 cells with IC50 of 4.1 μM. GK444 also reduces TGF-β1 induced COL1A1 mRNA levels in primary normal human lung fibroblasts. GK444 inhibits Bleomycin (HY-108345)-induced lung fibrosis in mice [1].
Snail/HDAC-IN-1 is a potent Snail/HDAC dual target inhibitor. Snail/HDAC-IN-1 displays potent inhibitory activity against HDAC1 with an IC50 of 0.405 μM and potent inhibition against Snail with a Kd of 0.18 μM. Snail/HDAC-IN-1 increases histone H4 acetylation in HCT-116 cells and decreases the expression of Snail protein to induce cell apoptosis[1].
HDAC-IN-7 (Chidamide impurity) is an impurity of Chidamide. Chidamide is a potent and orally bioavailable HDAC enzymes class I (HDAC1/2/3) and class IIb (HDAC10) inhibitor.
Tinostamustine (EDO-S101) is a pan HDAC inhibitor; inhibits HDAC6, HDAC1, HDAC2 and HDAC3 with IC50 values of 6 nM, 9 nM, 9 nM and 25 nM, respectively [1].
BRD2492 (compound 6d) is a potent, selective HDAC1 and HDAC2 inhibitor with IC50s of 13.2 nM and 77.2 nM, respecrtively. BRD2492 exhibits >100-fold selectivity for HDAC1/2 over selectivity over HDAC3 and HDAC6. BRD2492 inhibits breast cancer cell lines growth with IC50s of 1.01 μM and 11.13 μM for T-47D and MCF-7 cells, respectively [1].
CRA-026440 hydrochloride is a potent, broad-spectrum HDAC(HDAC) inhibitor. The Ki values against recombinant HDAC isoenzymes HDAC1, HDAC2, HDAC3, HDAC6, HDAC8, and HDAC10 are 4 nM, 14 nM, 11 nM, 15 nM, 7 nM, and 20 nM respectively. CRA-026440 hydrochloride shows antitumor and antiangiogenic activities [1]. CRA-026440 (hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Resminostat hydrochloride is a potent inhibitor of HDAC1, HDAC3 and HDAC6, with mean IC50 values of 42.5, 50.1, 71.8 nM, respectively, and shows less potent activities against HDAC8, with an IC50 of 877 nM.
Parthenolide is a sesquiterpene lactone found in the medicinal herb Feverfew. Parthenolide exhibits anti-inflammatory activity by inhibiting NF-κB activation; also inhibits HDAC1 protein without affecting other class I/II HDACs.
HDAC/BET-IN-1 displays submicromolar inhibitory activity against HDAC1 and 6 (IC50 = 0.163 μM and 0.067 μM), and BRD4 (Ki = 0.076 μM), and possess potent antileukemia activity.
Ricolinostat (ACY-1215) is a potent and selective HDAC6 inhibitor, with an IC50 of 5 nM. ACY-1215 also inhibits HDAC1, HDAC2, and HDAC3 with IC50s of 58, 48, and 51 nM, respectively.
Tubacin is a potent and selective inhibitor of HDAC6, with an IC50 value of 4 nM and approximately 350-fold selectivity over HDAC1. Tubacin also inhibits metallo-β-lactamase domain-containing protein 2 (MBLAC2).
m-Carboxycinnamic acid bishydroxamide is a potent HDAC inhibitor, exhibiting ID50 values of 10 and 70 nM in vitro for HDAC1 and HDAC3, respectively [1]. m-Carboxycinnamic acid bishydroxamide also induces apoptosis and suppresses tumor growth .
HDAC6-IN-14 is a highly selective HDAC6 (HDAC) inhibitor with an IC50 of 42 nM. HDAC6-IN-14 displays >100-fold selectivity over HDAC1/HDAC2/HDAC3/HDAC4 [1].
ACY-738 is a potent, selective and orally-bioavailable HDAC6 inhibitor, with an IC50 of 1.7 nM; ACY-738 also inhibits HDAC1, HDAC2, and HDAC3, with IC50s of 94, 128, and 218 nM.
Abexinostat (CRA 024781) is a novel pan-HDAC inhibitor mostly targeting HDAC1 with Ki of 7 nM. Abexinostat also inhibits metallo-β-lactamase domain-containing protein 2 (MBLAC2) hydrolase activity with an EC50 below 10 nM [1] .
Entinostat-d4 is the deuterium labeled Entinostat[1]. Entinostat is an oral and selective class I HDAC inhibitor, with IC50s of 243 nM, 453 nM, and 248 nM for HDAC1, HDAC2, and HDAC3, respectively[2].
DNMT/HDAC-IN-1 (Compund 15a) is a dual DNMT and HDAC inhibitor with IC50 values for HDAC1 and HDAC6 are 56.84 nM and 17.39 nM respectively. DNMT/HDAC-IN-1 can induce apoptosis and be used in tumor research.
mTOR/HDAC-IN-1 (Compound 50) is a selective mTOR and HDAC dual inhibitor with IC50 values of 0.49 and 0.91 nM against mTOR and HDAC1, respectively. mTOR/HDAC-IN-1 can be studied as an anti-cancer agent [1].
PI3K/HDAC-IN-3 (36) is a PI3K and HDAC dual inhibitor, with IC50 values of 0.23 nM and 172 nM for PI3Kα and HDAC1, respectively. PI3K/HDAC-IN-3 (36) suppresses AKT phosphorylation and increased H3 acetylation in MV4-11 cells. PI3K/HDAC-IN-3 (36) exhibits significant and dose-dependent anticancer efficacy in a MV4-11 xenograft model [1].
CM-675 is a dual phosphodiesterase 5 (PDE5) and class I histone deacetylases-selective inhibitor, with IC50 values of 114 nM and 673 nM for PDE5 and HDAC1, respectively. CM-675 has potential to treat Alzheimer’s disease [1].
KPZ560 is a potent inhibitor of HDAC1 and HDAC2, with IC50s of 12 nM and 68 nM, respectively. KPZ560 can increase in the spine density of granule neuron dendrites of mice and inhibitor cell growth of breast cancer cell line MCF [1].
PARP/HDAC-IN-1 (compound B102) is a potent dual inhibitor of PARP and HDAC. PARP/HDAC-IN-1 inhibits PARP1, PARP2 and HDAC1 with IC50s of 19.01, 2.13, 1690 nM, respectively [1].
SKLB-23bb is a potent and selective inhibitor for HDAC6 with an IC50 of 17 nM and shows 25-fold and 200-fold selectivity relative to HDAC1 (IC50=422 nM) and HDAC8 (IC50=3398 nM), respectively.
Resminostat (RAS2410; 4SC-201) is a potent inhibitor of HDAC1, HDAC3 and HDAC6, with mean IC50 values of 42.5, 50.1, 71.8 nM, respectively, and shows less potent activities against HDAC8, with an IC50 of 877 nM.
Tucidinostat-d4 is the deuterium labeled Tucidinostat. Tucidinostat is a potent and orally bioavailable HDAC enzymes class I (HDAC1/2/3) and class IIb (HDAC10) inhibitor, with IC50s of 95, 160, 67 and 78 nM, respectively[1].
HDAC6-IN-27 (compound 8C) is a HDAC inhibitor with IC50 vales of 15.9 nM 136.5 nM and 6180.2 nM for HDAC6, HDAC8 and HDAC1, respectively. HDAC6-IN-27 shows potent antiparasitic effects [1].
HDAC-IN-52 is a pyridine-containing HDAC inhibitor, with IC50s of 0.189, 0.227, 0.440 and 0.446 μM for HDAC1, HDAC2, HDAC3, and HDAC10, respectively. HDAC-IN-52 can be used for the research of cancer [1].
BG45 is a potent HDAC3 inhibitor with IC50 values of 0.289, 2, 2.2 and ﹥20 μM for HDAC3, HDAC1, HDAC2 and HDAC6, respectively. BG45 selectively targets multiple myeloma (MM) cells and induces caspase-dependent apoptosis [1] .
Citarinostat (ACY241) is a second generation potent, orally active and high-selective HDAC6 inhibitor with an IC50 of 2.6 nM (IC50s of 35 nM, 45 nM, 46 nM and 137 nM for HDAC1, HDAC2, HDAC3 and HDAC8, respectively). Citarinostat has anticancer effects [1].
HDAC-IN-27 (Compound 11h) is a potent, selective and orally active HDAC Class I inhibitor, with IC50 values ranging from 0.43 nM to 3.01 nM for HDAC1-3. HDAC-IN-27 shows anti-acute myeloid leukemia (AML) activity [1].
HDAC-IN-41 (Compound 7c) is a selective, orally active class I HDAC inhibitor with IC50 values of 0.62, 1.46 and 0.62 μM against HDAC1, HDAC2 and HDAC3, respectively. HDAC-IN-41 shows NO releasing activity [1].
HDAC-IN-56 ((S)-17b) is an orally active class I histone deacetylase (HDAC) inhibitor with IC50 values of 56.0 ± 6.0, 90.0 ± 5.9, 422.2 ± 105.1, >10000 nM for HDAC1, HDAC2, HDAC3, and HDAC4-11, respectively. HDAC-IN-56 has potent inhibitory activity while strongly increasing intracellular levels of acetylhistone H3 and P21 and effectively inducing G1 cell cycle arrest and apoptosis.HDAC-IN-56 has antitumor activity [1].
KH16 is a potent and low nanomolar HDAC inhibitor. KH16 is against class I HDACsHDAC1, HDAC2, and HDAC3, with IC50 ?values ranging from 6 to 34 nM. KH16 induces cell apoptosis and is against tumor cells with various gene expression patterns [1].
HDAC-IN-68 (Compound 29) is a potent HDAC inhibitor that disrupts microtubule structure and inhibits tumor growth. HDAC-IN-68 significantly inhibits class I HDACs (HDAC1, HDAC2, HDAC3) with IC50 values of 5.1, 11.5 and 8.8 nM, respectively [1].
YSR734 (Compound 21) is a covalent HDAC inhibitor with IC50 values of 110 nM, 154 nM, and 143 nM for HDAC1, HDAC2, and HDAC3, respectively. YSR734 can induce apoptosis in leukemia cells. YSR734 can induce myoblast differentiation and is used in the study of Duchenne muscular dystrophy [1].
Domatinostat tosylate (4SC-202) is a selective class I HDAC inhibitor with IC50 of 1.20 μM, 1.12 μM, and 0.57 μM for HDAC1, HDAC2, and HDAC3, respectively. It also displays inhibitory activity against Lysine specific demethylase 1(LSD1).
PTACH (NCH-51) is a potent HDAC inhibitor with IC50s of 48 nM, 32 nM, and 41 nM for HDAC1, HDAC4, and HDAC6, respectively. PTACH exerts potent growth inhibition against various cancer cells (EC50s of 1.1-9.1 µM) [1] .
APHA Compound 8 (Compound 4) is a histone deacetylase (HDAC) inhibitor. APHA Compound 8 has antimouse HDAC1 activity with an IC50 value of 0.78 μM. APHA Compound 8, as antiproliferative and cytodifferentiating agent on MEL cells, shows dose-dependent growth inhibition and hemoglobin accumulation effects [1].
NB512 (compound 39a) is a dual inhibitor for BET and HDAC, which exhibits a efficient binding affinity with BRD4 bromodomains and HDAC1/2, with EC50s of 100-400 nM. NB512 exhibits an anti-proliferative activity towards cancer cells PaTu8988T and NMC [1].
HDAC-IN-71 (Compound 17q) is a potent HDAC inhibitor with IC50 values of 12.6, 14.1, 20, 3, and 72 nM for HDAC1, HDAC2, HDAC3, HDAC6, and HDAC10, respectively. HDAC-IN-71 induces apoptosis and can be used in cancer research [1].
Domatinostat (4SC-202 free base) is a selective class I HDAC inhibitor with IC50 of 1.20 μM, 1.12 μM, and 0.57 μM for HDAC1, HDAC2, and HDAC3, respectively. It also displays inhibitory activity against Lysine specific demethylase 1(LSD1).
CAY10603 (BML-281) is a potent and selective HDAC6 inhibitor, with an IC50 of 2 pM; CAY10603 (BML-281) also inhibits HDAC1, HDAC2, HDAC3, HDAC8, HDAC10, with IC50s of 271, 252, 0.42, 6851, 90.7 nM.
Quisinostat dihydrochloride (JNJ-26481585 dihydrochloride) is an orally available, potent pan-HDAC inhibitor with IC50s of 0.11 nM, 0.33 nM, 0.64 nM, 0.46 nM, and 0.37 nM for HDAC1, HDAC2, HDAC4, HDAC10 and HDAC11, respectively. Quisinostat dihydrochloride has a broad spectrum antitumoral activity [1].
HDAC-IN-33 is a potent HDAC inhibitor with IC50s of 24, 46, and 47 nM for HDAC1, HDAC2 and HDAC6, respectively. HDAC-IN-33 possesses potent antiproliferation activities against tumor cells. HDAC-IN-33 shows potent antitumor efficacy in vivo That trigger antitumor immunity [1].
LMK-235 is a potent and selective HDAC4/5 inhibitor, inhibits HDAC5, HDAC4, HDAC6, HDAC1, HDAC2, HDAC11 and HDAC8, with IC50s of 4.22 nM, 11.9 nM, 55.7 nM, 320 nM, 881 nM, 852 nM and 1278 nM, respectively, and is used in cancer research.
HDAC-IN-32 is a potent HDAC inhibitor with IC50s of 5.2, 11, and 28 nM for HDAC1, HDAC2 and HDAC6, respectively. HDAC-IN-32 possesses potent antiproliferation activities against tumor cells. HDAC-IN-32 shows potent antitumor efficacy in vivo That trigger antitumor immunity [1].
c-Met/HDAC-IN-3 (Compound 15f) is a dual c-Met and HDAC inhibitor with IC50 values of 12.50 nM and 26.97 nM against c-Met and HDAC1, respectively. c-Met/HDAC-IN-3 induces apoptosis and cause cell cycle arrest in G2/M phase [1].
HDAC2-IN-1 (Compound 17) is a brain penetrant, orally active, competitive HDAC2 inhibitor with an IC50 of 0.5 μM [1]. HDAC2-IN-1 also inhibits HDAC1 and HDAC8 with IC50s of 1.61 μM and 0.98 μM, respectively [1].
HDAC-IN-70 (compound 4c) is HDAC6 inhibitor with the IC50 values of 64.13 nM, 166 nM, 618 nM and 253 nM for HDAC6, HDAC1, HDAC2 and HDAC4, respectively. HDAC-IN-70 induces cell cycle arrest, apoptosis and necrotic. HDAC-IN-70 can be used for study of leukemia [1].
BRD73954 is a potent HDAC inhibitor and selectively inhibiting both HDAC6 and HDAC8 with IC50 values of 0.0036, 0.12, 9, 12, 23 µM for HDAC6, HDAC8, HDAC2, HDAC1 and HDAC3, respectively. BRD73954 decreases the levels of HDAC6, associated with upregulation of Ac-Tubulin [1].
Quisinostat (JNJ-26481585) is a potent, second-generation and orally active pan-HDAC inhibitor (HDACi), with IC50 values ranging from 0.11 nM to 0.64 nM for HDAC1, HDAC2, HDAC4, HDAC10 and HDAC11. Quisinostat has a broad spectrum antitumoral activity [1]. Quisinostat can induce autophagy in neuroblastoma cells .
JPS014 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS014 degrades class I histone deacetylase (HDAC). JPS014 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
JPS016 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS016 degrades class I histone deacetylase (HDAC). JPS016 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
JPS035 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS035 degrades class I histone deacetylase (HDAC). JPS035 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
JPS036 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS036 degrades class I histone deacetylase (HDAC). JPS036 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
HDAC-IN-42 (compound 14f) is a potent and selective HDAC inhibitor with IC50 values of 0.19 and 4.98 µM for HDAC1 and HDAC6, respectively. HDAC-IN-42 shows anticancer and anti-proliferative activity. HDAC-IN-42 induces apoptosis and cell cycle arrest at G2/M phase [1].
A2AAR/HDAC-IN-1 (compound 14c) is an orally active, potent and balanced A2AAR/HDAC dual inhibitor, with a Ki of 163.5 nM for A2AAR and an IC50 of 145.3 nM for HDAC1. A2AAR/HDAC-IN-1 shows anticancer activity [1].
JPS016 is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS016 degrades class I histone deacetylase (HDAC). JPS016 is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
HDAC6/8/BRPF1-IN-1 is a dual inhibitor of both HDAC6/8 and the bromodomain and PHD finger containing protein 1(BRPF1). HDAC6/8/BRPF1-IN-1 has inhibitory activity for HDAC1, HDAC6 and HDAC8 with IC50 values of 797 nM, 344 nM and 908 nM, respectively. HDAC6/8/BRPF1-IN-1 has inhibitory activity for BRPF1 with an Kd value of 175.2 nM. HDAC6/8/BRPF1-IN-1 can be used for the research of cancer [1].
Mocetinostat (MGCD0103) is a potent, orally active and isotype-selective HDAC (Class I/IV) inhibitor with IC50s of 0.15, 0.29, 1.66 and 0.59 μM for HDAC1, HDAC2, HDAC3 and HDAC11, respectively. Mocetinostat shows no inhibition on HDAC4, HDAC5, HDAC6, HDAC7, or HDAC8.
HDAC-IN-45 (Compound 14) is a small molecule HDAC inhibitor and has anticancer activity, also can forms a hydrogen
bond with residue Y303. HDAC-IN-45 (Compound 14) has substantial inhibitory effects towards HDAC1, 2 and 3 isoforms with IC50 values of 0.108, 0.585 and 0.563 μM respectively [1].
HDAC8/BRPF1-IN-1 (Compound 23a) is a dual inhibitor of HDAC8 and BRPF1 with an IC50 of 443 nM against human HDAC8 and a Kd of 67 nM against human BRPF1. HDAC8/BRPF1-IN-1 shows low in vitro activity against HDAC1 and 6 [1].
HDAC6-IN-33 (compound 6) is a selective and irreversible HDAC6 inhibitor with an IC50 of 193 nM. HDAC6-IN-33 shows no activity against HDAC1-4. HDAC6-IN-33 is a tight-binding HDAC6 inhibitor capable of inhibiting HDAC6 via a two-step slow-binding mechanism [1].
Tucidinostat (Chidamide) is a potent and orally bioavailable HDAC enzymes class I (HDAC1/2/3) and class IIb (HDAC10) inhibitor, with IC50s of 95, 160, 67 and 78 nM, less active on HDAC8 and HDAC11 (IC50s, 733 nM, 432 nM, respectively), and shows no effect on HDAC4/5/6/7/9 [1].
Suberoyl bis-hydroxamic acid (Suberohydroxamic acid; SBHA) is a competitive and cell-permeable HDAC1 and HDAC3 inhibitor with ID50 values of 0.25 μM and 0.30 μM, respectively [1].Suberoyl bis-hydroxamic acid renders MM cells susceptible to apoptosis and facilitates the mitochondrial apoptotic pathways .Suberoyl bis-hydroxamic acid can be used for the study of medullary thyroid carcinoma (MTC) .
IHCH-3064 is a dual-acting compounds targeting Adenosine A2A Receptor and HDAC. IHCH-3064 exhibits potent binding to A2AR (Ki=2.2 nM) and selective inhibition of HDAC1 (IC50=80.2 nM), with good antiproliferative activity against tumor cell lines in vitro. IHCH-3064 is a tumor immunotherapeutic agent.
NMDAR/HDAC-IN-1 (Compound 9d) is a dual NMDAR and HDAC inhibitor with a Ki of 0.59 μM for NMDAR and IC50 values of 2.67, 8.00, 2.21, 0.18 and 0.62 μM for HDAC1, HDAC2, HDAC3, HDAC6 and HDAC8, respectively. NMDAR/HDAC-IN-1 efficiently penetrates the blood brain barrier [1].
JPS014 TFA is a benzamide-based Von Hippel-Lindau (VHL) E3-ligase proteolysis targeting chimeras (PROTAC). JPS014 TFA degrades class I histone deacetylase (HDAC). JPS014 TFA is potent HDAC1/2 degrader correlated with greater total differentially expressed genes and enhanced apoptosis in HCT116 cells [1].
Fimepinostat (CUDC-907) potently inhibits class I PI3Ks as well as classes I and II HDAC enzymes with an IC50 of 19/54/39 nM and 1.7/5.0/1.8/2.8 nM for PI3Kα/PI3Kβ/PI3Kδ and HDAC1/HDAC2/HDAC3/HDAC10 , respectively.
Romidepsin (FK 228) is a Histone deacetylase (HDAC) inhibitor with anti-tumor activities. Romidepsin (FK 228) inhibits HDAC1, HDAC2, HDAC4, and HDAC6 with IC50s of 36 nM, 47 nM, 510 nM and 1.4 μM, respectively [1]. Romidepsin (FK 228) is produced by Chromobacterium violaceum, induces cell G2/M phase arrest and apoptosis .
NKL 22 (compound 4b) is a potent and selective inhibitor of histone deacetylases (HDAC), with an IC50 of 199 and 69 nM for HDAC1 and HDAC3, respectively. NKL 22 exhibits selectivity over HDAC2/4/5/7/8 (IC50≥1.59 μM). NKL 22 ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice [1] .
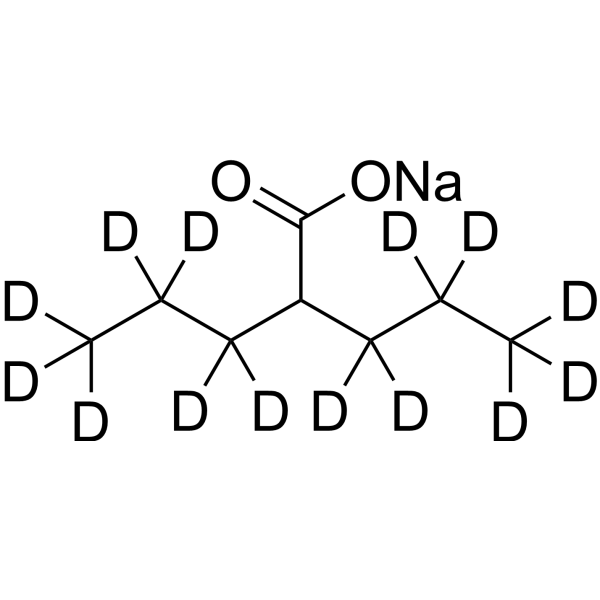
Valproic acid-d14 (sodium) is deuterium labeled Valproic acid (sodium). Valproic acid sodium salt (Sodium Valproate) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium salt activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches.
A2AAR/HDAC-IN-2 is a potent A2AAR/HDAC dual inhibitor, with good binding affinity for A2AAR (Ki=10.3 nM) and good inhibitory activity against HDAC1 (IC50=18.5 nM). A2AAR/HDAC-IN-2 can be used in study of antitumor [1].
MC2590 is a potent pyridine-containing histone deacetylase (HDAC) inhibitor. MC2590 is a inhibitor of HDAC1-3, -6, -8, and -10 (class I/IIb-selective inhibitor) with IC50s of 0.015 μM-0.156 μM. MC2590 also inhibits HDAC isoforms HDAC4, HDAC5, HDAC7, HDAC9, HDAC11 with IC50s of 1.35 μM-3.98 μM. MC2625 induces G2/M cell cycle arrest and modulates pro- and anti-apoptotic microRNAs towards apoptosis induction [1].
PI3K/HDAC-IN-2 is a potent dual PI3K/HDAC inhibitor with IC50s of 226 nM, 279 nM, 467 nM, 29 nM for PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ, respectively, and IC50s of 1.3 nM, 3.4 nM, 972 nM, 17 nM, 12 nM for HDAC1, HDAC2, HDC4, HDAC6, HDAC8, respectively. PI3K/HDAC-IN-2 exhibits PI3Kδ and class I and IIb HDAC selectivity. PI3K/HDAC-IN-2 has remarkable anticancer effects [1].
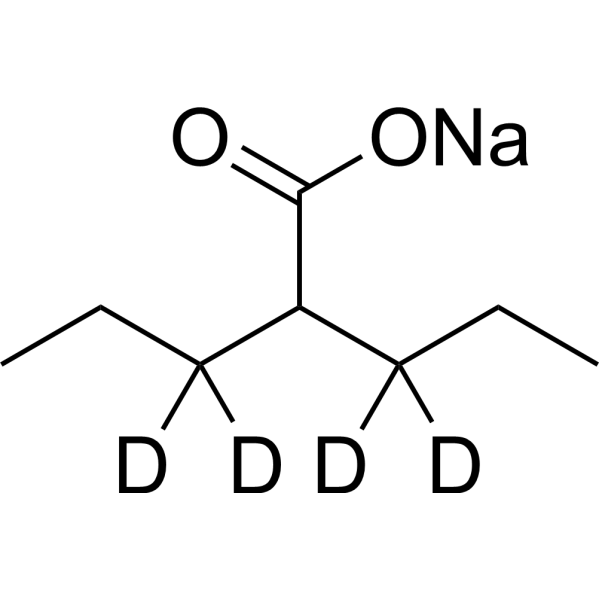
Valproic acid-d7 (sodium) is the deuterium labeled Valproic acid (sodium salt). Valproic acid sodium salt (Sodium Valproate) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium salt activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
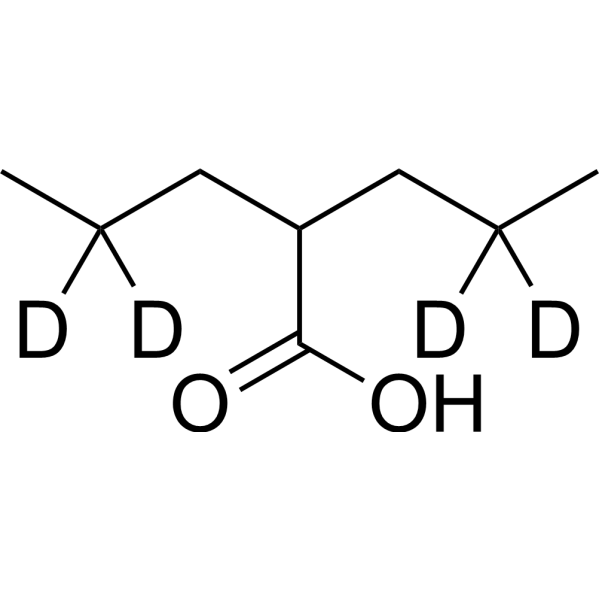
Valproic acid-d4 (sodium) is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches.
HDAC-IN-51 is a potent histone deacetylase (HDAC) inhibitor with IC50 values of 0.32, 0.353, 0.431, 0.515, and 85.4 μM for HDAC10, HDAC1, HDAC2, HDAC3 and HDAC11, respectively. HDAC-IN-51 induces cell cycle arrest and apoptosis, modulating cell cycle-/apoptosis-related miRNAs expression. HDAC-IN-51 can be used in research of cancer [1].
CDK/HDAC-IN-3 is an orally active HDACs/CDKs dual inhibitor. CDK/HDAC-IN-3 has potent and selective inhibition of CDK9, CDK12, CDK13, HDAC1, HDAC2 and HDAC3 with IC50 values of 98.32 nM, 98.85 nM, 100 nM, 62.12 nM, 93.28nM and 82.87 nM. CDK/HDAC-IN-3 can be used for the acute myeloid leukemia (AML) [1].
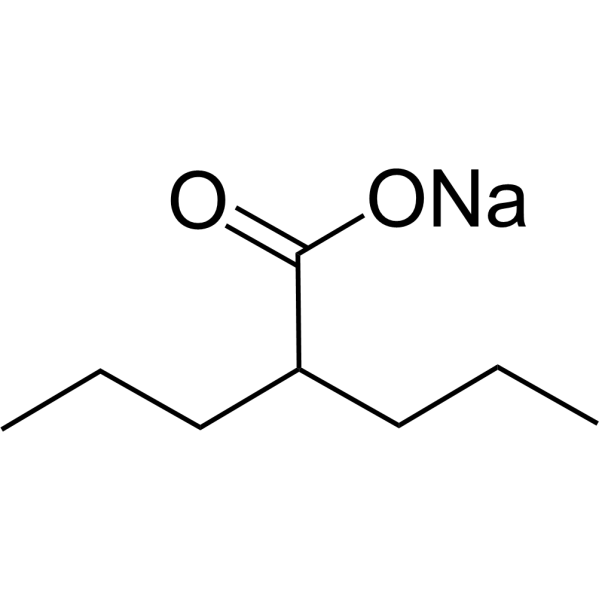
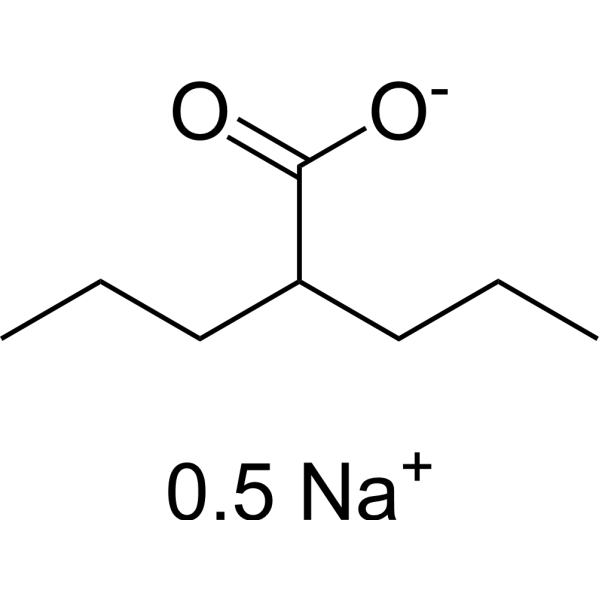
Valproic acid (Sodium Valproate) sodium is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
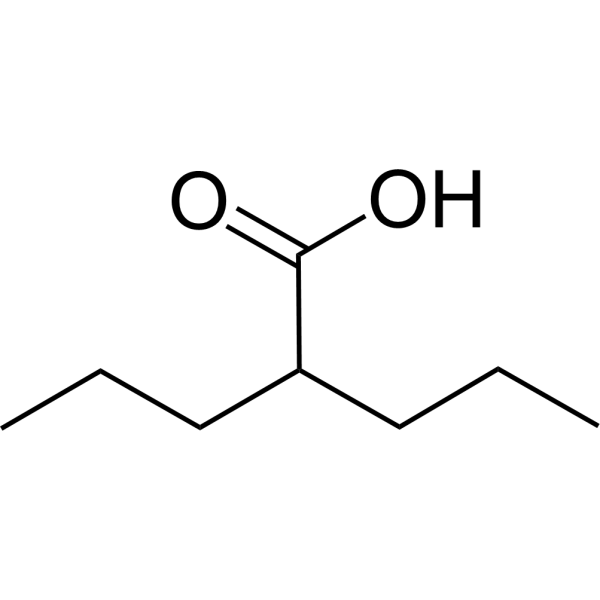
Valproic acid (VPA) is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
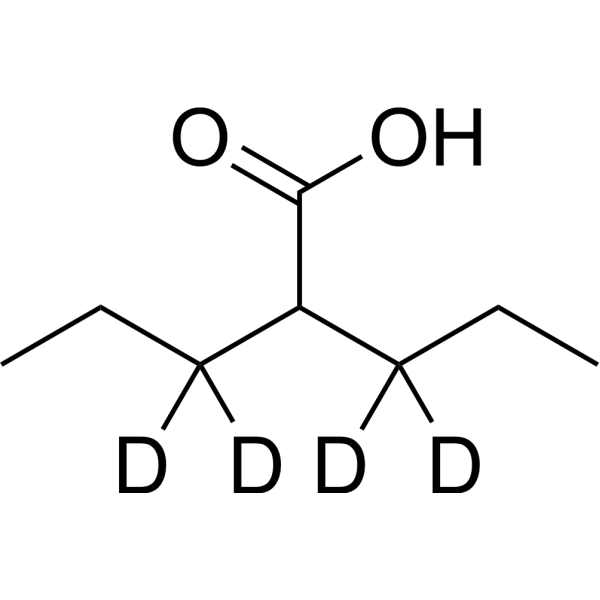
Valproic acid-d4-1 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
MPT0E028 is an orally active and selective HDAC inhibitor with IC50s of 53.0 nM, 106.2 nM, 29.5 nM for HDAC1, HDAC2 and HDAC6, respectively [1]. MPT0E028 reduces the viability of B-cell lymphomas by inducing apoptosis and possesses potent direct Akt targeting ability and reduces Akt phosphorylation in B-cell lymphoma. MPT0E028 has good anticancer activity .
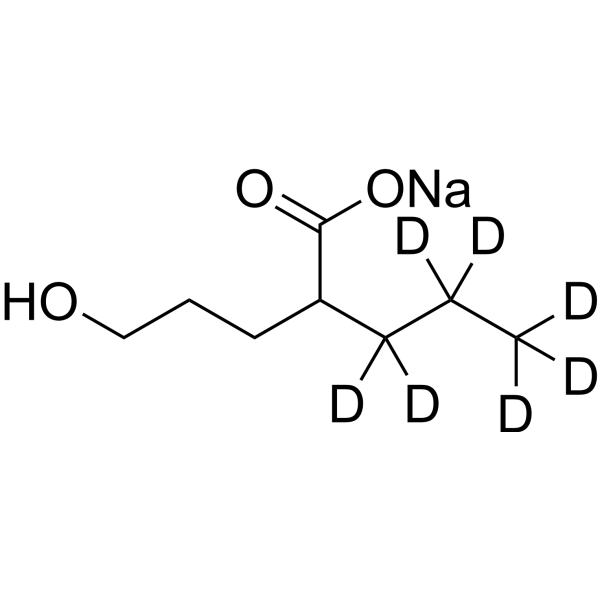
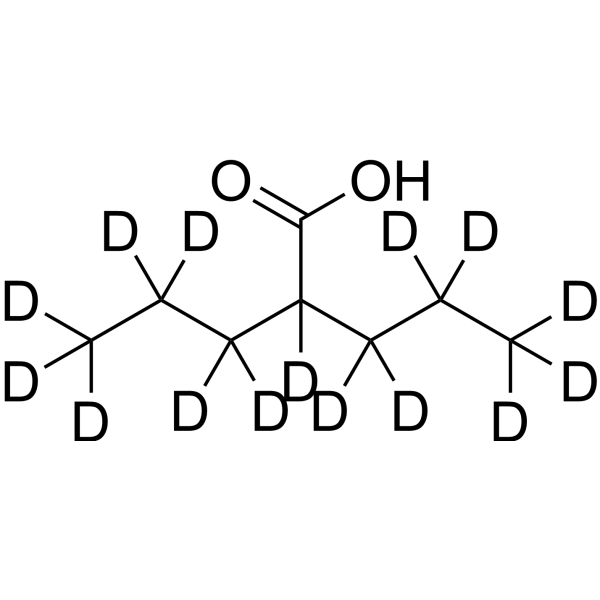
Valproic acid-d15 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
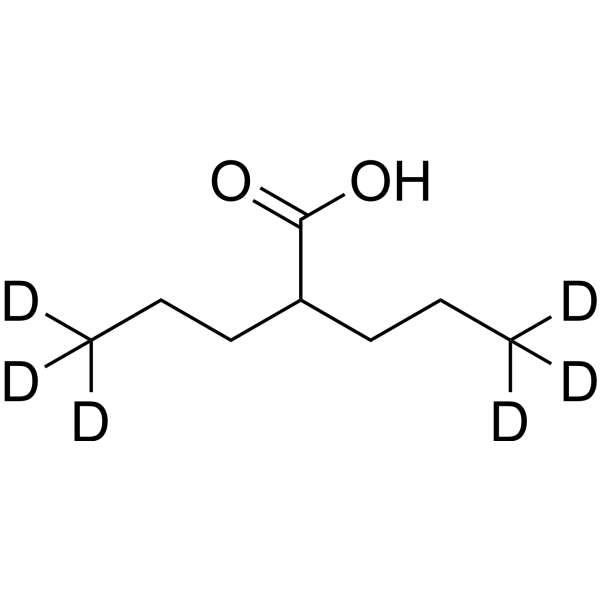
Valproic acid-d4 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
Valproic acid-d6 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
HDACs/mTOR Inhibitor 1 is a dual HDACs and mTOR inhibitor, with IC50s of 0.19 nM, 1.8 nM, 1.2 nM for HDAC1, HDAC6, mTOR, respectively. HDACs/mTOR Inhibitor 1 stimulates cell cycle arrest in G0/G1 phase and induces tumor cell apoptosis with low toxicity in vivo. HDACs/mTOR Inhibitor 1 can be used in the research of hematologic malignancies [1] .
1-Naphthohydroxamic acid (Compound 2) is a potent and selective HDAC8 inhibitor with an IC50 of 14 μM. 1-Naphthohydroxamic acid is more selectively for HDAC8 than class I HDAC1 and class II HDAC6 (IC50 >100 μM). 1-Naphthohydroxamic acid does not increase global histone H4 acetylation and also does not reduce total intracellular HDAC activity [1][2].1-Naphthohydroxamic acid can induce tubulin acetylation .
PHD2/HDACs-IN-1 is a potent PHD2/HDACs hybrid inhibitor (IC50s of 1.15 μM, 19.75 μM, 26.60 μM and 15.98 μM for PHD2, HDAC1, HDAC2 and HDAC6, respectively). PHD2/HDACs-IN-1 is a low-toxicity renoprotective agent for research of cisplatin-induced acute kidney injury (AKI) [1].
HDAC-IN-37 is a potent HDAC inhibitor with IC50s of 0.0551 μM, 1.24 μM, 0.948 μM and 34.2 μM for HDAC1, HDAC3, HDAC8 and HDAC6, respectively. HDAC-IN-37 induces histone acetylation in a slow-off manner. HDAC-IN-37 prevents cell transition from G1 phase to S phase and induces early cell apoptosis[1].
HDAC-IN-34 (compound 27) is a potent HDAC inhibitor, with IC50 values of 0.022 and 0.45 μM for HDAC1 and HDAC6, respectively. HDAC-IN-34 can bind to DNA and cause DNA damage. HDAC-IN-34 causes cells apoptosis through p53 signaling pathway. HDAC-IN-34 exhibits significant anti-proliferation effect against HCT-116 cells, with an IC50 of 1.41 μM [1].
HD-TAC7 is a potent PROTAC HDAC degrader with IC50 values of 3.6 μM, 4.2 μM and 1.1 μM for HDAC1, HDAC2 and HDAC3, respectively. HD-TAC7 can decreases NF-κB p65 in RAW 264.7 macrophages. HD-TAC7 can be used for the research of inflammatory diseases like asthma and chronic obstructive pulmonary disease (COPD) [1].
HDAC6-IN-13 (Compound 35m) is a potent, highly selective, orally active HDAC6 inhibitor with an IC50 of 0.019 μM. HDAC6-IN-13 also inhibits HDAC1, HDAC2 and HDAC3 with IC50s of 1.53, 2.06 and 1.03 μM, respectively. HDAC6-IN-13 shows significant BBB permeability and anti-inflammatory activity [1].
HDAC-IN-63 (Compound 63) is a dual FLT3/HDAC inhibitor (IC50: 0.844 and 30.0 nM for FLT3 and HDAC1 respectively). HDAC-IN-63 inhibits MV4-11 cell proliferation (IC50: 92 nM. HDAC-IN-63 induces apoptosis and arrests cell cycle in MV4-11 cells. HDAC-IN-63 can be used for research of acute myeloid leukemia (AML) [1].
HDAC6-IN-19 (Compound 14g) is a HDAC6 inhibitor (IC50: 2.68 nM). HDAC6-IN-19 also inhibits HDAC1, HDAC2 and HDAC3 with IC50s of 61.6 nM, 98.7 nM and 103 nM. HDAC6-IN-19 potently inhibits multiple cancer cell proliferation, including leukemia, colon cancer, melanoma, and breast cancer cell lines [1].
BRD3308 is a highly selective HDAC3 inhibitor with an IC50 of 54 nM. BRD3308 is 23-fold selectivity for HDAC3 over HDAC1 (IC50 of 1.26 μM) or HDAC2 (IC50 of 1.34 μM). BRD3308 suppresses pancreatic β-cell apoptosis induced by inflammatory cytokines or glucolipotoxic stress, and increases functional insulin release. BRD3308 activates HIV-1 transcription and disrupts HIV-1 latency [1] .
CRA-026440 is a potent, broad-spectrum HDAC inhibitor. The Ki values against recombinant HDAC isoenzymes HDAC1, HDAC2, HDAC3, HDAC6, HDAC8, and HDAC10 are 4, 14, 11, 15, 7, and 20 nM respectively. CRA-026440 shows antitumor and antiangiogenic activities [1]. CRA-026440 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
HDAC/Top-IN-1 is an orally active and pan HDAC/Top dual inhibitor with IC50s of 0.036 μM, 0.14 μM, 0.059 μM, 0.089 μM and 9.8 μM for HDAC1, HDAC2, HDAC3, HDAC6 and HDAC8. HDAC/Top-IN-1 efficiently induces apoptosis with S cell-cycle arrest in HEL cells. HDAC/Top-IN-1 has exhibits excellent in vivo antitumor efficacy [1].
HDAC-IN-31 is a potent, selective and orally active HDAC inhibitor with IC50s of 84.90, 168.0, 442.7, >10000 nM for HDAC1, HDAC2, HDAC3, HDAC8, respectively. HDAC-IN-31 induces apoptosis and cell cycle arrests at G2/M phase. HDAC-IN-31 shows good antitumor efficacy. HDAC-IN-31 has the potential for the research of diffuse large B-cell lymphoma [1].
HDAC-IN-39 (compound 16c) is a potent HDAC inhibitor, with IC50 values of 1.07 μM (HDAC1),1.47 μM (HDAC2), and 2.27 μM (HDAC3), respectively. HDAC-IN-39 also significantly inhibits microtubule polymerization. HDAC-IN-39 induces cell cycle arrest at the G2/M phase. HDAC-IN-39 displays promising anticancer activity against resistant cancer cells [1].
Top/HDAC-IN-1 (Compound 29b) is a topoisomerase/HDAC dual inhibitor with IC50s of 18, 230, 790, 87, and 5250 nM for HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8, respectively. Top/HDAC-IN-1 exhibits potent antitumor activities against the HCT116 cell line with the IC50 of 180 nM. Top/HDAC-IN-1 efficiently induces apoptosis with G2 cell cycle arrest in HCT116 cells [1].
Valproic acid (VPA) sodium (2:1) is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium (2:1) activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium (2:1) is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
HDAC-IN-46 (compound 12c) is a potent HDAC inhibitor with an IC50 value of 0.21 μM and 0.021 μM for HDAC1 and HDAC6, respectively. HDAC-IN-46 upregulates p-p38, and downregulates Bcl-xL and cyclin D1 in MDA-MB-231 cells. HDAC-IN-46 induces significant G2 phase arrest and apoptosis. HDAC-IN-46 can be used for researching triple-negative breast cancer (TNBC) [1].
SHP2/HDAC-IN-1 is a dual allosteric SHP2/HDAC inhibitor with IC50 values of 20.4 nM (SHP2) and 25.3 nM (HDAC1) respectively. SHP2/HDAC-IN-1 triggers efficient antitumor immunity by activating T cells, enhancing the antigen presentation function and promoting cytokine secretion. SHP2/HDAC-IN-1 can be used in the research of cancer immunoresearch [1].
JNJ-16241199 is an orally active, selective hydroxamate-based histone deacetylase (HDAC) inhibitor, with the IC50 of 3.3 nM and 23 nM for HDAC1 and HDAC8, respectively. JNJ-16241199 induces histone 3 acetylation and strongly increases the expression of p21 waf1, cip1 in A2780 ovarian carcinoma cells. JNJ-16241199 induces cell apoptosis and shows anticancer activity in a broad spectrum of human malignancies. JNJ-16241199 can be used for cancer study [1].
HDAC-IN-47 is an orally active inhibitor of histone deacetylase(HDAC), with IC50s of 19.75 nM (HDAC1), 5.63 nM (HDAC2), 40.27 nM (HDAC3), 57.8 nM (HDAC2), 302.73 nM (HDAC8), respectively. HDAC-IN-47 inhibits autophagy and induces apoptosis via the Bax/Bcl-2 and caspase-3 pathways. HDAC-IN-47 arrests cell cycle at G2/M phase, and shows anti-tumor efficacy in vivo [1].
HDAC-IN-53 is an orally active, and selective HDAC1-3 inhibitor with IC50 values of 47 nM, 125 nM, and 450 nM, respectively. HDAC-IN-53 does not inhibit class II HDACs (HDAC4, 5, 6, 7, 9; IC50>10 μM). HDAC-IN-53 induces caspase-dependent apoptosis. HDAC-IN-53 significantly inhibits the growth of human tumor xenografts in nude mice and murine tumor growth in immune-competent mice bearing MC38 colon cancer [1].
c-Met/HDAC-IN-2 is a highly potent c-Met and HDAC dual inhibitor with IC50s of 18.49 nM and 5.40 nM for HDAC1 and c-Met, respectively. c-Met/HDAC-IN-2 has antiproliferative activities against certain cancer cell lines. c-Met/HDAC-IN-2 can cause G2/M-phase arrest and induce apoptosis in HCT-116. c-Met/HDAC-IN-2 can be used for researching anti-cancer resistance [1].
MC1742 is a potent HDAC inhibitor, with IC50s of 0.1 μM, 0.11 μM, 0.02 μM, 0.007 μM, 0.61 μM, 0.04 μM and 0.1 μM for HDAC1, HDAC2, HDAC3, HDAC6, HDAC8, HDAC10 and HDAC11, respectively. MC1742 can increase acetyl-H3 and acetyl-tubulin levels and inhibits cancer stem cells growth. MC1742 can induce growth arrest, apoptosis, and differentiation in sarcoma CSC [1].
Crebinostat is a potent histone deacetylase (HDAC) inhibitor with IC50 values of 0.7 nM, 1.0 nM, 2.0 nM and 9.3 nM for HDAC1, HDAC2, HDAC3 and HDAC6, respectively. Crebinostat potently induces acetylation of both histone H3 and histone H4 as well as enhances the expression of the cAMP response element-binding protein (CREB) target gene Egr1. Crebinostat increases the density of synapsin-1 punctae along dendrites in cultured neurons. Crebinostat can modulate chromatin-mediated neuroplasticity and exhibits enhanced memory in mice [1].
HDAC6-IN-3 (Compound 14), an antiprostate cancer agent, is a potent, orally active HDAC6 inhibitor with IC50s ranging from 0.02-1.54 μM for HDAC1/2/3/6/8/10. HDAC6-IN-3 is also an effective MAO-A (IC50=0.79 μM) and LSD1 inhibitor [1]. HDAC6-IN-3 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Tubulin/HDAC-IN-2 (Compound II-19k) is a dual inhibitor of Tubulin and HDAC, with an IC50 of 0.403 μM, 0.591μM, 3.552μM, 0.459μM for HDAC1/2/3/6. Tubulin/HDAC-IN-2 blocks cell cycle arrest at G2 phase, induces cell apoptosis. Tubulin/HDAC-IN-2 inhibits the growth of hematoma and solid tumor cells, reduces tumor metastasis, and also inhibits tumor growth in a liver tumor allograft mouse model [1].
FNDR-20123 is a safe, first-in-class, and orally active anti-malarial HDAC inhibitor with IC50s of 31 nM and 3 nM for Plasmodium and human HDAC, respectively. FNDR-20123 exerts anti-malarial activity against Plasmodium falciparum asexual stage (IC50=41 nM) and sexual blood stage (IC50=190 nM for male gametocytes). FNDR-20123 inhibits HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8 (IC50=25/29/2/11/282 nM, respectively.) and inhibits Class III HDAC isoforms at nanomolar concentrations [1].
HDAC-IN-50 is a potent and orally active FGFR and HDAC dual inhibitor with IC50 values of 0.18, 1.2, 0.46, 1.4, 1.3, 1.6, 2.6, 13 nM for FGFR1, FGFR2, FGFR3, FGFR4, HDAC1, HDAC2, HDAC6, HDAC8, respectively. HDAC-IN-50 induces Apoptosis and cell cycle arrest at G0/G1 phase. HDAC-IN-50 decreases the expression of pFGFR1, pERK, pSTAT3. HDAC-IN-50 shows anti-tumor activity [1].
Tubulin/HDAC-IN-4 (compound 9n) is a dual Tubulin and HDAC inhibitor with IC50 values of 0.73, 0.43, 0.62, 2.34 µM for HDAC1, HDAC2, HDAC6, HDAC7, respectively. Tubulin/HDAC-IN-4 inhibits the tubulin polymerization by targeting the colchicine binding site. Tubulin/HDAC-IN-4 induces apoptosis and cell cycle arrest at G2/M phase. Tubulin/HDAC-IN-4 induces a significant elevation of intracellular ROS levels. Tubulin/HDAC-IN-4 shows anti-angiogenesis activity and anticancer activity [1].
FNDR-20123 free base is a safe, first-in-class, and orally active anti-malarial HDAC inhibitor with IC50s of 31 nM and 3 nM for Plasmodium and human HDAC, respectively. FNDR-20123 free base exerts anti-malarial activity against Plasmodium falciparum asexual stage (IC50=41 nM) and sexual blood stage (IC50=190 nM for male gametocytes). FNDR-20123 free base inhibits HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8 (IC50=25, 29, 2, 11, and 282 nM, respectively) and inhibits Class III HDAC isoforms at nanomolar concentrations [1].
Tacedinaline (N-acetyldinaline) is an inhibitor of the histone deacetylase (HDAC) with IC50s of 0.9, 0.9, 1.2 μM for recombinant HDAC 1, 2 and 3 respectively.
Pimelic Diphenylamide 106 is a slow, tight-binding inhibitor of class I HDAC (HDAC 1, 2, and 3, with IC50 values of 150 nM , 760nM, and 370 nM, respectively), demonstrating no activity against class II HDACs.
SIS17 is a mammalian histone deacetylase 11 (HDAC 11) inhibitor with an IC50 value of 0.83 μM, inhibits the demyristoylation HDAC11 substrate, serine hydroxymethyl transferase 2, without inhibiting other HDACs .
BRD9757 is a potent, capless and selective HDAC6 inhibitor with an IC50 of 30 nM. BRD9757 shows excellent selectivity toward HDAC6 versus the class I (>20-fold) and class II (>400-fold) HDACs .
(S)-Trichostatin A ((S)-TSA) is a HDAC6-selective inhibitor with IC50s of 9.88 nM and 11.1 nM for Zebrafish HDAC6 and Human HDAC6, respectively. (S)-Trichostatin A weakly inhibits other human HDACs .
SAHA-OH is a selective HDAC6 inhibitor (IC50=23 nM), shows a 10- to 47-fold selectivity for HDAC6 compared to HDAC 1, 2, 3, and 8. SAHA-OH shows anti-inflammatory activity, and attenuates macrophage apoptosis[1].
Psammaplin A, a marine metabolite, is a potent inhibitor of HDAC and DNA methyltransferases. Psammaplin A ia a highly potent and selective DAC1 inhibitor with an IC50 of 0.9 nM. Psammaplin A possess the antimicrobial effect on the Gram-positive bacteria and inhibits DNA synthesis and DNA gyrase activity. Antitumor Activity [1] .
HDAC-IN-43 is a potent HDAC 1/3/6 inhibitor with IC50 values of 82, 45, and 24 nM, respectively. HDAC-IN-43 is a weak PI3K/mTOR inhibitors with IC50 values of 3.6 and 3.7 μM, respectively. HDAC-IN-43 shows broad anti-proliferative activity [1].
CHDI-390576, a potent, cell permeable and CNS penetrant class IIa histone deacetylase (HDAC) inhibitor with IC50s of 54 nM, 60 nM, 31 nM, 50 nM for class IIa HDAC4, HDAC5, HDAC7, HDAC9, respectively, shows >500-fold selectivity over class I HDACs (1, 2, 3) and ~150-fold selectivity over HDAC8 and the class IIb HDAC6 isoform [1].
MC2625 is a potent pyridine-containing histone deacetylase (HDAC) inhibitor. MC2625 show selective HDAC3 and HDAC6 inhibition with IC50s of 80 nM and 11 nM. MC2625 increases acetyl-H3 and acetyl-tubulin levels and inhibits cancer stem cells (CSCs) growth by apoptosis induction [1] .
PIM-1/HDAC-IN-1 (compound 4d) is a PIM-1 inhibitor, with an IC50 of 343.87 nM. PIM-1/HDAC-IN-1 has strong inhibitory activity and selectivity against HDAC 1 and HDAC 6, with IC50 values of 63.65 and 62.39 nM, respectively. PIM-1/HDAC-IN-1 exhibits apoptosis inducing potential in MCF-7 cell lines. PIM-1/HDAC-IN-1 shows pre-G1 apoptosis and cell cycle arrest at G2/M phase [1].
Parthenolide is a sesquiterpene lactone found in the medicinal herb Feverfew. Parthenolide exhibits anti-inflammatory activity by inhibiting NF-κB activation; also inhibits HDAC1 protein without affecting other class I/II HDACs.
Valproic acid (Sodium Valproate) sodium is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
Valproic acid (VPA) is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
Valproic acid (VPA) sodium (2:1) is an orally active HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium (2:1) activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium (2:1) is used in the treatment of epilepsy, bipolar disorder, metabolic disease, HIV infection and prevention of migraine headaches [1] .
Psammaplin A, a marine metabolite, is a potent inhibitor of HDAC and DNA methyltransferases. Psammaplin A ia a highly potent and selective DAC1 inhibitor with an IC50 of 0.9 nM. Psammaplin A possess the antimicrobial effect on the Gram-positive bacteria and inhibits DNA synthesis and DNA gyrase activity. Antitumor Activity [1] .
Studies have confirmed that the histone deacetylase 1 (HDAC1) protein is a key enzyme that deacetylates lysine residues on core histones (H2A, H2B, H3, H4). This process establishes an epigenetic repressive signature that affects transcription, cell cycle, and developmental events. Histone deacetylase 1/HDAC1 Protein, Human (His-SUMO) is the recombinant human-derived Histone deacetylase 1/HDAC1 protein, expressed by E. coli , with N-SUMO, N-6*His labeled tag. The total length of Histone deacetylase 1/HDAC1 Protein, Human (His-SUMO) is 482 a.a., with molecular weight of ~71-74 kDa.
Entinostat-d4 is the deuterium labeled Entinostat[1]. Entinostat is an oral and selective class I HDAC inhibitor, with IC50s of 243 nM, 453 nM, and 248 nM for HDAC1, HDAC2, and HDAC3, respectively[2].
Valproic acid-d14 (sodium) is deuterium labeled Valproic acid (sodium). Valproic acid sodium salt (Sodium Valproate) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium salt activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches.
Tucidinostat-d4 is the deuterium labeled Tucidinostat. Tucidinostat is a potent and orally bioavailable HDAC enzymes class I (HDAC1/2/3) and class IIb (HDAC10) inhibitor, with IC50s of 95, 160, 67 and 78 nM, respectively[1].
Valproic acid-d7 (sodium) is the deuterium labeled Valproic acid (sodium salt). Valproic acid sodium salt (Sodium Valproate) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid sodium salt activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
Valproic acid-d4 (sodium) is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches.
Valproic acid-d4-1 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
Valproic acid-d15 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
Valproic acid-d4 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
Valproic acid-d6 is the deuterium labeled Valproic acid. Valproic acid (VPA; 2-Propylpentanoic Acid) is an HDAC inhibitor, with IC50 in the range of 0.5 and 2 mM, also inhibits HDAC1 (IC50, 400 μM), and induces proteasomal degradation of HDAC2. Valproic acid activates Notch1 signaling and inhibits proliferation in small cell lung cancer (SCLC) cells. Valproic acid sodium salt is used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches[1][2].
HDAC1 Antibody is a non-conjugated and Rabbit origined monoclonal antibody about 55 kDa, targeting to HDAC1. It can be used for WB,IHC-P,ICC assays with tag free, in the background of Human, Mouse.
HDAC6-IN-3 (Compound 14), an antiprostate cancer agent, is a potent, orally active HDAC6 inhibitor with IC50s ranging from 0.02-1.54 μM for HDAC1/2/3/6/8/10. HDAC6-IN-3 is also an effective MAO-A (IC50=0.79 μM) and LSD1 inhibitor [1]. HDAC6-IN-3 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Inquiry Online
Your information is safe with us. * Required Fields.