Delivery of temperature sensitive items including proteins and kits will be paused on 6/19 for the Juneteenth holiday. For urgent orders please contact customer service.
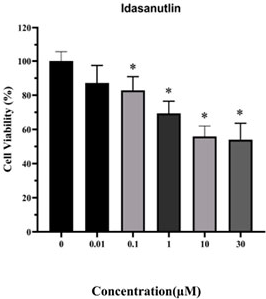
Idasanutlin (RG7388) is an orally bioavailable MDM2 inhibitor with an IC50 of 6 nM. Idasanutlin disrupts MDM2-p53 binding, stabilizes and activates p53, triggering cell cycle arrest, apoptosis, and reduced cancer cell viability. Idasanutlin reduces EGFR protein expression and phosphorylation, suppresses downstream SHP2, MEK1/2, ERK1/2, AKT, mTOR, p70(S6K1), and S6 signaling. Idasanutlin induces mitochondrial ROS production, drives p38 MAPK phosphorylation, upregulates NOXA, and mediates caspase-3-dependent apoptosis and gasdermin E-mediated pyroptosis. Idasanutlin can be used for the research of TP53-mutant non-small cell lung cancer, T-cell acute lymphoblastic leukemia, colorectal carcinoma, melanoma, diffuse large B-cell lymphoma, mantle cell lymphoma, non-Hodgkin lymphoma, severe fever with thrombocytopenia syndrome, neuroblastoma, acute lymphoblastic leukemia, relapsed or refractory acute myeloid leukemia, osteosarcoma, solid tumors, and hematological tumors.
Nur für Forschungszwecke. Wir verkaufen nicht an Patienten.
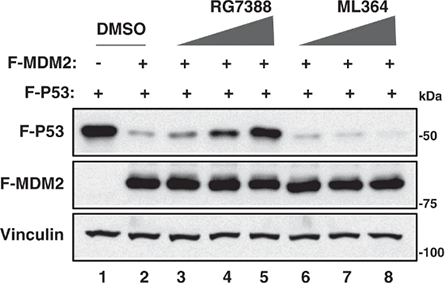
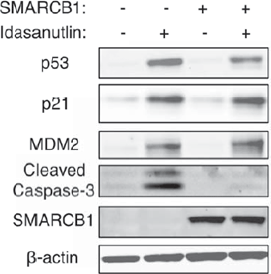
Immunoblot for p53 pathway responses to 壹dasanutlin treatment (1 μM for 24h) in TTC642 cells reexpressing SMARCB1. Images are representative of three biological replicates.
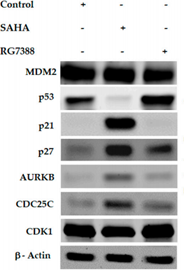
Effect of SAHA and RG7388 treatments on Cell Cycle related Protein levels in MCF-7 cells. Figure shows upregulation of p21, p27, AURKB and CDC25C levels after treatment with 7.5 µM concentration of SAHA and p53 upregulation after RG7388 treatment.
Idasanutlin (RG7388) is an orally bioavailable MDM2 inhibitor with an IC50 of 6 nM. Idasanutlin disrupts MDM2-p53 binding, stabilizes and activates p53, triggering cell cycle arrest, apoptosis, and reduced cancer cell viability. Idasanutlin reduces EGFR protein expression and phosphorylation, suppresses downstream SHP2, MEK1/2, ERK1/2, AKT, mTOR, p70(S6K1), and S6 signaling. Idasanutlin induces mitochondrial ROS production, drives p38 MAPK phosphorylation, upregulates NOXA, and mediates caspase-3-dependent apoptosis and gasdermin E-mediated pyroptosis. Idasanutlin can be used for the research of TP53-mutant non-small cell lung cancer, T-cell acute lymphoblastic leukemia, colorectal carcinoma, melanoma, diffuse large B-cell lymphoma, mantle cell lymphoma, non-Hodgkin lymphoma, severe fever with thrombocytopenia syndrome, neuroblastoma, acute lymphoblastic leukemia, relapsed or refractory acute myeloid leukemia, osteosarcoma, solid tumors, and hematological tumors[1][2][3][4][5][6][7][8][9][10][11][12].
Antiproliferative activity against human SJSA1 cells assessed as inhibition of EdU incorporation after 1 hr by Click-iT EdU HCS assay in presence of 10% human serum
Antiproliferative activity against human SJSA1 cells assessed as inhibition of EdU incorporation after 1 hr by Click-iT EdU HCS assay in presence of 10% human serum
Adezmapimod (SB 203580) (HY-10256) (40 μM; 1 h pre-treatment, 6 h treatment) prevents Idasanutlin (60 μM; 6 h)-induced apoptosis and pyroptosis in TP53mutant NSCLC cell lines HCC827, NCI-H23, PC9, and NCI-H1975, reversing cell death and restoring colony-forming ability[1]. Idasanutlin (60 μM; 6 h) remodels the tumor microenvironment in TP53mutant NSCLC cell lines HCC827 and PC9 by upregulating immune-related gene expression, increasing pro-inflammatory cytokine secretion, and reducing PD-L1 expression[1]. Idasanutlin (30-90 nM; 24-72 h) induces p53-dependent growth inhibition, apoptosis, and upregulation of pro-apoptotic p53 target genes BAX and BBC3 in wildtype TP53 MOLT-3 human T-ALL cells, while TP53 knockout MOLT-3 cells are completely resistant[2]. Idasanutlin (1.5 μM; 24-72 h) induces modest growth inhibition, p53 target gene upregulation, apoptosis, and G1 cell cycle arrest in wildtype TP53 DFCI12 T-ALL PDX cells[2]. Idasanutlin (1.5 μM; 16 h) activates the p53 pathway and upregulates p53 target genes in wildtype TP53 DFCI12 T-ALL PDX cells[2]. Idasanutlin (0.5-5 μM; 8-24 h) increases p53, p21, and (in CT26 cells) MDM2 protein levels in p53wtB16-F10 and CT26 cells, but not in p53mutMC38 cells[3]. Idasanutlin (0.05-5 μM; 8-24 h) dose-dependently activates p53 transcriptional activity, inducing a 2-3-fold luminescence increase in p53wt B16-F10 and CT26 cells and a 6-fold increase in U-2 OS cells, but has no effect on p53mut MC38 cells[3]. Idasanutlin (5 days) reduces cell viability in a p53-dependent manner, with EC50 values of 0.186 μM for B16-F10 cells, 1.492 μM for CT26 cells, 49.665 μM for MC38 cells, and 0.046 μM for U-2 OS cells[3]. Idasanutlin (0.05-5 μM; 24 h) induces dose-dependent cell cycle arrest in p53wt B16-F10, CT26, and U-2 OS cells, reducing S-phase cell fractions, with a stronger response in U-2 OS cells[3]. Idasanutlin (0.05-5 μM; 24-72 h) induces dose-dependent caspase 3/7 activation in B16-F10 cells, but does not activate caspase 3/7 in CT26 cells[3]. Idasanutlin (0.5-2 μM; 5 days) dose-dependently decreases the proliferation of activated human T cells co-cultured with U-2 OS cells[3]. Idasanutlin (1-40 μM) exhibits no cellular toxicity in HeLa cells at concentrations ≤10 μM[5]. Idasanutlin (1-40 μM) potently inhibits SFTSV replication in HeLa cells with an IC50 of approximately 5 μM[5]. Idasanutlin (10 μM) activates p53 and Apaf-1 accumulation, induces apoptosis, and inhibits SFTSV replication in SFTSV-infected HeLa cells, while having no such effects in uninfected HeLa cells[5]. Idasanutlin (10 nM-1 μM; 72 h) acts as a resensitizing agent for Venetoclax (HY-15531)-resistant KCNR and SJNB12 human neuroblastoma cells, demonstrating greater efficacy in resistant cells under venetoclax pressure than in non-resistant cells[6]. Idasanutlin (25 nM-400 nM (KCNR); 62.5 nM-1 μM (SJNB12); 15.5 nM-1 μM (FACS); 72 h) induces p21-mediated growth arrest in non-resistant and Venetoclax-resistant KCNR and SJNB12 human neuroblastoma cells, and BAX-mediated apoptosis in Venetoclax-resistant KCNR and SJNB12 human neuroblastoma cells treated with venetoclax[6]. Idasanutlin (1 nM-100 μM; 24 h-72 h) potently induces dose- and time-dependent cell death in ABC, GCB, Rituximab (HY-P9913)-sensitive, and Rituximab-resistant DLBCL cell lines with 48-hour IC50 values ranging from 0.7 μM to 63.07 μM[7]. Idasanutlin (1 μM) reduces mitochondrial membrane potential in ABC (TMD8, U2932) and GCB (VAL, OCILy2, DHL4, ROS50) DLBCL cell lines[7]. Idasanutlin (1 μM; 24 h) decreases MDM2 expression in ABC (TMD8, U2932) and GCB (VAL, OCILy2, DHL4, ROS50) DLBCL cell lines, decreases XIAP expression in VAL and DHL4 GCB DLBCL cell lines, and increases PUMA expression in these DLBCL cell lines[7]. Idasanutlin (48 h) induces significant apoptosis in high-risk adult ALL patient-derived samples after 48 hours of treatment at respective EC50 concentrations, as shown by increased annexin-V positivity (mean 59.0%) and >3-fold higher cleaved PARP levels[8]. Idasanutlin (RG7388) potently and selectively inhibits proliferation of wild-type p53 human cancer cell lines (SJSA1, RKO, HCT116) with an average IC50 of 0.03 μM and a selectivity ratio of 344 relative to mutant p53 cell lines (SW480, MDA-MB435)[10]. Idasanutlin (RG7388) induces dose-dependent p53 stabilization, cell cycle arrest, and apoptosis in wild-type p53cancer cells via a nongenotoxic p53 activation mechanism[10]. Idasanutlin (RG7388) (0.5-2 μM; 20 h) activates the p53 pathway in SJSA1 osteosarcoma cells, increasing p53, MDM2, and p21 protein levels after 20 h of incubation at concentrations of 0.5 μM, 1 μM, and 2 μM[10]. Idasanutlin (0.01-30 μM; 5 days) potently inhibits the viability of p53 wild-type cancer cell lines (including SJSA, RKO, HCT116, H460, A375, SK-MEL-5) with an average IC90 of 300 nM, while showing minimal activity against p53-mutant lines (SW480, MDA435, HeLa)[11]. Idasanutlin (300 nM-1.8 μM; 16-hour treatment) induces sustained apoptosis in SJSA osteosarcoma cells after a single 16-hour treatment, with 1.8 μM producing a maximal response of 48% annexin V positivity at 48 hours post-washout, eliminating the need for continuous drug exposure[11]. Idasanutlin (0.3-1.8 μM; 16-hour treatment) activates the p53 pathway in SJSA osteosarcoma cells, with p53 protein remaining elevated for at least 48 hours after a single 16-hour 1.8 μM treatment, supporting sustained antitumor activity without continuous dosing[11]. Idasanutlin (0.6-2000 nM; 72 h) dose-dependently reduces viability in p53 wild-type MV4-11 (relative IC50 55 nM), MOLM-13 (relative IC50 35 nM), and OCI-AML-3 (relative IC50 164 nM) AML cell lines, but has no effect on p53 mutant HL-60 AML cells[12]. Idasanutlin (0.6-2000 nM; 72 h) induces apoptosis in a dose-dependent manner in p53 wild-type MV4-11 (relative IC50 203 nM) and MOLM-13 (relative IC50 102 nM) AML cell lines, has minimal apoptotic activity in OCI-AML-3 cells, and has no effect on p53 mutant HL-60 AML cells[12]. Idasanutlin (60 nM; 72 h) induces G1 cell cycle arrest in MV4-11 and MOLM-13 AML cell lines, with apoptosis only evident after cells have gone through at least two cell cycles[12]. Idasanutlin (100 nM; 6 h) induces significant changes in gene expression in MOLM-13 AML cells, activating the TP53 pathway and inhibiting the CCND1 pathway to drive G1 cell cycle arrest[12]. Idasanutlin (100 nM; 7-16 h) increases p53 protein levels, induces caspase-3 cleavage (after 16 h), and downregulates Mcl-1 expression over time in MOLM-13 and MV4-11 AML cell lines[12].
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.
p.o.; daily on a 5-days-on 2-days-off schedule; 14 days
Result:
Induced a significant decrease in T-ALL burden in 3 of 4 tested xenografts. Demonstrated synergistic tumor burden suppression. Significantly increased overall survival in the combination treatment group compared to single-agent or vehicle controls.
Animal Model:
BALB/c (6- to 10-week-old; syngeneic subcutaneous colorectal carcinoma model via injection of 1×106 CT26 cells into a single flank)[3]
Dosage:
50 mg/kg; 100 mg/kg; 200 mg/kg
Administration:
p.o.; daily; 14 days
Result:
Slowed tumor growth in 2/8 mice at 50 mg/kg monotherapy. Significantly reduced tumor size when combined with anti-PD-1 at 50 mg/kg, with only 2/8 mice developing tumors ≥1000 mm3. Led to 6/8 mice being sacrificed before day 15, with all tumors reaching ≥1000 mm3 at 100 mg/kg monotherapy. Led to 4/8 mice surviving to day 15, with all tumors reaching ≥1000 mm3 at 200 mg/kg monotherapy. Caused a significant decrease in the proportion of TCR-positive T cells within the blood lymphocyte population between day 0 and day 7 at 200 mg/kg monotherapy, with the most pronounced decrease when combined with anti-PD-1. Did not affect PD-1 expression on CD4+ or CD8+ T cells across all doses of monotherapy. Showed no significant changes in CD69+ and CD25+ T cell populations across all groups.
Animal Model:
C57BL/6 (syngeneic subcutaneous melanoma model via injection of B16-F10 cells into a single flank)[3]
Dosage:
100 mg/kg; 200 mg/kg
Administration:
p.o.; daily; 14 days
Result:
Provided some tumor growth control at 200 mg/kg monotherapy. Produced a significant therapeutic effect when combined with anti-PD-1 at 200 mg/kg. Showed no therapeutic effects at 100 mg/kg alone or combined with anti-PD-1.
Animal Model:
SCID beige (female, bearing established subcutaneous human DoHH-2 tumors)[4]
Dosage:
30 mg/kg
Administration:
p.o.; days 13-17, 20-24, 27-29
Result:
Achieved 56% tumor growth inhibition (TGI) with a normalized percent tumor control rate (npTCR) of 0.48.
Animal Model:
SCID beige (female, bearing established subcutaneous human Z-138 tumors)[4]
Dosage:
100 mg/kg (days 18-22); 80 mg/kg (days 25-36)
Administration:
p.o.; days 18-22, 25-36
Result:
Achieved 67% tumor growth inhibition (TGI) with a normalized percent tumor control rate (npTCR) of 0.43.
25 mg/kg/day (monotherapy; combination with 100 mg/kg/day venetoclax)
Administration:
p.o.; daily; up to three weeks
Result:
Resulted in an average tumor volume change of 186% after three weeks, compared with 512% for vehicle control. In combination with venetoclax, resulted in an average tumor volume change of -80%, with 1/6 complete remissions, 2/6 very good partial responses, and 3/6 partial regressions. Increased BAX levels in tumors, with more pronounced upregulation in combination-treated tumors. Mice receiving delayed combination therapy (one week venetoclax monotherapy followed by two weeks combination treatment) showed slightly less favorable responses, with 1 very good partial response, 3 partial regressions, and 3 no responses.
25 mg/kg/day (monotherapy; combination with 100 mg/kg/day venetoclax)
Administration:
p.o.; daily; three weeks
Result:
Resulted in an average tumor volume change of 138% after three weeks, compared with 236% for vehicle control. In combination with venetoclax, resulted in an average tumor volume change of 97%. No statistically significant differences in tumor volume change were observed between treatment groups and vehicle control. BIM was already bound to MCL-1 in untreated xenografts, and complex levels increased upon combination treatment.
25 mg/kg/day (monotherapy; combination with 100 mg/kg/day venetoclax)
Administration:
p.o.; daily; 10 days
Result:
Resulted in an average tumor volume change of 96% after 10 days, compared with 597% for vehicle control, and led to 1/4 stable disease. In combination with venetoclax, resulted in an average tumor volume change of -72%, with 5/6 partial regressions. Upregulated p53, MDM2, and BAX levels, and combination therapy increased cleaved PARP levels, indicating enhanced apoptosis.
25 mg/kg/day (monotherapy; combination with 100 mg/kg/day venetoclax)
Administration:
p.o.; daily; 4 days
Result:
Resulted in an average tumor volume change of 185% after 4 days, compared with 216% for vehicle control. In combination with venetoclax, resulted in an average tumor volume change of 178%. No statistically significant differences in tumor volume change were observed between treatment groups and vehicle control, and no treatment responses were seen. Combination therapy led to enhanced PARP cleavage, and idasanutlin treatment upregulated p53 and MDM2 levels.
Achieved 88% tumor growth inhibition with an AUC of 23 μg·h/mL. Achieved greater than 100% tumor growth inhibition with an AUC of 29 μg·h/mL. Resulted in mean tumor volumes near baseline through day 32 post-tumor cell implant.
Animal Model:
Crl:NU-Foxn1nu (female, 10-12 weeks old, implanted with human SJSA osteosarcoma cells)[11]
p.o.; daily (21 days, 28 days, 5 days); once weekly (21 days, 28 days); twice weekly (21 days); 5 days on / 23 days off (28-day cycle); 2 days on / 5 days off (4 cycles over 28 days); single dose
Result:
Achieved 11% tumor growth inhibition (TGI), 0 partial regressions (PR), and 0% increase in lifespan (ILS) at 1.11 mg/kg daily. Achieved 50% TGI, 0 PR, and 25% ILS at 3.33 mg/kg daily. Achieved 77% TGI, 1 PR, and 25% ILS at 10 mg/kg daily. Achieved >100% TGI, 9 PR, and 125% ILS at 30 mg/kg daily for 21 days. Achieved >100% TGI, 7 PR, and 127% ILS at 30 mg/kg daily for 28 days. Achieved 79% TGI, 1 PR, and 32% ILS at 50 mg/kg once weekly. Achieved 96% TGI, 3 PR, and 57% ILS at 50 mg/kg twice weekly. Achieved >100% TGI, 6 PR, and 127% ILS at 80 mg/kg 5 days on/23 days off. Achieved >100% TGI, 9 PR, and 188% ILS at 100 mg/kg 2 days on/5 days off. Achieved >100% TGI, 7 PR, and 162% ILS at 200 mg/kg once weekly (split dose). Produced statistically significant increase in caspase 3/7 luminescence (max at 48 hours post-dose), increased cleaved PARP-1 (cPARP-1) levels, and reduced Ki-67 positive nuclei at 48 hours post-dose at single 80 mg/kg dose compared to vehicle. Produced statistically significant increase in caspase 3/7 luminescence (max at 48 hours post-dose), maximal increase in cPARP-1 levels, and maximal reduction in Ki-67 positive nuclei at 48 hours post-dose at single 200 mg/kg dose compared to vehicle. Produced statistically significant increase in caspase 3/7 luminescence and cPARP-1 levels, plus reduction in Ki-67 positive nuclei, with maximal effects observed on day 3 of dosing at 80 mg/kg daily for 5 days compared to vehicle. Showed statistical equivalence between 30 mg/kg daily and 50 mg/kg twice weekly; between 50 mg/kg once weekly and 10 mg/kg daily; and between multiple intermittent regimens and 30 mg/kg daily.
Achieved 30% tumor growth inhibition relative to vehicle control, with a treatment-to-control ratio (TCR) of 0.5 and 95% confidence interval (CI) of 0.26-0.98.
Resulted in a median survival of 52 days post-inoculation, corresponding to a 35% increase in lifespan (ILS) relative to vehicle control (median survival 38.5 days).\nResulted in a median survival of 23 days post-inoculation, corresponding to a 10% increase in lifespan (ILS) relative to vehicle control (median survival 21 days).
Room temperature in continental US; may vary elsewhere.
Speicherung
Powder
-20°C
3 years
4°C
2 years
In solvent
-80°C
1 year
-20°C
6 months
Lösungsmittel & Löslichkeit
In Vitro:
DMSO : ≥ 45 mg/mL (73.00 mM; Hygroscopic DMSO has a significant impact on the solubility of product, please use newly opened DMSO)
*"≥" means soluble, but saturation unknown.
Preparing Stock Solutions
ConcentrationSolventMass
1 mg
5 mg
10 mg
1 mM
1.6221 mL
8.1106 mL
16.2211 mL
5 mM
0.3244 mL
1.6221 mL
3.2442 mL
10 mM
0.1622 mL
0.8111 mL
1.6221 mL
View the Complete Stock Solution Preparation Table
*Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles. Storage method and period of stock solution: -80°C, 1 year; -20°C, 6 months. When stored at -80°C, please use it within 1 year. When stored at -20°C, please use it within 6 months.
For the following dissolution methods, please ensure to first prepare a clear stock solution using an In Vitro approach and then sequentially add co-solvents:
To ensure reliable experimental results, the clarified stock solution can be appropriately stored based on storage conditions. As for the working solution for in vivo experiments, it is recommended to prepare freshly and use it on the same day. The percentages shown for the solvents indicate their volumetric ratio in the final prepared solution. If precipitation or phase separation occurs during preparation, heat and/or sonication can be used to aid dissolution.
Protocol 1
Add each solvent one by one: 10% DMSO 90% Corn Oil
Solubility: ≥ 2.5 mg/mL (4.06 mM); Clear solution
This protocol yields a clear solution of ≥ 2.5 mg/mL (saturation unknown). If the continuous dosing period exceeds half a month, please choose this protocol carefully.
Taking 1 mL working solution as an example, add 100 μLDMSO stock solution (25.0 mg/mL) to 900 μLCorn oil, and mix evenly.
For the following dissolution methods, please prepare the working solution directly.
It is recommended to prepare fresh solutions and use them promptly within a short period of time. The percentages shown for the solvents indicate their volumetric ratio in the final prepared solution.
If precipitation or phase separation occurs during preparation,
heat and/or sonication can be used to aid dissolution.
Protocol 1
Add each solvent one by one: 0.5% HPMC/1% Tween-80 in Saline water
Solubility: 10 mg/mL (16.22 mM); Suspended solution; Need ultrasonic
In Vivo Dissolution Calculator
Please enter the basic information of animal experiments:
Dosage
mg/kg
Animal weight (per animal)
g
Dosing volume (per animal)
μL
Number of animals
Recommended: Prepare an additional quantity of animals to account for potential losses during experiments.
Please enter your animal formula composition:
%
DMSO+
%
+
%
Tween-80
+
%
Saline
Recommended: Keep the proportion of DMSO in working solution below 2% if your animal is weak.
The co-solvents required include: DMSO,
. All of co-solvents are available by MedChemExpress (MCE).
, Tween 80. All of co-solvents are available by MedChemExpress (MCE).
Calculation results:
Working solution concentration:
mg/mL
Method for preparing stock solution:
mg
drug dissolved in
μL
DMSO (Stock solution concentration: mg/mL).
The concentration of the stock solution you require exceeds the measured solubility. The following solution is for reference only. If necessary, please contact MedChemExpress (MCE).
Method for preparing in vivo working solution for animal experiments: Take
μL DMSO stock solution, add
μL .
μL , mix evenly, next add
μL Tween 80, mix evenly, then add
μL Saline.
Dissolve 0.9 g sodium chloride in ddH₂O and dilute to 100 mL to obtain a clear Saline solution
If the continuous dosing period exceeds half a month, please choose this protocol carefully.
Please ensure that the stock solution in the first step is dissolved to a clear state, and add co-solvents in sequence. You can use ultrasonic heating (ultrasonic cleaner, recommended frequency 20-40 kHz), vortexing, etc. to assist dissolution.
*Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles. Storage method and period of stock solution: -80°C, 1 year; -20°C, 6 months. When stored at -80°C, please use it within 1 year. When stored at -20°C, please use it within 6 months.
Species cross-reactivity must be investigated individually for each product. Many human cytokines will produce a nice response in mouse cell lines, and many mouse proteins will show activity on human cells. Other proteins may have a lower specific activity when used in the opposite species.
Customer Validation
Immunoblot for p53 pathway responses to 壹dasanutlin treatment (1 μM for 24h) in TTC642 cells reexpressing SMARCB1. Images are representative of three biological replicates.
Customer Validation
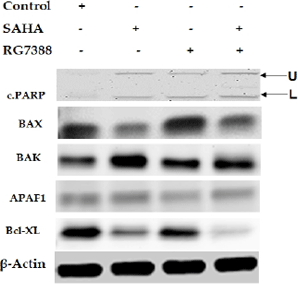
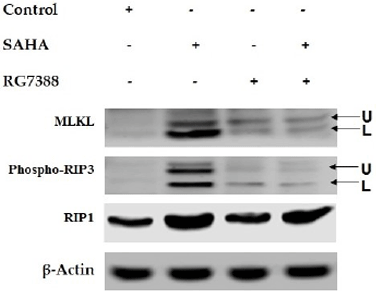
Modulation of apoptosis-related protein expressions in MCF-7 cells after SAHA (7.5 μM), RG7388 (2.0 μM) and SAHA + RG7388 treatment for 24 h.
Customer Validation
Modulation of necroptosis-related protein expressions in MCF-7 cells after (7.5 μM), RG7388 (2.0 μM) and SAHA + RG7388 treatment for 24 h
Customer Validation
Effect of SAHA and RG7388 treatments on Cell Cycle related Protein levels in MCF-7 cells. Figure shows upregulation of p21, p27, AURKB and CDC25C levels after treatment with 7.5 µM concentration of SAHA and p53 upregulation after RG7388 treatment.
Thanks, your subscription has been confirmed. You will hear from us soon.
Submission failed, please try again later.
Produkte sind chemische Reagenzien, die nur für Forschungszwecke bestimmt sind und nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
MedChemExpress values your privacy and your trust is important to us. We use cookies to enhance your website experience. Some cookies are necessary to run the website.
Privacy and Cookie Policy




















 Powered by Bioz
Powered by Bioz




