


Molecular Dynamics Simulation
Molecular dynamics is a computer simulation method based on Newtonian mechanics and integrating various disciplines such as physics, mathematics and chemistry, which is used to study the motions and interactions of molecular systems and to predict the behavior and structural properties of molecular systems. Through computer molecular simulation, researchers are able to understand the motion and biological functions of biomolecules and protein-small molecule interaction mechanisms at the molecular level. Currently, MD has been widely used in the fields of biomedicine, physics, chemistry and materials science, and is a powerful complement to theoretical calculations and experiments.
Structure Setting Main
Parameter Molecular Dynamics
Simulation Data Analysis Giving the corresponding
molecular force field Setting of box size and
pH values Optimizing the system
energy
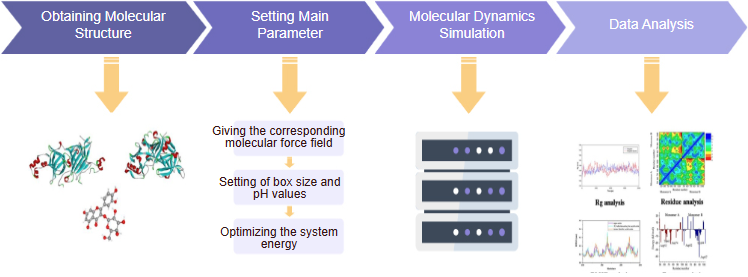
Figure 1. Diagram of molecular dynamics simulation (Food Chem. 2023 Mar 30;405(Pt A):134824.)
Analysis Items
- • Skeleton fluctuation: RMSD, RMSF
- • Interaction analysis: hydrogen bond network, salt bridge, Contact-Map, etc.
- • Conformational transformation: PCA, Energy-landscape plotting
- • Hot residues: Alanine-scanning, Energy-decomposition
- • Conformational sampling: cluster analysis, dominant conformation identification
- • Binding free energy: MM-PBSA, TI, FEP
- • Physicochemical properties: Energy, volume, pressure, temperature, density monitoring
Service Advantages
-

One-stop service
from molecular
docking to kinetic
simulation -

High performance
computer servers -

Professional
molecular simulation
and drug design
team -

Diversified analyses
available -
Competitive
price advantage -

High standard of
data privacy
management
If you have any questions, please do not hesitate to contact us via email [email protected].