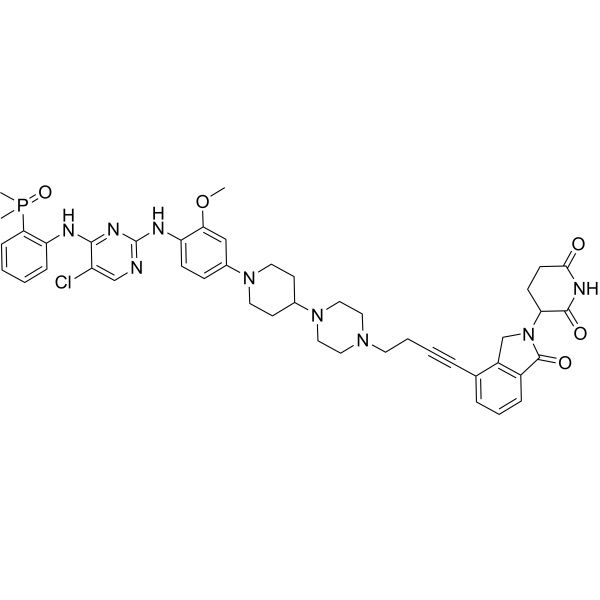
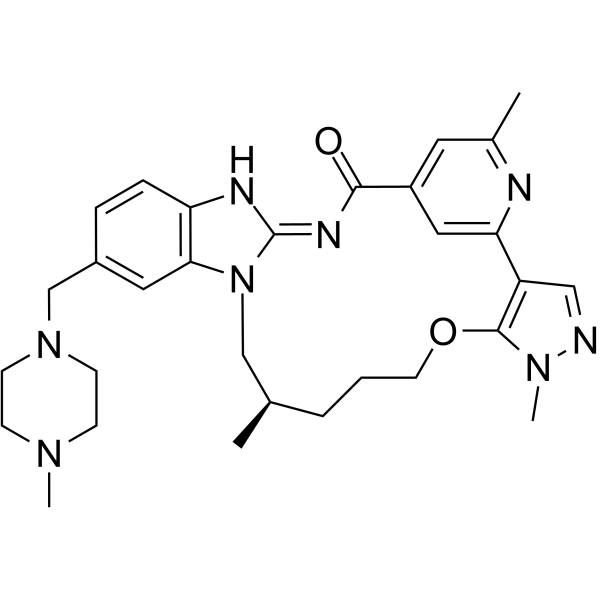
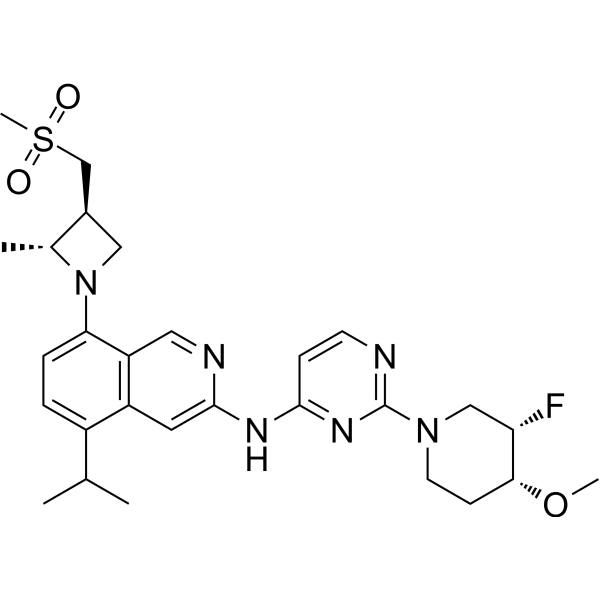
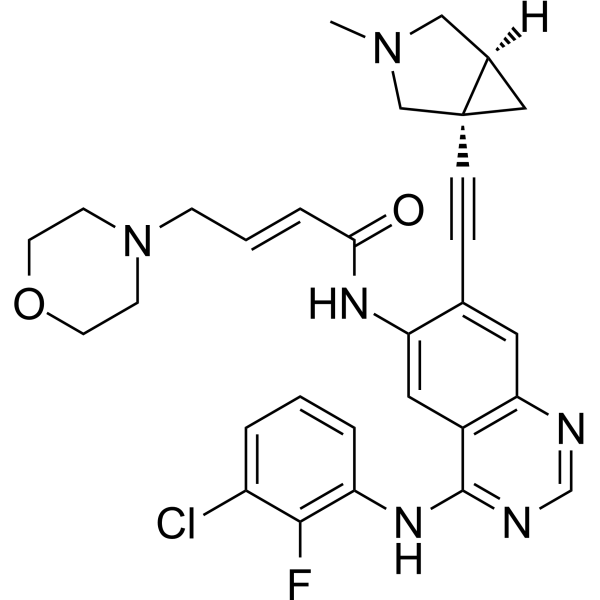
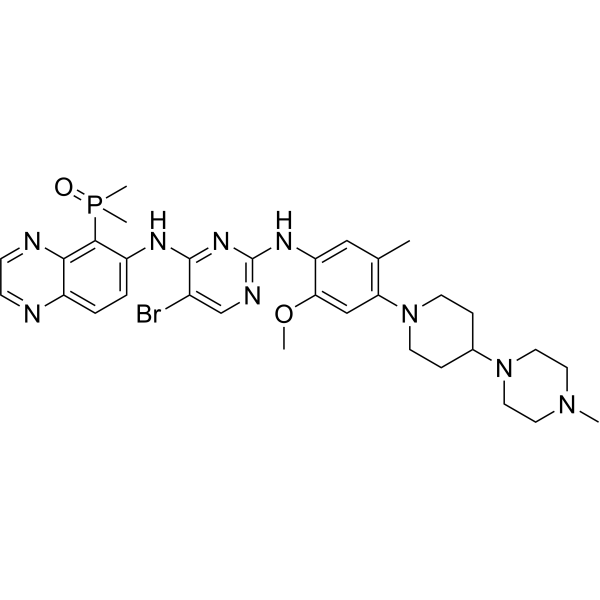
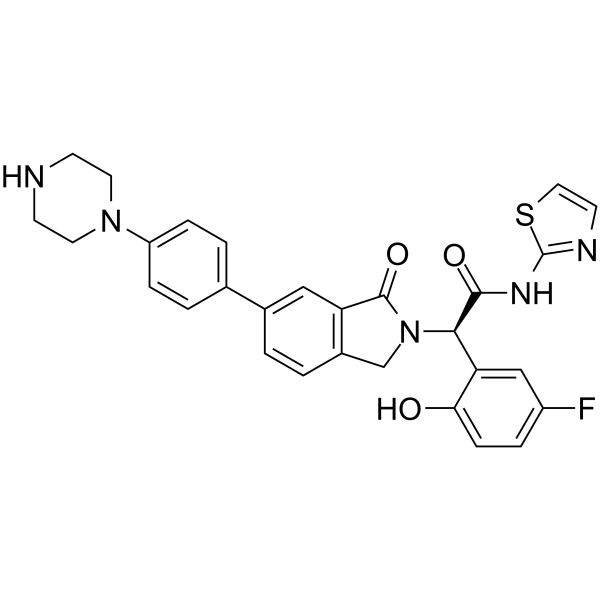
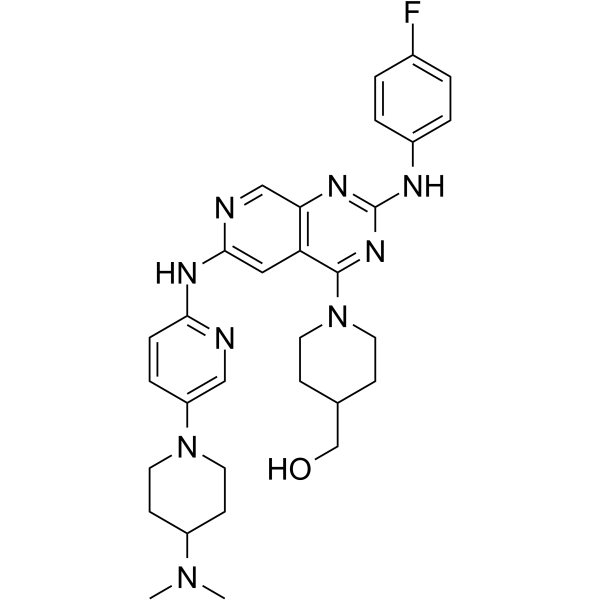
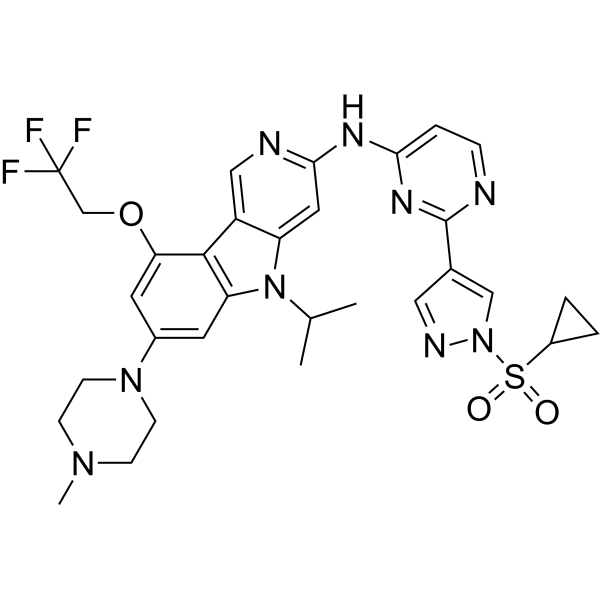
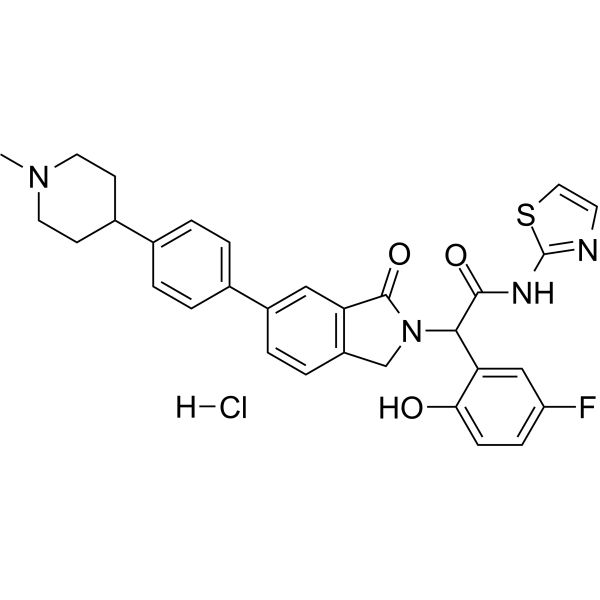
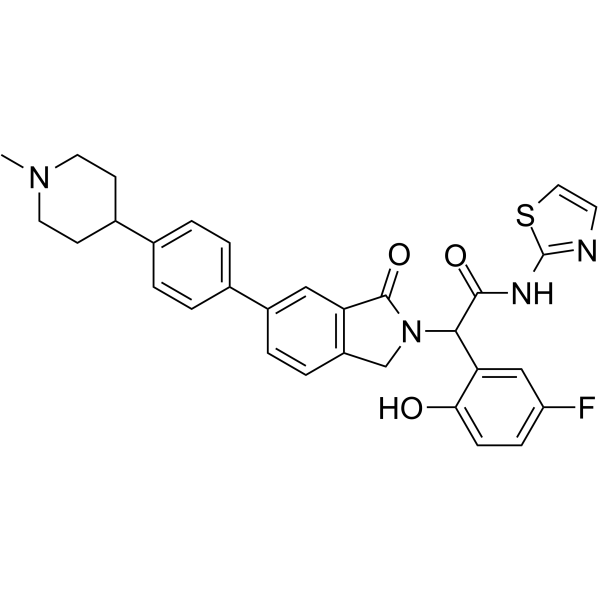
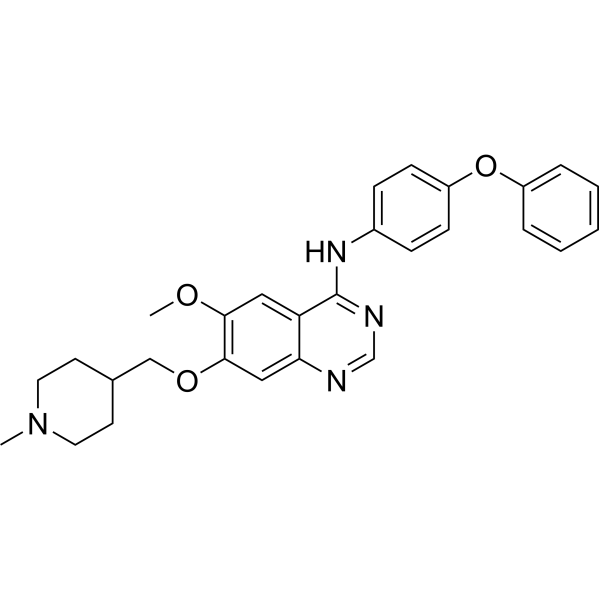
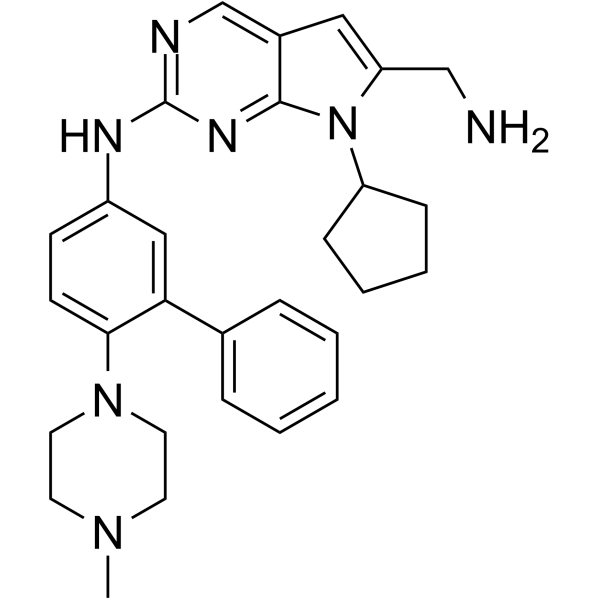
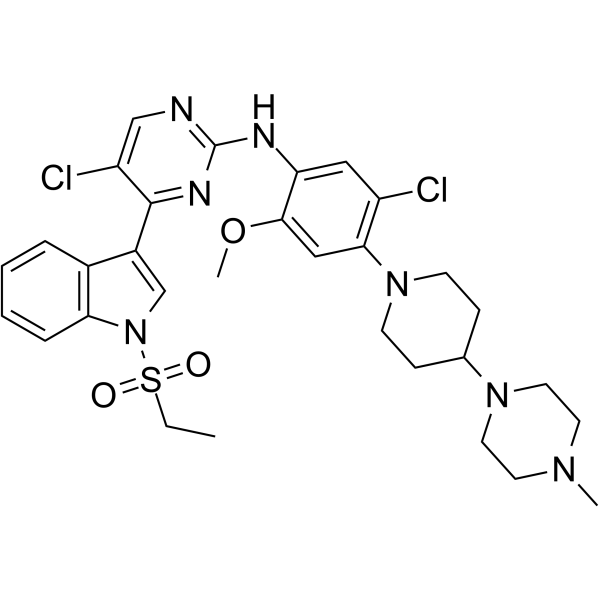
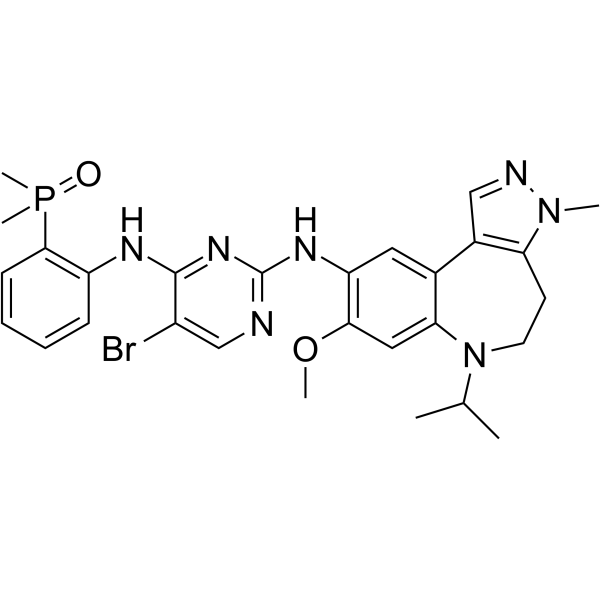
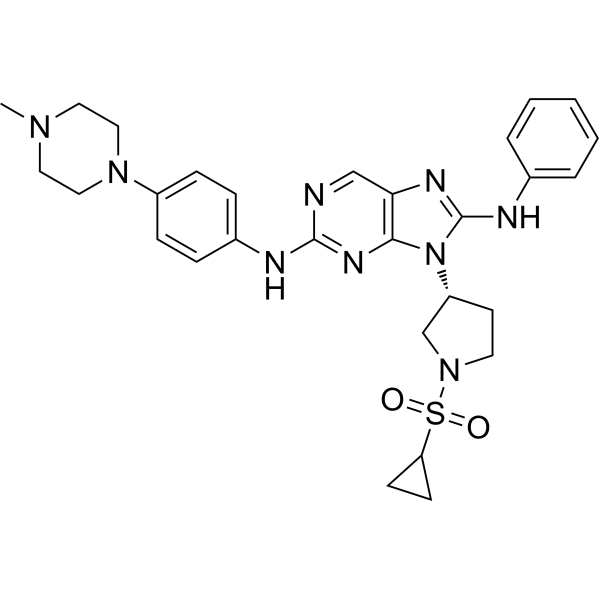
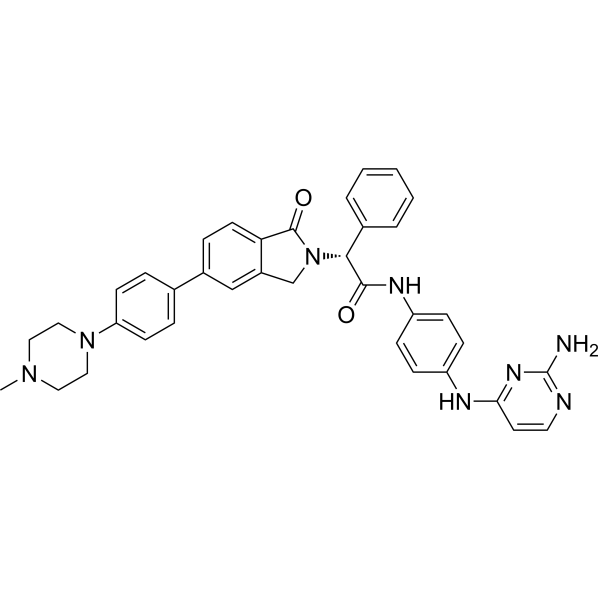
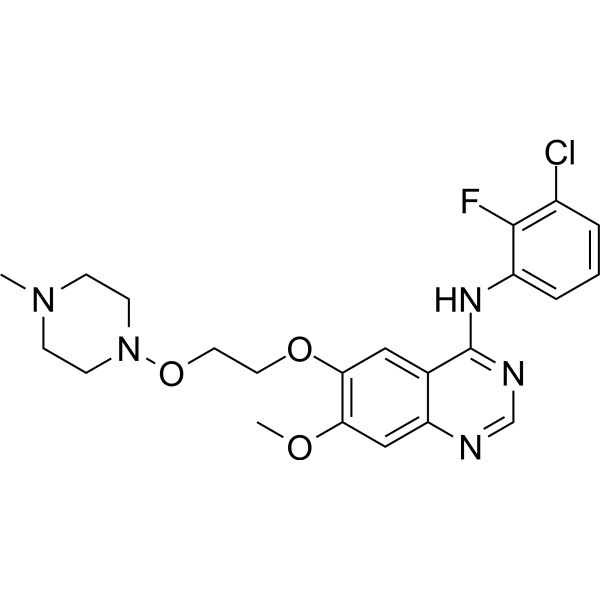
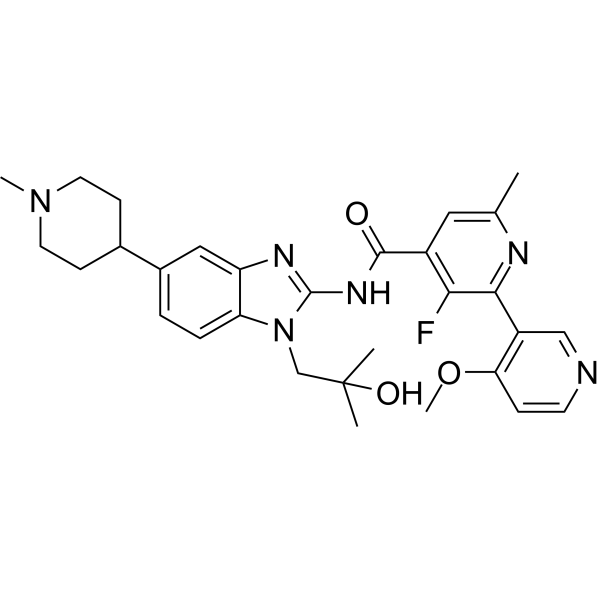
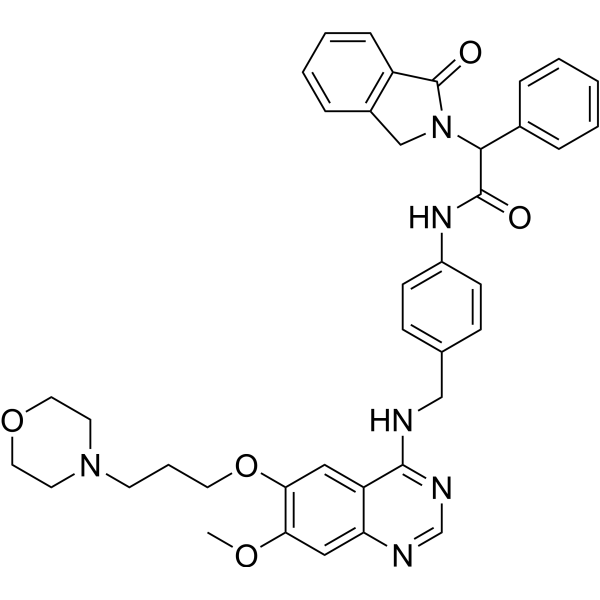
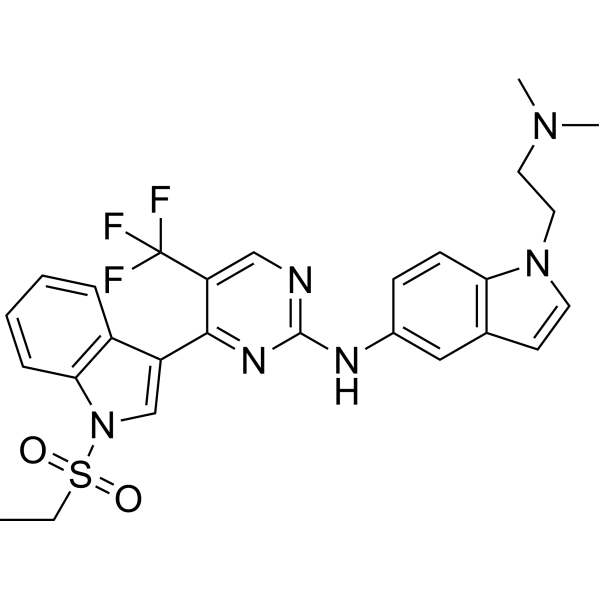
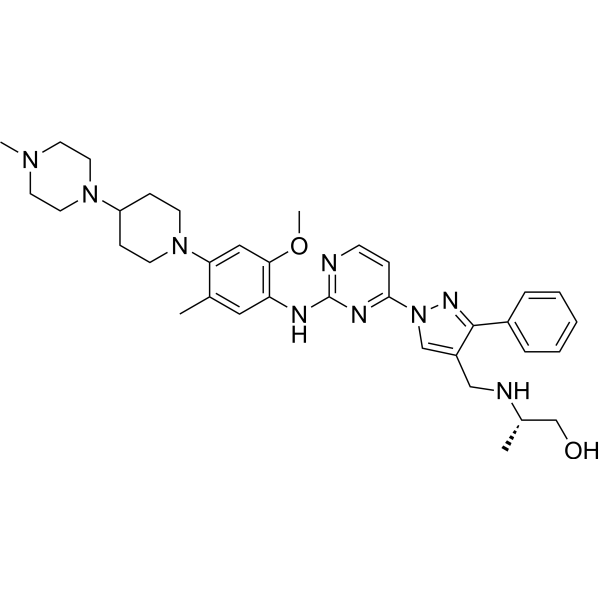
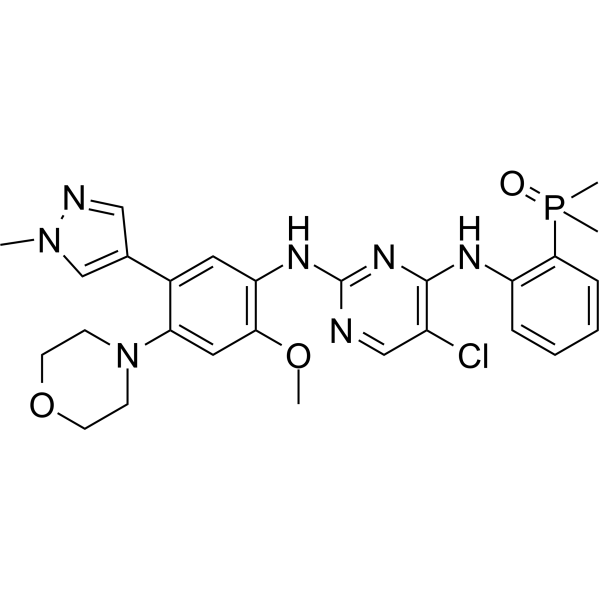
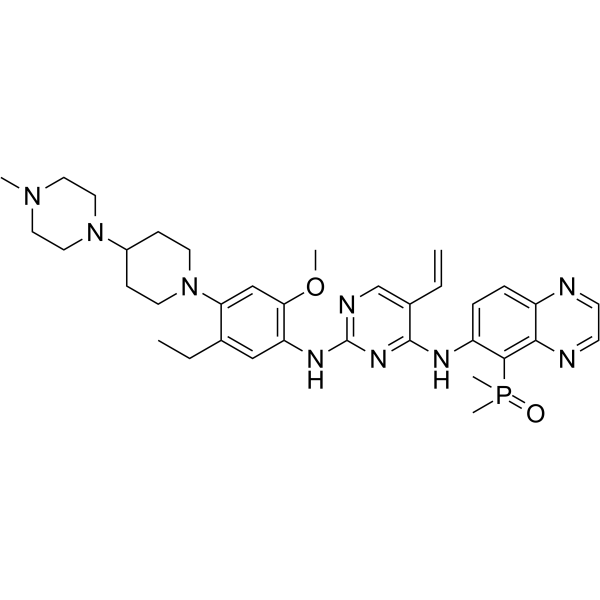
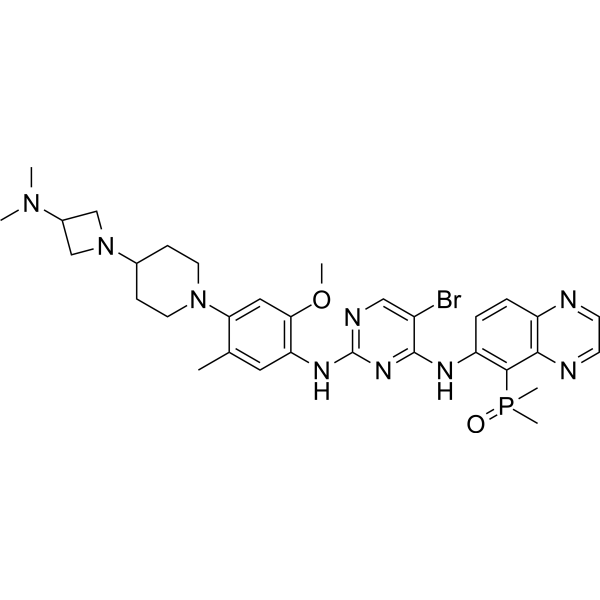
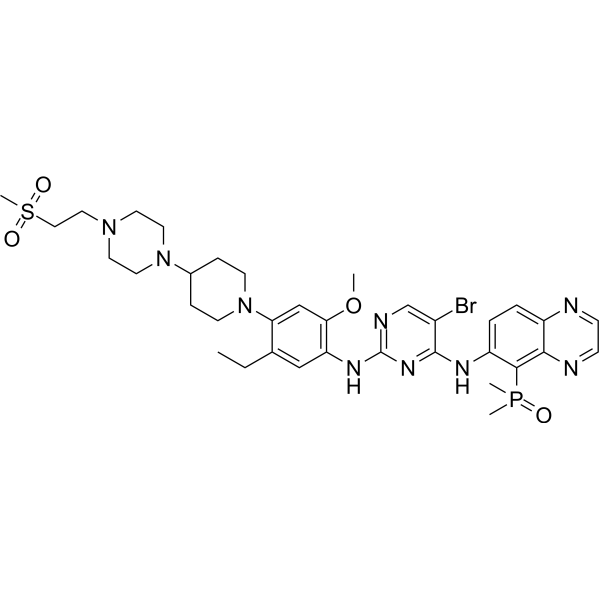
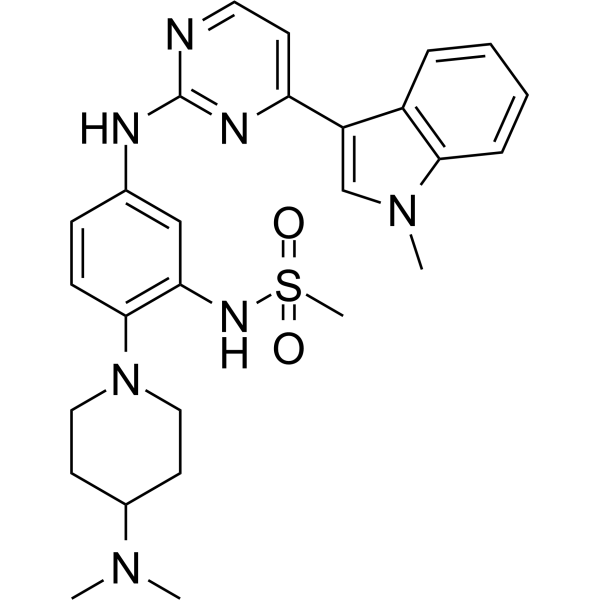
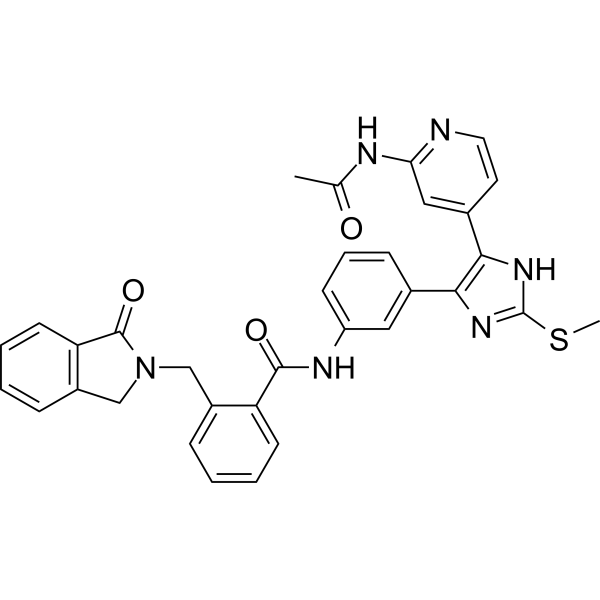
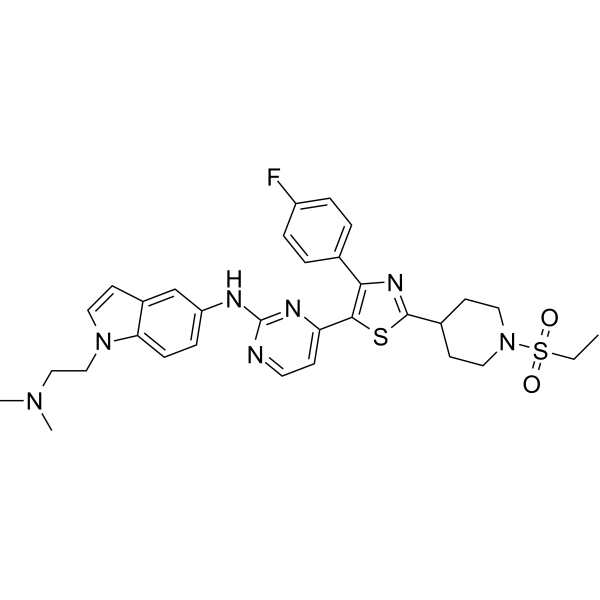
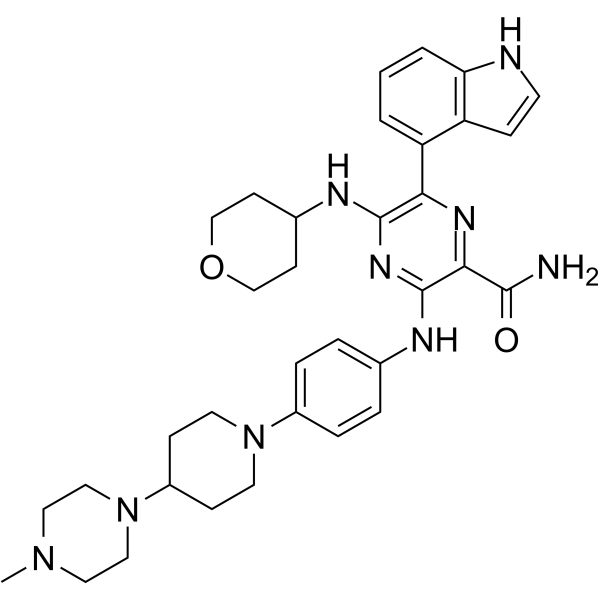
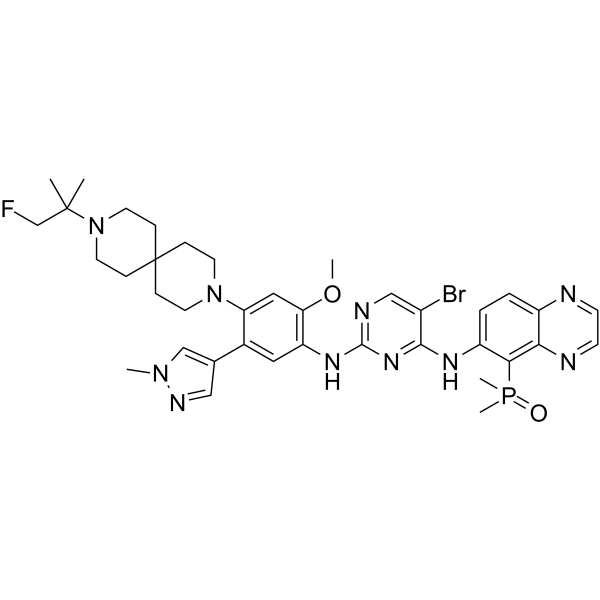
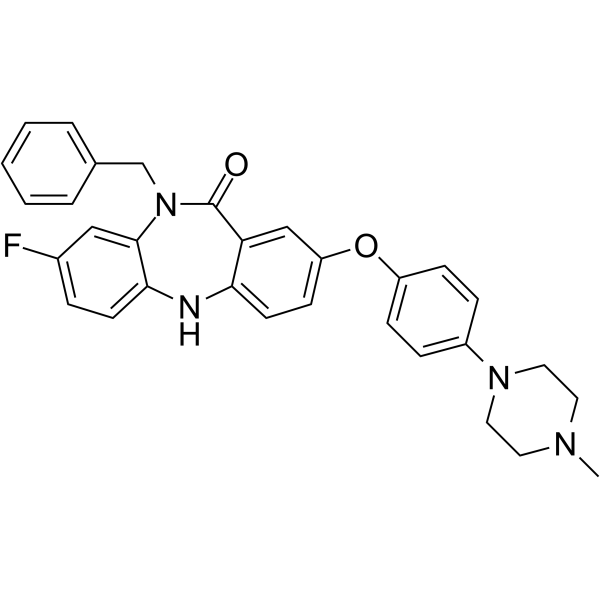
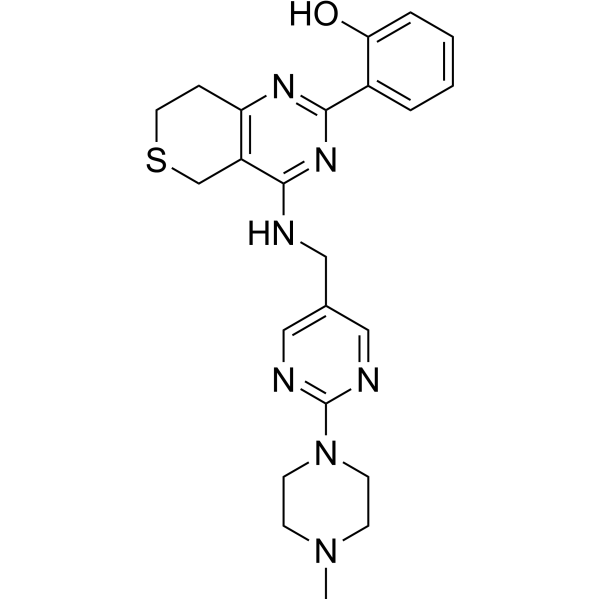
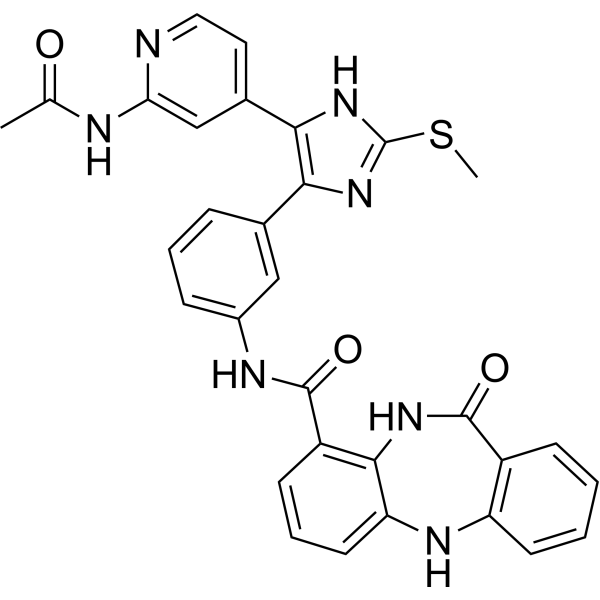
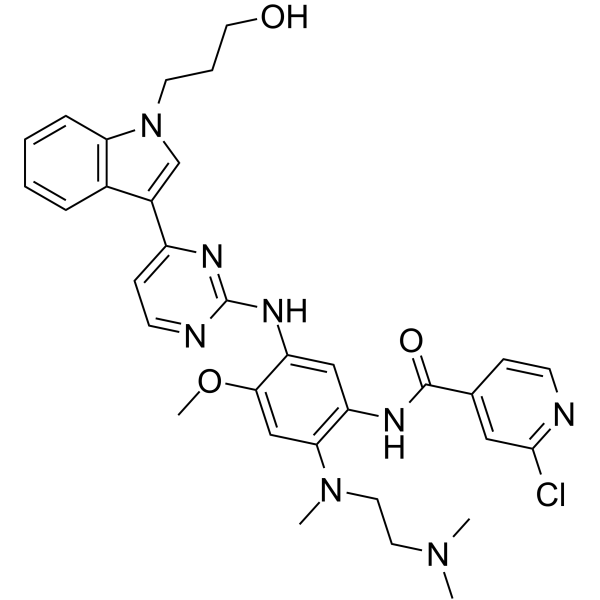
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
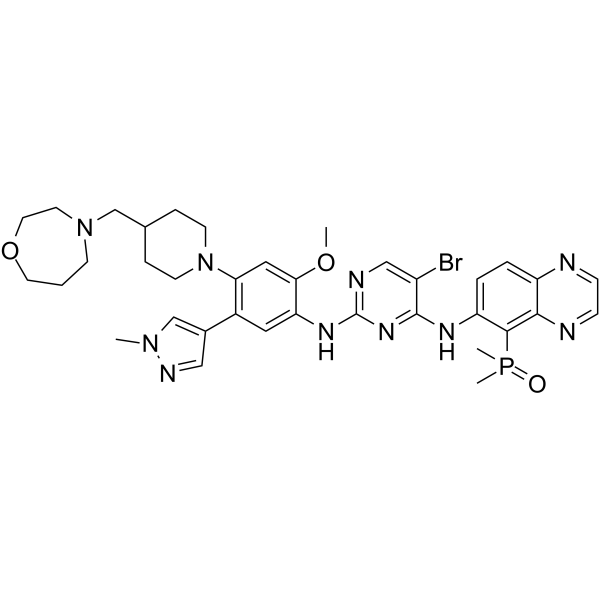
HJM-561 is a selective and effective orally active EGFRPROTAC degrader. HJM-561 is able to overcome the triple EGFR mutations that are resistant to Osimertinib (HY-15772). HJM-561 exhibits potent degradation of EGFR Del19/T790M/C797S (DC50: 9.2 nM) and L858R/T790M/C797S (DC50: 5.8 nM), and has anti-tumor activity (pink: EGFR ligand (HY-12857); blue: CRBN ligand (HY-A0003); black: linker) .
BI-4020 is a fourth-generation, orally active, and non-covalent EGFR tyrosine kinase inhibitor. BI-4020 inhibits not only the triple mutant EGFR del19 T790M C797S variant (IC50=0.2 nM in BaF3 cell lines) but also the double mutant EGFR del19 T790M and primary mutant EGFR del19 (IC50=1 nM). BI-4020 also shows activity against EGFR wt (IC50=190 nM). BI-4020 shows high kinome selectivity and good DMPK properties .
BLU-945 is a potent, highly selective, reversible and orally active epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKIs). BLU-945 can effectively inhibit EGFR with L858R and/or exon 19 deletion mutation, T790M mutation and C797S mutation. BLU-945 can be used for the research of lung cancer including non-small cell lung cancer (NSCLC) .
Cyclopropylboronic acid is an intermediate. Cyclopropylboronic acid can be used to synthesize EGFR kinase inhibitor (Compound 25). Compound 25 has antiproliferative effects on EGFR mutant (EGFR Δ19del/T790M/C797S) cells. Cyclopropylboronic acid can be used in lung cancer research .
Silevertinib (BDTX-1535) is an irreversible, brain-penetrant, selective and orally active EGFR inhibitor with wild-type EGFR-sparing. Silevertinib targets key EGFR resistance mutations, including the kinase domain (C797S,L718Q, G724S, S768I), extracellular domain (EGFRvIII, A289X), and EGFR amplification. Silevertinib exerts anti-tumor activity with well tolerated in vivo. Silevertinib can be used for non-small cell lung cancer (NSCLC) and glioblastoma (GBM) research .
EGFR-IN-7 is a potent, selective and orally active EGFR kinase inhibitor. EGFR-IN-7 has inhibitory effect for for EGFR (WT) and EGFR (mutant C797S/T790M/L858R) with IC50 values of 7.92 nM and 0.218 nM, respectively. EGFR-IN-7 can be used for the research of various cancers .
JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 0.26 nM for EGFRL858R/T790M. JBJ-04-125-02 can inhibit cancer cell proliferation and EGFRL858R/T790M/C797S signaling. JBJ-04-125-02 has anti-tumor activities .
PROTAC EGFR degrader 9 (Compound C6) is an orally active CRBN-based PROTAC EGFR degrader. PROTAC EGFR degrader 9 exhibits a DC50 of 10.2 nM and a Kd of 240.2 nM against EGFRL858R/T790M/C797S. PROTAC EGFR degrader 9 exhibits potent degradation activity against various EGFR mutants, while sparing the EGFRWT. (Blue: CRBN ligand (HY-A0003), Black: linker (HY-161613); Pink: EGFR inhibitor (HY-161537)) .
CH7233163 is a noncovalent ATP-competitive inhibitor for EGFR-Del19/T790M/C797S. CH7233163 can overcome Osimertinib (HY-15772)-Resistant EGFR-Del19/T790M/C797S mutation. CH7233163 blocks the EGFR phosphorylation in the Del19/T790M/C797S_NIH3T3 cells. CH7233163 has antitumor activities .
JND3229 is a reversible EGFRC797S inhibitor with IC50 values of 5.8, 6.8 and 30.5 nM for EGFRL858R/T790M/C797S, EGFRWT and EGFRL858R/T790M, respectively. JND3229 has good anti-proliferative activity and can effectively inhibit tumour growth in vivo. JND3229 can be used in cancer research, especially in non-small cell carcinoma .
BI-4732 is an orally active, reversible, ATP-competitive EGFR inhibitor with blood-brain barrier penetration. BI-4732 inhibits the kinase activity of EGFR L858R, T790M and C797S with IC50 values of 1 nM while sparing EGFR wild-type. BI-4732 inhibits EGFR and reduces the phosphorylation of AKT, ERK, and S6K. BI-4732 demonstrates excellent intracranial anti-tumor efficacy in YU-1097 xenograft model harboring EGFR_E19del/T790M/C797S. BI-4732 can be used for the study of non-small cell lung cancer (NSCLC) .
PROTAC EGFR degrader 14 is a potent and selective EGFR PROTACdegrader with a DC50 of about 2.9 nM and a Dmax of 93.1% for EGFRL858R/T790M/C797S. PROTAC EGFR degrader 14 selectively induces EGFRC797S degradation through a VHL and proteasome-dependent manner and downregulated EGFR-associated transcriptome and exhibits good selectivity over EGFRWT. PROTAC EGFR degrader 14 induces cell cycle arrest and apoptosis and significantly inhibits tumor growth. PROTAC EGFR degrader 14 can be used for the study of nonsmall cell lung cancer (NSCLC) (Pink: Target protein ligand: (HY-143337); Blue: E3 ligand (HY-125845); Black: Linker (HY-W004688)) .
DDC-01-163 is an allosteric PROTAC degrader targeting EGFR. DDC-01-163 is dependent on the ubiquitin–proteasome system. DDC-01-163 can selectively inhibit the proliferation of L858R/T790M (L/T) mutant Ba/F3 cells. DDC-01-163 is effective against Osimertinib (HY-15772)-resistant cells with L/T/C797S and L/T/L718Q EGFR mutations. DDC-01-163 exhibits enhanced anti-proliferative activity against L858R/T790M EGFR-Ba/F3 cells when combined with the ATP-site EGFR inhibitor Osimertinib. DDC-01-163 can be used for the study of non-small cell lung cancer .
EGFR-IN-95 (compound 5j) is an 2,4-diaminonicotinamide derivative. EGFR-IN-95 has potent inhibitory activity against EGFR del19/T790M/C797S and L858R/T790M/C797S .
BBT-176 is an orally effective EGFR inhibitor. BBT-176 has inhibitory activity against multiple EGFRC797S mutant cell lines. BBT-176 is commonly used in cancer research .
JBJ-09-063 TFA is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 TFA effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 TFA is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 TFA can be used for researching EGFR-mutant lung cancer .
JBJ-09-063 hydrochloride is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 hydrochloride effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 hydrochloride is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 hydrochloride can be used for researching EGFR-mutant lung cancer .
EGFR-IN-98 (Compound 4c) is a EGFR inhibitor. The IC50 values of L858R/T790M/C797S and Del19/T790M/C797S are 0.277 μM and 0.089 μM, respectively. EGFR-IN 98 can be used in the study of tumors .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFRC797S Recombinant Human Active Protein Kinase is a recombinant EGFRC797S protein that can be used to study EGFRC797S-related functions .
EGFR-IN-1671 is a selective EGFR inhibitor with an IC50 of 0.19 nM. EGFR-IN-167 exhibits good potency against various EGFR mutants (IC50 = 0.109 nM, 0.75 nM and <0.05 nM against EGFR (L858R), EGFR (C797S) and EGFR (del19), respectively). EGFR-IN-1671 covalently engages the catalytically conserved lysine of EGFR in live mammalian cells. EGFR-IN-1671 demonstrates excellent anti-proliferative activity by inhibiting EGFR autophosphorylation. EGFR-IN-1671 can be used for the study of non-small-cell lung carcinomas (NSCLC), glioblastoma and many solid tumors .
EGFR-IN-172 is a EGFR inhibitor. EGFR-IN-172 effectively inhibits the proliferation of non-small cell lung cancer (NSCLC) cells carrying the L858R, T790M and C797S drug-resistant mutations. EGFR-IN-172 inhibits EGFR phosphorylation, induces cell cycle arrest and apoptosis. EGFR-IN-172 can be used for the study of NSCLC .
JBJ-09-063 is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFR L858R, EGFR L858R/T790M, EGFR L858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 can be used for researching EGFR-mutant lung cancer .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFRC797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFRC797S/L858R protein that can be used to study EGFRC797S/L858R-related functions .
EMI1 is an EGFRex19del/T790M/C797S and EGFRL858R/T790M/C797S inhibitor. EMI1 can be used for the research of mutant EGFR-associated, drug-resistant non-small-cell lung cancer (NSCLC) .
EGFR-IN-126 (compound 9d) is a potent inhibitor of EGFRL858R/T790M/C797S, with the IC50 value of 0.005 μM. EGFR-IN-126 shows antitumor effects in vivo and in vitro .
EGFR/C797S-IN-1 is a potent EGFR-C797S inhibitor with an IC50 value of 0.128 µM. EGFR/C797S-IN-1 shows anti-proliferative activity and anti-tumor activity. EGFR/C797S-IN-1 inhibits the expression of p-EGFR in a dose-dependent manner .
EGFR-IN-47 is a potent and orally active EGFRL858R/T790M/C797S inhibitor with an IC50 of 0.01 μM. EGFR-IN-47 induces cell cycle attest and cell apoptosis. EGFR-IN-47 has the potential for the research of NSCLC .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFRd746-750/C797S Recombinant Human Active Protein Kinase is a recombinant EGFRd746-750/C797S protein that can be used to study EGFR d746-750/C797S-related functions .
EGFR-IN-176 is an orally active and ATP-competitive EGFR mutant inhibitor (particularly C797S-mediated EGFR triple mutant). EGFR-IN-176 effectively inhibits subsequent AKT signaling and induces apoptosis in Ba/F3 and PC-9 cells expressing EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S. EGFR-IN-176 selectively inhibits EGFR signaling in cell lines harboring EGFR triple mutation and shows no inhibitory effect against A431 cells that express wild-type EGFR. EGFR-IN-176 can effectively inhibit the enzymatic activity of ALK (IC50 < 0.5 nM). EGFR-IN-176 can be used for the study of non-small cell lung cancer (NSCLC) .
EGFR-IN-69 (compound 17g) is a potent EGFR inhibitor, with IC50 values of 4.3, 6.6 and 25.6 nM against EGFRL858R/T790M/C797S, EGFRL858R/T790M, and EGFR19del/T790M/C797S, respectively. EGFR-IN-69 can be used for non-small-cell-lung-cancer (NSCLC) research .
EGFR-IN-177 is a potent EGFR inhibitor with IC50s of 0.32, 1.04, 0.65, 0.67, 0.48, 0.55 and 0.38 nM against EGFR L858R, EGFR L858R/T790M, EGFRL858R/T790M/C797S, EGFR D746-750, EGFRD746-750/C797S, EGFR D770_N77linsNPG, and EGFR WT. EGFR-IN-177 inhibits lung cancer proliferation and EGFR phosphorylation. EGFR-IN-177 can be used for the study of cancers such as non-small cell lung cancer .
EGFR-IN-30 is a potent EGFR inhibitor with IC50s of 1-10 nM, <1 nM for EGFR (WT), EGFR (L858R/T790M/C797S), respectively. EGFR-IN-30 has potential for cell proliferative diseases, such as cancer research .
ZSH-2117 is a covalent and selective EGFRPROTAC degrader with a DC50 of 45 nM in Ba/F3-EGFRL858R/T790M/C797S cells. ZSH-2117 significantly inhibits cell proliferation and reduces the downstream EGFR signaling proteins level of AKT and ERK. ZSH-2117 effectively inhibits tumor growth in Ba/F3-EGFRL858R/T790M/C797S xenograft mice model . Pink: EGFR ligand (HY-175162); Blue: NEDD4 ligase ligand (HY-175159); Black: linker
EGFR-IN-179 (Compound 8d) is an EGFR inhibitor (IC50: 0.068 μM for EGFR L858R/T790M/C797S; 2.56 μM for EGFR-WT-TK). EGFR-IN-179 has anticancer activity against non-small cell lung cancer .
EGFR-IN-11 is a fourth-generation EGFR-tyrosine kinase inhibitor (EGFR-TKI) with an IC50 of 18 nM for triple mutant EGFRL858R/T790M/C797S. EGFR-IN-11 significantly suppresses the EGFR phosphorylation, induce the apoptosis, and arrest cell cycle at G0/G1 .
EGFR-IN-164 (Compound 4) is a selective and covalent allosteric EGFR inhibitor. EGFR-IN-164 significantly inhibits the activity of EGFRL858R/T790M/C797S kinase (IC50: 48.1 nM) and proliferation of of EGFR-mutant cells. EGFR-IN-164 can be used for drug resistance of cancer research .
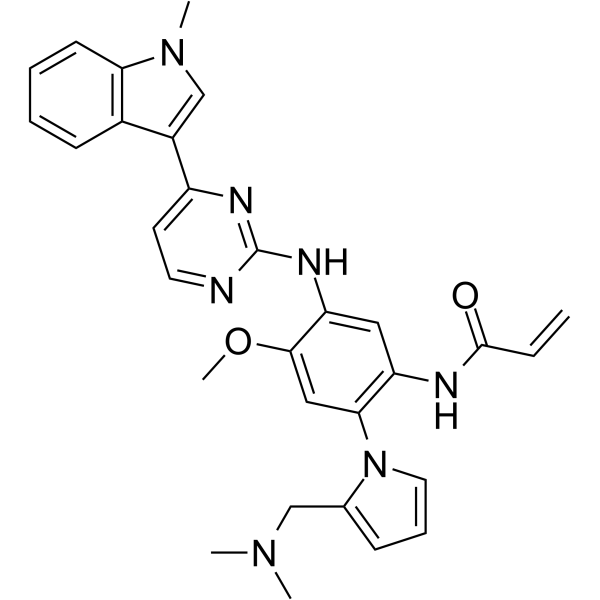
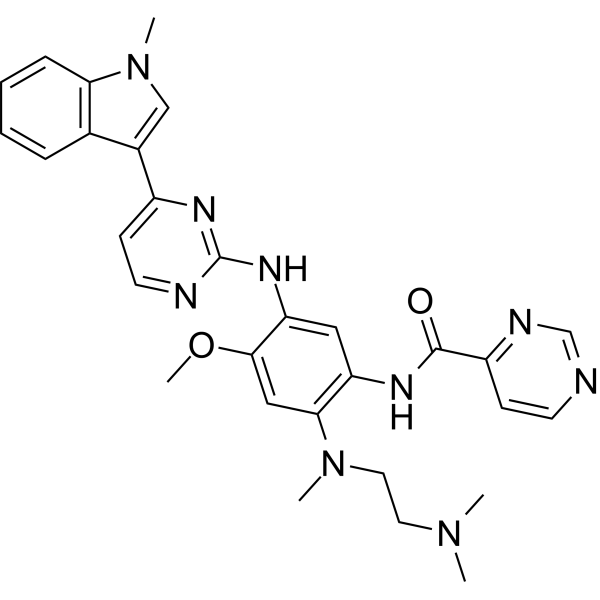
EGFR-IN-173 is an orally active, pan-mutant EGFR tyrosine kinase inhibitor that targets EGFR 19del, L858R/T790M and C797S triple-mutations, potently inhibiting EGFR19del/T790M/C797S with an IC50 of 1.19 nM while showing over 100-fold selectivity for mutant over wild-type EGFR (IC50 = 19.362 μM against WT). EGFR-IN-173 significantly inhibits cell migration, induces apoptosis in non-small cell lung cancer (NSCLC) cells. EGFR-IN-173 inhibits EGFR phosphorylation and suppresses the downstream pathways (MAPK/ERK, AKT, STAT3). EGFR-IN-173 exhibits antitumor efficacy in NSCLC and Ba/F3 xenograft models. EGFR-IN-173 can be used for NSCLC research .
EGFR-IN-181 is an orally active, potent, brain-penetrant EGFRL858R/T790M/C797S triple mutations inhibitor (IC50 = 1.32 nM). EGFR-IN-181 can inhibit EGFR phosphorylation (p-EGFR) and phosphorylation of its downstream signaling proteins AKT (p-AKT) and ERK (p-ERK). EGFR-IN-181 can induce apoptosis and cause G2 phase arrest. EGFR-IN-181 can be used for the study of non-small cell lung cancer (NSCLC) and brain metastases .
PROTAC EGFR degrader 13 (compound 106) is an EGFR PROTAC degrader with a DC50 of <0.1 μM. PROTAC EGFR degrader 13 has proliferation inhibitory activity on cell Ba/F3-TEL-EGFR-T790M-L858R-C797S with an IC50 of 15.6 nM. PROTAC EGFR degrader 13 can be used for the study of EGFR-related diseases such as cancer (Pink: Target protein ligand; Blue: E3 ligand (HY-14658); Black: linker (HY-W262798); E3 ligand + linker: HY-W998306) .
EGFR-IN-97 (compound 6q) is a EGFR inhibitor. EGFR-IN-97 shows inhibitory activity against Ba/F3-EGFRL858R/T790M/C797S and Ba/F3-EGFRDel19/T790M/C797S cells, with IC50 values of 0.42 μM and 0.41 μM, respectively. EGFR-IN-97 can promote apoptosis of NCI-H1975-EGFRL858R/T790M/C797S cells at the concentration of 0.8 μM .
EGFR-IN-91 (compound 9) is an orally available EGFR inhibitor with blood-brain barrier penetrability. EGFR-IN-91 inhibits EGFRL858R/C797S and EGFRexon 19del/C797S, inducing tumor regression in xenograft (PDX) mouse models. EGFR-IN-91 has the potential to inhibit localized and metastatic non-small cell lung cancer (NSCLC) driven by EGFR mutants .
LS-106 is an orally active and potent inhibitor against epidermal growth factor receptor (EGFR) . LS-106 exhibits antitumor activities both in vitro and in vivo. LS-106 inhibits the kinase activities of EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S with IC50 values of 2.4 nmol/L and 3.1 nmol/L, respectively, which is more potent than Osimertinib (HY-15772). LS-106 induces Apoptosis, suppresses cell proliferation of tumor cells harboring EGFR19del/T790M/C797S and leas to significant tumor regression in a C797S-mutant xenograft model .
EGFR-IN-140 (Compound 31) is the inhibitor for EGFR, that inhibits EGFR wildtype and EGFR L858R/T790M/C797S mutant with Ki of 0.95 nM and 2.1 nM, and inhibits EGFR del19/T790M/C797S in Ba/F3 with an IC50 of 56.9 nM. EGFR-IN-140 exhibits antitumor efficacy in mouse model .
EGFR-IN-24 is a selective EGFR triple mutant inhibitor with >300 nM IC50 against human EGFRdel19/T790M/C797S,EGFRL858R/T790M/C797S, and EGFRWT. EGFR-IN-24 can be used for the research of non-small cell lung cancer .
EGFR kinase inhibitor 2 (compound A-7) is a potent EGFR inhibitor targeting EGFRL858R/T790M/C797S and EGFRDel19/T790M/C797S mutants. EGFR kinase inhibitor 2 has the potential to address acquired resistance in the treatment of non-small cell lung cancer .
EGFR mutant-IN-2 (Compound D51) is an EGFR mutant inhibitor. EGFR mutant-IN-2 inhibits the EGFRL858R/T790M/C797S mutant with an IC50 value of 14 nM. EGFR mutant-IN-2 inhibits the EGFRdel19/T790M/C797S mutant with an IC50 value of 62 nM. EGFR mutant-IN-2 has favorable PK parameters, safety properties, in vivo stability, and antitumor activity .
EGFR-IN-101 (I-10) is a 2-phenylamino pyrimidine derivative. EGFR-IN-101 is a EGFR inhibitor. The IC50 values for EGFRL858R/T790M/C797S and Ba/F3-EGFRL858R/T790M/C797S are 33.26 and 106.4 nM, respectively. EGFR-IN-101 can be used IN the study of non-small cell lung cancer (NSCLC) .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/T790M/C797S Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/T790M/C797S protein that can be used to study EGFR d746-750/T790M/C797S-related functions .
EGFR-IN-27 is a potent EGFR inhibitor with IC50s of <50 nM for EGFR Del, L858R, Del/T790M, L858R/T790M, Del/T790M/C797S, and L858R/T790M/C797S, respectively (WO2021249324A1, compound 511) .
EGFR-IN-82 (Cmpound 8a) is a potent and orally active EGFR inhibitor with IC50 values of 0.09 and 0.06 nM for EGFRL858R/T790M/C797S and EGFRDel19/T790M/C797S, respectively. EGFR-IN-82 has no significant effect on EGFRWT. EGFR-IN-82 has anti-proliferative activity and inhibits tumor formation in nude mice. EGFR-IN-82 can be used in non-small cell lung cancer research .
EGFR-IN-48 is a potent and orally active EGFR inhibitor with IC50s of 0.193 nM, 0.251 nM, 10.4 nM for EGFRd19/TM/CS, EGFRLR/TM/CS, EGFRWT, respectively. EGFR-IN-48 inhibits the proliferation of BaF3 EGFRdel19/T790M/C797S and PC-9 EGFRdel19/T790M/C797S cells with IC50s of 1.526, 66.7 nM, respectively .
Os30, a potent fourth-generation EGFR inhibitor, is a potent EGFRC797S-TK inhibitor with IC50 values of 18 nM and 113 nM for EGFRDel19/T790M/C797S TK and EGFRL858R/T790M/C797S TK, respectively. Os30 can suppress EGFR phosphorylation, arrest at G1 phase and induce the apoptosis of KC-0116 (BaF3-EGFRDel19/T790M/C797S) cells. Os30 shows potent antitumor efficacy on non-small cell lung cancer (NSCLC) with EGFmRC797S mutation .
EGFR-IN-90 (compound 34) is an orally active EGFR inhibitor. EGFR-IN-90 shows inhibitory activity against EGFRL858R/T790M/C797S with an IC50 of 5.1 nM and inhibits the proliferation of the H1975-TM cell line harboring EGFRL858R/T790M/C797S with an IC50 of 0.05 μM. EGFR-IN-90 and inhibits tumor growth in the H1975-TM xenograft tumor model .
EGFR-IN-117 (Compound 8h) exhibits inhibitory activity against EGFR mutation, targets the tumor environment, and induces apoptosis of cancer cells. EGFR-IN-117 inhibits proliferations of H1975, PC-9, and EGFR mutant cells BaF3-EGFRL858R/T790M/C797S and BaF3– C797S/Del19/T790M, with IC50 of 13 nM, 19 nM, 1.2 nM and 1.3 nM, respectively. EGFR-IN-117 exhibits antitumor efficacy in mouse models .
EGFR-IN-133 (Compound 24) is an inhibitor for EGFR, that inhibits the EGFR wildtype, L858R/T790M, d19/T790M, L858R/T790M/C797S, and d19/T790M/C797S mutans with IC50 of 0.1, 0.044, 0.036, 0.04, and 0.054 nM. EGFR-IN-133 exhibits good pharmacokinetics characteristics with high oral exposure .
EGFR-IN-132 (Compound 23) is an inhibitor for EGFR, that inhibits the EGFR wildtype, L858R/T790M, d19/T790M, L858R/T790M/C797S, and d19/T790M/C797S mutans with IC50 of 1.6, 0.025, 0.019, 0.022, and 0.029 nM. EGFR-IN-132 exhibits good pharmacokinetics characteristics with high oral exposure .
EGFR-IN-22 is a potent EGFR inhibitor with IC50s of 4.91 nM and 0.54 nM for wild type EGFR and EGFRL858R/T790M/C797S, respectively (CN112538072A, compound 243) .
EGFR-IN-120 (Compound 11eg) is an orally active EGFR inhibitor. EGFR-IN-120 inhibits EGFRL858R/T790M/C797S with an IC50 value of 0.053 μM, and has a relatively weak effect on EGFRWT (IC50: 1.05 μM). EGFR-IN-120 inhibits the phosphorylation of EGFR and main downstream effectors (STAT3, AKT, and Erk). EGFR-IN-120 induces cell cycle arrest and cell apoptosis in EGFR mutant cells. EGFR-IN-120 inhibits the proliferation of the NSCLC cells harboring EGFRL858R/T790M/C797S with an IC50 of 0.052 μM .
EGFR-IN-23 is a potent EGFR TKI (tyrosine kinase inhibitor) with an IC50 of 8.05 nM for BaF3/EGFR-DEL19/T790M/C797S cell (WO2021244502A1, compound 8) .
EGFR-IN-89 (compound 13k) is a potent, fourth-generation EGFR mutation inhibitor with an IC50 of 10.1 nM against Del19/T790M/C797S mutations. EGFR-IN-89 shows higher selectivity over wild type .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/T790M/C797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/T790M/C797S/L858R protein that can be used to study EGFR d746-750/T790M/C797S/L858R-related functions .
EGFR-IN-78 (compound A5),a 2-aminopyrimidine derivative,is a reversible inhibitor of EGFRC797S-TK,and also an inducer of apoptosis. EGFR-IN-78 shows anti-proliferative activity,inhibits EGFR phosphorylation and arrests cell cycle at G2/M phase .
EGFR mutant-IN-1, a 5-methylpyrimidopyridone derivative, is a potent and selective EGFRL858R/T790M/C797S mutant inhibitor with an IC50 of 27.5 nM, while being a significantly less potent for EGFRWT (IC50 >1.0 μM) .
EGFR-IN-127 is an ATP-competitive EGFR inhibitor with IC50s of 136.3 nM and 161. 2 nM for EGFRdel19 and EGFRdel19/T790M/C797S, respectively. EGFR-IN-127 has the potential for the study of non-small-cell lung cancer (NSCLC) .
DA-0157 is the orally active inhibitor for EGFR and ALK that overcomes drug-resistant mutations of EGFRC797S and ALK in NSCLC) cells. DA-0157 inhibits the proliferation of Ba/F3-EGFR Del19/T790M/C797S (IC50 = 6.9 nM), Ba/F3-EGFR WT (IC50 = 0.83 μM), Ba/F3-EML4-ALK-L1196M (IC50 = 5.5 nM), and Ba/F3-EML4-ALK (IC50 = 7.4 nM). DA-0157 inhibits CYP2D6 with IC50 of 5.26 μM. DA-0157 exhibits antitumor efficacy in mouse models .
Mutated EGFR-IN-3 (compound 3) is a potent, ATP-competitive and highly selective allosteric dibenzodiazepinone inhibitor of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with IC50 values of 12 nM and 13 nM, respectively .
EGFR-IN-161 (Compound DD-8) is a potent and reversible inhibitor of L858R/T790M/C797S mutant EGFR kinases, with an IC50 of 0.87 nM. EGFR-IN-161 can induce apoptosis process, G1-phase arrestation, and migration inhibition in tumor cells .
EGFR-IN-104 (Compound A23) is an effective inhibitor of EGFR, with IC50 values of 0.33 μM and 0.133 μM against EGFRL858R/T790M and EGFRDel19/T790M/C797S, respectively. EGFR-IN-104 exhibits anticancer activity both in vitro and in vivo .
EGFR-IN-180 (Compound L15) is an EGFR inhibitor. EGFR-IN-180 shows inhibitory activity against EFGR and EGFR harboring the L858R/T790M/C797S triple drug-resistant mutation, with IC50 values of 80.96 nM and 16.43 nM, reapectively. EGFR-IN-180 can be used for the study of non-small cell lung cancer (NSCLC) .
EGFR-IN-175 is an orally active and selective EGFRL858R/T790M/C797S inhibitor with an IC50 of 18.94 nM. EGFR-IN-175 can induce cell apoptosis and cause G1 phase arrest. EGFR-IN-175 can downregulate p-EGFR, p-AKT, and p-ERK expression. EGFR-IN-175 can be used for the research of cancer, such as lung cancer .
DS06652923 is an orally active EGFR triple mutation inhibitor. DS06652923 has a growth inhibition effect on Ba/F3 EGFR del19/T90M/C797S cells, with a GI50 value of 9.4 nM. DS06652923 can lead to tumor regression in Ba/F3 xenograft models .
EGFR-IN-61 (compound 22a) is a potent EGFR kinase inhibitor, with IC50 values of 42 nM (L858R/T790 M), 137 nM (L858R/T790 M/C797S), and 743 nM (WT), respectively. EGFR-IN-61 shows antiproliferative activity against A549 and H1975 cell lines, with IC50 values of 2.14 and 1.82 μM, respectively .
LSD1/EGFR-IN-1 (compound L-1) is a potent inhibitor of LSD1, EGFRT790M/L858R and EGFRL858R/T790M/C797S, with IC50s of 6.24 and 2.06 and 5.01 μM, respectively. LSD1/EGFR-IN-1 plays an important role in cancer research .
BI-8128 is a potent EGFR inhibitor, with IC50 values of 12, 6.7, 22, 10, and 3 nM against wild-type, T790M, C797S, T790M/C797S, and L858R/T790M/C797S mutant EGFR, respectively. BI-8128 significantly inhibits the proliferation of Ba/F3 and PC-9 drug-resistant mutant cells. BI-8128 is applicable for the research of non-small cell lung cancer .
EGFR-IN-160 (Compound R12) is an EGFR inhibitor (IC50 values are 1.62, 0.49 and 0.98 μM for EGFRWT, EGFRT790M and EGFRL858R/T790M/C797S, respectively). EGFR-IN-160 induces G2/M and S phase arrest and apoptosis in NCI-H522 cells, and has anticancer activity. EGFR-IN-160 has antioxidant effects against DPPH (IC50: 12.11 µM) and H2O2 (IC50: 8.89 µM) .
EGFR-IN-139 (compound PD 18) is an epidermal growth factor receptor (EGFR) inhibitor, with IC50s of 12.88 (wild type), 10.84 (L858R/T790M), 42.68 (L858R/T790M/C797S) nM, respectively. EGFR-IN-139 displays strong anticancer activity against A549 and H1975 cancer cell lines, which are highly expressed EGFR. EGFR-IN-139 has a strong selectivity to cancer cells. EGFR-IN-139 can be used for nonsmall cell lung cancer (NSCLC) research[1].
EGFR-IN-62 (compound 9h) is a potent and reversible EGFR kinase inhibitor, with IC50 values of 10 nM (L858R/T790 M), 29 nM (WT), and 242 nM (L858R/T790 M/C797S), respectively. EGFR-IN-62 shows antiproliferative activity against A549 and H1975 cell lines, with IC50 values of 2.53 and 1.56 μM, respectively. EGFR-IN-62 induces dose-dependent apoptosis process, G1/G0-phase arrestation, and the inhibition of motility on A549 and/or H1975 cell lines .
EGFR-IN-203 is a EGFR kinase domain inhibitor targeting EGFRT790M/C797S/V948R. EGFR-IN-203 stably binds to the allosteric pocket of EGFR in an inactive conformation. EGFR-IN-203 can be used in research related to cancers such as non-small cell lung cancer .
EGFR-IN-189 (Compound 7) is a selective covalent epidermal growth factor receptor(EGFR) inhibitor with an IC50 of 200 nM. EGFR-IN-189 can effectively inhibit the C797S mutant type of EGFR. EGFR-IN-189 can inhibit cancer cells proliferation and inhibit EGFR phosphorylation and downstream ERK1/2 phosphorylation. EGFR-IN-189 can be used for research of nonsmall cell lung cancer .
EGFR-IN-213 is a selective inhibitor of EGFRL858R/T790M/C797S with a human IC50 of 0.48 nM. EGFR-IN-213 acts as an antiproliferative agent, inducing apoptosis and cell cycle arrest, and inhibiting colony formation, cell migration, and tube formation. EGFR-IN-213 can be used for the research of non-small cell lung cancer, chronic myeloid leukemia, gastric cancer, prostate cancer .
EGFR-IN-208 is an allosteric mutant EGFRL858R/T790M and EGFRL858R/T790M/C797S inhibitor, with IC50 values of 3.06 μM and 1.08 μM, respectively. EGFR-IN-208 binds to the allosteric site of EGFR and inhibits EGFR phosphorylation. EGFR-IN-208 induces apoptosis and exhibits antiproliferative effects in cancer cells. EGFR-IN-208 can be used in research related to non-small cell lung cancer .
EGFR-IN-152 (compound D4) is a potent EGFR tyrosine kinase inhibitor, exhibiting potent EGFRL858R/T790M/C797S inhibition activity (IC50 = 40 nM). EGFR-IN-152 induces G0/G1 phase cell cycle arrest and apoptosis, thereby inhibiting colony formation and cell proliferation of non-small cell lung cancer (NSCLC) cells. EGFR-IN-152 can be used for NSCLC research .
EGFR-IN-209 (Compound 6g) is an orally active, selective EGFRDel19/T790M/C797S inhibitor with an IC50 of 0.056 μM. EGFR-IN-209 blocks the phosphorylation of EGFR and its downstream effector molecules (pMAPK, pAKT). EGFR-IN-209 induces Apoptosis. EGFR-IN-209 exhibits antitumor activity against Osimertinib (HY-15772)-resistant non-small cell lung cancer. EGFR-IN-209 can be used in research related to non-small cell lung cancer .
EGFR-IN-201 is a potent EGFR inhibitor, with an IC50 of 0.091 μM against wild-type EGFR; for mutant EGFR variants, the IC50 values of EGFRT790M, EGFRL858R and EGFRC797S are 0.147 μM, 0.221 μM and 0.703 μM, respectively. EGFR-IN-201 inhibits EGFR downstream signaling proteins AKT1 (IC50 = 0.225 μg/mL) and ERK1 (IC50 = 0.705 μg/mL). EGFR-IN-201 induces G0/G1 cell cycle arrest, apoptosis and low-level necrosis in cancer cells. EGFR-IN-201 is applicable to research on cancers such as colon cancer .
EGFR-IN-197 is an EGFR inhibitor with IC50 values of 19.5 nM and 12.0 nM against EGFRL858R/T790M and EGFRL858R/T790M/C797S, respectively. EGFR-IN-197 arrests the cell cycle of NCI-H1975 cells at the G2/M phase, while inhibiting their proliferation, colony formation and migration; it also inhibits mitochondrial translocation and upregulates mitochondrial H2S levels. EGFR-IN-197 disrupts anti-apoptotic signaling pathways by regulating apoptosis-related proteins; it induces DNA damage and activates pro-apoptotic pathways to trigger apoptosis. EGFR-IN-197 can be used in studies related to non-small cell lung cancer (NSCLC) .
SG-55 is a selective, noncompetitive and orally active AKR1C3 inhibitor with an IC50 of 5 nM and a Ki of 10 nM. SG-55 shows >2000-fold selectivity for AKR1C3 over AKR1C1, AKR1C2, and AKR1C4 (> 10 μM). SG-55 increases the ratio of reduced/oxidized nicotinamide adenine dinucleotide phosphate (NADPH/NADP +), decreases the ratio of reduced/oxidized glutathione (GSH/GSSG), and induces DNA double-strand breaks. SG-55 can overcome Osimertinib (HY-15772) resistance mediated by EGFRC797S triple mutation in non-small cell lung cancer (NSCLC) .
EGFR/VEGFR2-IN-12 (compound 11a) is a dual EGFR/VEGFR2 inhibitor, with an IC50 value of 64 nM against human EGFR and an IC50 value of 74 nM against human VEGFR2. EGFR/VEGFR2-IN-12 inhibits the phosphorylation of EGFR and VEGFR2, induces cell cycle arrest at the G1/S phase, activates apoptotic pathways, promotes PARP-1 cleavage, exhibits low micromolar antiproliferative activity, and shows much higher selectivity for cancer cells than normal cells. EGFR/VEGFR2-IN-12 is applicable for cancer-related research .
Cyclopropylboronic acid is an intermediate. Cyclopropylboronic acid can be used to synthesize EGFR kinase inhibitor (Compound 25). Compound 25 has antiproliferative effects on EGFR mutant (EGFR Δ19del/T790M/C797S) cells. Cyclopropylboronic acid can be used in lung cancer research .
The EGFR protein is a receptor tyrosine kinase that can bind to a variety of ligands, such as EGF, TGFA, AREG, epigen, BTC, epiregulin, and HBEGF, to initiate signaling cascades that mediate cellular responses. This involves receptor dimerization, autophosphorylation and recruitment of adapter proteins such as GRB2, activating downstream pathways such as RAS-RAF-MEK-ERK, PI3-kinase-AKT, PLCgamma-PKC and STAT. EGFR Protein, Human (C797S, T790M, L858R, sf9, GST) is the recombinant human-derived EGFR, expressed by Sf9 insect cells, with N-GST labeled tag.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
MedchemExpress Validation 03
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
MedchemExpress Validation 04
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedChemExpress values your privacy and your trust is important to us. We use cookies to enhance your website experience. Some cookies are necessary to run the website.
Privacy and Cookie Policy