


Cobicistat
Based on 12 publication(s) in Google Scholar
Cobicistat is a potent and selective inhibitor of cytochrome P450 3A (CYP3A) enzymes with IC50s of 30-285 nM. Cobicistat is a pharmacokinetic enhancer which increases the overall absorption of several HIV medications.
Nur für Forschungszwecke. Wir verkaufen nicht an Patienten.
- Reinheit: 99.49%
- CAS. Nr.: 1004316-88-4
- Formel: C40H53N7O5S2
- Molecular Weight:776.02
-
Speicherung:Powder -20°C, 3 years , 4°C, 2 years ; In solvent -80°C, 1 year , -20°C, 6 months

To place orders, for customer services and technical support, please contact: MedChemExpress USA
Tel: 609-228-6898 E-mail: [email protected] [email protected]



-
 Biologische Aktivität
Biologische Aktivität
-
Chemical Information
-
Lösungsmittel & Löslichkeit
- Protokoll
- Reinheit & Dokumentation
- Verweise
-
 Help & FAQs
Help & FAQs
-


Anti-Infection Compound Library
HY-L002
-
Metabolism/Protease Compound Library
HY-L012
-
FDA-Approved Drug Library
HY-L022
-
Anti-Cancer Compound Library
HY-L025
-
Antiviral Compound Library
HY-L027
-
Anti-Aging Compound Library
HY-L034
-
Drug Repurposing Compound Library
HY-L035
-
Antioxidant Compound Library
HY-L037
-
Oxygen Sensing Compound Library
HY-L045
-
Anti-COVID-19 Compound Library
HY-L052
-
Orally Active Compound Library
HY-L061
-
FDA Approved & Pharmacopeial Drug Library
HY-L066
-
Drug-Induced Liver Injury (DILI) Compound Library
HY-L076
-
Mitochondria-Targeted Compound Library
HY-L089
-
Rare Diseases Drug Library
HY-L102
-
EMA-Approved Drug Library
HY-L116
-
FDA-Approved Anticancer Drug Library
HY-L122
-
Human Metabolite Library
HY-L123
-
Heterocyclic Compound Library
HY-L138
-
Metabolic Enzyme Compound Library
HY-L146
-
Highly Selective Inhibitors Library
HY-L158
-
Multi-Target Compound Library
HY-L176
-
Radioprotector Library
HY-L178
-
Bioactive Compound Library Max
HY-L181
-
MCE Bioactive Compound Library
HY-L001V
-
Drug Repurposing Compound Library Plus
HY-L035P
-
FDA-Approved Drug Library Plus
HY-L022P
-
FDA-Approved Drug Library Mini
HY-L022M
-
Bioactive Compound Library
HY-L001
-
High-Throughput Bioactive Compound Library
HY-L205
-
Mass Spectrometry Human Metabolite Library
HY-L215
-
Nephrotoxicity Compound Library
HY-L229
-
Classic FDA-Approved Drug Library
HY-L261


Publications Citing Use of MedChemExpress (MCE) Cobicistat
More- ACS Nano. 2025 Sep 30;19(38):33879-33890. [Abstract]
- Acta Pharm Sin B. 2021 Jun;11(6):1607-1616. [Abstract]
- Acta Pharm Sin B. 2021 Mar;11(3):810-822. [Abstract]
- J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
- Eur J Med Chem. 2020 Oct 15;204:112626. [Abstract]
- Eur J Med Chem. 2020 Aug 15;200:112427. [Abstract]
- Toxicol Sci. 2021 Apr 27;181(1):58-67. [Abstract]
- Expert Opin Drug Metab Toxicol. 2019 Nov;15(11):975-984. [Abstract]
- Cancer Res Commun. 2021 Nov;1(2):79-89. [Abstract]
- Viruses. 2020 Apr 16;12(4):452. [Abstract]
- Chemrxiv. 2021, Jun 10.
- Drug Metab Lett. 2016;10(2):111-23. [Abstract]
 Customer Validation & Images
Customer Validation & Images

-
Cell Proliferation/Viability Assay
-
WB
-
In Vivo Imaging
-
Histological Imaging/Staining
-
Flow Cytometry


Biologische Aktivität
|
CYP3 |
HIV-1 |
In HIV-1 protease enzymatic assay and antiviral cellular assays. Cobicistat is inactive against HIV-1 protease (IC50>30 μM) . And Cobicistat has no inhibitory effect against HIV replication in a multicycle 5-day MT-2 HIV infection assay (EC50>30 μM). In assays using MT-2 cells, Cobicistat exhibits minimal cytotoxicity, with a CC50 value above 80 μM[1]. The mode of inhibition of human CYP3A by Cobicistat and Ritonavir shares the same mechanism of action for the inhibition of CYP3A. It shows its inhibitory effects on CYP3A may involve directly at the heme group of the CYP3A enzyme[1]. The minimal adverse effects of Cobicistat in these assays suggest a lower potential for toxicity related to altered lipid metabolism. In the lipid accumulation assay with the human adipocytes, Ritonavir shows a clear effect with an EC50 of 16 μM. However, Cobicistat exhibits no effect at a concentration up to 30 μM[1]. In the glucose uptake assay with mouse adipocytes, Ritonavir shows a pronounced effect at the concentration of 10 μM. In contrast, the effects on glucose uptake by Cobicistat (10 μM) is significantly less[1].
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.
| NCT Number | Sponsor | Condition | Start Date |
Phase
|
|---|---|---|---|---|
| NCT01329991 | Plexxikon| | 2011-05 | PHASE1 |


Chemical Information
-
CAS. Nr. 1004316-88-4
-
Appearance Solid
-
Molecular Weight 776.02
-
Formel C40H53N7O5S2
-
Color White to yellow
-
SMILES
CN(C(N[C@@H](CCN1CCOCC1)C(N[C@@H](CC2=CC=CC=C2)CC[C@@H](NC(OCC3=CN=CS3)=O)CC4=CC=CC=C4)=O)=O)CC5=CSC(C(C)C)=N5
-
Synonyms
GS-9350
-
Versand
Room temperature in continental US; may vary elsewhere.
-
Speicherung
Powder -20°C 3 years 4°C 2 years In solvent -80°C 1 year -20°C 6 months


Publications (12)
-
Journal Impact Factor
-
Most Recent
-
ACS Nano
Endolysosomal Sequestration Effects Controlled Release of BRAF Paradox Breaker Nanoparticles. [Abstract]2025 Sep 30;19(38):33879-33890. PMID: 40954128 -
Acta Pharm Sin B
Can remdesivir and its parent nucleoside GS-441524 be potential oral drugs? An in vitro and in vivo DMPK assessment. [Abstract]2021 Jun;11(6):1607-1616. PMID: 34221871 -
Acta Pharm Sin B
Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. [Abstract]2021 Mar;11(3):810-822. PMID: 33777683 -
J Exp Clin Cancer Res
CYP3A5 promotes glioblastoma stemness and chemoresistance through fine-tuning NAD+/NADH ratio. [Abstract]2025 Jan 3;44(1):3. PMID: 39754188
Cobicistat purchased from MedChemExpress. Usage Cited in: J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
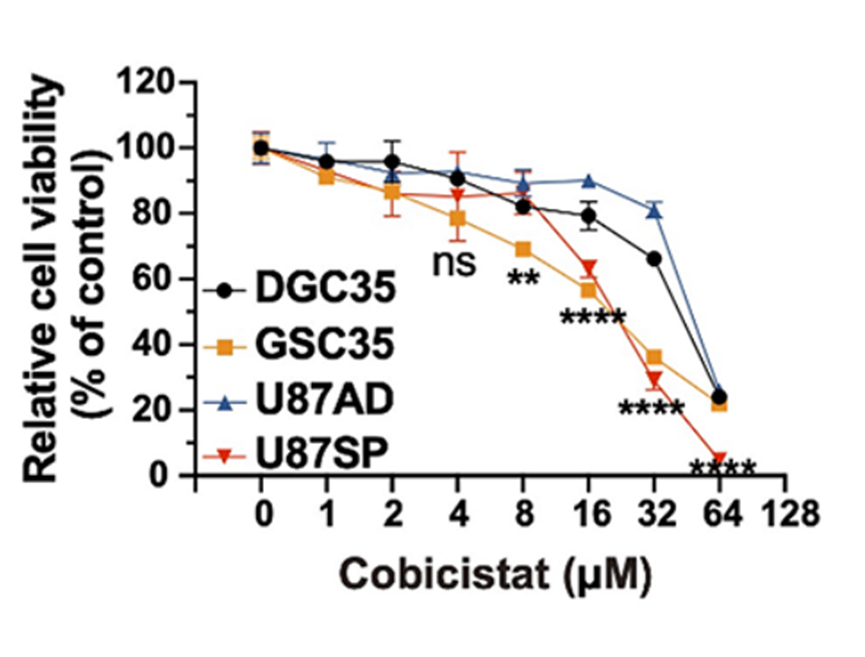
Cell viability assay with the increasing concentrations of Cobicistat (Cobi) (1, 2, 4, 8, 16, 32, 64, 128 μM).
Cobicistat purchased from MedChemExpress. Usage Cited in: J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
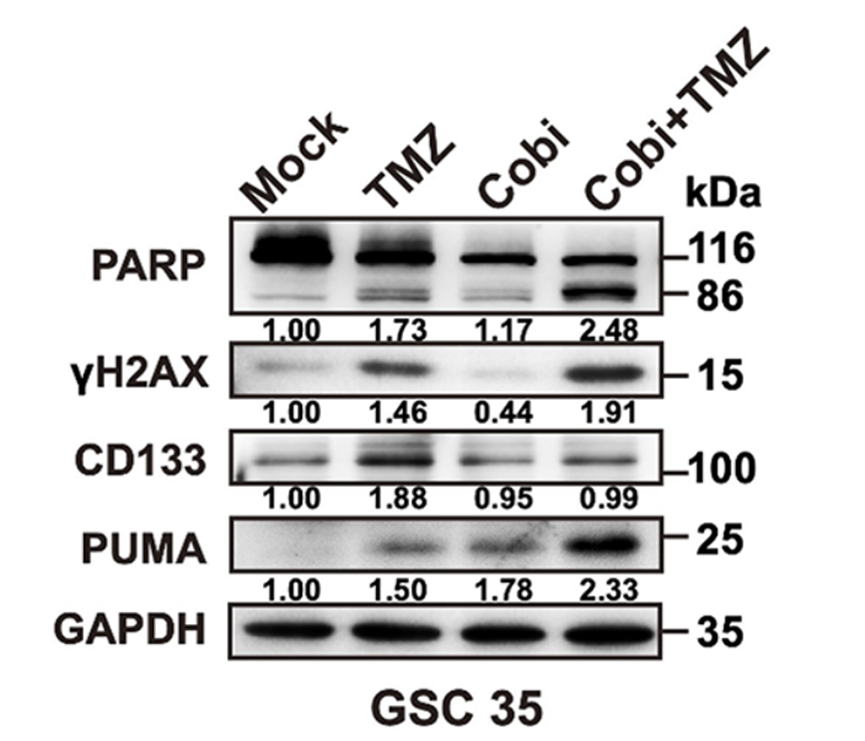
Immunoblots of cleaved PARP, γH2AX, CD133, and PUMA in GSCs treated with Cobicistat (Cobi) and TMZ.
Cobicistat purchased from MedChemExpress. Usage Cited in: J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
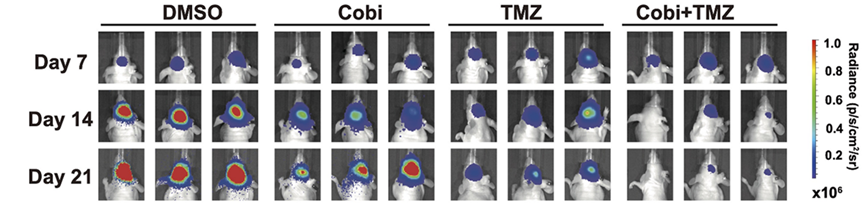
Representative images of bioluminescence intensities of nude mice intracranially implanted with U87MG GSCs in treatment of DMSO, TMZ (5 mg/kg), Cobicistat (Cobi) (10 mg/kg) or TMZ (5 mg/kg) + Cobi (10 mg/kg) by gavage.
Cobicistat purchased from MedChemExpress. Usage Cited in: J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
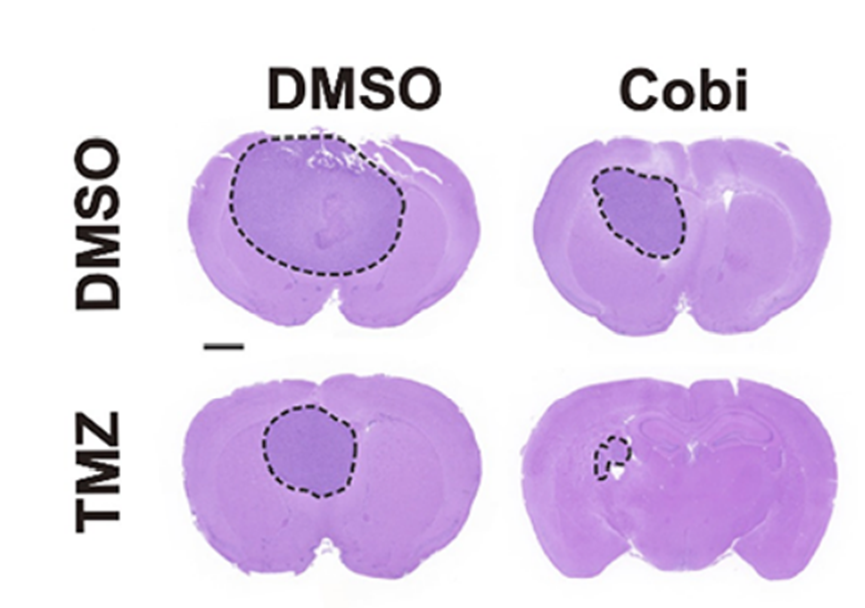
Representative images of H&E staining of tumor sections in treatment of DMSO, TMZ (5 mg/kg), Cobicistat (Cobi) (10 mg/kg) or TMZ (5 mg/kg) + Cobi (10 mg/kg) by gavage.
Cobicistat purchased from MedChemExpress. Usage Cited in: J Exp Clin Cancer Res. 2025 Jan 3;44(1):3. [Abstract]
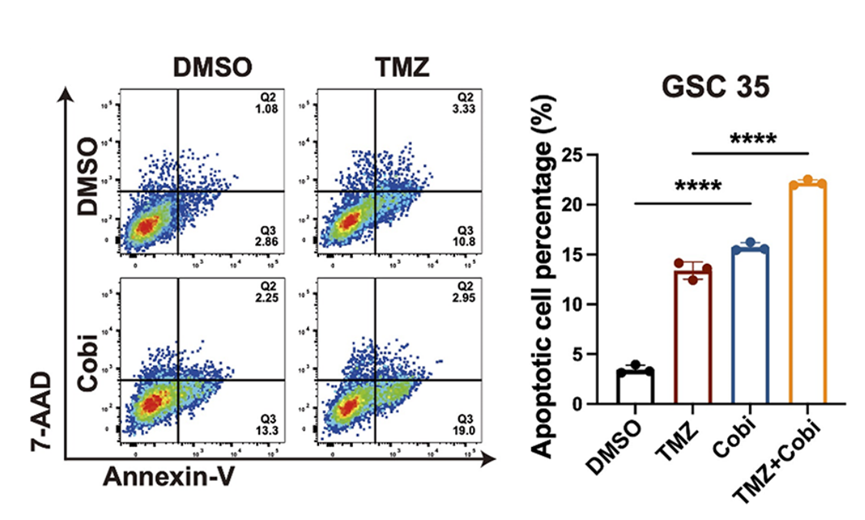
Representative images and quantification of cell apoptosis measured by flow cytometry in treatment of DMSO, TMZ (5 mg/kg), Cobicistat (Cobi) (10 mg/kg) or TMZ (5 mg/kg) + Cobi (10 mg/kg) by gavage.
-
Eur J Med Chem
2020 Oct 15;204:112626. PMID: 32814250 -
Eur J Med Chem
2020 Aug 15;200:112427. PMID: 32438252 -
Toxicol Sci
2021 Apr 27;181(1):58-67. PMID: 33629115 -
Expert Opin Drug Metab Toxicol
In vitro- in vivo correlation of the drug-drug interaction potential of antiretroviral HIV treatment regimens on CYP1A1 substrate riociguat. [Abstract]2019 Nov;15(11):975-984. PMID: 31619082 -
Cancer Res Commun
2021 Nov;1(2):79-89. PMID: 34950932 -
Viruses
Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. [Abstract]2020 Apr 16;12(4):452. PMID: 32316297 -
-
Drug Metab Lett
2016;10(2):111-23. PMID: 26935921


Lösungsmittel & Löslichkeit
DMSO : 250 mg/mL (322.16 mM; Need ultrasonic; Hygroscopic DMSO has a significant impact on the solubility of product, please use newly opened DMSO)
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 1 year; -20°C, 6 months. When stored at -80°C, please use it within 1 year. When stored at -20°C, please use it within 6 months.
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 1 year; -20°C, 6 months. When stored at -80°C, please use it within 1 year. When stored at -20°C, please use it within 6 months.
Konzentration (Stammlösung) × Volumen (Stammlösung) = Konzentration (Ziellösung) × Volumen (Ziellösung)
Select the appropriate dissolution method based on your experimental animal and administration route.
- For the following dissolution methods, please ensure to first prepare a clear stock solution using an In Vitro approach and then sequentially add co-solvents:
- To ensure reliable experimental results, the clarified stock solution can be appropriately stored based on storage conditions. As for the working solution for In Vivo experiments, it is recommended to prepare freshly and use it on the same day.
- The percentages shown for the solvents indicate their volumetric ratio in the final prepared solution. If precipitation or phase separation occurs during preparation, heat and/or sonication can be used to aid dissolution.
Add each solvent one by one: 10% DMSO 40% PEG300 5% Tween-80 45% Saline
Solubility: ≥ 2.08 mg/mL (2.68 mM); Clear solution
This protocol yields a clear solution of ≥ 2.08 mg/mL (saturation unknown).
Taking 1 mL working solution as an example, add 100 μL DMSO stock solution (20.8 mg/mL) to 400 μL PEG300, and mix evenly; then add 50 μL Tween-80 and mix evenly; then add 450 μL Saline to adjust the volume to 1 mL.
Preparation of Saline: Dissolve 0.9 g sodium chloride in ddH₂O and dilute to 100 mL to obtain a clear Saline solution.
Add each solvent one by one: 10% DMSO 90% (20% SBE-β-CD in Saline)
Solubility: ≥ 2.08 mg/mL (2.68 mM); Clear solution
This protocol yields a clear solution of ≥ 2.08 mg/mL (saturation unknown).
Taking 1 mL working solution as an example, add 100 μL DMSO stock solution (20.8 mg/mL) to 900 μL 20% SBE-β-CD in Saline, and mix evenly.
Preparation of 20% SBE-β-CD in Saline (4°C, storage for one week): 2 g SBE-β-CD powder is dissolved in 10 mL Saline, completely dissolve until clear.
Please enter the basic information of animal experiments:
-
-
-
-
Recommended: Prepare an additional quantity of animals to account for potential losses during experiments.
Please enter your animal formula composition:
-
%DMSO +
Recommended: Keep the proportion of DMSO in working solution below 2% if your animal is weak.
-
%+
-
+%Tween-80 + +
-
%Saline +
The co-solvents required include: DMSO, . All of co-solvents are available by MedChemExpress (MCE). , Tween 80. All of co-solvents are available by MedChemExpress (MCE).
Working solution concentration: 0.22 mg/mL
Method for preparing stock solution: mg drug dissolved in μL DMSO. Stock solution concentration: mg/mL.
1. Take μL DMSO stock solution;
2. Add μL .
μL , mix evenly;
3. Then add μL Tween 80, mix evenly;
4. Then add μL
Please ensure that the stock solution in the first step is dissolved to a clear state, and add co-solvents in sequence. You can use ultrasonic heating (ultrasonic cleaner, recommended frequency 20-40 kHz), vortexing, etc. to assist dissolution.

Protokoll
Inhibition of human cytochrome P450 activities is determined in duplicate in pooled human hepatic microsomal fractions following current scientific and regulatory guidelines. Reaction conditions are linear with respect to incubation time and hepatic microsomal protein concentration. Substrates are present at concentrations equal to or less than their respective Km values determined under the same reaction conditions. Metabolite and/or substrate concentrations are determined using specific, internal standard controlled HPLC MS/MS assays. For reactions monitoring metabolite formation there is less than 20% consumption of substrate during the reaction. Unless otherwise noted microsomal fraction, diluted in potassium phosphate buffer, is preincubated with substrate and inhibitor for 5 min at 37°C and the reaction initiated by the addition of an NADPH generating system followed by further incubation at 37°C with shaking. Enzyme-selective positive control inhibitors are tested in parallel. At appropriate times aliquots of the mixture are removed and the reaction terminated by addition to a mixture of methanol and acetonitrile containing the respective internal standard. After centrifugation aliquots of the supernatant are subjected to HPLC-MS/MS analysis.
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.
Five-fold serial dilutions of the tested compounds are prepared in triplicate in 96-well plates. MT-2 cells are added to plates at a density of 20,000/well in a final assay volume of 200 μL. After a 5-day incubation at 37°C, the cytotoxic effect is determined using a cell viability assay. One hundred μL media is removed from each well and replaced with 100 μL of phosphate-buffered saline containing 1.7 mg/mL XTT and 5 μg/mL PMS. Following 1-hour incubation at 37°C, 20 μL of 2% Triton X- 100 is added to each well and absorbance is read at 450 nm with a background subtraction at 650 nm. The data are plotted as cell viability vs. drug concentration. Cell viability is expressed as a percentage of the signal from untreated samples (0% cytotoxicity) after the subtraction of signal from samples treated with 10 μM of Podophyllotoxin (100% cytotoxicity). The CC50 value is calculated from the inhibition plots as the concentration of drug which inhibits cell proliferation by 50%.
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.


Reinheit & Dokumentation
-
Data Sheet (279 KB)
-
SDS (393 KB)
- English - EN (393 KB)
- Français - FR (393 KB)
- Deutsch - DE (393 KB)
- Norwegian - NO (393 KB)
- Español - ES (393 KB)
- Swedish - SV (393 KB)
- Italian - IT (393 KB)
- Korean - KR (393 KB)
- Portuguese - PT (393 KB)
-
Handling Instructions (2659 KB)
Verweise


Complete Stock Solution Preparation Table
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 1 year; -20°C, 6 months. When stored at -80°C, please use it within 1 year. When stored at -20°C, please use it within 6 months.
| Optional Solvent | Concentration Solvent Mass | 1 mg | 5 mg | 10 mg | 25 mg |
|---|---|---|---|---|---|
| DMSO | 1 mM | 1.2886 mL | 6.4431 mL | 12.8863 mL | 32.2157 mL |
| 5 mM | 0.2577 mL | 1.2886 mL | 2.5773 mL | 6.4431 mL | |
| 10 mM | 0.1289 mL | 0.6443 mL | 1.2886 mL | 3.2216 mL | |
| 15 mM | 0.0859 mL | 0.4295 mL | 0.8591 mL | 2.1477 mL | |
| 20 mM | 0.0644 mL | 0.3222 mL | 0.6443 mL | 1.6108 mL | |
| 25 mM | 0.0515 mL | 0.2577 mL | 0.5155 mL | 1.2886 mL | |
| 30 mM | 0.0430 mL | 0.2148 mL | 0.4295 mL | 1.0739 mL | |
| 40 mM | 0.0322 mL | 0.1611 mL | 0.3222 mL | 0.8054 mL | |
| 50 mM | 0.0258 mL | 0.1289 mL | 0.2577 mL | 0.6443 mL | |
| 60 mM | 0.0215 mL | 0.1074 mL | 0.2148 mL | 0.5369 mL | |
| 80 mM | 0.0161 mL | 0.0805 mL | 0.1611 mL | 0.4027 mL | |
| 100 mM | 0.0129 mL | 0.0644 mL | 0.1289 mL | 0.3222 mL |


 Powered by Bioz
Powered by Bioz