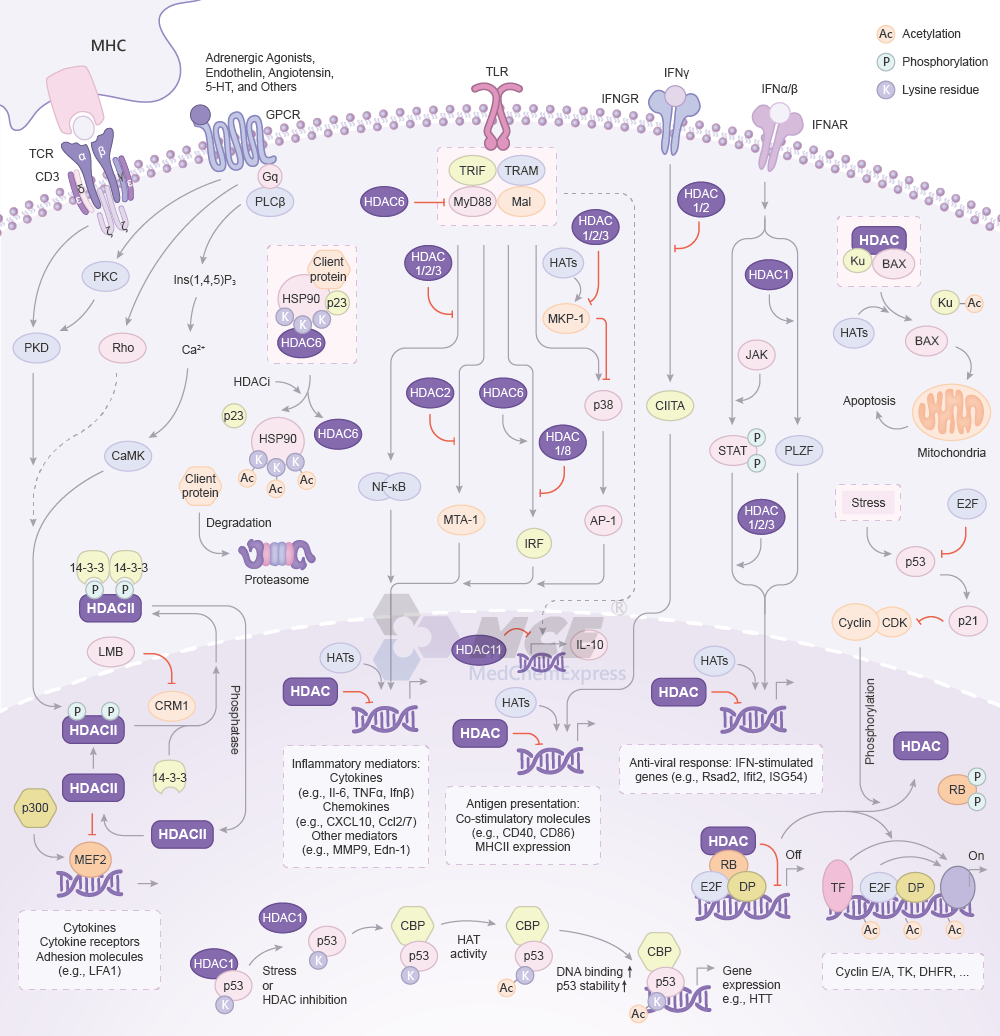
HDAC Signaling Pathway
TCR, GPCR and HDAC II interaction: Diverse agonists act through G-protein-coupled receptors (GPCRs) to activate the PKC-PKD axis, CaMK, Rho, or MHC binding to antigens stimulates TCR to activate PKD, leading to phosphorylation of class II HDACs. Phospho-HDACs dissociate from MEF2, bind 14-3-3, and are exported to the cytoplasm through a CRM1-dependent mechanism. CRM1 is inhibited by leptomycin B (LMB). Release of MEF2 from class II HDACs allows p300 to dock on MEF2 and stimulate gene expression. Dephosphorylation of class II HDACs in the cytoplasm enables reentry into the nucleus[1].
TLR: TLR signaling is initiated by ligand binding to receptors. The recruitment of TLR domain-containing adaptor protein MyD88 is repressed by HDAC6, whereas NF-κB and MTA-1 can be negatively regulated by HDAC1/2/3 and HDAC2, respectively. Acetylation by HATs enhance MKP-1 which inhibits p38-mediated inflammatory responses, while HDAC1/2/3 inhibits MKP-1 activity. HDAC1 and HDAC8 repress, whereas HDAC6 promotes, IRF function in response to viral challenge. HDAC11 inhibits IL-10 expression and HDAC1 and HDAC2 represses IFNγ-dependent activation of the CIITA transcription factor, thus affecting antigen presentation[2][3].
IRNAR: IFN-α/β induce activation of the type I IFN receptor and then bring the receptor-associated JAKs into proximity. JAK adds phosphates to the receptor. STATs bind to the phosphates and then phosphorylated by JAKs to form a dimer, leading to nuclear translocation and gene expression. HDACs positively regulate STATs and PZLF to promote antiviral responses and IFN-induced gene expression[2][3].
Cell cycle: In G1 phase, HDAC, Retinoblastoma protein (RB), E2F and polypeptide (DP) form a repressor complex. HDAC acts on surrounding chromatin, causing it to adopt a closed chromatin conformation, and transcription is repressed. Prior to the G1-S transition, phosphorylation of RB by CDKs dissociates the repressor complex. Transcription factors (TFs) gain access to their binding sites and, together with the now unmasked E2F activation domain. E2F is then free to activate transcription by contacting basal factors or by contacting histone acetyltransferases, such as CBP, that can alter chromatin structure[4].
The function of non-histone proteins is also regulated by HATs/HDACs. p53: HDAC1 impairs the function of p53. p53 is acetylated under conditions of stress or HDAC inhibition by its cofactor CREB binding protein (CBP) and the transcription of genes involved in differentiation is activated. HSP90: HSP90 is a chaperone that complexes with other chaperones, such as p23, to maintain correct conformational folding of its client proteins. HDAC6 deacetylates HSP90. Inhibition of HDAC6 would result in hyperacetylated HSP90, which would be unable to interact with its co-chaperones and properly lead to misfolded client proteins being targeted for degradation via the ubiquitin-proteasome system[5][6].
Reference:
[1]. Vega RB, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5.Mol Cell Biol. 2004 Oct;24(19):8374-85.
[2]. Shakespear MR, et al. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011 Jul;32(7):335-43.
[3]. Suliman BA, et al. HDACi: molecular mechanisms and therapeutic implications in the innate immune system.Immunol Cell Biol. 2012 Jan;90(1):23-32.
[4]. Brehm A, et al. Retinoblastoma protein meets chromatin.Trends Biochem Sci. 1999 Apr;24(4):142-5.
[5]. Butler R, et al. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders.Nat Rev Neurosci. 2006 Oct;7(10):784-96
[6]. Minucci S, et al. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer.Nat Rev Cancer. 2006 Jan;6(1):38-51.