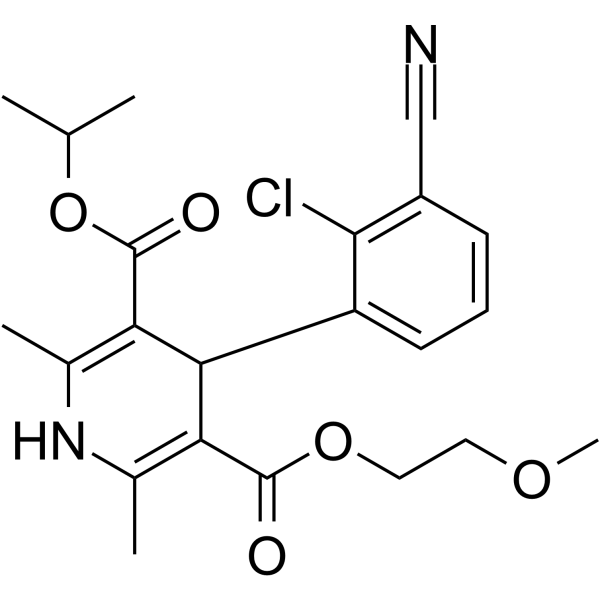
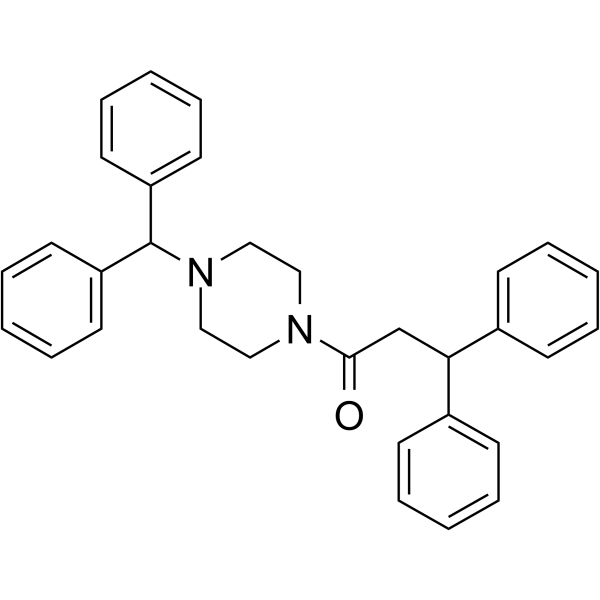
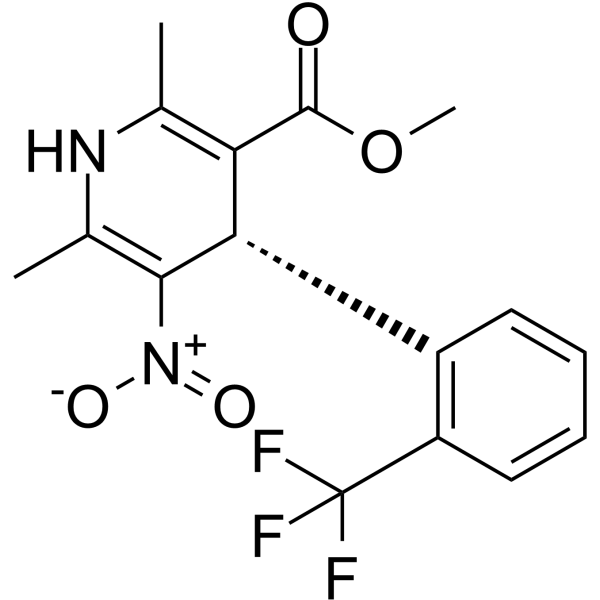
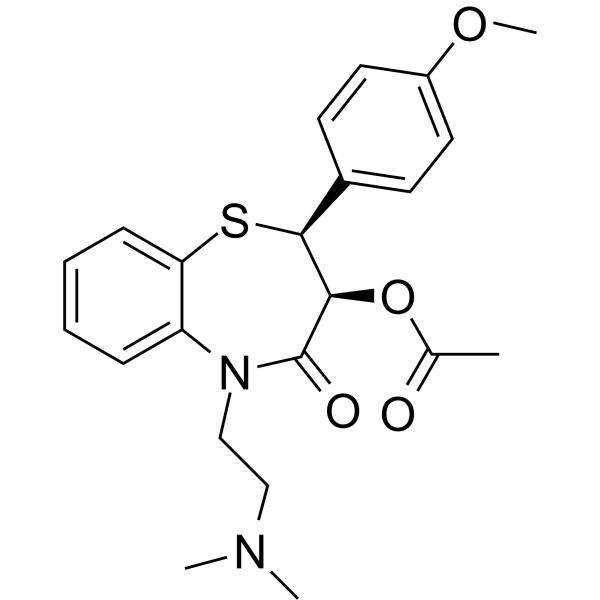
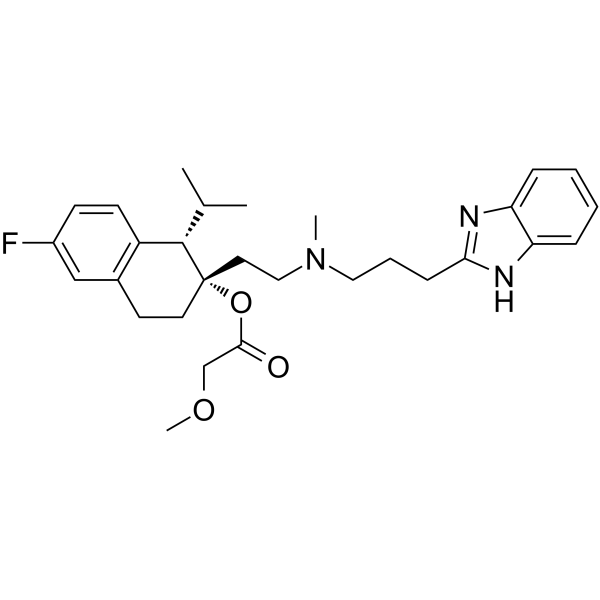
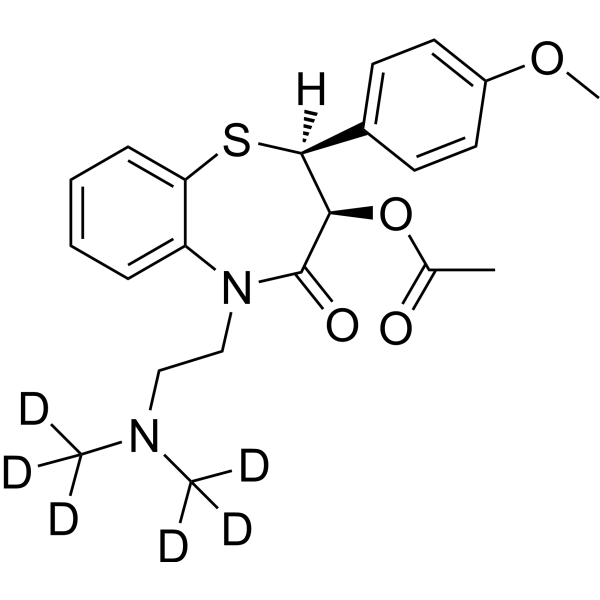
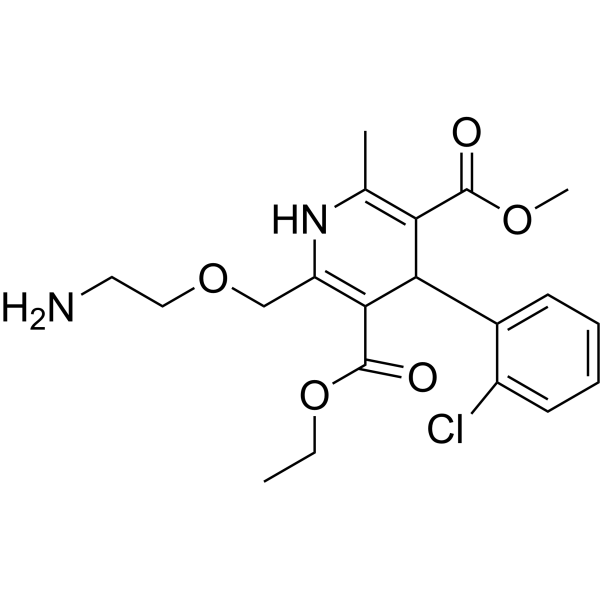
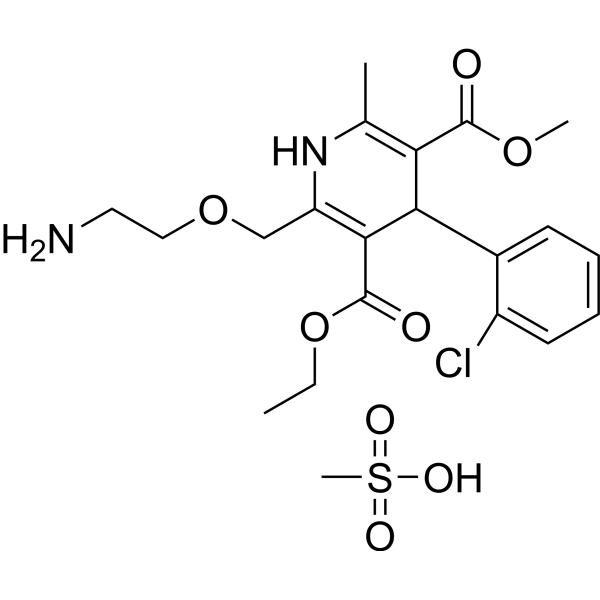
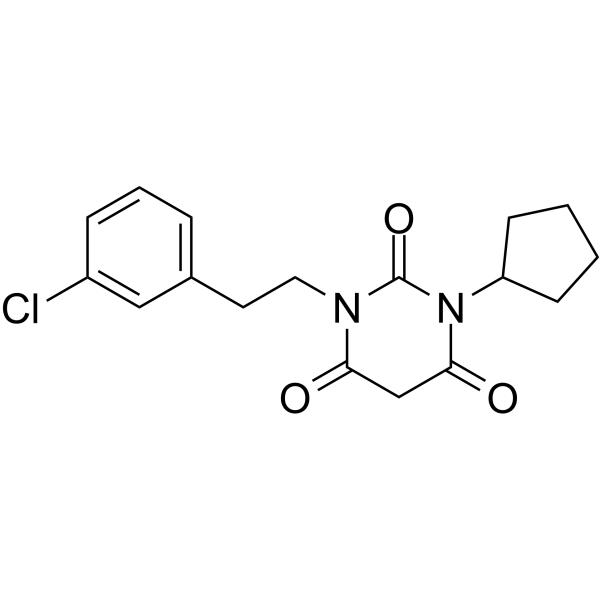
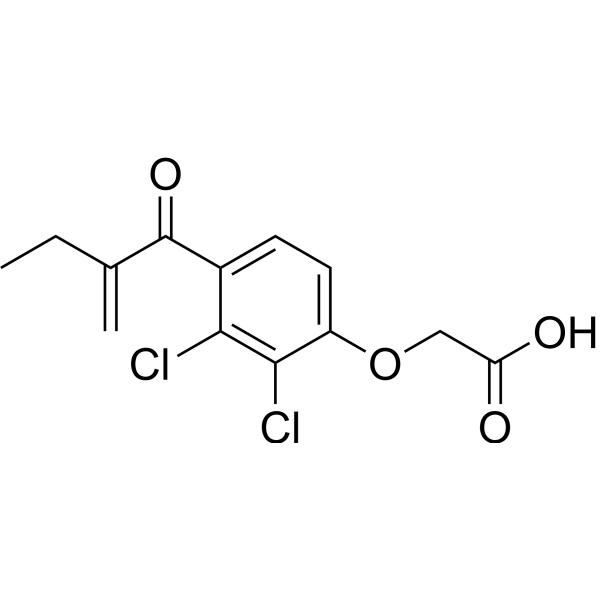
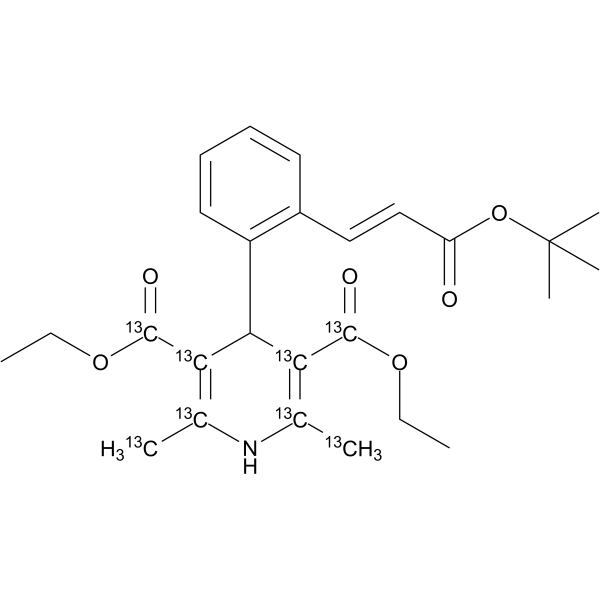
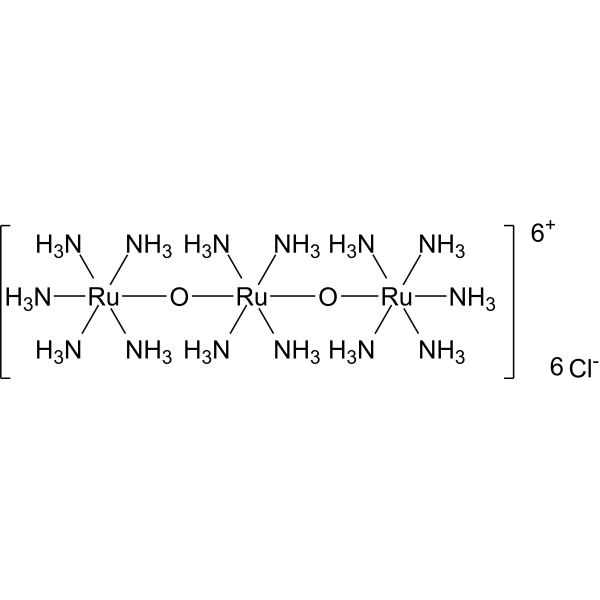From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
Bay K 8644 ((±)-Bay K 8644) is a racemate consisting of two isomers (R)-(+)-Bay-K-8644 and (S)-(-)-Bay-K-8644 . Bay K 8644 is a L-type Ca 2+ channel agonist with an EC50 of 17.3 nM. Bay K 8644 increases Ca 2+ influx through sarcolemmal Ca 2+ channels by increasing the open time of the channel. Bay K 8644 has vasoconstrictive effects .
SR33805 is a potent Ca 2+ channel antagonist, with EC50s of 4.1 nM and 33 nM in depolarized and polarized conditions, respectively. SR33805 blocks L-type but not T-type Ca 2+ channels. SR33805 can be used for the research of acute or chronic failing hearts .
Nemadipine-A is a specific inhibitor of the EGL-19 L-typeCa 2+ channel . Nemadipine-A, a cell-permeable L-type calcium channel inhibitor, sensitizes TRAIL-resistant cancer cells to this ligand .
Levobetaxolol is a potent and high affinity β-adrenergic antagonist with IC50 values of 33.2, 2970, 709 nM for guinea pig atrial β1, tracheal β2 and rat colonic β3 receptors, respectively. Levobetaxolol reduces IOP (intraocular pressure). Levobetaxolol exhibits a micromolar affinity for L-type Ca21-channels. Levobetaxolol decreases the effects of ischaemia/reperfusion injury in rats. Levobetaxolol has the potential for the research of glaucoma .
Ethacrynic acid D5 is a deuterium labeled Ethacrynic acid. Ethacrynic acid is a diuretic. Ethacrynic acid is an inhibitor of glutathione S-transferases (GSTs). Ethacrynic acid is a potent inhibitor of NF-kB-signaling pathway, and also modulates leukotriene formation. Ethacrynic acid also inhibits L-type voltage-dependent and store-operated calcium channel, leading to relaxation of airway smooth muscle (ASM) cells. Ethacrynic acid has anti-inflammatory properties that reduces the retinoid-induced ear edema in mice .
Kurtoxin is a selective Cav3 (T-type) voltage-gated Ca 2+ channel gating inhibitor with a Kd of 15 nM for Cav3.1 (α1G T-type) Ca 2+ channel. Kurtoxin can interact with high affinity with native neuronal high-threshold L-type, N-type, and P-type Ca 2+ channels in central and peripheral neurons. Kurtoxin also shows cross-reactivity with voltage-gated Na + channel .
Isradipine (PN 200-110) is an orally active L-type calcium channel blocker. Isradipine, as a powerful peripheral vasodilator, is a dihydropyridine calcium antagonist with selective actions on the heart as well as the peripheral circulation. Isradipine is a potentially viable neuroprotective agent for Parkinson's disease .
Catharanthine ((+)-3,4-Didehydrocoronaridine), a constituent of anticancer vinca alkaloids, inhibits voltage-operated L-type Ca 2+ channel (VOCC). Catharanthine has IC50s of 220 μM and 8 μM for VOCC currents in cardiomyocytes and vascular smooth muscle cells (VSMCs), respectively. Catharanthine lowers blood pressure (BP), heart rate (HR). Catharanthine has anti-cancer activity .
Norverapamil hydrochloride ((±)-Norverapamil hydrochloride), an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor .
Norverapamil ((±)-Norverapamil), an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor .
Fluspirilene is a non-competitive antagonist of L-type calcium channels with an IC50 of 0.03 μM. Fluspirileneis a long-acting injectable depot antipsychotic agent used for schizophrenia.
(Rac)-MEM 1003 is the racemate of MEM 1003. MEM 1003, a dihydropyridine compound, is a potent L-typeCa 2+ channel antagonist and has the potential for Alzheimer’s disease research .
NP118809 is a potent N-type calcium channel blocker, with an IC50 of 0.11 μM; also less potently inhibits L-type calcium channel with an IC50 of 12.2 μM.
Diltiazem is an orally active L-type Ca 2+ channel blocker. Diltiazem shows antihypertensive and antiarrhythmic effects. Diltiazem can be used for the research of cardiac arrhythmia, hypertension, and angina pectoris .
Tribendimidine is an orally active, broad-spectrum anthelmintic agent, with particularly high activity against A. lumbricoides and N. americanus. Tribendimidine is also an L-typenicotinic acetylcholine receptor (nAChR) agonist .
Norverapamil-d7 (hydrochloride) is a deuterium labeled Norverapamil. Norverapamil ((±)-Norverapamil), an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor[1][2].
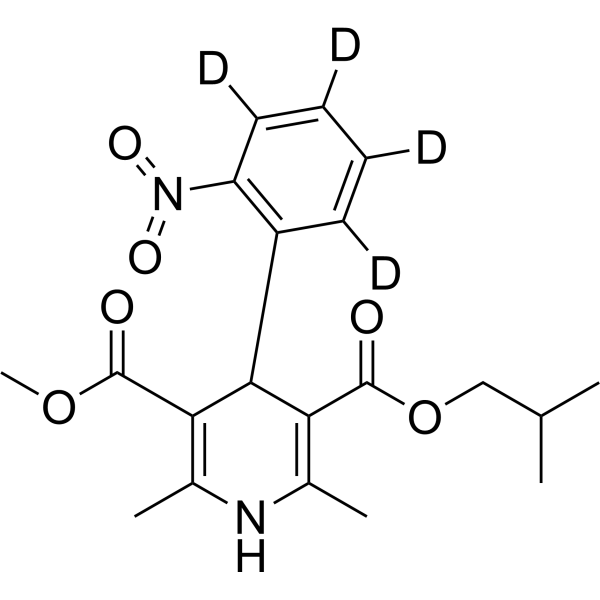
Nisoldipine-d6 is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552; Sular) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with an IC50 of 10 nM.
Norverapamil-d7 is a deuterium labeled Norverapamil ((±)-Norverapamil). Norverapamil, an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor[1][2].
Diltiazem malate is a potent and orally active L-type calcium channel inhibitor. Diltiazem malate shows antihypertensive and antiarrhythmic effects. Diltiazem malate can be used for the research of cardiac arrhythmia, hypertension, and angina pectoris .
FluoBar1 is an imaging fluorescence probe modified by a barbiturate ligand with fluorescent coumarin. FluoBar1 can monitor L-type voltage-gated calcium channels (LTCC) in living cells in real time for the study of neurological diseases .
Mibefradil dihydrochloride (Ro 40-5967 dihydrochloride) is a calcium channel blocker with moderate selectivity for T-type Ca 2+ channels (IC50s of 2.7 μM and 18.6 μM for T-type and L-type currents, respectively) .
Pinaverium bromide is an L-typecalcium channel blocker with selectivity for the gastrointestinal tract, effectively relieves pain, diarrhea and intestinal discomfort, provides good therapeutic efficacies without significant adverse effects on Irritable bowel syndrome (IBS) patients .
Calcicludine is a protein toxin from the venom of the green mamba Dendroaspis angusticeps that inhibits high-voltage-activated calcium channel, especially L-typecalcium channel with the IC50 of 88 nM. Calcicludine has role in excitatory synaptic transmission .
Mibefradil (Ro 40-5967) is a calcium channel blocker with moderate selectivity for T-type Ca 2+ channels displaying IC50s of 2.7 μM and 18.6 μM for T-type and L-type currents, respectively .
Diltiazem-d6 is the deuterium labeled Diltiazem. Diltiazem is an orally active L-type Ca2+ channel blocker, with antihypertensive and antiarrhythmic effects. Diltiazem can be used for the research of cardiac arrhythmia, hypertension, and angina pectoris[1][2][3].
Amlodipine, an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine can be used for the research of high blood pressure and cancer .
Amlodipine mesylate, an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine mesylate can be used for the research of high blood pressure and cancer .
Amlodipine maleate is a dihydropyridine calcium channel blocker, acts as an orally active antianginal agent. Amlodipine maleate blocks the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine maleate can be used for the research of high blood pressure and cancer .
Drotaverine (hydrochloride) is a type4 cyclic nucleotide phosphodiesterase (PDE4) inhibitor and an L-type voltage-dependent calcium channel (L-VDCC) blocker, blocks the degradation of 3',5'-cyclic adenosine monophosphate. Drotaverine (hydrochloride) exhibits in vivo antispasmodic efficacy without anticholinergic effects.
Felodipine, a dihydropyridine, is a potent, vasoselective calcium channel antagonist. Felodipine lowers blood pressure (BP) by selective action on vascular smooth muscle, especially in the resistance vessels. Felodipine, an anti-hypertensive agent, induces autophagy. Felodipine can cross the blood-brain barrier .
Amlodipine besylate (Amlodipine benzenesulfonate), an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine besylate can be used for the research of high blood pressure and cancer .
Ruthenium red (Ammoniated ruthenium oxychloride) is a polycationic dye widely used for electron microscopy (EM) of cells, tissues and vegetative bacteria. Ruthenium red strongly reacts with phospholipids and fatty acids and binds to acidic mucopolysaccharides. Ruthenium red is a L-type calcium current (ICa) blocker .
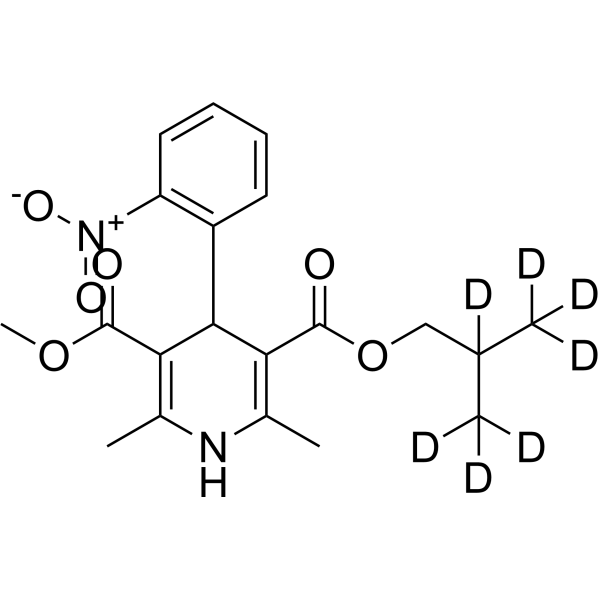
Nisoldipine-d4 (BAY-k 5552-d4) is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with IC50 of 10 nM[1][2].
Nisoldipine-d7 (BAY-k 5552-d7) is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with IC50 of 10 nM[1][2].
AVE-0118 is a nonselective Kv1.5 blocker with an IC50 of 1.1 μM. AVE-0118 is a multichannel inhibitor with weak, micromolar activity against Kv1.5 and other ion channels. It is inactive against IKs, IKATP, and L-type Ca+ channels .
Barnidipine hydrochloride (Mepirodipine hydrochloride) is an L-type calcium antagonist (CaA) with high affinity for [ 3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors . Barnidipine hydrochloride (Mepirodipine hydrochloride) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action .
Barnidipine (Mepirodipine) is an L-type calcium antagonist (CaA) with high affinity for [ 3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors . Barnidipine (Mepirodipine) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action .
Urolithin C, a gut-microbial metabolite of Ellagic acid, is a glucose-dependent activator of insulin secretion. Urolithin C is a L-type Ca 2+ channel opener and enhances Ca 2+ influx. Urolithin C induces cell apoptosis through a mitochondria-mediated pathway and also stimulates reactive oxygen species (ROS) formation .
Pinaverium bromide-d4 is deuterium labeled Pinaverium bromide. Pinaverium bromide is an L-type calcium channel blocker with selectivity for the gastrointestinal tract, effectively relieves pain, diarrhea and intestinal discomfort, provides good therapeutic efficacies without significant adverse effects on Irritable bowel syndrome (IBS) patients[1].
Amlodipine-d4 (maleate) is the deuterium labeled Amlodipine maleate. Amlodipine maleate is a dihydropyridine calcium channel blocker, acts as an orally active antianginal agent. Amlodipine maleate blocks the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine maleate can be used for the research of high blood pressure and cancer[1][2][3].
KMH-233, a potent, reversible and selective l-type amino acid transporter 1 (LAT1) inhibitor, inhibits the uptake of LAT1 substrate, l-leucin (IC50=18 μM) as well as cell growth. KMH-233 significantly potentiates the efficacy of Bestatin and Cisplatin even at low concentrations (25 μM) .
Etripamil (MSP-2017) is a short-acting L-type calcium-channel antagonist, can be used for the research of Paroxysmal Supraventricular Tachycardia (PSVT). Etripamil (MSP-2017) slows atrioventricular nodal conduction and prolongs atrioventricular nodal refractory periods by inhibiting calcium ion influx through the calcium slow channels in the atrioventricular node cells .
Drotaverine-d10 (hydrochloride) is the deuterium labeled Drotaverine hydrochloride. Drotaverine hydrochloride is a type 4 cyclic nucleotide phosphodiesterase (PDE4) inhibitor and an L-type voltage-dependent calcium channel (L-VDCC) blocker, blocks the degradation of 3',5'-cyclic adenosine monophosphate. Drotaverine hydrochloride exhibits in vivo antispasmodic efficacy without anticholinergic effects[1][2].
Isradipine-d6 is the deuterium labeled Isradipine[1]. Isradipine (PN 200-110) is an orally active L-type calcium channel blocker. Isradipine, as a powerful peripheral vasodilator, is a dihydropyridine calcium antagonist with selective actions on the heart as well as the peripheral circulation. Isradipine is a potentially viable neuroprotective agent for Parkinson's disease[2][3][4].
Amlodipine-d4 is a deuterium labeled Amlodipine (HY-B0317). Amlodipine, an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine can be used for the research of high blood pressure and cancer .
Amlodipine-1,1,2,2-d4 (maleate) is the deuterium labeled Amlodipine. Amlodipine, an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine can be used for the research of high blood pressure and cancer[1][2][3].
Amlodipine-d4 (besylate) is the deuterium labeled Amlodipine besylate. Amlodipine besylate (Amlodipine benzenesulfonate), an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine besylate can be used for the research of high blood pressure and cancer[1][2][3].
Dronedarone (SR 33589), a derivative of amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone is a substrate for and a moderate inhibitor of CYP3A4 .
Flufenamic acid is a non-steroidal anti-inflammatory agent, inhibits cyclooxygenase (COX), activates AMPK, and also modulates ion channels, blocking chloride channels and L-type Ca 2+ channels, modulating non-selective cation channels (NSC), activating K + channels. Flufenamic acid binds to the central pocket of TEAD2 YBD and inhibits both TEAD function and TEAD-YAP-dependent processes, such as cell migration and proliferation.
Isradipine-d3 (PN 200-110-d3) is the deuterium labeled Isradipine. Isradipine (PN 200-110) is an orally active L-type calcium channel blocker. Isradipine, as a powerful peripheral vasodilator, is a dihydropyridine calcium antagonist with selective actions on the heart as well as the peripheral circulation. Isradipine is a potentially viable neuroprotective agent for Parkinson's disease[1][2][3].
ILS-920 is a nonimmunosuppressive Rapamycin analog with reduced immunosuppressive activity and potent neuroprotective activity. ILS-920 binds selectively to the immunophilin FKBP52 and to the β1-subunit of L-type voltage-gated calcium channels (VGCC). ILS-920 shows 200-fold selectivity for FKBP52 versus FKBP12 .
BBT is an enhancer of impaired glucose-stimulated insulin secretion (GSIS). BBT exhibits anti-hyperglycemia activity, and protects β-cells from cytokine- or streptozotocin (STZ)-induced cell death in type 2 diabetes models. BBT acts function via cAMP/PKA and long-lasting (L-type) voltage-dependent Ca2+ channel/CaMK2 pathway .
Amlodipine (besylate) (Standard) is the analytical standard of Amlodipine (besylate). This product is intended for research and analytical applications. Amlodipine besylate (Amlodipine benzenesulfonate), an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine besylate can be used for the research of high blood pressure and cancer .
VU 0240551 is a potent neuronal K-Cl cotransporter KCC2 inhibitor (IC50=560 nM) and is selective versus NKCC1. VU 0240551 also inhibits hERG and L-type Ca 2+ channels. VU 0240551 attenuates GABA-induced hyperpolarization of P cells, produces a positive shift in the P cell GABA reversal potential and enhances P cell synaptic transmission .
Barnidipine-d5 (hydrochloride) is the deuterium labeled Barnidipine hydrochloride. Barnidipine hydrochloride (Mepirodipine hydrochloride) is an L-type calcium antagonist (CaA) with high affinity for [3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors[1].Barnidipine hydrochloride (Mepirodipine hydrochloride) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action[2].
Ethacrynic acid (Etacrynic acid sodium) sodium is a diuretic. Ethacrynic acid sodium is an inhibitor of glutathione S-transferases (GSTs). Ethacrynic acid sodium is a potent inhibitor of NF-kB-signaling pathway, and also modulates leukotriene formation. Ethacrynic acid sodium also inhibits L-type voltage-dependent and store-operated calcium channel, leading to relaxation of airway smooth muscle (ASM) cells. Ethacrynic acid sodium has anti-inflammatory properties that reduces the retinoid-induced ear edema in mice .
Lacidipine is an orally active and highly selective L-type calcium channel blocker that acts on smooth muscle calcium channels, primarily dilates peripheral arteries, reduces peripheral resistance, and has long-lasting anti-hypertensive activity. Lacidipine protects HKCs from apoptosis induced by ATP depletion and recovery by modulating the caspase-3 pathway. Lacidipine can be used in studies of hypertension, atherosclerosis and acute kidney injury (AKI) .
ω-Agatoxin TK, a peptidyl toxin of the venom of Agelenopsis aperta, is a potent and selective P/Q type Ca 2+ channel blocker. ω-Agatoxin TK inhibits the high K + depolarisation-induced rise in internal Ca 2+ in cerebral isolated nerve endings with an IC50 of of 60 nM. ω-Agatoxin TK has no effect on L-type, N-type, or T-type calcium channels .
Dronedarone-d6 (hydrochloride) is the deuterium labeled Dronedarone. Dronedarone hydrochloride, a derivative of Amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone hydrochloride is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone hydrochloride is a substrate for and a moderate inhibitor of CYP3A4[1][2][3][4].
Flufenamic acid-d4 is deuterium labeled Flufenamic acid. Flufenamic acid is a non-steroidal anti-inflammatory agent, inhibits cyclooxygenase (COX), activates AMPK, and also modulates ion channels, blocking chloride channels and L-type Ca 2+ channels, modulating non-selective cation channels (NSC), activating K+ channels. Flufenamic acid binds to the central pocket of TEAD2 YBD and inhibits both TEAD function and TEAD-YAP-dependent processes, such as cell migration and proliferation.
Flufenamic acid- 13C6 is the 13C6 labeled Flufenamic acid. Flufenamic acid is a non-steroidal anti-inflammatory agent, inhibits cyclooxygenase (COX), activates AMPK, and also modulates ion channels, blocking chloride channels and L-type Ca 2+ channels, modulating non-selective cation channels (NSC), activating K+ channels. Flufenamic acid binds to the central pocket of TEAD2 YBD and inhibits both TEAD function and TEAD-YAP-dependent processes, such as cell migration and proliferation.
(R)-KMH-233 is the isomer of KMH-233 (HY-120139), and can be used as an experimental control. KMH-233, a potent, reversible and selective l-type amino acid transporter 1 (LAT1) inhibitor, inhibits the uptake of LAT1 substrate, l-leucin (IC50=18 μM) as well as cell growth. KMH-233 significantly potentiates the efficacy of Bestatin and Cisplatin even at low concentrations (25 μM) .
Barnidipine-d5 is the deuterium-labeled Barnidipine (HY-107322A). Barnidipine-d5 (Mepirodipine) is an L-type calcium antagonist (CaA) with high affinity for [ 3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors . Barnidipine-d5 (Mepirodipine) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action .
CaV1.3 antagonist-1 is a potent and highly selective CaV1.3 L-type calcium channel (LTCC) antagonist with an IC50 of 1.7 μM. CaV1.3 antagonist-1 inhibits CaV1.3 LTCC >600-fold more potently than CaV1.2 LTCC. CaV1.3 antagonist-1, a cyclopentyl derivative, has the potential for Parkinson's disease research .
Ethacrynic acid (Standard) is the analytical standard of Ethacrynic acid. This product is intended for research and analytical applications. Ethacrynic acid (Etacrynic acid) is a diuretic. Ethacrynic acid is an inhibitor of glutathione S-transferases (GSTs). Ethacrynic acid is a potent inhibitor of NF-kB-signaling pathway, and also modulates leukotriene formation. Ethacrynic acid also inhibits L-type voltage-dependent and store-operated calcium channel, leading to relaxation of airway smooth muscle (ASM) cells. Ethacrynic acid has anti-inflammatory properties that reduces the retinoid-induced ear edema in mice .
Lacidipine- 13C8 is the deuterium labeled Lacidipine[1]. Lacidipine is an orally active and highly selective L-type calcium channel blocker that acts on smooth muscle calcium channels, primarily dilates peripheral arteries, reduces peripheral resistance, and has long-lasting anti-hypertensive activity. Lacidipine protects HKCs from apoptosis induced by ATP depletion and recovery by modulating the caspase-3 pathway. Lacidipine can be used in studies of hypertension, atherosclerosis and acute kidney injury (AKI)[2][3].
ω-Agatoxin IVA is a potent, selective P/Q type Ca 2+ (Cav2.1) channel blocker with IC50s of 2 nM and 90 nM for P-type and Q-type Ca 2+ channels, respectively. ω-Agatoxin IVA (IC50, 30-225 nM) inhibits glutamate exocytosis and calcium influx elicited by high potassium. ω-Agatoxin IVA also blocks the high potassium-induced release of serotonin and norepinephrine. ω-Agatoxin IVA has no effect on L-type or N-type calcium channels .
ω-Agatoxin IVA TFA is a potent, selective P/Q type Ca 2+ (Cav2.1) channel blocker with IC50s of 2 nM and 90 nM for P-type and Q-type Ca 2+ channels, respectively. ω-Agatoxin IVA TFA (IC50, 30-225 nM) inhibits glutamate exocytosis and calcium influx elicited by high potassium. ω-Agatoxin IVA TFA also blocks the high potassium-induced release of serotonin and norepinephrine. ω-Agatoxin IVA TFA has no effect on L-type or N-type calcium channels .
Minocycline hydrochloride is an orally active, potent and BBB-penetrated semi-synthetic tetracycline antibiotic. Minocycline hydrochloride is a hypoxia-inducible factor (HIF)-1α inhibitor. Minocycline hydrochloride shows anti-cancer, anti-inflammatory, and glutamate antagonist effects. Minocycline hydrochloride reduces glutamate neurotransmission and shows neuroprotective properties and antidepressant effects. Minocycline hydrochloride inhibits bacterial protein synthesis through binding with the 30S subunit of the bacterial ribosome, resulting in a bacteriostatic effect .
Minocycline is an orally active, potent and BBB-penetrated semi-synthetic tetracycline antibiotic. Minocycline is a hypoxia-inducible factor (HIF)-1α inhibitor. Minocycline shows anti-cancer, anti-inflammatory, and glutamate antagonist effects. Minocycline reduces glutamate neurotransmission and shows neuroprotective properties and antidepressant effects. Minocycline inhibits bacterial protein synthesis through binding with the 30S subunit of the bacterial ribosome, resulting in a bacteriostatic effect .
Ruthenium red (Ammoniated ruthenium oxychloride) is a polycationic dye widely used for electron microscopy (EM) of cells, tissues and vegetative bacteria. Ruthenium red strongly reacts with phospholipids and fatty acids and binds to acidic mucopolysaccharides. Ruthenium red is a L-type calcium current (ICa) blocker .
FluoBar1 is an imaging fluorescence probe modified by a barbiturate ligand with fluorescent coumarin. FluoBar1 can monitor L-type voltage-gated calcium channels (LTCC) in living cells in real time for the study of neurological diseases .
Kurtoxin is a selective Cav3 (T-type) voltage-gated Ca 2+ channel gating inhibitor with a Kd of 15 nM for Cav3.1 (α1G T-type) Ca 2+ channel. Kurtoxin can interact with high affinity with native neuronal high-threshold L-type, N-type, and P-type Ca 2+ channels in central and peripheral neurons. Kurtoxin also shows cross-reactivity with voltage-gated Na + channel .
Calcicludine is a protein toxin from the venom of the green mamba Dendroaspis angusticeps that inhibits high-voltage-activated calcium channel, especially L-typecalcium channel with the IC50 of 88 nM. Calcicludine has role in excitatory synaptic transmission .
ω-Agatoxin TK, a peptidyl toxin of the venom of Agelenopsis aperta, is a potent and selective P/Q type Ca 2+ channel blocker. ω-Agatoxin TK inhibits the high K + depolarisation-induced rise in internal Ca 2+ in cerebral isolated nerve endings with an IC50 of of 60 nM. ω-Agatoxin TK has no effect on L-type, N-type, or T-type calcium channels .
ω-Agatoxin IVA is a potent, selective P/Q type Ca 2+ (Cav2.1) channel blocker with IC50s of 2 nM and 90 nM for P-type and Q-type Ca 2+ channels, respectively. ω-Agatoxin IVA (IC50, 30-225 nM) inhibits glutamate exocytosis and calcium influx elicited by high potassium. ω-Agatoxin IVA also blocks the high potassium-induced release of serotonin and norepinephrine. ω-Agatoxin IVA has no effect on L-type or N-type calcium channels .
ω-Agatoxin IVA TFA is a potent, selective P/Q type Ca 2+ (Cav2.1) channel blocker with IC50s of 2 nM and 90 nM for P-type and Q-type Ca 2+ channels, respectively. ω-Agatoxin IVA TFA (IC50, 30-225 nM) inhibits glutamate exocytosis and calcium influx elicited by high potassium. ω-Agatoxin IVA TFA also blocks the high potassium-induced release of serotonin and norepinephrine. ω-Agatoxin IVA TFA has no effect on L-type or N-type calcium channels .
Catharanthine ((+)-3,4-Didehydrocoronaridine), a constituent of anticancer vinca alkaloids, inhibits voltage-operated L-type Ca 2+ channel (VOCC). Catharanthine has IC50s of 220 μM and 8 μM for VOCC currents in cardiomyocytes and vascular smooth muscle cells (VSMCs), respectively. Catharanthine lowers blood pressure (BP), heart rate (HR). Catharanthine has anti-cancer activity .
Calcicludine is a protein toxin from the venom of the green mamba Dendroaspis angusticeps that inhibits high-voltage-activated calcium channel, especially L-typecalcium channel with the IC50 of 88 nM. Calcicludine has role in excitatory synaptic transmission .
Urolithin C, a gut-microbial metabolite of Ellagic acid, is a glucose-dependent activator of insulin secretion. Urolithin C is a L-type Ca 2+ channel opener and enhances Ca 2+ influx. Urolithin C induces cell apoptosis through a mitochondria-mediated pathway and also stimulates reactive oxygen species (ROS) formation .
The PKLR protein is a key enzyme in cellular metabolism that acts as a pyruvate kinase, converting phosphoenolpyruvate to pyruvate. This process is an integral part of glycolysis, the fundamental pathway for cellular energy production. PKLR Protein, Human (HEK293, His) is the recombinant human-derived PKLR protein, expressed by HEK293 , with C-6*His labeled tag. The total length of PKLR Protein, Human (HEK293, His) is 574 a.a., with molecular weight of ~62.0 kDa.
The CACNA2D1 protein, particularly its α-2/δ subunit, plays a key role in regulating calcium current density and activation/deactivation kinetics of voltage-dependent calcium channels. It contributes to excitation-contraction coupling, coordinating cellular processes. CACNA2D1 Protein, Human (His-SUMO) is the recombinant human-derived CACNA2D1 protein, expressed by E. coli , with N-6*His, N-SUMO labeled tag. The total length of CACNA2D1 Protein, Human (His-SUMO) is 141 a.a., with molecular weight of ~32.3 kDa.
The CACNA1C protein is the alpha-1C subunit of voltage-gated calcium channels that generate L-type calcium currents that are critical for calcium influx and sarcoplasmic release. Its role in excitation-contraction coupling is critical for cardiac development, rhythm regulation, and smooth muscle cell contraction. CACNA1C Protein, Pig (Cell-Free, His) is the recombinant pig-derived CACNA1C protein, expressed by E. coli Cell-free , with N-10*His labeled tag. The total length of CACNA1C Protein, Pig (Cell-Free, His) is 169 a.a., with molecular weight of 22.3 kDa.
CACNG1 Protein, a regulatory subunit of the voltage-gated calcium channel, contributes to L-type calcium currents in skeletal muscle and modulates channel inactivation kinetics. Collaborating with CACNA1S, CACNB1 or CACNB2, and CACNA2D1, CACNG1 orchestrates a balanced 1:1:1:1 ratio channel assembly, influencing the voltage-gated calcium channel's functionality in skeletal muscle. CACNG1 Protein, Mouse (Cell-Free, His) is the recombinant mouse-derived CACNG1 protein, expressed by E. coli Cell-free , with N-10*His labeled tag. The total length of CACNG1 Protein, Mouse (Cell-Free, His) is 223 a.a., with molecular weight of 26.6 kDa.
PKLR Protein, a pyruvate kinase, plays a crucial role in glycolysis, converting phosphoenolpyruvate to pyruvate and generating ATP. As a key enzyme, PKLR mediates the final steps of glycolysis, contributing to energy production. Its catalytic activity influences pyruvate, a central metabolite with implications for cellular processes, including energy metabolism and the tricarboxylic acid (TCA) cycle. PKLR's significance in glycolytic flux underscores its essential role in cellular energy homeostasis, presenting potential for therapeutic interventions in metabolic pathways. PKLR Protein, Rat (P. pastoris, His) is the recombinant rat-derived PKLR protein, expressed by P. pastoris, with N-6*His labeled tag. The total length of PKLR Protein, Rat (P. pastoris, His) is 574 a.a., with molecular weight of 64.2 kDa.
Norverapamil-d7 (hydrochloride) is a deuterium labeled Norverapamil. Norverapamil ((±)-Norverapamil), an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor[1][2].
Nisoldipine-d6 is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552; Sular) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with an IC50 of 10 nM.
Norverapamil-d7 is a deuterium labeled Norverapamil ((±)-Norverapamil). Norverapamil, an N-demethylated metabolite of Verapamil, is a L-type calcium channel blocker and a P-glycoprotein (P-gp) function inhibitor[1][2].
Diltiazem-d6 is the deuterium labeled Diltiazem. Diltiazem is an orally active L-type Ca2+ channel blocker, with antihypertensive and antiarrhythmic effects. Diltiazem can be used for the research of cardiac arrhythmia, hypertension, and angina pectoris[1][2][3].
Nisoldipine-d4 (BAY-k 5552-d4) is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with IC50 of 10 nM[1][2].
Nisoldipine-d7 (BAY-k 5552-d7) is the deuterium labeled Nisoldipine. Nisoldipine(BAY-k 5552) is a calcium channel blocker belonging to the dihydropyridines class, specific for L-type Cav1.2 with IC50 of 10 nM[1][2].
Pinaverium bromide-d4 is deuterium labeled Pinaverium bromide. Pinaverium bromide is an L-type calcium channel blocker with selectivity for the gastrointestinal tract, effectively relieves pain, diarrhea and intestinal discomfort, provides good therapeutic efficacies without significant adverse effects on Irritable bowel syndrome (IBS) patients[1].
Amlodipine-d4 (maleate) is the deuterium labeled Amlodipine maleate. Amlodipine maleate is a dihydropyridine calcium channel blocker, acts as an orally active antianginal agent. Amlodipine maleate blocks the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine maleate can be used for the research of high blood pressure and cancer[1][2][3].
Drotaverine-d10 (hydrochloride) is the deuterium labeled Drotaverine hydrochloride. Drotaverine hydrochloride is a type 4 cyclic nucleotide phosphodiesterase (PDE4) inhibitor and an L-type voltage-dependent calcium channel (L-VDCC) blocker, blocks the degradation of 3',5'-cyclic adenosine monophosphate. Drotaverine hydrochloride exhibits in vivo antispasmodic efficacy without anticholinergic effects[1][2].
Isradipine-d6 is the deuterium labeled Isradipine[1]. Isradipine (PN 200-110) is an orally active L-type calcium channel blocker. Isradipine, as a powerful peripheral vasodilator, is a dihydropyridine calcium antagonist with selective actions on the heart as well as the peripheral circulation. Isradipine is a potentially viable neuroprotective agent for Parkinson's disease[2][3][4].
Amlodipine-d4 is a deuterium labeled Amlodipine (HY-B0317). Amlodipine, an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine can be used for the research of high blood pressure and cancer .
Amlodipine-d4 (besylate) is the deuterium labeled Amlodipine besylate. Amlodipine besylate (Amlodipine benzenesulfonate), an antianginal agent and an orally active dihydropyridine calcium channel blocker, works by blocking the voltage-dependent L-type calcium channels, thereby inhibiting the initial influx of calcium. Amlodipine besylate can be used for the research of high blood pressure and cancer[1][2][3].
Isradipine-d3 (PN 200-110-d3) is the deuterium labeled Isradipine. Isradipine (PN 200-110) is an orally active L-type calcium channel blocker. Isradipine, as a powerful peripheral vasodilator, is a dihydropyridine calcium antagonist with selective actions on the heart as well as the peripheral circulation. Isradipine is a potentially viable neuroprotective agent for Parkinson's disease[1][2][3].
Barnidipine-d5 (hydrochloride) is the deuterium labeled Barnidipine hydrochloride. Barnidipine hydrochloride (Mepirodipine hydrochloride) is an L-type calcium antagonist (CaA) with high affinity for [3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors[1].Barnidipine hydrochloride (Mepirodipine hydrochloride) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action[2].
Dronedarone-d6 (hydrochloride) is the deuterium labeled Dronedarone. Dronedarone hydrochloride, a derivative of Amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone hydrochloride is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone hydrochloride is a substrate for and a moderate inhibitor of CYP3A4[1][2][3][4].
Flufenamic acid-d4 is deuterium labeled Flufenamic acid. Flufenamic acid is a non-steroidal anti-inflammatory agent, inhibits cyclooxygenase (COX), activates AMPK, and also modulates ion channels, blocking chloride channels and L-type Ca 2+ channels, modulating non-selective cation channels (NSC), activating K+ channels. Flufenamic acid binds to the central pocket of TEAD2 YBD and inhibits both TEAD function and TEAD-YAP-dependent processes, such as cell migration and proliferation.
Flufenamic acid- 13C6 is the 13C6 labeled Flufenamic acid. Flufenamic acid is a non-steroidal anti-inflammatory agent, inhibits cyclooxygenase (COX), activates AMPK, and also modulates ion channels, blocking chloride channels and L-type Ca 2+ channels, modulating non-selective cation channels (NSC), activating K+ channels. Flufenamic acid binds to the central pocket of TEAD2 YBD and inhibits both TEAD function and TEAD-YAP-dependent processes, such as cell migration and proliferation.
Barnidipine-d5 is the deuterium-labeled Barnidipine (HY-107322A). Barnidipine-d5 (Mepirodipine) is an L-type calcium antagonist (CaA) with high affinity for [ 3H] initrendipine binding sites (Ki=0.21 nmol/l), has selective action against CaA receptors . Barnidipine-d5 (Mepirodipine) is an antihypertensive agent and acts by the reduction of peripheral vascular resistance secondary to its vasodilatory action .
Lacidipine- 13C8 is the deuterium labeled Lacidipine[1]. Lacidipine is an orally active and highly selective L-type calcium channel blocker that acts on smooth muscle calcium channels, primarily dilates peripheral arteries, reduces peripheral resistance, and has long-lasting anti-hypertensive activity. Lacidipine protects HKCs from apoptosis induced by ATP depletion and recovery by modulating the caspase-3 pathway. Lacidipine can be used in studies of hypertension, atherosclerosis and acute kidney injury (AKI)[2][3].