Search Result
Results for "
Gi signaling
" in MedChemExpress (MCE) Product Catalog:
2
Biochemical Assay Reagents
3
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-112779
-
|
|
Endogenous Metabolite
|
Infection
|
|
Pertussis Toxin is a protein-based AB5-type exotoxin produced by the bacterium Bordetella pertussis, which causes whooping cough. Pertussis Toxin inhibits G protein-coupled receptor (GPR) signaling through Gi proteins .
|
-

-
- HY-114364
-
|
|
Endogenous Metabolite
P2Y Receptor
|
Metabolic Disease
|
|
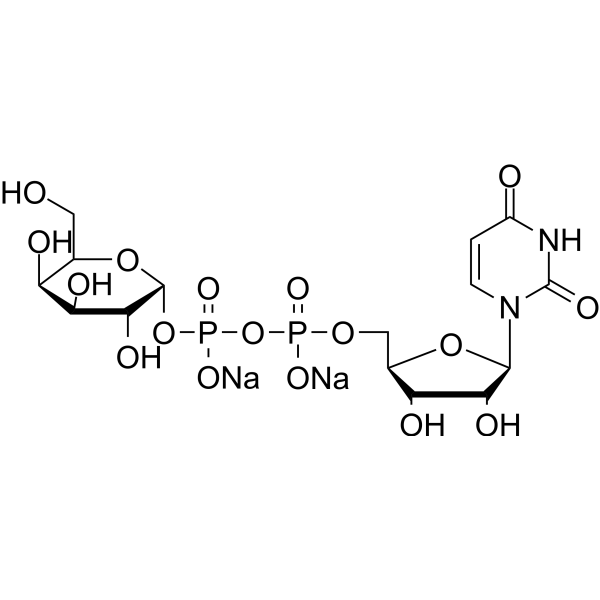
UDP-Galactose disodium is a monosaccharide and a key glycosyl donor molecule in cells that participates in nucleotide sugar metabolism. UDP-Galactose disodium is the natural agonist of the P2Y14 receptor coupled to Gi proteins in the immune system (IC50 = 0.67 μM, hP2Y14). UDP-Galactose disodium can be used to study cell signal transduction and substance metabolism .
|
-
-
- HY-P99643
-
|
UCB6114
|
Dan family
|
Cancer
|
|
Ginisortamab (UCB6114) is a fully human IgG4P anti Gremlin-1 monoclonal antibody, with mean IC50 values of 8.2 nM and 9 nM against human and mouse gremlin-1, respectivly. Ginisortamab inhibits gremlin-1 antagonism of BMP signaling pathways. Ginisortamab has the potential for the research of gastrointestinal (GI) tumors .
|
-
-
- HY-119272
-
EF24
2 Publications Verification
|
ERK
Caspase
NF-κB
Apoptosis
p38 MAPK
Reactive Oxygen Species (ROS)
|
Inflammation/Immunology
Cancer
|
|
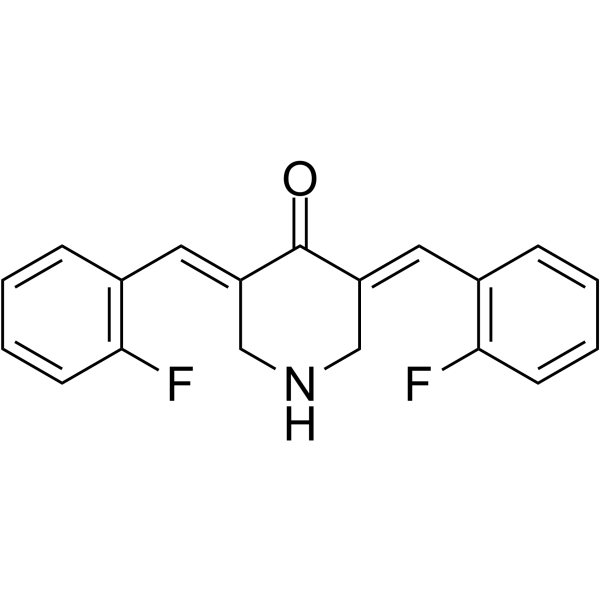
EF24, a curcumin analogue, is an NF-kB inhibitor with great anti-tumor efficacy and oral bioavailability via deactivation of the MAPK/ERK signaling pathway in oral squamous cell carcinoma (OSCC). EF24 is active against melanoma and breast cancer cell lines with GI50 values of 0.7 μM and 0.8 μM, respectively. EF24 induces cell cycle arrest and apoptosis in MDA-MB-231 human breast cancer cells and DU-145 human prostate cancer cells. EF24 increases the levels of activated caspase 3 and 9, and decreases the phosphorylated forms of MEK1 and ERK .
|
-
-
- HY-B0194A
-
|
|
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
Tizanidine hydrochloride, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine hydrochloride primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine hydrochloride has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine hydrochloride can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine hydrochloride can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI) .
|
-
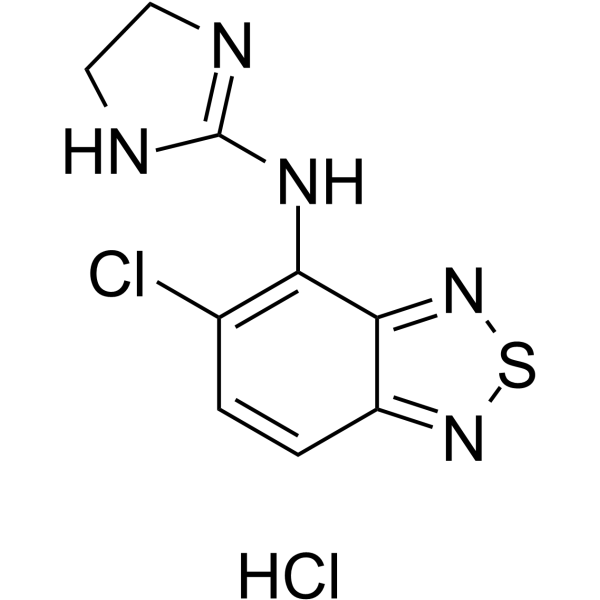
-
- HY-B0194
-
|
|
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
Tizanidine, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI) .
|
-
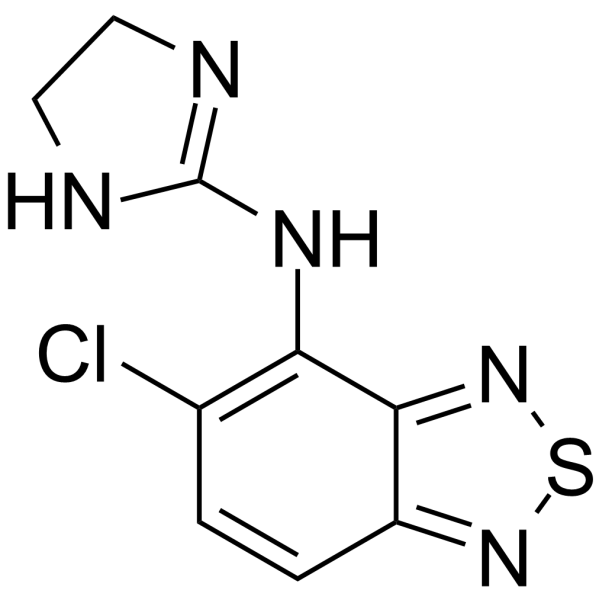
-
- HY-145404
-
|
|
Opioid Receptor
|
Metabolic Disease
|
|
Mitragynine pseudoindoxyl is a potent orally active agonist of the μ-opioid receptor (MOR-1, Ki=0.8 nM) and an antagonist of the δ-opioid receptor (DOR-1, Ki=3.0 nM). Mitragynine pseudoindoxyl has moderate affinity for the κ-opioid receptor (KOR-1, Ki=24 nM) and does not recruit β-arrestin-2, acting through G protein-mediated signaling pathways without β-arrestin-2-related activation. Mitragynine pseudoindoxyl produces potent analgesic activity through a mixed μ-agonist/δ-antagonist mechanism, with low side effects such as physical dependence, respiratory depression, and constipation, and no rewarding or aversive behaviors. Mitragynine pseudoindoxyl reduces hyperactivity, inhibits GI transit, and enhances characteristics, making it a potential analgesic .
|
-

-
- HY-103211
-
L748337
1 Publications Verification
|
Adrenergic Receptor
|
Cardiovascular Disease
Metabolic Disease
Cancer
|
|
L748337 is a potent β3-adrenergic receptor antagonist and displays selectivity over β1 and β2 receptors. The Ki values of L748337 for β3-, β2- and β1-adrenoceptors are 4.0 nM, 204 nM and 390 nM, respectively . L748337 couples predominantly to Gi to activate MAPK signaling and increases phosphorylation of Erk1/2 with pEC50 value of 11.6 . L748337 can be used for the research of cancer, nonalcoholic fatty liver disease (NAFLD), and cardiovascular related diseases .
|
-
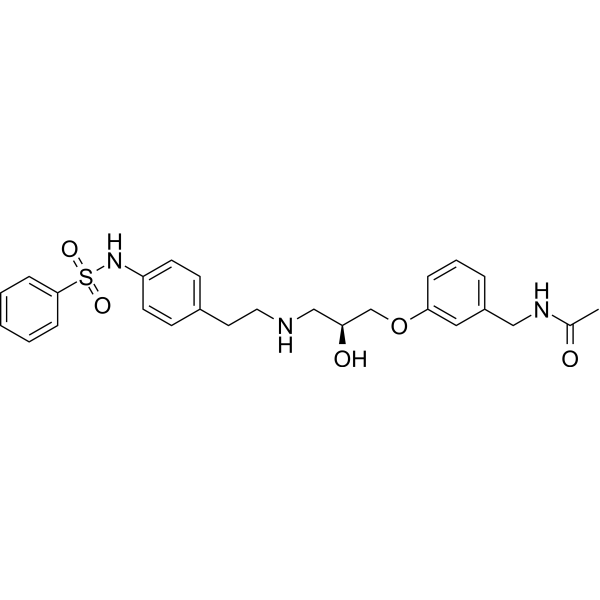
-
- HY-150524
-
|
|
Endogenous Metabolite
P2Y Receptor
|
Metabolic Disease
|
|
UDP-Galactose is a monosaccharide and a key glycosyl donor molecule in cells that participates in nucleotide sugar metabolism. UDP-Galactose is the natural agonist of the P2Y14 receptor coupled to Gi proteins in the immune system (IC50 = 0.67 μM, hP2Y14). UDP-Galactose can be used to study cell signal transduction and substance metabolism .
|
-
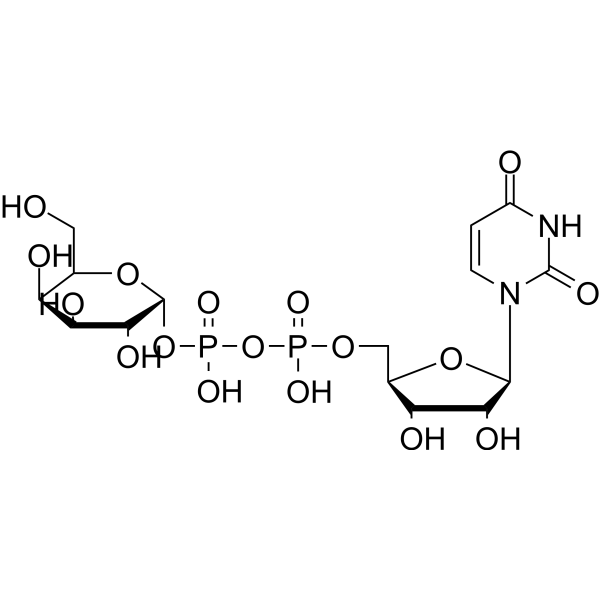
-
- HY-111385
-
|
|
Dopamine Receptor
5-HT Receptor
Arrestin
|
Neurological Disease
|
|
UNC9994 hydrochloride is a functionally selective, β-arrestin–biased dopamine D2 receptor (D2R) agonist that selectively activates β-arrestin recruitment and signaling. UNC9994 hydrochloride shows a binding affinity with a Ki of 79 nM for D2R. UNC9994 hydrochloride is also an antagonist of Gi-regulated cAMP production and partial agonist for D2R/β-arrestin-2 interactions. UNC9994 hydrochloride shows antipsychotic-like activity .
|
-
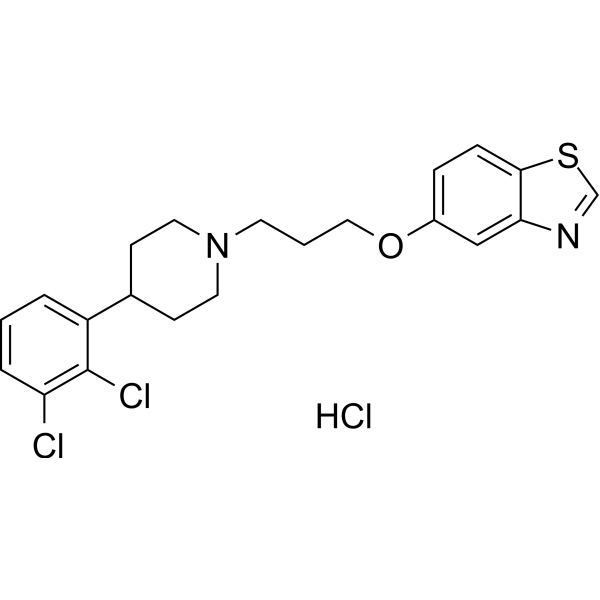
-
- HY-107024
-
|
OGP(10-14); Historphin
|
Src
|
Others
|
|
Osteogenic Growth Peptide (10-14) (OGP(10-14)), the C-terminal truncated pentapeptide of osteogenic growth peptide (OGP), retains the full OGP-like activity. Osteogenic Growth Peptide (10-14) is responsible for the binding to the OGP receptor and activates an intracellular Gi-protein-MAP kinase signaling pathway. Osteogenic Growth Peptide (10-14) is a potent mitogen and stimulator of osteogenesis and hematopoiesis. Osteogenic Growth Peptide (10-14) acts as a Src inhibitor .
|
-
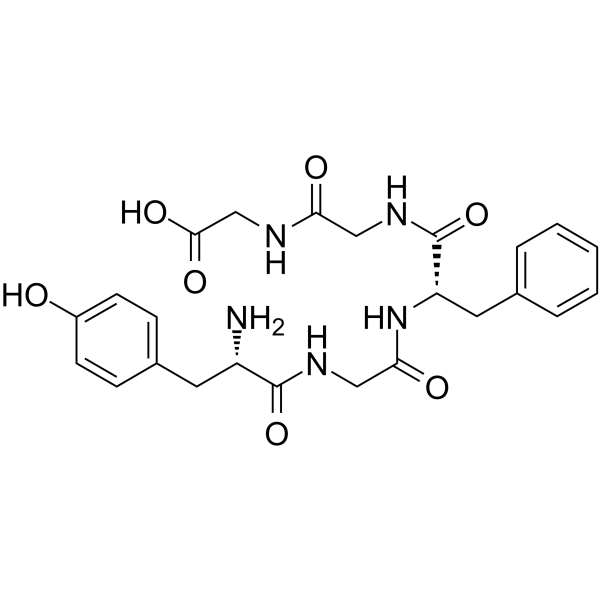
-
- HY-108648A
-
|
2-Methylthioadenosine diphosphate; 2-Methylthio-ADP
|
P2Y Receptor
|
Neurological Disease
|
|
2-MeSADP is a P2Y receptor agonist, with an EC50 of 5 nM for human P2Y12, EC50 values of 19 nM and 13 nM for human P2Y13, and an EC50 of 6.2 nM for mouse P2Y13. It shows slightly higher selectivity for P2Y12 over human P2Y13. 2-MeSADP triggers increases in intracellular calcium levels, Gi-mediated cAMP inhibition, adenylate cyclase inhibition and downstream signal transduction. 2-MeSADP can be used in research related to glaucoma .
|
-

-
- HY-171978
-
|
|
Adrenergic Receptor
|
Cardiovascular Disease
|
|
LM-189 is a β2-adrenergic receptor (β2AR) ligand and G protein-biased modulator with a human β2AR Ki of 0.063 nM.LM-189 promotes β2AR coupling to Gαs and Gαi heterotrimers, stabilizes distinct β2AR conformations including a TM6 outward state, and increases β2AR ICL2 dynamics.LM-189 restricts β2AR ligand-binding pocket conformational heterogeneity, stabilizes polar ligand-receptor interaction networks, and exhibits bias toward Gαi signaling over Gαs signaling.LM-189 enabled cryo-EM structural characterization of the β2AR-Gi complex.LM-189 can be used for the research of congestive heart failure .
|
-

-
- HY-162702
-
|
|
PROTACs
Androgen Receptor
|
Cancer
|
|
AZ‘3137 is an orally active PROTAC-type androgen receptor (AR) degrader with a DC50 value of 22 nM. AZ‘3137 can degrade L702H mutant AR (DC50 of 92 nM). AZ'3137 can inhibit cell proliferation of LNCaP, with a GI50 value of 74 nM. AZ'3137 can inhibit AR signaling and tumor growth in prostate cancer mice (Pink: AR Ligand (HY-172954); Blue: CRBN ligand (HY-172955); Black: linker (HY-W262798); E3 Ligand+Linker: HY-172956) .
|
-

-
- HY-156025
-
|
|
Hydroxycarboxylic Acid Receptor (HCAR)
|
Inflammation/Immunology
|
|
HCAR2 agonist 1 (Compound 9n) is a Gi protein-biased allosteric modulator of HCAR2. HCAR2 agonist 1 activates the Gi protein signaling pathway. HCAR2 agonist 1 shows anti-inflammatory effect, and reduces mRNA level of pro-inflammatory cytokine (TNF-α, IL-1β, IL-6, and MCP-1). HCAR2 agonist 1 enhances anti-inflammatory effects of orthosteric agonists in the mouse model of colitis .
|
-
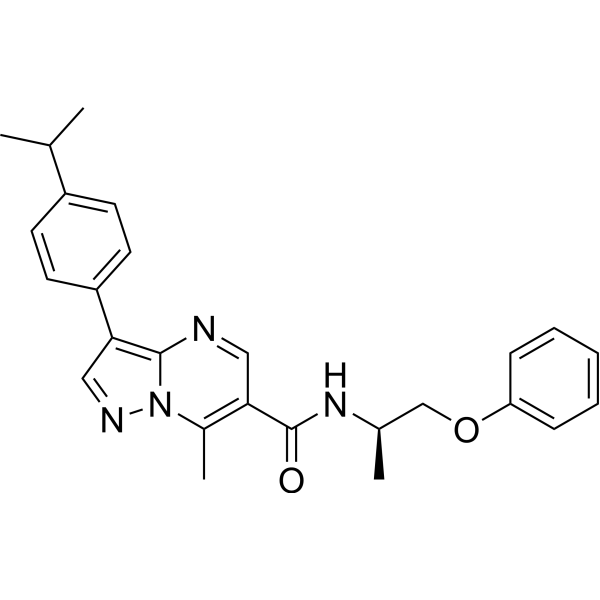
-
- HY-N0927
-
|
Tetrahydrocolumbamine; (S)-Tetrahydrocolumbamine
|
Dopamine Receptor
|
Others
Neurological Disease
|
|
(-)-Isocorypalmine (Tetrahydrocolumbamine; (S)-Tetrahydrocolumbamine) is a dopamine receptor ligand and modulator capable of crossing the blood-brain barrier. (-)-Isocorypalmine exhibits receptor selectivity, acting as a partial agonist of dopamine D1 and D5 receptors (coupled via Gs proteins), as well as an antagonist of D2, D3 and D4 receptors (by blocking Gi protein-mediated signaling). (-)-Isocorypalmine shows no significant binding to various non-dopamine receptors, ion channels and transporters. (-)-Isocorypalmine is metabolically stable in vivo, effectively inhibits spontaneous and cocaine-induced hyperlocomotion, sensitization and conditioned place preference in mice, and does not induce addictive place preference when administered alone. (-)-Isocorypalmine can be used in addiction research .
|
-
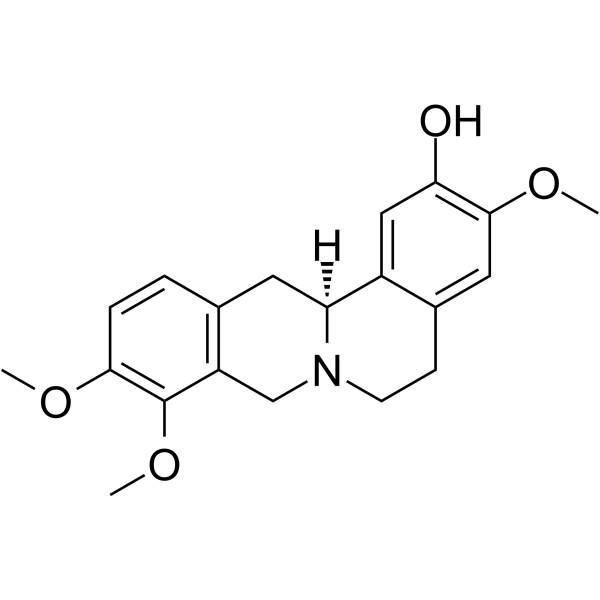
-
- HY-175231
-
|
|
5-HT Receptor
|
Neurological Disease
|
|
ST171 is a bitopic 5-HT1AR agonist with an Ki of 0.41 nM. ST171 selectively activates Gi/o signaling pathway and inhibits 5-HT1AR-mediated cAMP accumulation without Gs activation and marginal β-arrestin recruitment. T171 reduces hypersensitivity in chronic neuropathic and inflammatory pain mice model. ST171 can be used for pain research .
|
-

-
- HY-170027
-
|
|
HIF/HIF Prolyl-Hydroxylase
AMPK
|
Cancer
|
|
LW1564 is an inhibitor for HIF-1α with an IC50 of 1.2 µM in HepG2. LW1564 inhibits mitochondrial respiration, reduces ATP production, stimulates HIF-1α degradation, and inhibits proliferation of various cancer cells with GI50 of 0.4-4.6 μM. LW1564 activates AMPK signaling pathway and inhibits lipid synthesis. LW1564 exhibits antitumor in HepG2 xenograft mouse model .
|
-

-
- HY-B0194S
-
|
|
Isotope-Labeled Compounds
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
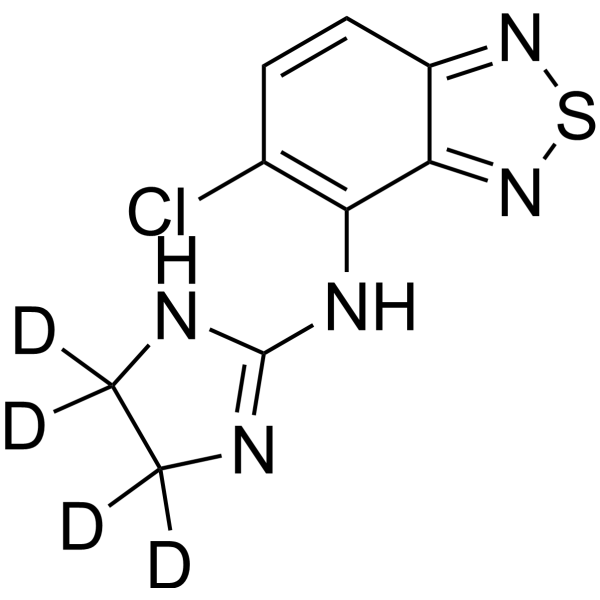
Tizanidine-d4 is the deuterium labeled Tizanidine (HY-B0194). Tizanidine, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-
-
- HY-P5358
-
|
|
Protease Activated Receptor (PAR)
|
Others
|
|
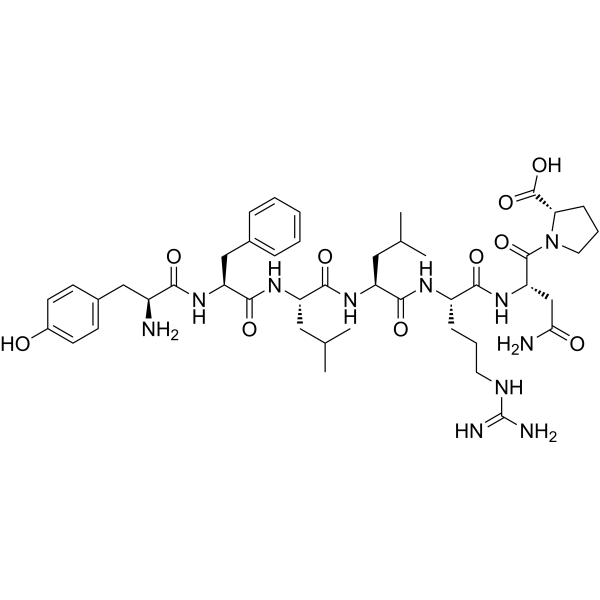
YFLLRNP is a biological active peptide. (a partial agonist of PAR-1. YFLLRNP selectively active G12/13 signaling pathway without activating Gq or Gi pathways at low concentrations. YFLLRNP (60 μM))
|
-
-
- HY-114364S
-
|
|
Isotope-Labeled Compounds
|
Others
|
|
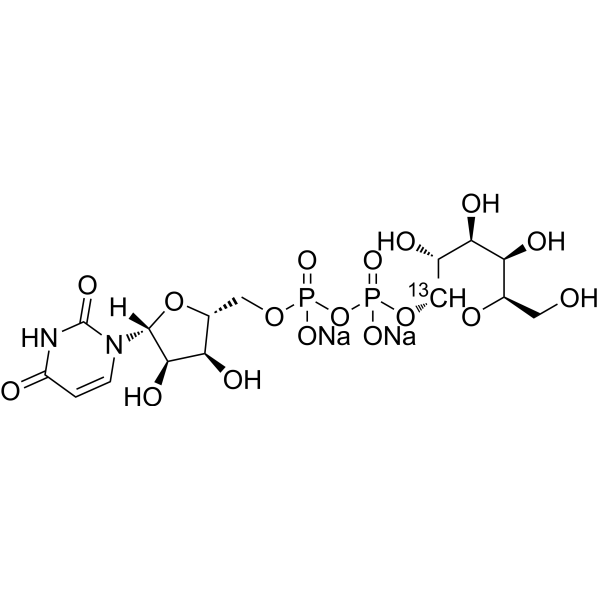
UDP-α-D-Galactose- 13C disodium is the 13C labeled UDP-α-D-Galactose disodium. UDP-Galactose disodium is a monosaccharide and a key glycosyl donor molecule in cells that participates in nucleotide sugar metabolism. UDP-Galactose disodium is the natural agonist of the P2Y14 receptor coupled to Gi proteins in the immune system (IC50 = 0.67 μM, hP2Y14). UDP-Galactose disodium can be used to study cell signal transduction and substance metabolism .
|
-
-
- HY-121140
-
|
|
Free Fatty Acid Receptor
|
Metabolic Disease
|
|
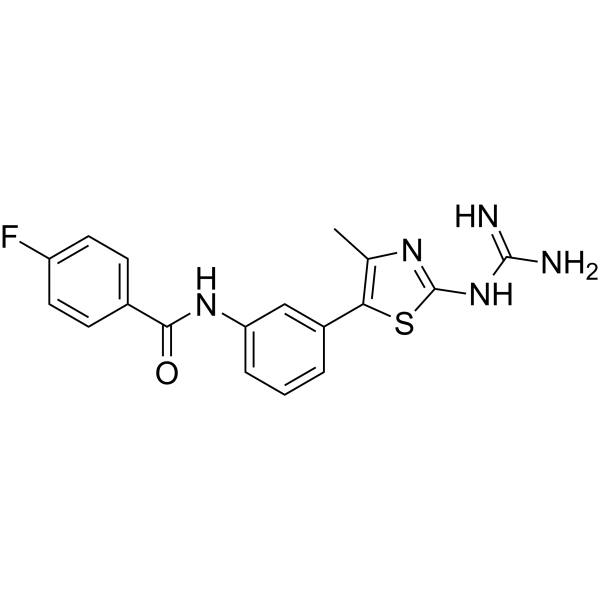
AZ1729 is a potent free fatty acid 2 receptor (FFA2) activator, acting as a direct allosteric agonist and as a positive allosteric modulator. AZ1729 increases the activity of the endogenously produced short chain fatty acid propionate in Gi-mediated pathways, but not at those transduced by Gq/G11. AZ1729 induces inhibition of isoproterenol-induced lipolysis in mouse adipocytes. AZ1729 also can Induce migration of human neutrophils. AZ1729 can be used for researching the signaling pathways of the physiological roles of FFA2 .
|
-
-
- HY-175298
-
|
|
STAT
Src
Apoptosis
|
Cancer
|
|
STAT-3/c-Src-IN-1 (Compound 12d) is a dual STAT-3 (IC50=0.844 μM) and c-Src (IC50=0.268 μM) inhibitor. STAT-3/c-Src-IN-1 blocks tumor cell proliferation, migration, and angiogenesis signaling pathways. STAT-3/c-Src-IN-1 exhibits potent anti-proliferative activity against melanoma (SK-MEL-2) and CNS cancer (SNB-75) cell lines (GI50=-5.75 μM and -5.63 μM), inducing tumor cell apoptosis via G0/G1 and G2/M phase cell cycle arrest. STAT-3/c-Src-IN-1 is promising for research of melanoma and glioblastoma .
|
-

-
- HY-168997
-
|
|
Cannabinoid Receptor
|
Neurological Disease
Inflammation/Immunology
|
|
CB1R agonist 1 is a potent, CNS-penetrant, and selective Cannabinoid-1 receptor (CB1R) agonist with a Ki of 0.95 nM, modulating Gi/o signaling. CB1R agonist 1 exhibits high selectivity over CB2R and other off-target GPCRs, including GPR55, GPR18, and GPR119. CB1R agonist 1 induces analgesia and hypothermia, and potentiates opioid analgesia in mice. CB1R agonist 1 can be used for the research of pain .
|
-

-
- HY-W744699
-
|
(+)-Larixol
|
Src
ERK
Akt
|
Inflammation/Immunology
|
|
Larixol is an fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Larixol can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Larixol inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM), cathepsin G release (IC50: 2.76 μM), and chemotaxis. Larixol improves neutrophil hyperactivation and reduces inflammation or tissue damage. A series of Larixol derivatives were found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-
-
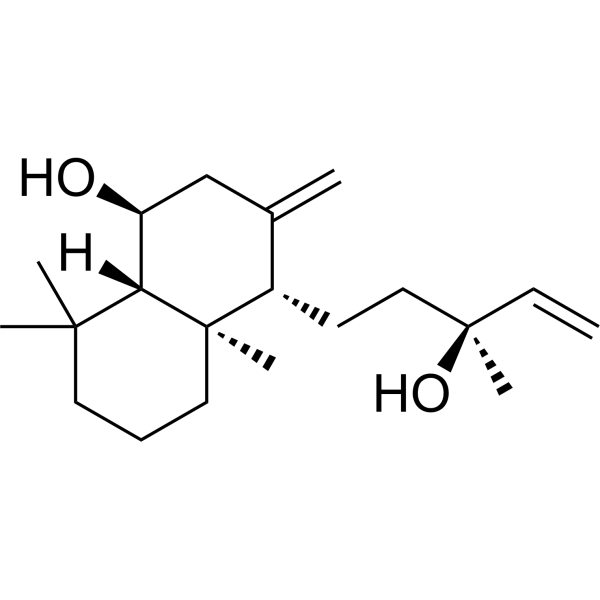
- HY-116445
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
UNC9975 is a D2R agonist that displays signaling bias via β-arrestin–ergic signaling and a simultaneously antagonist of Gi-regulated cAMP production and partial agonist for D2R/β-arrestin-2 interactions. UNC9975 can be utilized in antipsychotic research .
|
-

-
- HY-172966
-
|
|
EGFR
|
Cancer
|
|
EG31 is an EGFR inhibitor. EG31 effectively inhibits the proliferation of triple-negative breast cancer (TNBC) cells (GI50 values of MDA-MB-231 and Hs578T cells are 498.90 nM and 740.73 nM, respectively) and induces apoptosis by inhibiting the EGFR signaling pathway. EG31 still maintains antiproliferative activity against 5-fluorouracil (5-FU)-resistant TNBC cells (GI50: 519.5 nM). EG31 can be used to study TNBC resistance .
|
-

-
- HY-168925
-
|
|
Wnt
PARP
β-catenin
|
Cancer
|
|
EXQ-2d is the inhibitor for tankyrase and inhibits TNKS1 and TNKS2 with an IC50 of 48.8 nM and 13.8 nM (pIC50=7.31 and 7.86). EXQ-2d inhibits WNT/β-catenin signaling pathway with IC50 of 515 nM. EXQ-2d exhibits anti-proliferative activity in cancer cells COLO 320DM and RKO with GI50 of 4.9 μM and 77 μM .
|
-

-
- HY-145900
-
|
|
MDM-2/p53
|
Cancer
|
|
S100A2-p53-IN-1 (compound 51) is a S100A2-p53 interactions inhibitor. S100A2 is a Ca 2+ binding protein with implications in cell signaling and is known to be upregulated in pancreatic cancer. S100A2-p53-IN-1 can inhibit the growth of the MiaPaCa-2 pancreatic cancer cell line (GI 50 of 1.2-3.4 μM) .
|
-
-
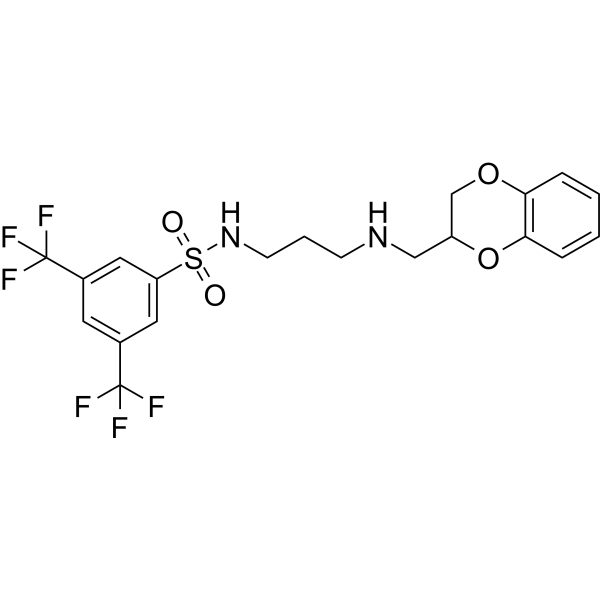
- HY-173039
-
|
|
Microtubule/Tubulin
Keap1-Nrf2
Reactive Oxygen Species (ROS)
Apoptosis
|
Cancer
|
|
α-Tubulin polymerization-IN-1 (Compound 8l) is an inhibitor for α-Tubulin polymerization. α-Tubulin polymerization-IN-1 modulates the NRF2/KEAP-1 signaling pathway, induces ROS generation in PC-3 cell, thereby inducing apoptosis in PC-3. α-Tubulin polymerization-IN-1 inhibits the proliferation of PC-3 cell with a GI50 of 0.17 µM, arrests the cell cycle at G2/M phase. α-Tubulin polymerization-IN-1 exhibits antitumor efficacy in mouse model .
|
-

-
- HY-W806047
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
BRD4 Inhibitor-37 is a compound with anticancer activity that has inhibitory activity against BRD4. BRD4 Inhibitor-37 has an IC50 of approximately 0.05-0.1 μM in binding assays and shows a GI50 of 0.1-0.3 μM in cell-based assays. The effect of BRD4 Inhibitor-37 on c-Myc, a downstream protein of BRD4, has been validated, demonstrating its ability to intervene in this signaling pathway. BRD4 Inhibitor-37 exhibits selectivity among five different bromodomain proteins, enhancing its potential as a BET protein inhibitor .
|
-

-
- HY-W745090
-
|
|
Formyl Peptide Receptor (FPR)
Src
ERK
Akt
p38 MAPK
|
Others
|
|
Isomaltulose monohydrate is a fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Isomaltulose monohydrate can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Isomaltulose monohydrate inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM) , cathepsin G release (IC< sub>50: 2.76 μM) and chemotaxis. Isomaltulose monohydrate can improve excessive activation of neutrophils and reduce inflammation or tissue damage. A series of derivatives of Isomaltulose monohydrate are found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-
-
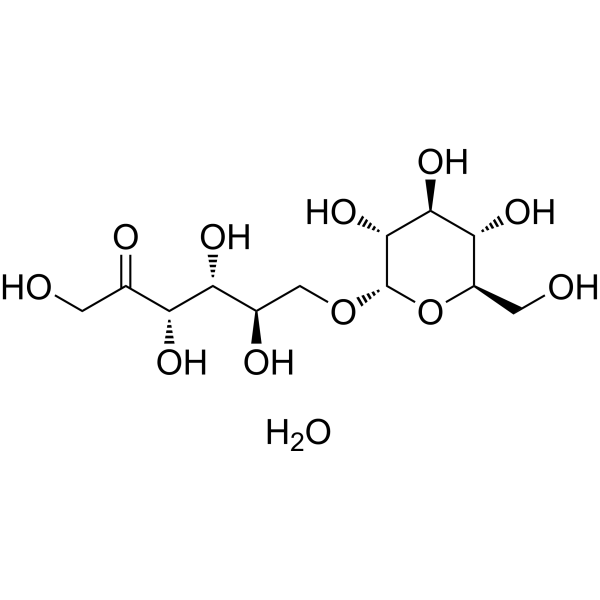
- HY-B0194R
-
|
|
Reference Standards
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
Tizanidine (Standard) is the analytical standard of Tizanidine (HY-B0194). This product is intended for research and analytical applications. Tizanidine, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-

-
- HY-B0194AR
-
|
|
Reference Standards
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
Tizanidine hydrochloride (Standard) is the analytical standard of Tizanidine hydrochloride (HY-B0194A). This product is intended for research and analytical applications. Tizanidine hydrochloride, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine hydrochloride primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine hydrochloride has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine hydrochloride can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine hydrochloride can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-

-
- HY-B0194AS
-
|
|
Isotope-Labeled Compounds
Adrenergic Receptor
Apoptosis
Akt
Wnt
β-catenin
|
Neurological Disease
Endocrinology
Cancer
|
|
Tizanidine-d4 hydrochloride is deuterium labeled Tizanidine hydrochloride (HY-B0194A). Tizanidine hydrochloride, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine hydrochloride primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine hydrochloride has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine hydrochloride can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine hydrochloride can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-
-
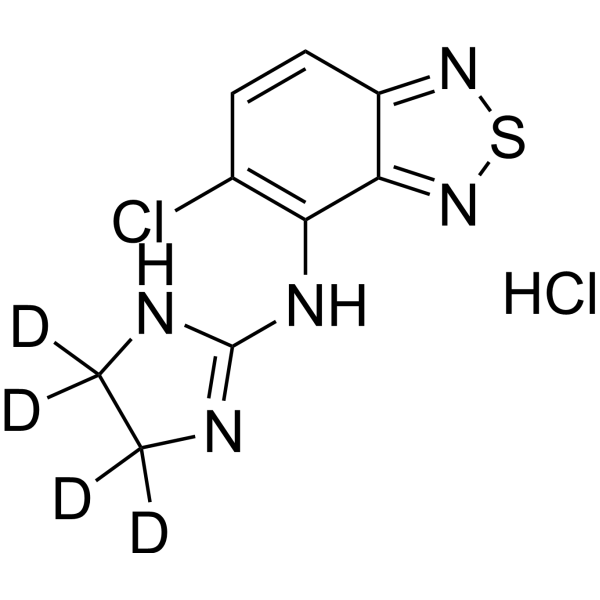
- HY-179721
-
|
|
Cholecystokinin Receptor
|
Neurological Disease
|
|
CCKBR agonist-2 (Compound z-44) is a Gi-preferring CCKBR agonist. CCKBR agonist-2 effectively activates the CCKBR-Gi signaling pathway (EC50 = 0.27 nM), but has almost no activity on Gq and Gs signaling pathways. CCKBR agonist-2 shows no significant protective effect in the mouse Alzheimer's disease model, proving that the simple activation of the Gi signal pathway does not play a dominant role in the improvement of cognitive function .
|
-

-
- HY-47412
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
Cariprazine impurity 1 is a dopamine D2 receptor (D2R) agonist. Cariprazine impurity 1 modulates D2R-mediated Gi/o signaling pathway to inhibit cAMP production, and regulates D2R-mediated β-arrestin2 recruitment pathway .
|
-

-
- HY-179451
-
|
|
Apoptosis
p38 MAPK
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Apoptosis inducer 53 is an apoptosis inducer. Apoptosis inducer 53 can inhibit proliferation of human tumor cell lines (A549, HeLa, SW1573, T-47D, WiDr) with GI50 values of 2.5-9.1 μM. Apoptosis inducer 53 can induce cancer cells apoptosis and reduce colony formation. Apoptosis inducer 53 can activate p38α MAPK signaling and exerts anti-inflammatory effect. Apoptosis inducer 53 also shows anti-Leishmania donovani activity. Apoptosis inducer 53 can be used for the researches of cancer, infection and inflammation .
|
-

-
- HY-181878
-
|
Z7149
|
Serotonin Transporter
Adrenergic Receptor
5-HT Receptor
|
Neurological Disease
|
Z8779877149 (Z7149) is a blood-brain barrier-permeable multi-target ligand that targets SERT (Ki=198 nM), α2A adrenergic receptor (Ki=180 nM; EC50=440 nM) and 5-HT2A receptor (EC50=172 nM, Emax=76%). Z8779877149 inhibits 5-HT reuptake and activates Gi and Gq protein signaling pathways, respectively. Z8779877149 effectively alleviates pain responses as well as depression- and anxiety-like behaviors, while exhibiting favorable safety without inducing sedation or motor impairment. Z8779877149 is available for the research of pain, depression and anxiety disorders .
|
-

-
- HY-179381
-
|
|
FGFR
|
Cancer
|
|
FGFR4-IN-24 is a selective irreversible covalent FGFR4 inhibitor (IC50 = 1.2 nM). FGFR4-IN-24 shows much weaker activity against the other three FGFR family kinases (FGFR1-3). FGFR4-IN-24 inhibits the FGF19/FGFR4 signaling pathway, effectively suppressing the proliferation of the HuH-7 HCC cell line (GI50 = 17 nM). FGFR4-IN-24 exhibits significant antitumor activity in a HuH-7 mouse xenograft model. FGFR4-IN-24 can be used for the research of hepatocellular carcinoma .
|
-

| Cat. No. |
Product Name |
Type |
-
- HY-114364
-
|
|
Biochemical Assay Reagents
|
|
UDP-Galactose disodium is a monosaccharide and a key glycosyl donor molecule in cells that participates in nucleotide sugar metabolism. UDP-Galactose disodium is the natural agonist of the P2Y14 receptor coupled to Gi proteins in the immune system (IC50 = 0.67 μM, hP2Y14). UDP-Galactose disodium can be used to study cell signal transduction and substance metabolism .
|
-
- HY-W745090
-
|
|
Biochemical Assay Reagents
|
|
Isomaltulose monohydrate is a fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Isomaltulose monohydrate can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Isomaltulose monohydrate inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM) , cathepsin G release (IC< sub>50: 2.76 μM) and chemotaxis. Isomaltulose monohydrate can improve excessive activation of neutrophils and reduce inflammation or tissue damage. A series of derivatives of Isomaltulose monohydrate are found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-107024
-
|
OGP(10-14); Historphin
|
Src
|
Others
|
|
Osteogenic Growth Peptide (10-14) (OGP(10-14)), the C-terminal truncated pentapeptide of osteogenic growth peptide (OGP), retains the full OGP-like activity. Osteogenic Growth Peptide (10-14) is responsible for the binding to the OGP receptor and activates an intracellular Gi-protein-MAP kinase signaling pathway. Osteogenic Growth Peptide (10-14) is a potent mitogen and stimulator of osteogenesis and hematopoiesis. Osteogenic Growth Peptide (10-14) acts as a Src inhibitor .
|
-
- HY-P5358
-
|
|
Protease Activated Receptor (PAR)
|
Others
|
|
YFLLRNP is a biological active peptide. (a partial agonist of PAR-1. YFLLRNP selectively active G12/13 signaling pathway without activating Gq or Gi pathways at low concentrations. YFLLRNP (60 μM))
|
| Cat. No. |
Product Name |
Target |
Research Area |
Image |
-
- HY-P99643
-
|
UCB6114
|
Dan family
|
Cancer
|
|
Ginisortamab (UCB6114) is a fully human IgG4P anti Gremlin-1 monoclonal antibody, with mean IC50 values of 8.2 nM and 9 nM against human and mouse gremlin-1, respectivly. Ginisortamab inhibits gremlin-1 antagonism of BMP signaling pathways. Ginisortamab has the potential for the research of gastrointestinal (GI) tumors .
|
-

(5)
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-114364
-
-
-
- HY-150524
-
-
-
- HY-N0927
-
|
Tetrahydrocolumbamine; (S)-Tetrahydrocolumbamine
|
Alkaloids
Structural Classification
Monophenols
other families
Classification of Application Fields
Other Diseases
Phenols
Plants
Isoquinoline Alkaloids
Disease Research Fields
Source Classification
|
Dopamine Receptor
|
|
(-)-Isocorypalmine (Tetrahydrocolumbamine; (S)-Tetrahydrocolumbamine) is a dopamine receptor ligand and modulator capable of crossing the blood-brain barrier. (-)-Isocorypalmine exhibits receptor selectivity, acting as a partial agonist of dopamine D1 and D5 receptors (coupled via Gs proteins), as well as an antagonist of D2, D3 and D4 receptors (by blocking Gi protein-mediated signaling). (-)-Isocorypalmine shows no significant binding to various non-dopamine receptors, ion channels and transporters. (-)-Isocorypalmine is metabolically stable in vivo, effectively inhibits spontaneous and cocaine-induced hyperlocomotion, sensitization and conditioned place preference in mice, and does not induce addictive place preference when administered alone. (-)-Isocorypalmine can be used in addiction research .
|
-
-
- HY-W744699
-
|
(+)-Larixol
|
Larix decidua Miller
Natural Products
Pinaceae
Plants
Source Classification
|
Src
ERK
Akt
|
|
Larixol is an fMLP inhibitor and also inhibits Src kinase, ERK1/2, p38 and AKT phosphorylation signals in immune regulation. Larixol can interfere with the interaction between the βγ subunit of the fMLP receptor Gi protein and its downstream molecules, thereby inhibiting fMLP-induced respiratory burst. Larixol inhibits fMLP (0.1 μM)-induced superoxide anion production (IC50: 1.98 μM), cathepsin G release (IC50: 2.76 μM), and chemotaxis. Larixol improves neutrophil hyperactivation and reduces inflammation or tissue damage. A series of Larixol derivatives were found to have inhibitory effects on FSGS-related TRPC6 functional mutants .
|
-
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-B0194S
-
|
|
|
Tizanidine-d4 is the deuterium labeled Tizanidine (HY-B0194). Tizanidine, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-
-
- HY-114364S
-
|
|
|
UDP-α-D-Galactose- 13C disodium is the 13C labeled UDP-α-D-Galactose disodium. UDP-Galactose disodium is a monosaccharide and a key glycosyl donor molecule in cells that participates in nucleotide sugar metabolism. UDP-Galactose disodium is the natural agonist of the P2Y14 receptor coupled to Gi proteins in the immune system (IC50 = 0.67 μM, hP2Y14). UDP-Galactose disodium can be used to study cell signal transduction and substance metabolism .
|
-
-
- HY-B0194AS
-
|
|
|
Tizanidine-d4 hydrochloride is deuterium labeled Tizanidine hydrochloride (HY-B0194A). Tizanidine hydrochloride, a skeletal muscle relaxant, is an orally effective central α2-adrenoceptor agonist (IC50 = 6.9 nmol). Tizanidine hydrochloride primarily exerts muscle relaxation effects by inhibiting the release of excitatory amino acids (glutamate and aspartate) from the presynaptic terminals of spinal cord interneurons. Tizanidine hydrochloride has anti-injury activity and can inhibit gastrointestinal (GI) transport. Tizanidine hydrochloride can inhibit the proliferation, migration, and invasion of lung cancer cells and induce cell apoptosis by upregulating Nischarin and inhibiting the AKT and Wnt3a/β-catenin signaling pathways. Tizanidine hydrochloride can be used to treat spasticity caused by diseases such as multiple sclerosis (MS), stroke, and spinal cord injury (SCI).
|
-
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent: