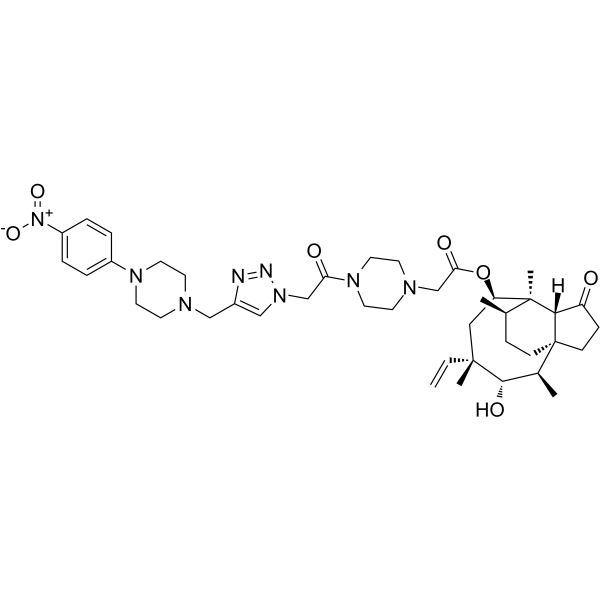
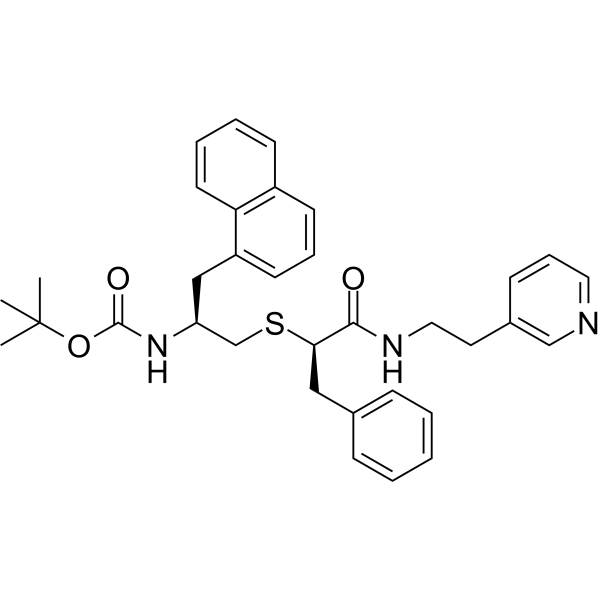
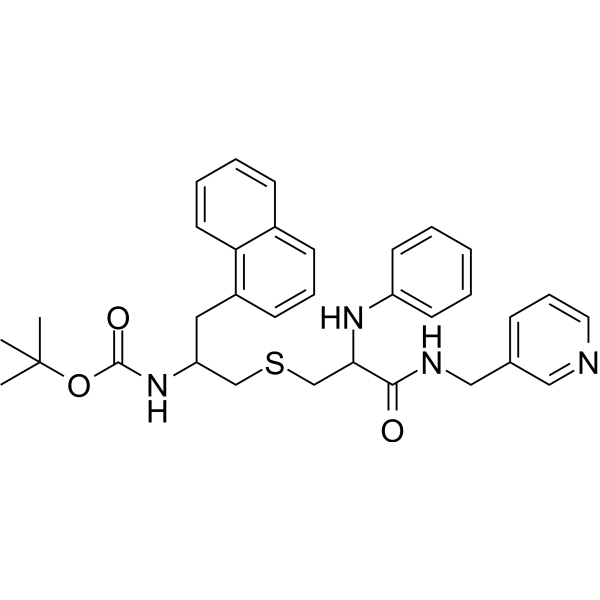
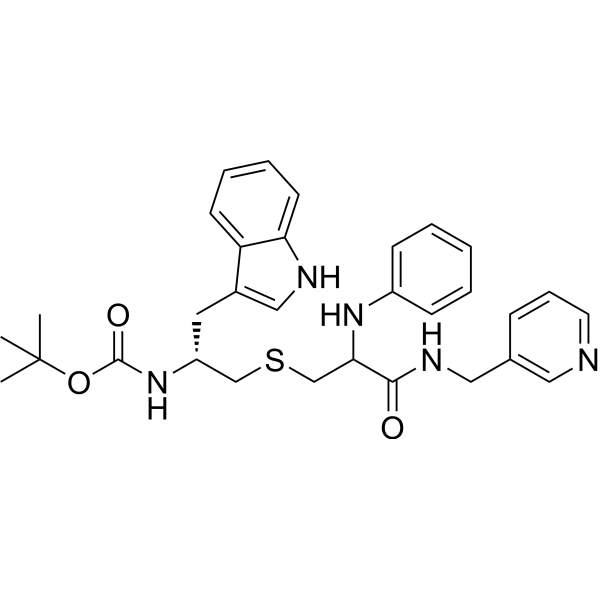
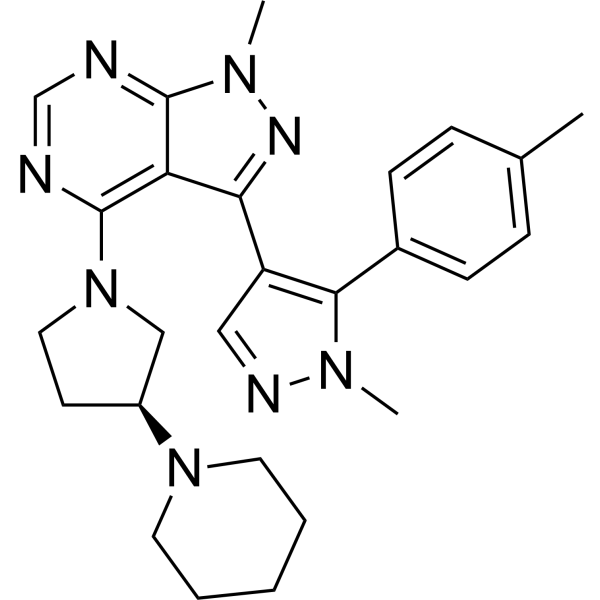
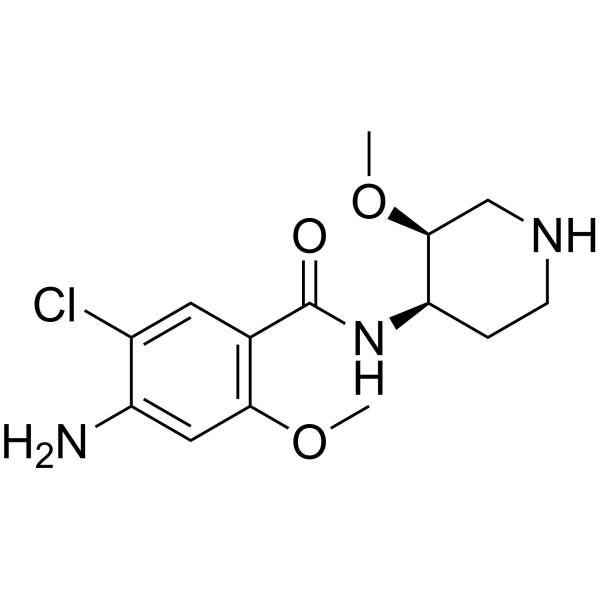
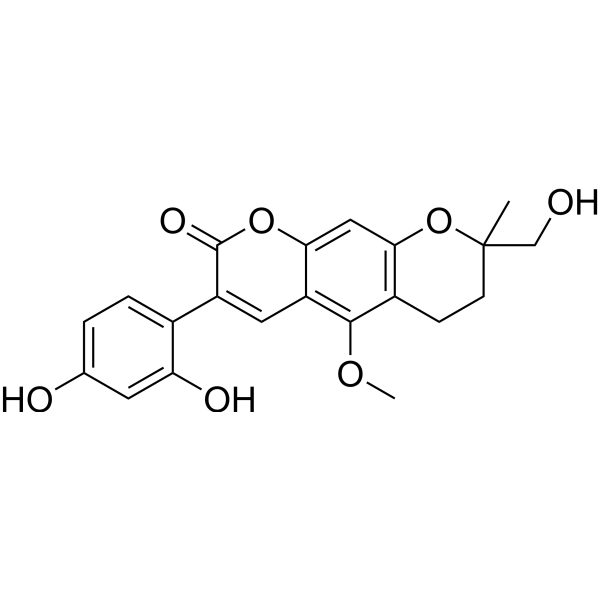
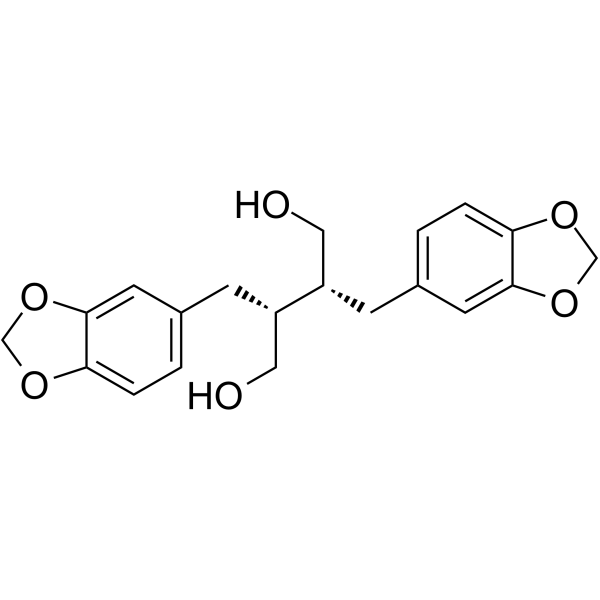
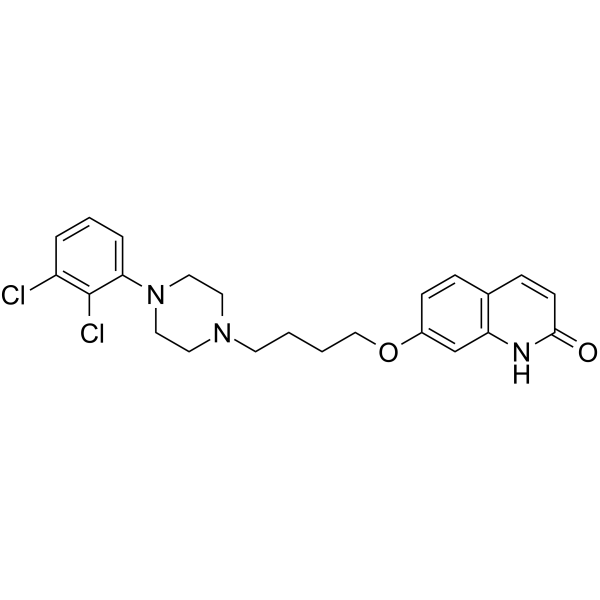
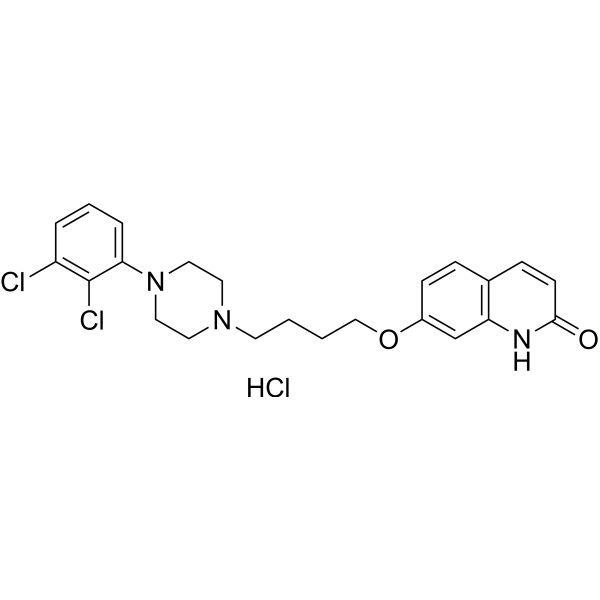
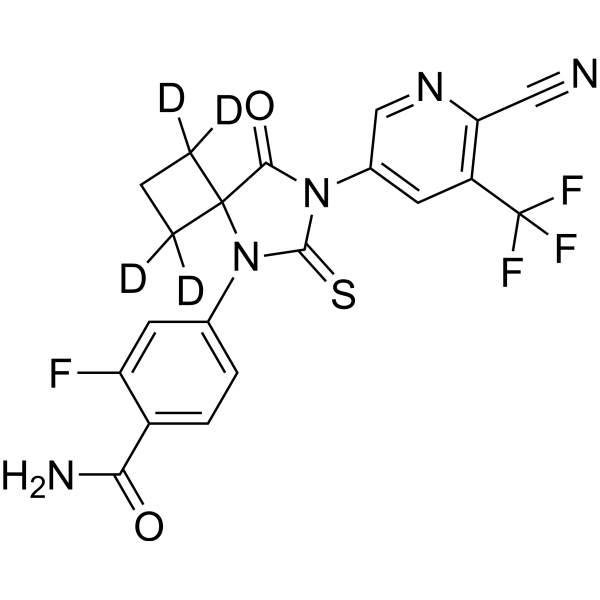
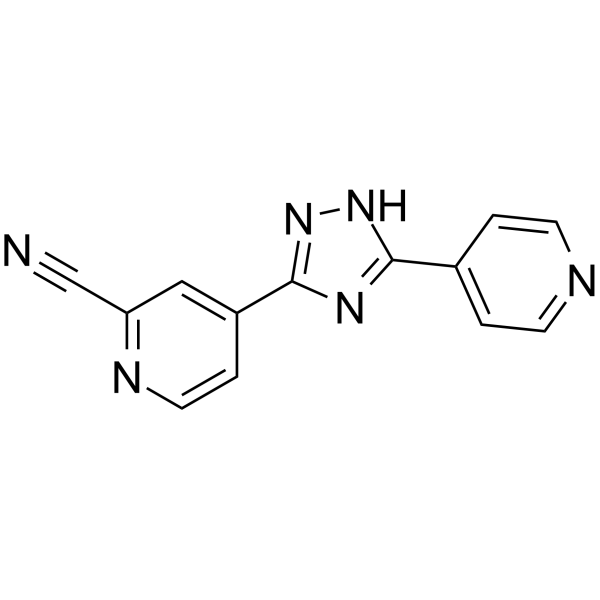
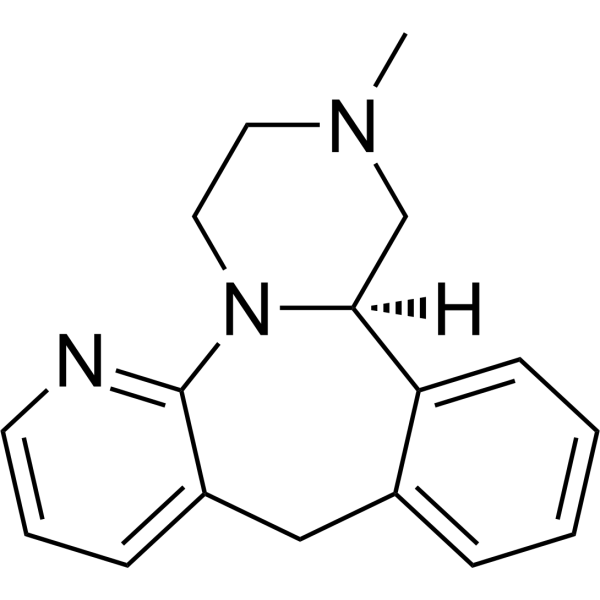
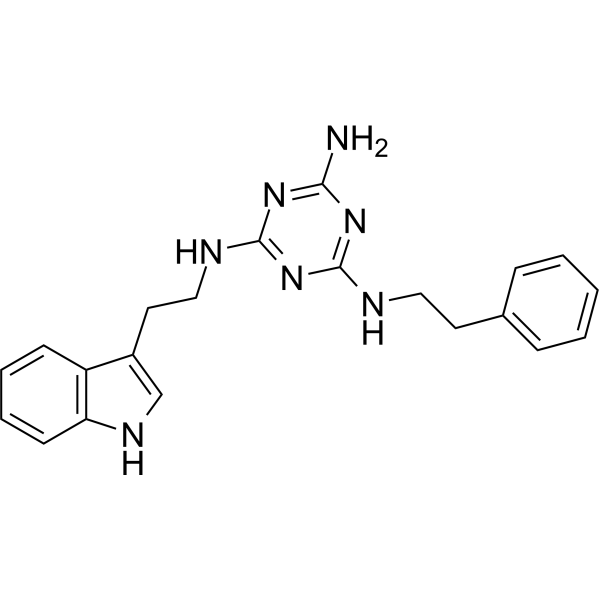
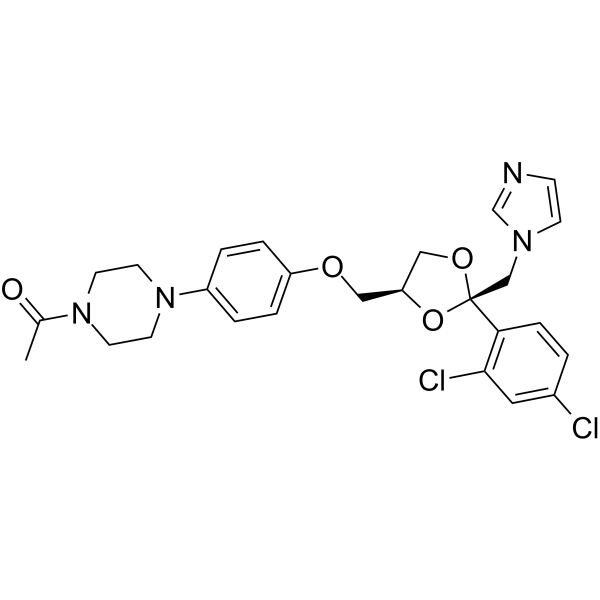
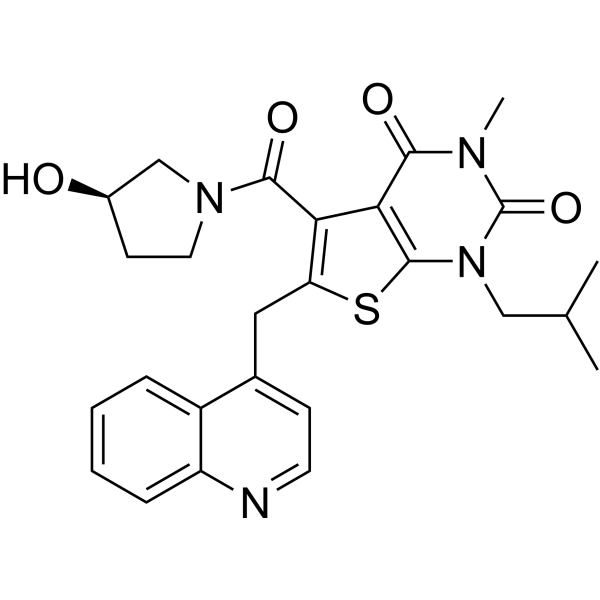
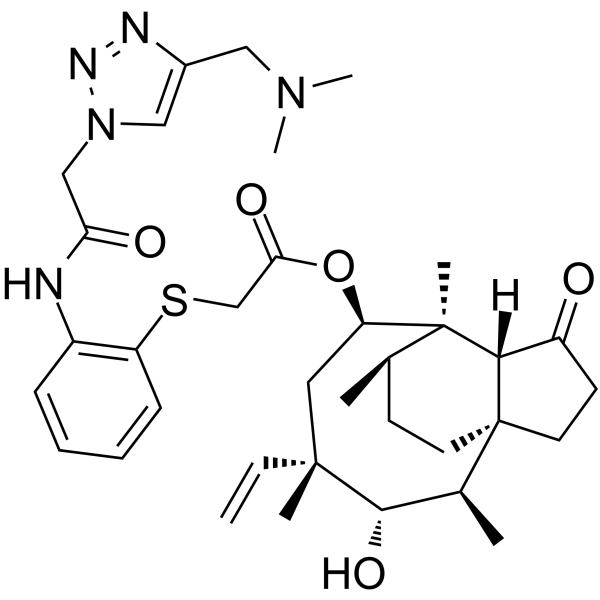
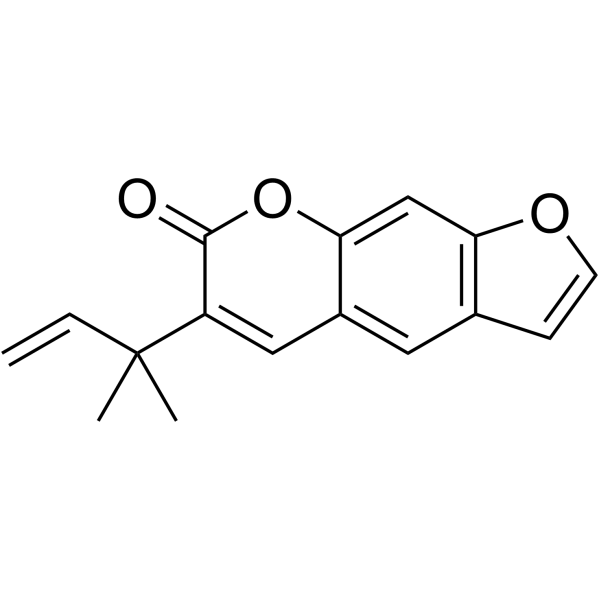
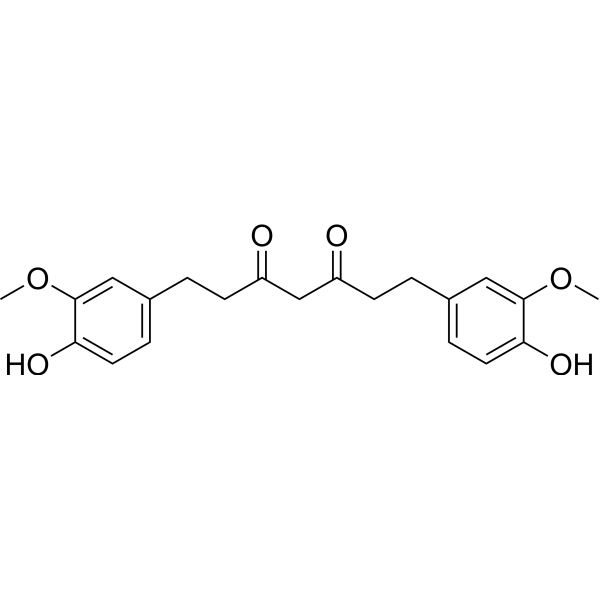
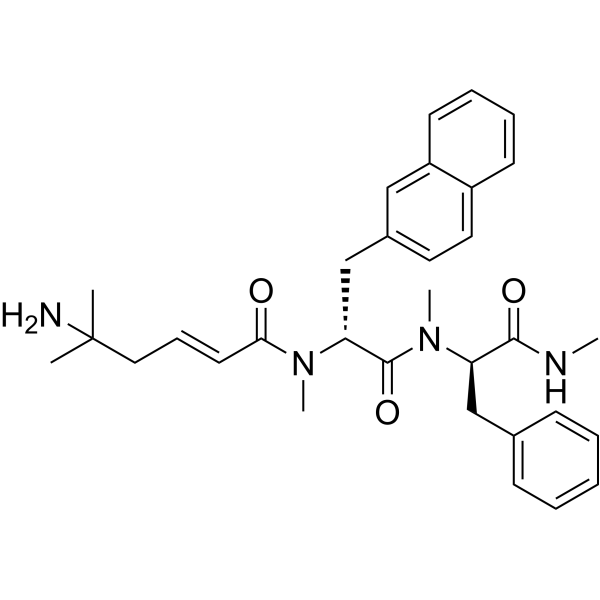
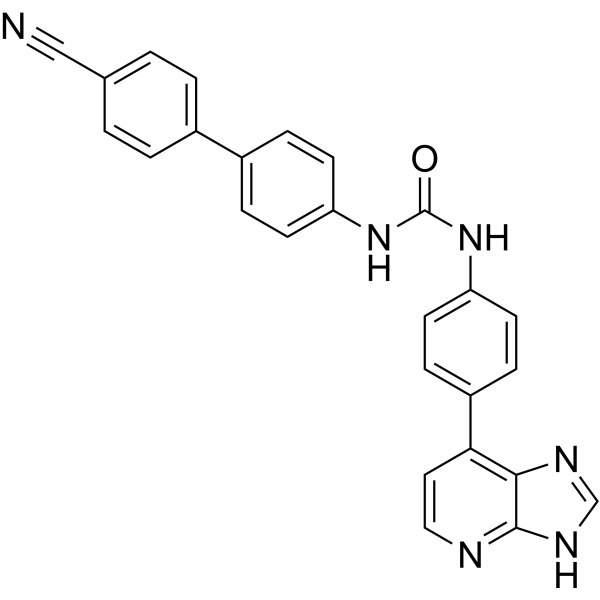
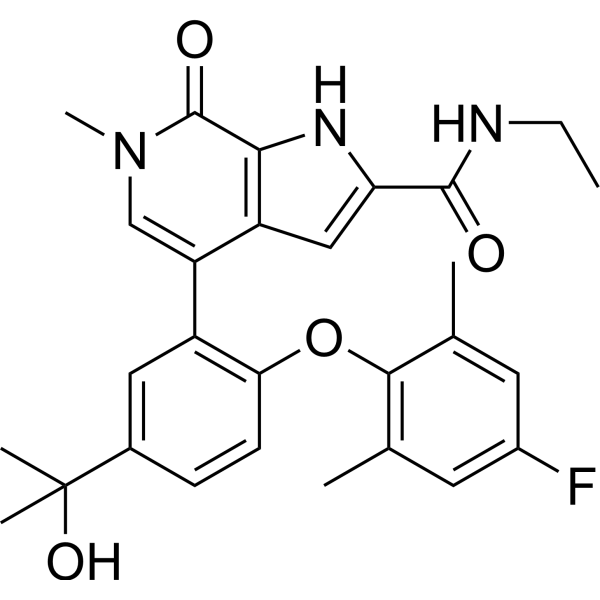
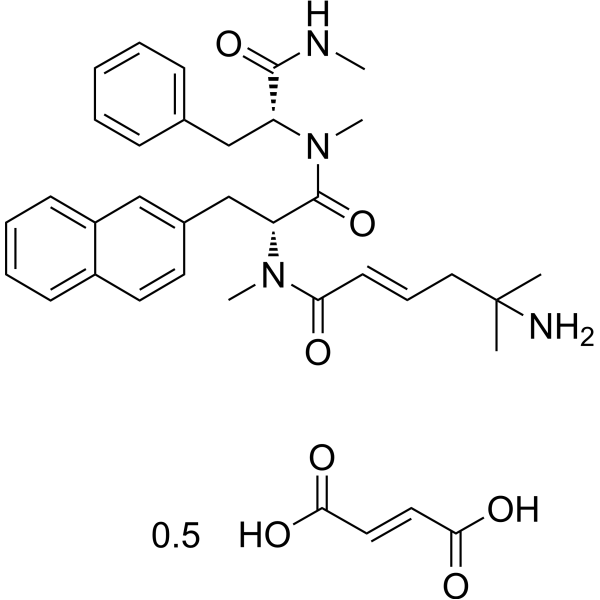
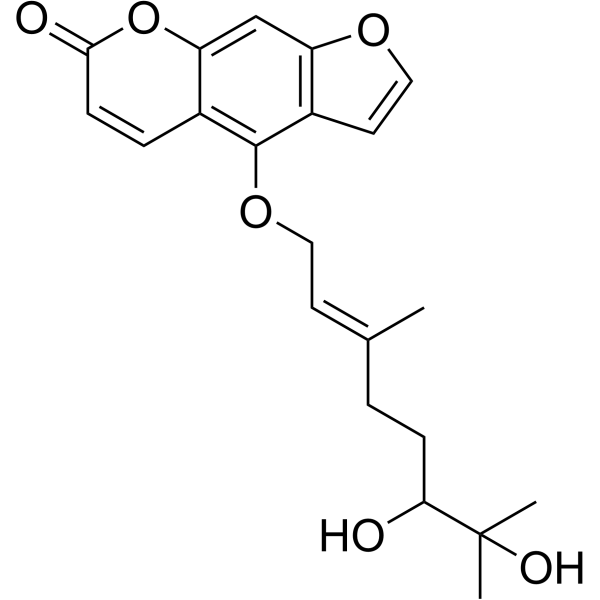
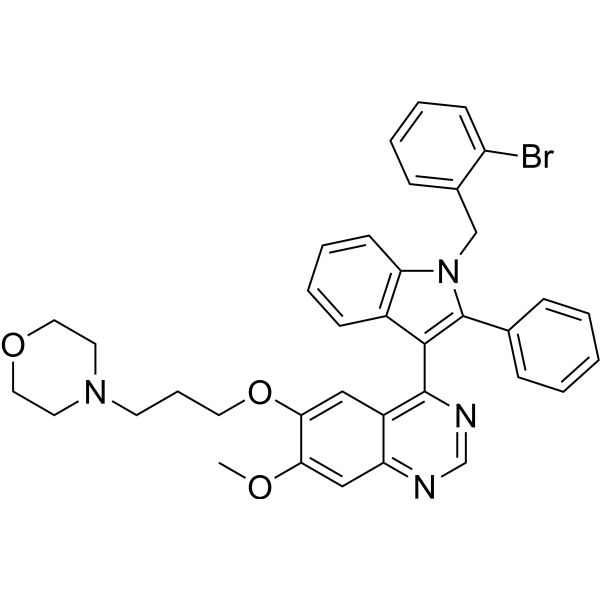
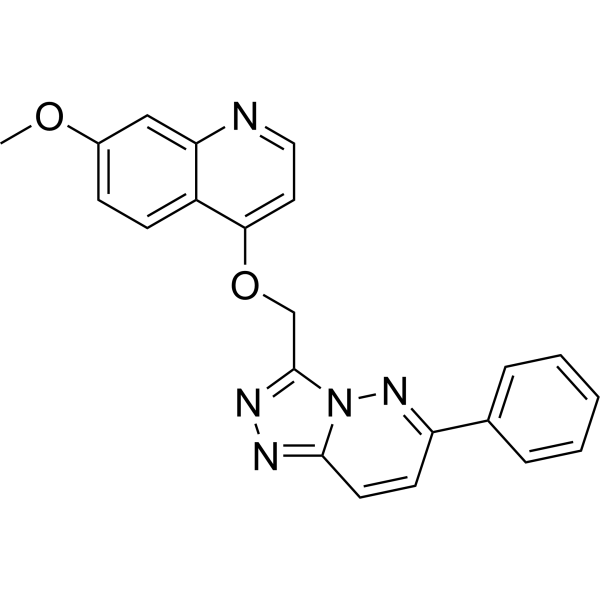
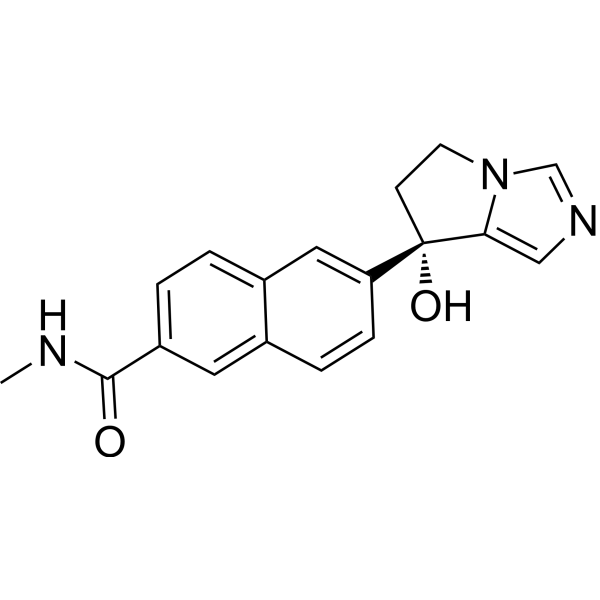
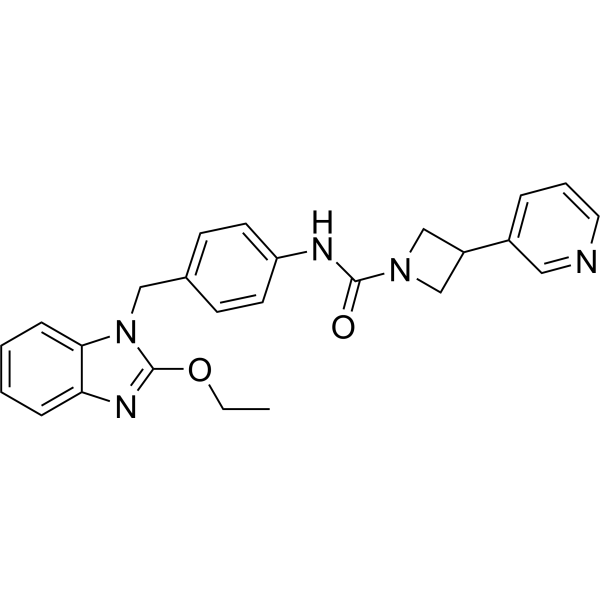
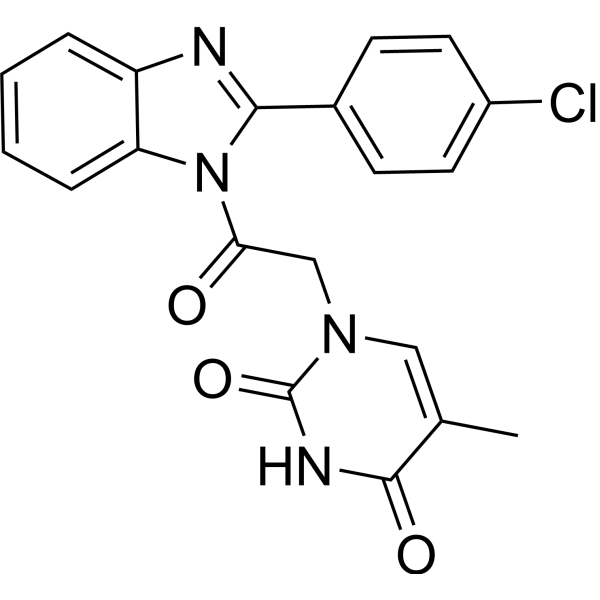
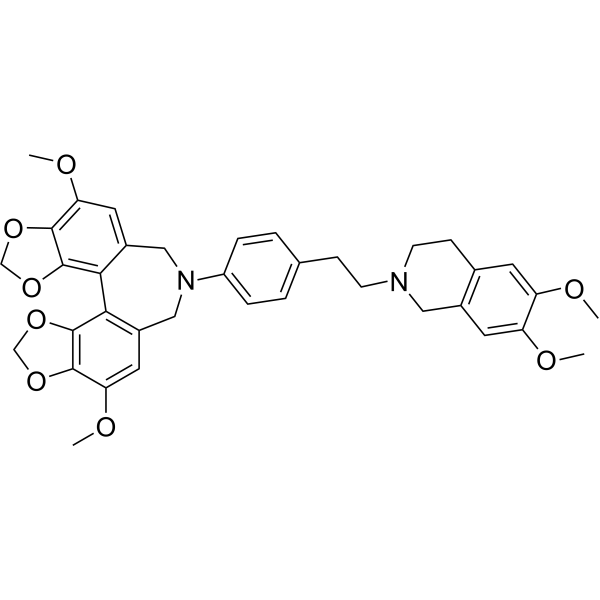
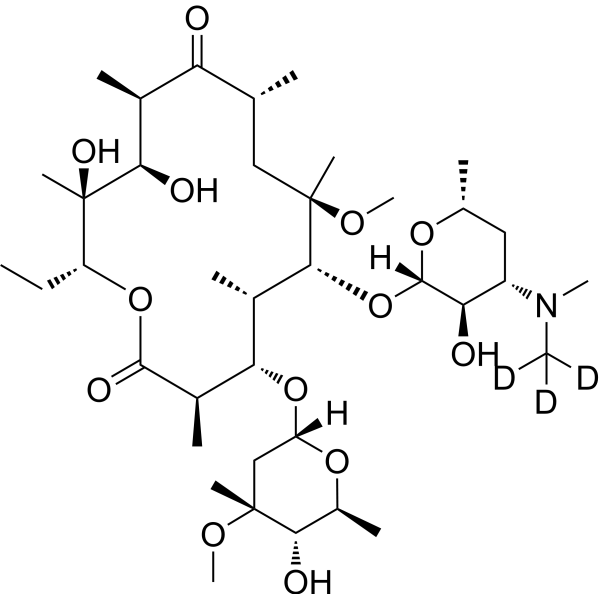
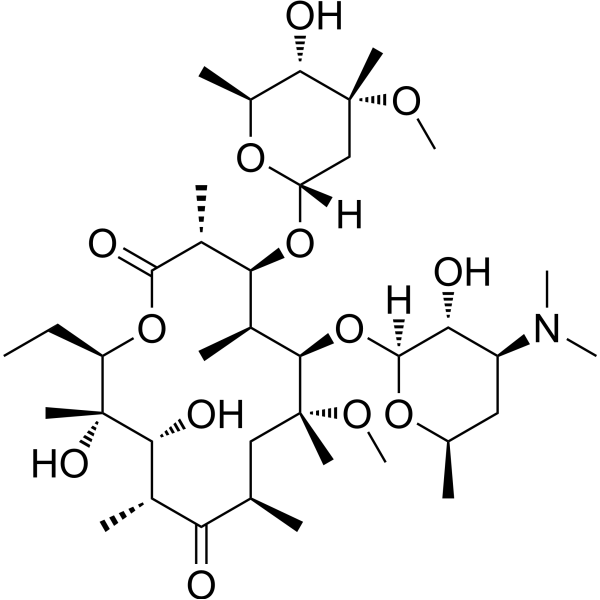
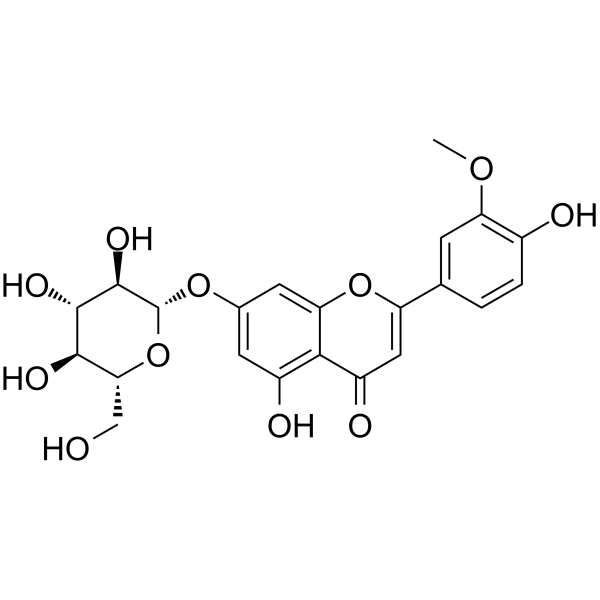
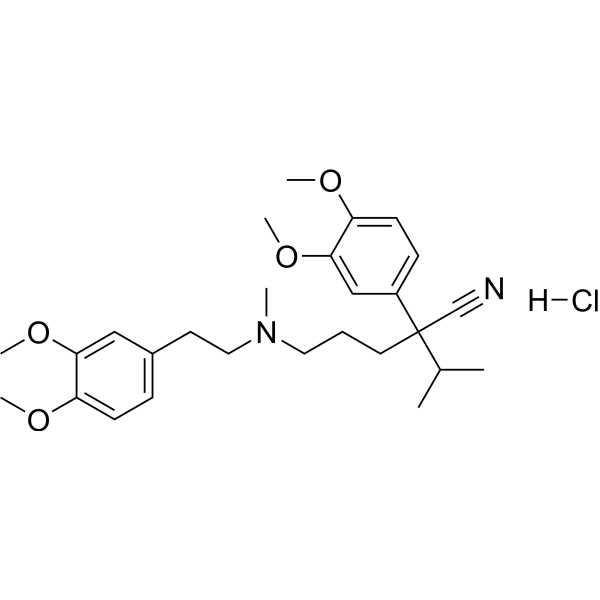
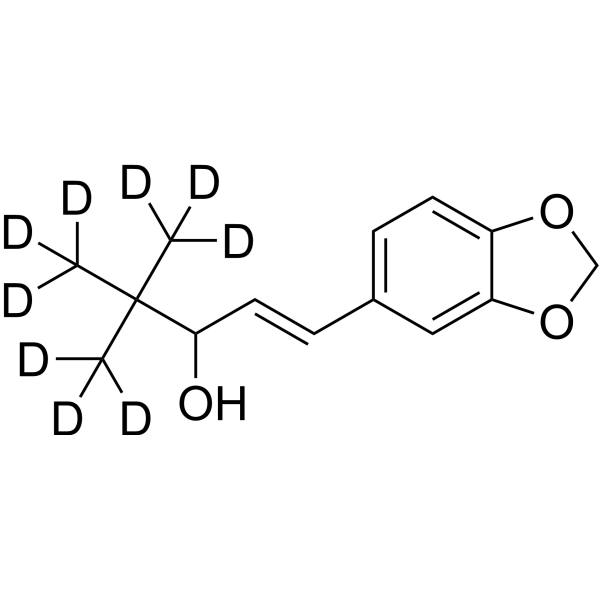
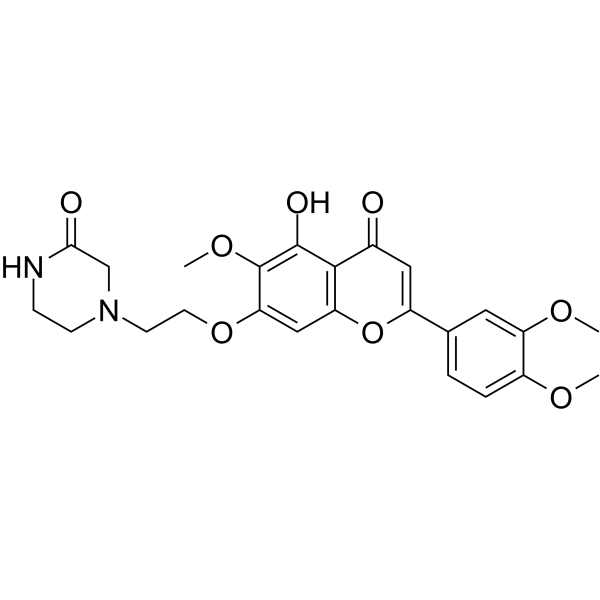
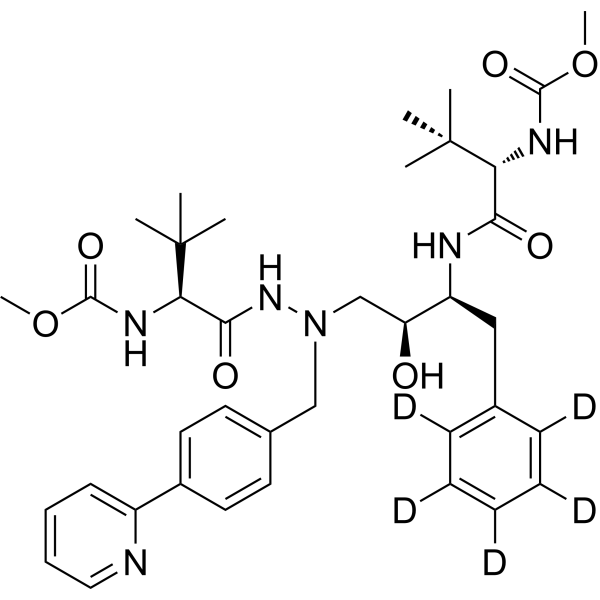
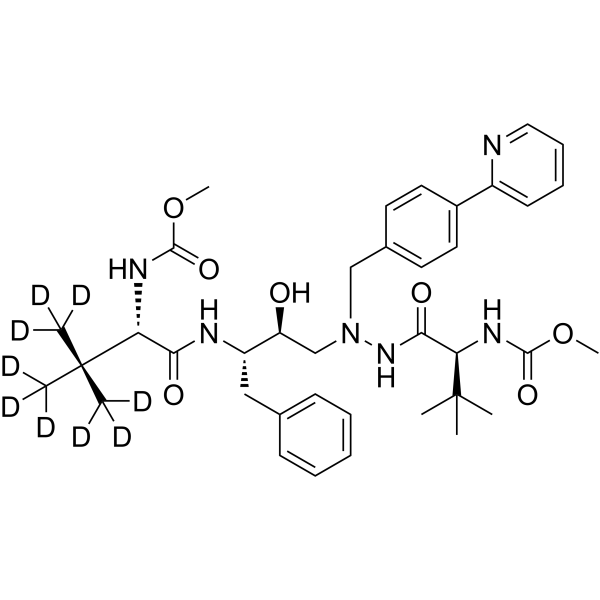
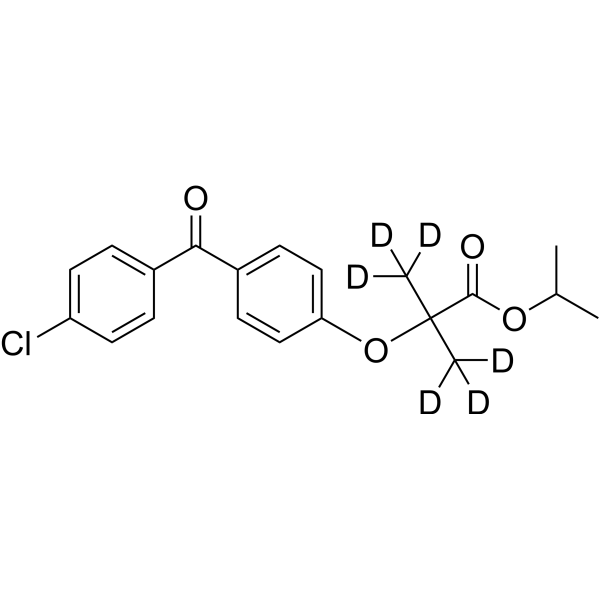
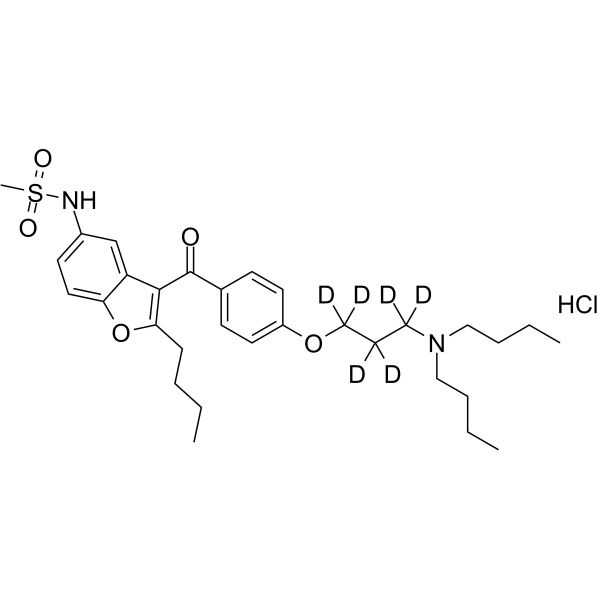
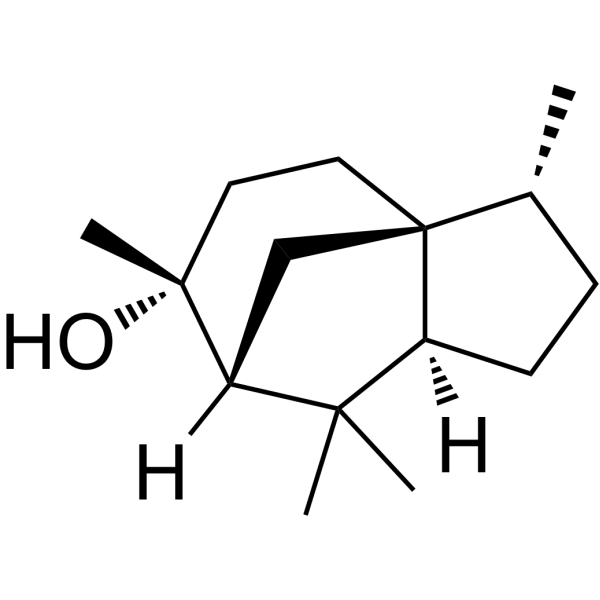
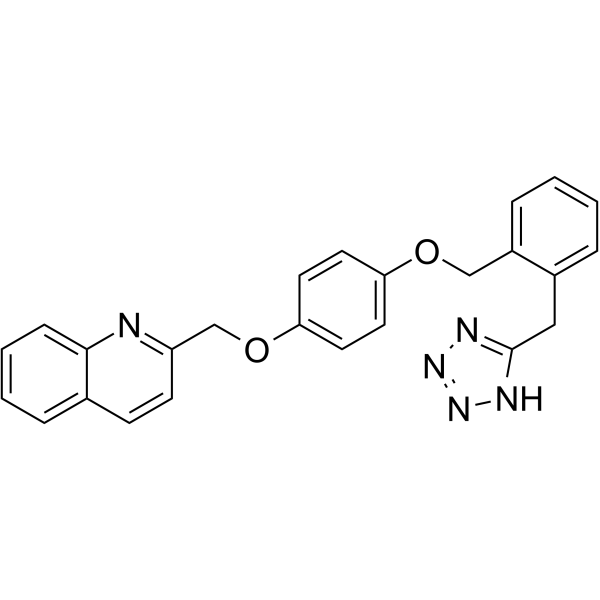
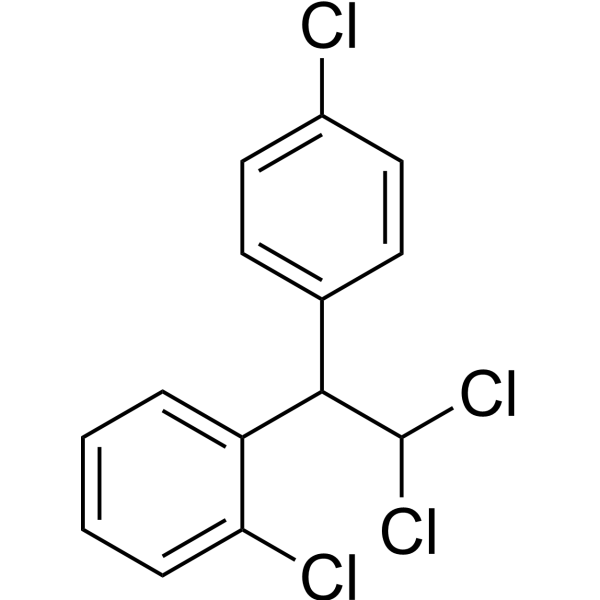
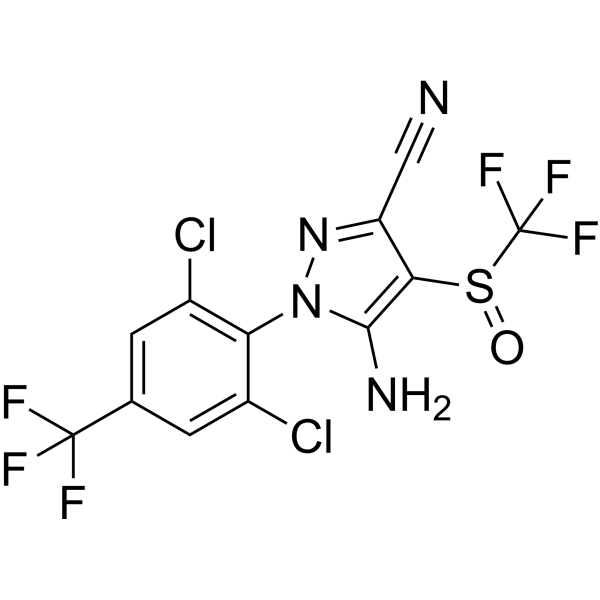
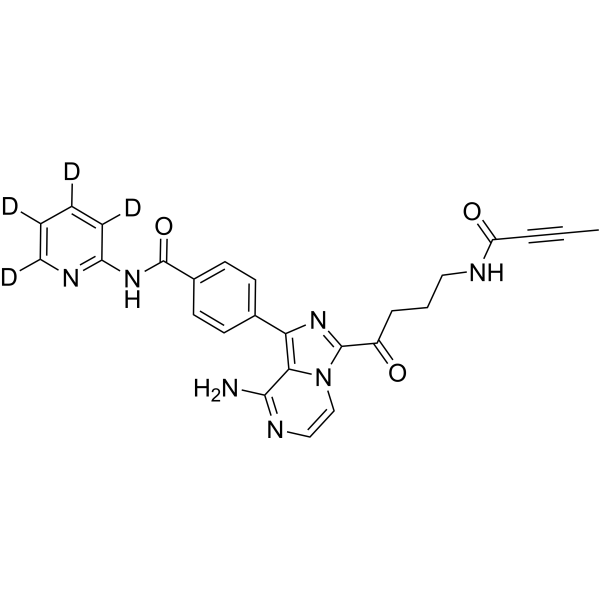
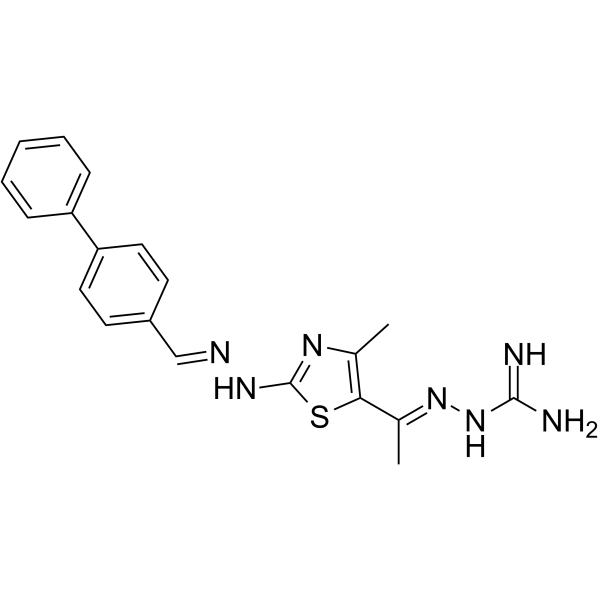
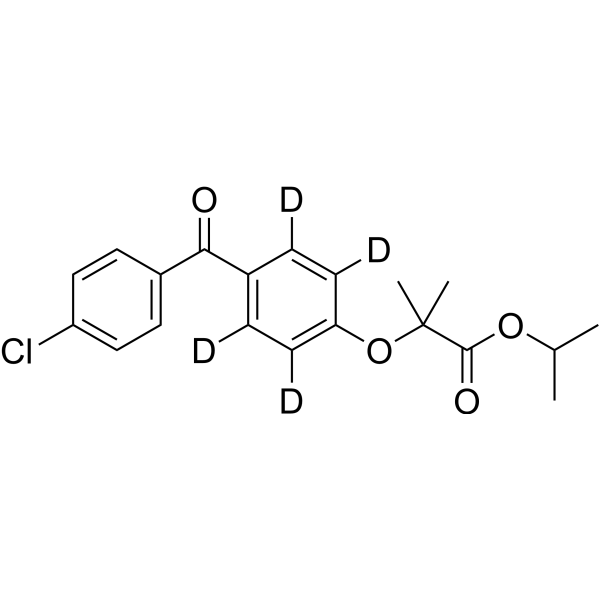
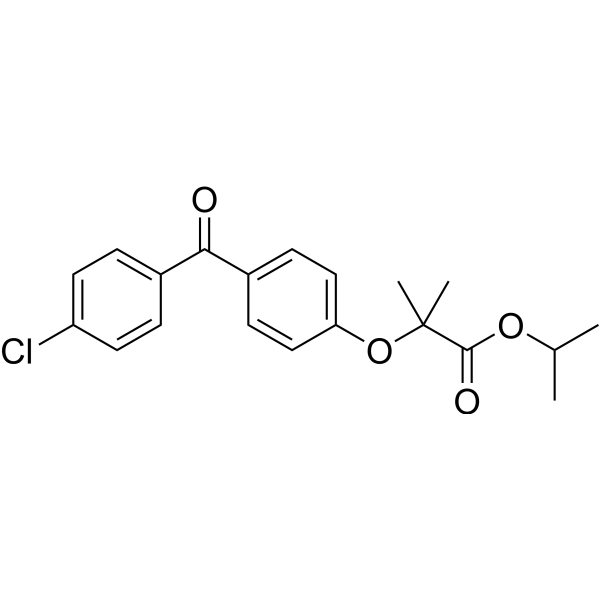
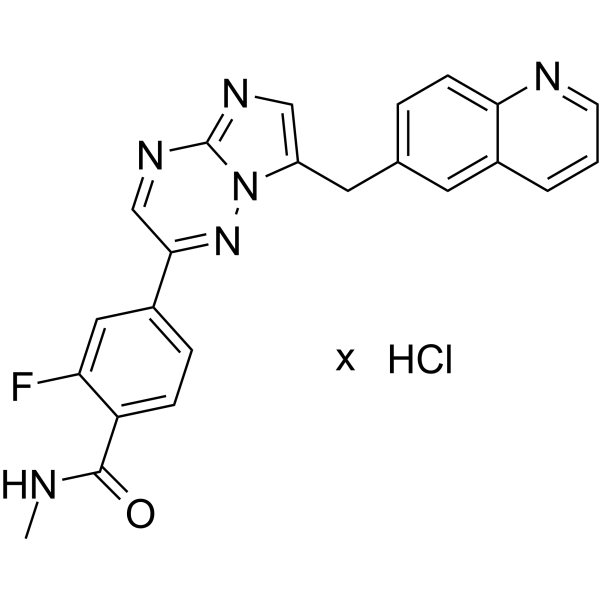
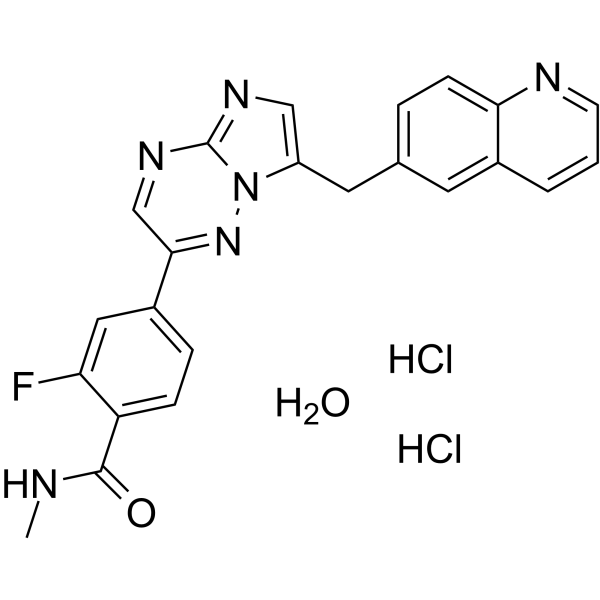
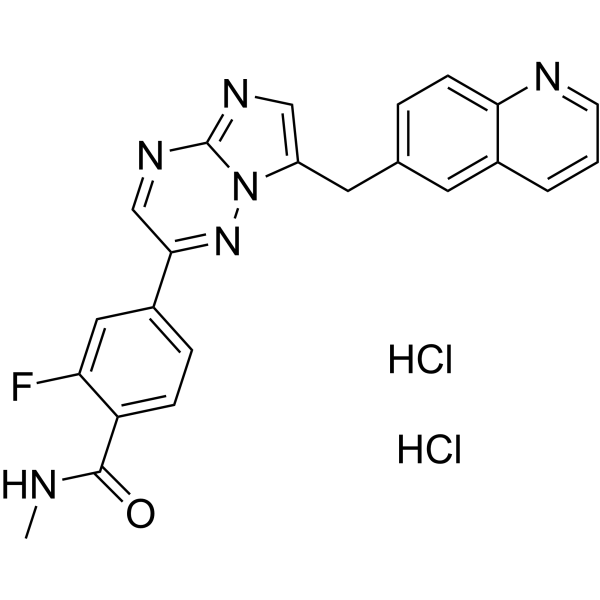
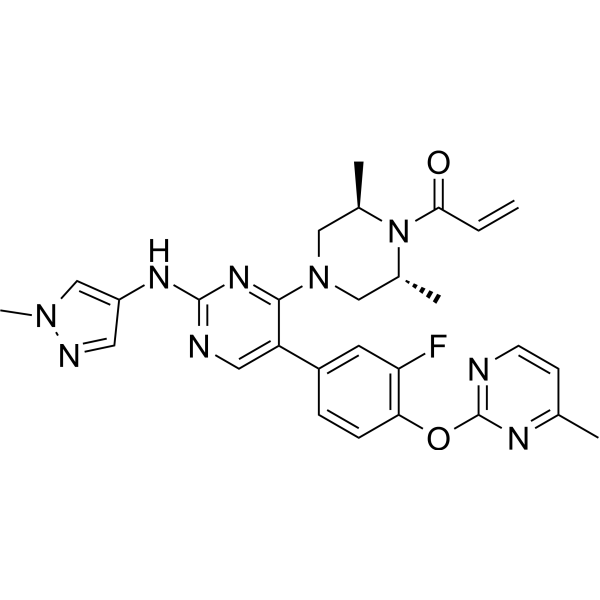
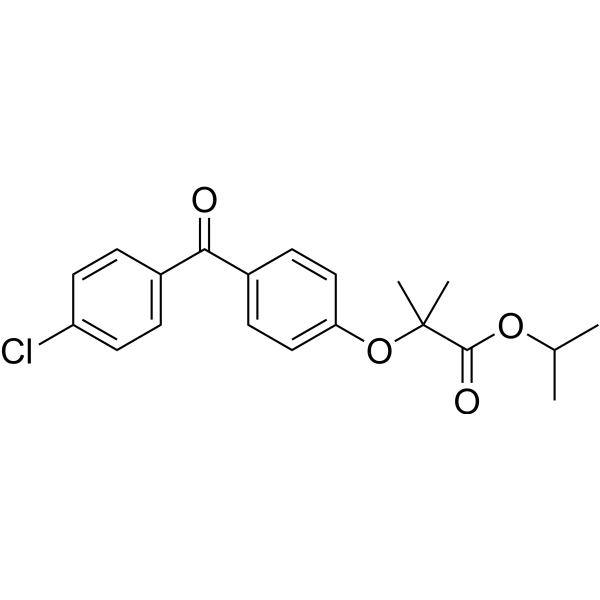
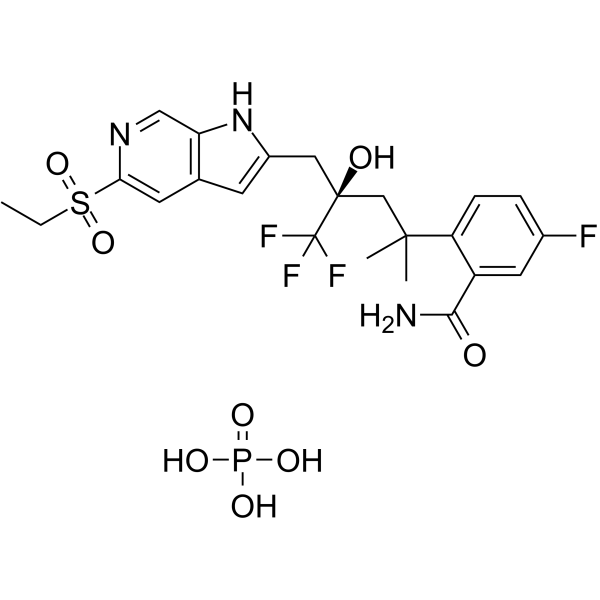
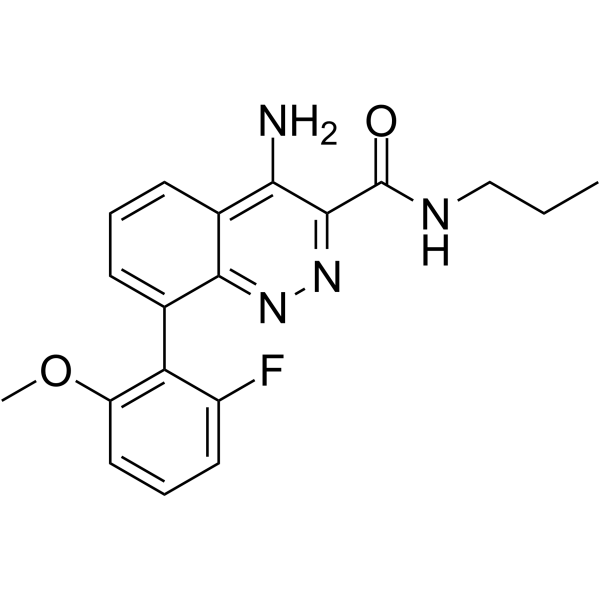
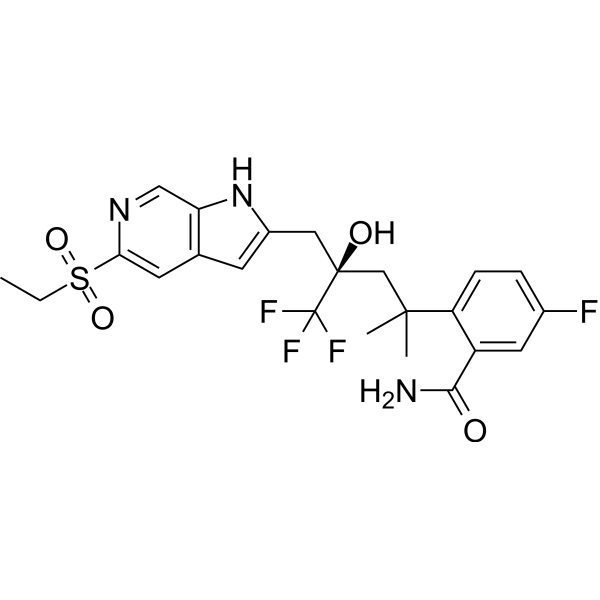
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
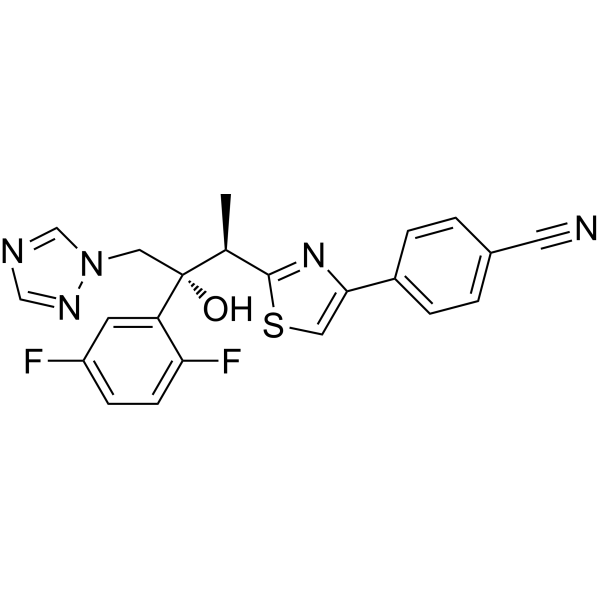
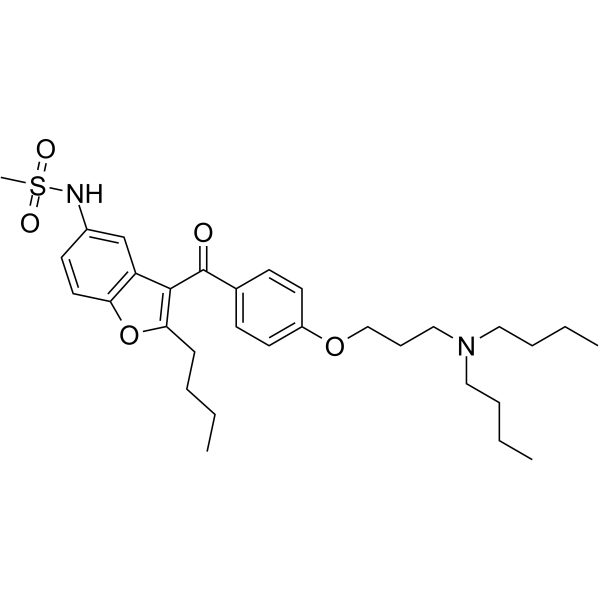
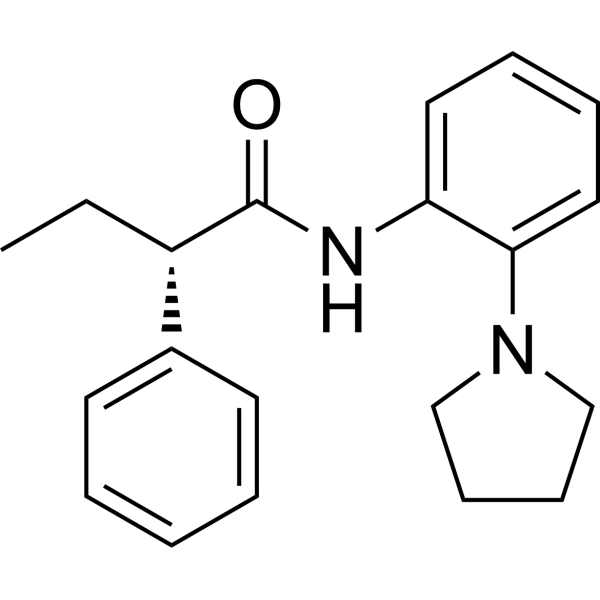
CYP3A4 enzyme-IN-1 (compound 59) is a potent antibacterial agent, with a MIC of 1 μg/mL for MRSA. CYP3A4 enzyme-IN-1 exhibits low to moderate inhibitory effects on CYP1A2, CYP2E1, CYP2D6, and CYP3A4 enzymes .
CYP3A4 Human Pre-designed siRNA Set A contains three designed siRNAs for CYP3A4 gene (Human), as well as a negative control, a positive control, and a FAM-labeled negative control.
CYP3A4-IN-3 is a high-affinity specific inhibitor of cytochrome P450 3A4 (CYP3A4) with the IC50 value of 0.075 μM. CYP3A4-IN-3 is a ritonavir analogue, but with a simpler structure and twice the inhibitory effect of ritonavir. CYP3A4-IN-3 is used as an antiviral agent and immunosuppressant .
CYP3A4-IN-2 is a specific inhibitor of cytochrome P450 3A4 (CYP3A4) with the IC50 value of 0.055 μM. CYP3A4-IN-2 is a ritonavir analogue with increased hydrophobicity of the R2 side group and stronger inhibitory effect compared to ritonavir. CYP3A4-IN-2 can be used as an antiviral agent and immunosuppressants .
CYP3cide (PF-4981517) is a potent, selective and time-dependent inhibitor of cytochrome P4503A4 (CYP3A4). The IC50 values for Midazolam 1’-hydroxylase activity are 0.03 μM, 17 μM, and 71 μM for CYP3A4, CYP3A5, and CYP3A7, respectively. CYP3cide can be used to distinguish the contributions of CYP3A4 versus CYP3A5 on agent metabolism .
Licopyranocoumarin is an isoflavonoid that shows CYP3A4 inhibitory activity with an IC50 of 32 μM. Licopyranocoumarin has potent neuroprotective activities .
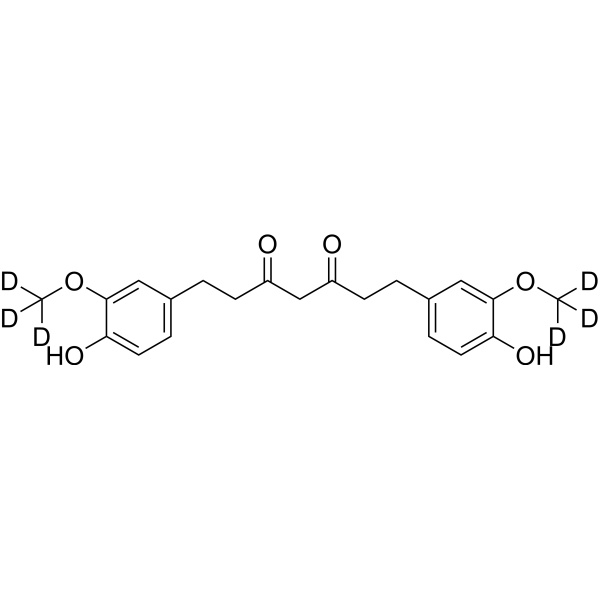
Tetrahydrocurcumin-d6 is a deuterium labeled Tetrahydrocurcumin. Tetrahydrocurcumin is a Curcuminoid which displays inhibitory activity for CYP2C9 and CYP3A4[1].
Dasatinib analog-1 (compound 5826) inhibits CYP3A4 viability with a Ki value of 5.4 μM. Dasatinib analog-1 blocks the formation of glutathione adducts .
N-Desmethyl Apalutamide is an active metabolite of Apalutamide. N-Desmethyl Apalutamide is a less potent antagonist of the androgen receptor and is responsible for one-third of the activity of Apalutamide. The formation of N-Desmethyl Apalutamide mediated predominantly by CYP2C8 and CYP3A4. N-Desmethyl Apalutamide is moderate to strong CYP3A4 and CYP2B6 inducer and has an excellent plasma-proteins bound concentration .
RO6889678 is a highly potent HBV capsid formation inhibitor with a complex absorption, distribution, metabolism, and excretion (ADME) profile. RO6889678 is a potent inducer of CYP3A4 and coregulated proteins in human hepatocytes. RO6889678 is metabolized by a combination of CYP3A4-mediated oxidation and UDP-glucuronosyltransferase UGT1A3- and UGT1A1-mediated direct glucuronidation .
Peucedanol is a non-competitive inhibitor of CYP3A4 with a Ki value of 4.07 μM and a competitive inhibitor of CYP1A2 and CYP2D6 with Ki values of 3.39 μM and 6.77 μM, respectively .
RPR132595A (NPC) hydrochloride is an active metabolite of CPT-11, which is generated by cytochrome P-450 3A4 (CYP3A4) and finally excreted through urine .
Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole .
Dehydroaripiprazole (OPC-14857) hydrochloride is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole hydrochloride. Dehydroaripiprazole hydrochloride has with antipsychotic activity equivalent to Aripiprazole .
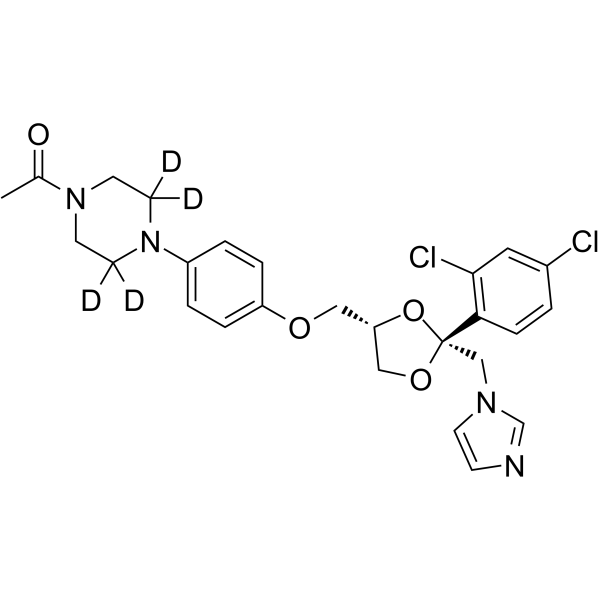
N-Desmethyl apalutamide-d4 is the deuterium-labeled N-Desmethyl-Apalutamide (HY-135331). N-Desmethyl Apalutamide is an active metabolite of Apalutamide. N-Desmethyl Apalutamide is a less potent antagonist of the androgen receptor and is responsible for one-third of the activity of Apalutamide. The formation of N-Desmethyl Apalutamide mediated predominantly by CYP2C8 and CYP3A4. N-Desmethyl Apalutamide is moderate to strong CYP3A4 and CYP2B6 inducer and has an excellent plasma-proteins bound concentration .
Topiroxostat (FYX-051) is a potent and orally active xanthine oxidoreductase (XOR) inhibitor with an IC50 value of 5.3 nM and a Ki value of 5.7 nM. Topiroxostat exhibits weak CYP3A4-inhibitory activity (18.6%). Topiroxostat has the potential for hyperuricemia treatment .
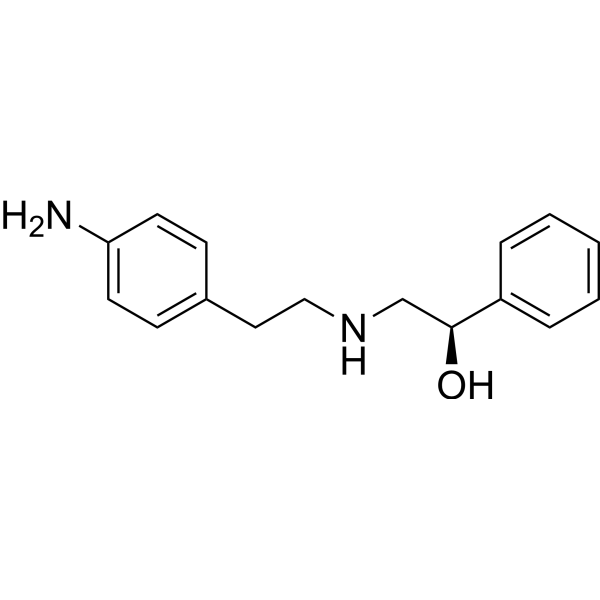
(R)-Mirtazapine ((R)-Org3770) is a R(−)-enantiomer of Mirtazapine with antinociceptive properties in an animal model of acute thermal nociception. (R)-Mirtazapine is a 5-HT3 receptor antagonist. (R)-Mirtazapine is mainly metabolized by CYP3A4 .
5-HT7 receptor ligand 1 (Compound 5c) is a 5-HT7 receptor ligand with a Ki of 8 nM. 5-HT7 receptor ligand 1 is not hepatotoxic and exhibit moderate potential interaction with other agents metabolized by CYP3A4 or CYP2D6 .
AR-C141990 (Compound 30) is a potent blocker of the monocarboxylate transporter MCT1 with a Ki of 4.8 nM. AR-C141990 (Compound 30) inhibits CYP3A4 and CYP2C9 with IC50 values of 16 μM .
Clarithromycin has a broad spectrum of antimicrobial activity. Clarithromycin inhibits the CYP3A4-catalyzed triazolam alpha-hydroxylation with the IC50 (Ki) value of 56 (43) μM . Clarithromycin significantly inhibits the HERG potassium current .Clarithromycin affects the autophagic flux by impairing the signaling pathway linking hERG1 and PI3K .
1β-Hydroxydeoxycholic acid (1β-OH-DCA), a secondary bile acid, is a CYP3A biomarker. Deoxycholic acid is specifically metabolized into 1β-Hydroxydeoxycholic acid by CYP3A4 and CYP3A7 using recombinant human CYP450 enzymes .
Carbosulfan-d18 is the deuterium labeled Carbosulfan. Carbosulfan inhibited relatively potently CYP3A4 and moderately CYP1A1/2 and CYP2C19 in pooled HLM (human livers). Carbosulfan activation is predominantly catalyzed in humans by CYP3A4.
RPR121056 (APC) is a metabolite of Irinotecan (CPT-11), which is generated by CYP3A4. Irinotecan (CPT-11) is an antineoplastic agent that inhibits topoisomerase type I, causing cell death, and is widely used in the treatment of colorectal cancer. Irinotecan also directly inhibits AChE .
Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research .
Verapamil ((±)-Verapamil) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil also inhibits CYP3A4. Verapamil has the potential for high blood pressure, heart arrhythmias and angina research .
(R)-Mirtazapine-d3 is a deuterium labeled (R)-Mirtazapine. (R)-Mirtazapine is a R(−)-enantiomer of Mirtazapine with antinociceptive properties in an animal model of acute thermal nociception. (R)-Mirtazapine is a 5-HT3 receptor antagonist. (R)-Mirtazapine is mainly metabolized by CYP3A4[1].
SOS1-IN-16 (Comp 54) is a selective inhibitor of SOS1 with an IC50 of 7.2 nM. SOS1-IN-16 has inhibitory activity of CYP3A4 when using testosterone as a substrate, with an IC50 of 8.9μM. SOS1-IN-16 can be used for cancer research .
2-Me PeER is a rhodamine dye-based fluorescent probe that detects CYP3A4 activity. In fluorescence-activated cell sorting (FACS) based on CYP3A4 activity, homogeneous and functional human induced pluripotent stem cell (hiPSC)-derived hepatocytes and intestinal epithelial cells can be obtained with the aid of 2-Me PeER .
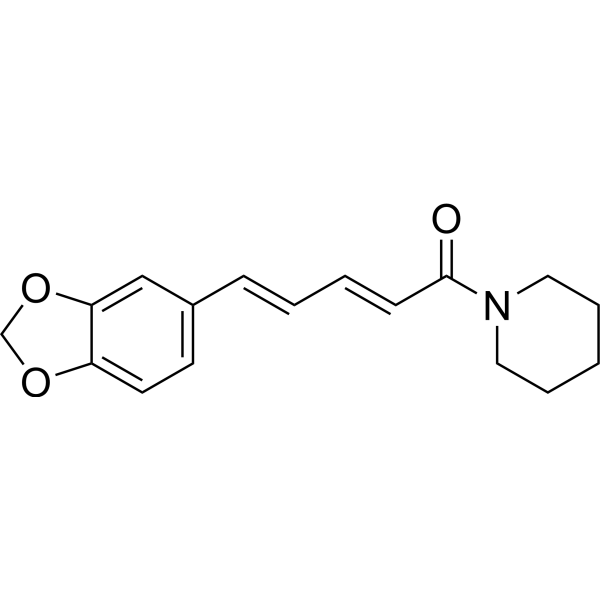
Antibacterial agent 102 (compound 32) possesses potent in vitro and in vivo antibacterial activity, with MICs < 0.5 μg/mL in Staphylococcus aureus (S. aureus). Antibacterial agent 102 also moderately inhibits CYP3A4 with an IC50 value of 6.148 μM. Antibacterial agent 102 can reduce Methicillin-resistant Staphylococcus aureus (MRSA) load in thigh infected mice .
Chalepensin, a furanocoumarin, is a competitive CYP2A6 inhibitor. Chalepensin also inhibits human CYP1A1, CYP1A2, CYP2A13, CYP2C9, CYP2D6, CYP2E1, and CYP3A4 to different extents .
Tetrahydrocurcumin is a Curcuminoid found in turmeric (Curcuma longa) that is produced by the reduction of Curcumin. Tetrahydrocurcumin inhibit CYP2C9 and CYP3A4.
SR9186 (ML368) is a potent CYP3A4 inhibitor with IC50 s for inhibition of midazolam → 1′hydroxymidazolam, testosterone → 6β-hydroxytestosterone, and vincristine → vincristine M1 of 9, 4, and 38 nM, respectively. SR-9186 inhibits liver-stage development of P. falciparum to block ivermectin metabolism .
ABBV-744 is a first-in-class, orally active and selective inhibitor of the BDII domain of BET family proteins with IC50 values ranging from 4 to 18 nM for BRD2, BRD3, BRD4 and BRDT. ABBV-744 is primarily metabolized by CYP3A4 with agent-like properties enable the investigation of its antitumor efficacy and tolerability .
Tabimorelin (NN703) hemifumarate is an orally active growth hormone (GH) secretagogue. Tabimorelin hemifumarate is also a potent inhibitor of CYP3A4 activity .
Kushenol K, a flavonoid antioxidant isolated from the roots of Sophora flavescens. Kushenol K is a cytochrome P-450 3A4 (CYP3A4) inhibitor with a Ki value of 1.35 μM . Kushenol K shows weak antiviral activity against HSV-2 (EC50 of 147 μM) . Kushenol K also inhibits the activity of SGLT1 and SGLT2 .
Revexepride is a highly selective 5-HT4 receptor agonist, and a potential inducer of CYP3A4 enzyme, used for the treatment of gastroesophageal reflux disease.
Pradefovir mesylate is a good substrate for liver CYP3A4. Pradefovir is converted to 9-(2-phosphonylmethoxyethyl)adenine (PMEA) in human liver microsomes with a Km of 60 μM.
Bergaptol is an inhibitor of debenzylation of the CYP3A4 enzyme with an IC50 of 24.92 μM. Recent studies have shown that it has anti-proliferative and anti-cancer properties .
Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment .
Verapamil-d7 is the deuterium labeled Verapamil (HY-14275). Verapamil ((±)-Verapamil) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil also inhibits CYP3A4. Verapamil has the potential for high blood pressure, heart arrhythmias and angina research .
Brexpiprazole S-oxide (DM-3411) is a main metabolite of Brexpiprazole and is metabolized by cytochrome P450 3A4 (CYP3A4). Brexpiprazole is an atypical antipsychotic agent and a partial agonist of human 5-HT1A and dopamine receptor with Kis of 0.12 nM and 0.3 nM, respectively. Brexpiprazole is also a 5-HT2A receptor antagonist with a Ki of 0.47 nM .
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
Piperine, a natural alkaloid isolated from Piper nigrum L, inhibits P-glycoprotein and CYP3A4 activities with an IC50 value of 61.94±0.054 μg/mL in HeLa cell.
Ethiprole is an insecticide.Metabolic sulfones are produced faster than Fipronil (HY-B0822) in CYP3A4-expressing cells and in vivo in mouse brain and liver.Ethiprole's sulfide, sulfoxide, sulfone and desulfinyl derivatives have better biological activity .
Kushenol M is a flavonoid from Sophora flavescens. Kushenol M is a cytochrome P450 (CYP) inhibitor, with IC50 values of 1.29 μM for CYP3A4 in human liver microsomes .
Casopitant mesylate (GW679769B) is a potent, selective, brain permeable and orally active neurokinin 1 (NK1) receptor antagonist. Casopitant mesylate is a second in the class of antiemetics that acts to antagonise the emetogenic effect of substance P. Casopitant mesylate is also a substrate and a weak-to-moderate inhibitor of CYP3A4. Casopitant mesylate can be used for chemotherapy-induced nausea and vomiting (CINV) and postoperative nausea and vomiting (PONV) .
(±)14,15-Epoxyeicosatrienoic acid ((±)14(15)-EET) is the Cytochrome P450 metabolite of arachidonic acid. While CYP3A4 may be involved in breast cancer cell growth, (±)14,15-Epoxyeicosatrienoic acid may promote mitosis and anchorage-dependent cloning through STAT3 affected by CYP3A4. (±)14,15-Epoxyeicosatrienoic acid exhibits STAT3-dependent cell growth promotion and may also participate in the autocrine/paracrine pathway that drives cell growth .
Ilaprazole sulfone is the major metabolite of Ilaprazole (HY-101664), is predominantly catalysed by CYP3A4/5. Ilaprazole (IY-81149) is an orally active proton pump inhibitor .
(R)-3-Hydroxy Midostaurin ((R)-CGP52421) is a potent kinases inhibitor. (R)-3-Hydroxy Midostaurin is a major metabolite of midostaurin (PKC412; HY-10230) undergoing by the hepatic CYP3A4 enzyme. (R)-3-Hydroxy Midostaurin has the potential for acute myeloid leukemia (AML) .
Cletoquine oxalate (Desethylhydroxychloroquine oxalate) is a major active metabolite of Hydroxychloroquine. Cletoquine oxalate is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine oxalate is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine oxalate has antimalarial effects and has the potential for autoimmune diseases treatment .
Verapamil-d6 (CP-16533-1-d6) hydrochloride is the deuterium labled Verapamil (hydrochloride) (HY-A0064). Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research .
Veledimex S enantiomer (INXN-1001 S enantiomer) is the S enantiomer of veledimex. Veledimex is an oral activator ligand for a proprietary gene therapy promoter system, and a moderate inhibitor of and substrate for CYP3A4/5 .
MCH-1 antagonist 1 is a potent melanin concentrating hormone (MCH-1) antagonist with a Ki of 2.6 nM. MCH-1 antagonist 1 also inhibits CYP3A4 with an IC50 of 10 μM.
Isavuconazole (BAL-4815) is a triazole proagent with antifungal activity against yeasts, molds, and dimorphic fungi. Isavuconazole inhibits ergosterol biosynthesis and results in the disruption of fungal membrane structure and function. Isavuconazole is a moderate inhibitor of CYP3A4.
BMS-819881 is a melaninconcentrating hormone receptor 1 (MCHR1) antagonist, which binds rat MCHR1 with a Ki of 7 nM. BMS-819881 also is selective and potent for CYP3A4 activity with an EC50 of 13 μM.
AWZ1066S is a highly potent, specific and orally active anti-Wolbachia agent with EC50 value of 121 nM. AWZ1066S also is a weak CYP2C9 inhibitor and a weak CYP3A4 inducer with IC50 values of 9.7 μM and 37 uM, respectively. AWZ1066S can be used for the research of tropical diseases such as Onchocerciasis (river blindness) and lymphatic filariasis (elephantiasis) .
XEN907 is a potent and spirooxindole blocker of NaV1.7, with an IC50 of 3 nM. XEN907 also inhibits CYP3A4 in a recombinant human enzyme assay. XEN907 can be used for the research of pain .
Oxiconazole (Ro 13-8996) is a broad spectrum anti-fungal agent which can inhibit the growth of Candida, Aspergillus and Trichophyton. Oxiconazole is also a highly efficacious activator of CYP3A4 transactivation, which could be antagonized by Rifampicin (HY-B0272) in a competitive manner. Oxiconazole exhibits inhibitory effect against colorectal cancer (CRC) via peroxiredoxin-2 (PRDX2)-mediated autophagy arrest .
Anticancer agent 111 (compound 11) is a potent cytochrome P450 inhibitor with an IC50 value of 3 μM for CYP3A4. Anticancer agent 111 has anticancer activity. Anticancer agent 111 can be used in research of cancer .
Norfluoxetine hydrochloride is an active N-demethylated metabolite of Fluoxetine. Fluoxetine is a selective serotonin (5-HT) reuptake inhibitor that is metabolized to Norfluoxetine hydrochloride by cytochrome P450 (CYP) 2D6, CYP2C19, and CYP3A4. Norfluoxetine hydrochloride inhibits 5-HT uptake and inhibits CaV3.3 T current (IC50 = 5 μM). Norfluoxetine hydrochloride has anticonvulsant activity .
7-Benzyloxy-4-(trifluoromethyl)coumarin (7-BFC) is a coumarin fluorescent substrate. 7-Benzyloxy-4-(trifluoromethyl)coumarin is a substrate for cDNA-expressed CYP1A2 and CYP3A4 and is metabolized to 7-hydroxy-4-trifluoromethylcoumarin (HFC). 7-Benzyloxy-4-(trifluoromethyl)coumarin is used for rapid CYP isoform metabolism and inhibition screening studies .
Brexpiprazole S-oxide-d8 is a deuterium labeled Brexpiprazole S-oxide. Brexpiprazole S-oxide is a main metabolite of Brexpiprazole and is metabolized by cytochrome P450 3A4 (CYP3A4). Brexpiprazole is an atypical antipsychotic agent and a partial agonist of human 5-HT1A and dopamine receptor with Kis of 0.12 nM and 0.3 nM, respectively. Brexpiprazole is also a 5-HT2A receptor antagonist with a Ki of 0.47 nM[1][2][3].
Topiroxostat-d4 is deuterium labeled Topiroxostat. Topiroxostat (FYX-051) is a potent and orally active xanthine oxidoreductase (XOR) inhibitor with an IC50 value of 5.3 nM and a Ki value of 5.7 nM. Topiroxostat exhibits weak CYP3A4-inhibitory activity (18.6%). Topiroxostat has the potential for hyperuricemia treatment[1][2].
Oxiconazole (Ro 13-8996) nitrate is a broad spectrum anti-fungal agent which can inhibit the growth of Candida, Aspergillus and Trichophyton. Oxiconazole nitrate is also a highly efficacious activator of CYP3A4 transactivation, which could be antagonized by Rifampicin (HY-B0272) in a competitive manner. Oxiconazole nitrate exhibits inhibitory effect against colorectal cancer (CRC) via peroxiredoxin-2 (PRDX2)-mediated autophagy arrest .
(S)-3-Hydroxy Midostaurin ((S)-CGP52421) is a potent kinases inhibitor with IC50 values of <400 nM for 13 kinases (VEGFR-2, TRK-A, FLT3, et). (S)-3-Hydroxy Midostaurin is a minor metabolite of midostaurin (PKC412; HY-10230) undergoing by the hepatic CYP3A4 enzyme. (S)-3-Hydroxy Midostaurin has the potential for acute myeloid leukemia (AML) .
Clobetasol propionate is a potent and selective CYP3A5 inhibitor with an IC50 of 0.206 μM. Clobetasol propionate has no inhibiting on CYP3A4 or other major CYPs. Clobetasol propionate is a corticosteroid and has the potential for psoriasis and other dermatoses research .
Rolapitant (SCH619734) is a potent, selective, long-acting and orally active neurokinin 1 (NK1) receptor antagonist with a Ki of 0.66 nM. Rolapitant does not interact with CYP3A4. Rolapitant shows potent anti-emetic activity in a ferret emesis model .
Rolapitant (SCH619734) hydrochloride is a potent, selective, long-acting and orally active neurokinin 1 (NK1) receptor antagonist with a Ki of 0.66 nM. Rolapitant hydrochloride does not interact with CYP3A4. Rolapitant hydrochloride shows potent anti-emetic activity in a ferret emesis model .
Rolapitant hydrochloride hydrate (SCH619734 hydrochloride hydrate) is a potent, selective, long-acting and orally active neurokinin 1 (NK1) receptor antagonist with a Ki of 0.66 nM. Rolapitant hydrochloride hydrate does not interact with CYP3A4. Rolapitant hydrochloride hydrate shows potent anti-emetic activity in a ferret emesis model .
YS-370 (compound 44) is a potent, high selective, and orally active inhibitor of P-glycoprotein (P-gp). YS-370 stimulates the P-gp ATPase activity and has moderate inhibition against CYP3A4. YS-370 effectively reverses multidrug resistance (MDR) to paclitaxel and colchicine in SW620/AD300 and HEK293T-ABCB1 cells. YS-370 in combination with paclitaxel achieves much stronger antitumor activity .
AMG-208 is an orally active c-Met/RON dual selective inhibitor with an IC50 of 9 nM for c-Met. AMG-208 is a CYP3A4 inhibitor with an IC50 of 32 μM. AMG-208 has anti-cancer activity .
Orteronel (TAK-700) is a highly selective inhibitor of human 17,20-lyase (CYP17) with IC50 of 38 nM, and exhibits >1000-fold selectivity over other CYPs such as 11-hydroxylase and CYP3A4 .
Nampt-IN-5 is a potent nicotinamide phosphoribosyltransferase (NAMPT) inhibitor. Nampt-IN-5 also inhibits CYP3A4 activity and has cellular IC50s of 0.7 nM and 3.9 nM against A2780 and COR-L23, respectively .
AS1810722 is an orally active and potent STAT6 inhibitor with an IC50 of 1.9 nM. AS1810722 shows a good profile of CYP3A4 inhibition. AS1810722, a derivative of fused bicyclic pyrimidine, has the potential for allergic diseases such as asthma and atopic diseases research .
Stiripentol (STP) is an anticonvulsant agent, which can inhibit N-demethylation of CLB to NCLB mediatated by CYP3A4 (noncompetitively) and CYP2C19 (competitively) with Ki of 1.59±0.07 and 0.516±0.065 μM and IC50 of 1.58 and 3.29 μM, respectively.
K777 is a potent, orally active and irreversible cysteine protease inhibitor. K777 is also a potent CYP3A4 inhibitor with an IC50 of 60 nM and a selective CCR4 antagonist featuring the potent chemotaxis inhibition. K777 irreversibly inhibits Cruzain, the major cysteine protease of Trypansoma cruzi, and cathepsins B and L. K777 is a broad-spectrum antiviral by targeting cathepsin-mediated cell entry. K777 inhibits SARS-CoV and EBOV pseudovirus entry with IC50 values of 0.68 nM and 0.87 nM, respectively .
Dehydroaripiprazole-d8 is deuterium labeled Dehydroaripiprazole. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole[1][2][3].
HIV-IN-9 (Compound 2b) is a HIV inhibitor (IC50: 6.65 μg/mL), and has high binding affinity with HIV-RT. HIV-IN-9 also inhibits CYP3A4, CYP1A2, CYP2C1, and CYP2D6 .
Clarithromycin-d3is the deuterium labeledClarithromycin(HY-17508) . Clarithromycin has a broad spectrum of antimicrobial activity. Clarithromycin inhibits the CYP3A4-catalyzed triazolam alpha-hydroxylation with the IC50 (Ki) value of 56 (43) μM . Clarithromycin significantly inhibits the HERG potassium current .Clarithromycin affects the autophagic flux by impairing the signaling pathway linking hERG1 and PI3K .
Clarithromycin (Standard) is the analytical standard of Clarithromycin. This product is intended for research and analytical applications. Clarithromycin has a broad spectrum of antimicrobial activity. Clarithromycin inhibits the CYP3A4-catalyzed triazolam alpha-hydroxylation with the IC50 (Ki) value of 56 (43) μM . Clarithromycin significantly inhibits the HERG potassium current .Clarithromycin affects the autophagic flux by impairing the signaling pathway linking hERG1 and PI3K .
Resorufin benzyl ether (BzRes), a fluorogenic enzyme substrate, can be used to detect CYP3A4 enzyme activity. Resorufin benzyl ether modified with a recognizing moiety boronate, can be used for ONOO - detection via a self-immolation mechanism. Ex/Em=530-570 nm/590 nm .
Dehydroaripiprazole (Standard) is the analytical standard of Dehydroaripiprazole. This product is intended for research and analytical applications. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole .
Verapamil-d3 (hydrochloride) is the deuterium labeled Verapamil hydrochloride. Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research[1][2][3].
Friedelin is isolated from isolated from the leaves of Maytenus ilicifolia(Mart). Friedelin is a noncompetitive inhibitor of CYP3A4 with IC50 and Kivalues of 10.79 μM and 6.16 μM, respectively. Friedelin is also a competitive inhibitor of CYP2E1 with IC50 and Ki values of 22.54 μM and 18.02 μM, respectively .
Thermopsoside is a flavone derivative isolated from Aspalathus linearis. Thermopsoside exhibits inhibitory effects on CYP450 isozymes with IC50 values of 6.0 μM, 9.5 μM, 12.0 μM, 32.0 μM, for CYP3A4, CYP2C19, CYP2D6 and CYP2C9, respectively .
Verapamil (hydrochloride) (Standard) is the analytical standard of Verapamil (hydrochloride). This product is intended for research and analytical applications. Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research .
Stiripentol-d9 is the deuterium labeled Stiripentol. Stiripentol (STP) is an anticonvulsant agent, which can inhibit N-demethylation of CLB to NCLB mediatated by CYP3A4 (noncompetitively) and CYP2C19 (competitively) with Ki of 1.59±0.07 and 0.516±0.065 μM and IC50 of 1.58 and 3.29 μM, respectively[1][2].
EMT inhibitor-2 (Compound 1) inhibits epithelial-mesenchymal transition (EMT) induced by substances such as IL-1β and TGF-β released from the immunocytes. EMT inhibitor-2 inhibits CYP3A4 testosteron and CYP2C9 with IC50s of 49.72 and 5.54 μM, respectively .
Hydroxy desmethyl Bosentan (Ro 64-105) is a Bosentan metabolism produced by the cytochrome P450 enzymes CYP2C9 and CYP3A4 in the liver . Bosentan is a competitive and dual antagonist of endothelin-1 (ET) for the ETA and ETB receptors with Ki of 4.7 nM and 95 nM in human SMC, respectively. Bosentan can be used in treatment of pulmonary arterial hypertension .
(S)-Ceralasertib ((S)-AZD6738) is extracted from patent WO2011154737A1, Compound II, exhibits an IC50 of 2.578 nM .
(S)-Ceralasertib is a potent and selective sulfoximine morpholinopyrimidine ATR inhibitor with excellent preclinical physicochemical and pharmacokinetic (PK) characteristics.
(S)-Ceralasertib is developed improving aqueous solubility and eliminates CYP3A4 time-dependent inhibition .
ACP-5862 is a major active, circulating, pyrrolidine ring-opened metabolite of Acalabrutinib with an IC50 of 5.0 nM for Bruton tyrosine kinase (BTK). ACP‐5862 is a weak time‐dependent inactivator of CYP3A4 and CYP2C8. Acalabrutinib is an orally active, irreversible, and highly selective BTK inhibitor, with an IC50 of 3 nM and EC50 of 8 nM .
Clobetasol propionate (Standard) is the analytical standard of Clobetasol propionate. This product is intended for research and analytical applications. Clobetasol propionate is a potent and selective CYP3A5 inhibitor with an IC50 of 0.206 μM. Clobetasol propionate has no inhibiting on CYP3A4 or other major CYPs. Clobetasol propionate is a corticosteroid and has the potential for psoriasis and other dermatoses research .
Dronedarone (SR 33589), a derivative of amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone is a substrate for and a moderate inhibitor of CYP3A4 .
Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration . Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp) . Atazanavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 3.49 μM .
Atazanavir (BMS-232632) sulfate, a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration . Atazanavir sulfate is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp) . Atazanavir sulfate is also a SARS-CoV 3CL pro inhibitor with an IC50 of
3.49 μM .
Curcumenol ((+)-Curcumenol) is a potent CYP3A4 inhibitor with an IC50 of 12.6 μM, which is one of constituents in the plants of medicinally important genus of Curcuma zedoaria, with neuroprotection, anti-inflammatory, anti-tumor and hepatoprotective activities. Curcumenol ((+)-Curcumenol) suppresses Akt-mediated NF-κB activation and p38 MAPK signaling pathway in LPS-stimulated BV-2 microglial cells .
S-F24 is an antifungal agent with excellent broad-spectrum. S-F24 inhibits CYP3A4 with an IC50 value of 0.4 μM. S-F24 displays a good safety profile with high selectivity, low hemolytic effects, and low tendency to induce resistance. S-F24 can be used for research on fungal infections .
Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
Xanthotoxol (8-Hydroxypsoralen) It is a kind of fragrant bean substance, and it is a CYP450 inhibitor. Xanthotoxol has anti-inflammatory, anti-inflammatory, and 5-HT antagonistic and protective effects. Xanthotoxol inhibited CYP3A4 sum CYP1A2 IC50s separation 7.43 μM sum 27.82 μM. Xanthotoxol can pass through MAPK and NF-κB, inhibiting inflammation.
Cletoquine-d4 is deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Cletoquine-d4-1 is the deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Nefazodone is an orally active phenylpiperazine antidepressant. Nefazodone can potently and selectively block postsynaptic 5-HT2A receptors, and moderately inhibit 5-HT and noradrenaline reuptake. Nefazodone can also relieve the adverse effects of stress on the the immune system of mice. Nefazodone has a high affinity for CYP3A4 isoenzyme, which indicates that it has certain risk of agent-agent interaction .
hCYP3A4-IN-1 (compound C6) is a potent, orally active hCYP3A4 inhibitor. hCYP3A4-IN-1 shows the IC50 values of 43.93 nM and 153.00 nM against hCYP3A4 in human liver microsomes (HLMs) and CHO-3A4 stably transfected cell line, respectively. hCYP3A4-IN-1 potently inhibits CYP3A4-catalyzed N-ethyl-1,8-naphthalimide (NEN) hydroxylation in a competitive manner (Ki = 30.00 nM) .
Atazanavir-d5 is the deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
Atazanavir-d9 is the deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
Fenofibrate-d6 is the deuterium labeled Fenofibrate. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
Dronedarone-d6 (hydrochloride) is the deuterium labeled Dronedarone. Dronedarone hydrochloride, a derivative of Amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone hydrochloride is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone hydrochloride is a substrate for and a moderate inhibitor of CYP3A4[1][2][3][4].
Aβ42-IN-1, compound 1v, is a novel, potent and orally active γ-secretase modulator (GSM). Aβ42-IN-1 potently reduced Aβ42 levels with an IC50 value of 0.091 µM without CYP3A4 inhibition. Aβ42-IN-1 shows a sustained pharmacokinetic profile.
Simvastatin acid-d6 (ammonium)mis the deuterium labeled Simvastatin acid ammonium. Simvastatin ammonium is an active metabolite of simvastatin lactone mediated by CYP3A4/5 in the intestinal wall and liver (pKa=5.5). Simvastatin ammonium reduces indoxyl sulfate-mediated reactive oxygen species and modulates OATP3A1 expression in cardiomyocytes and HEK293 cells transfected with the OATP3A1 gene[1].
Ticlopidine (PCR 5332), an antithrombotic proagent, acts as an allosteric, noncompetitive inhibitor of CD39 with the IC50 of 81.7 μM. Ticlopidine blocks several NTPDase isoenzymes with IC50s of 170 μM and 149 μM for NTPDase2 and NTPDase3, respectively. Ticlopidine is an inhibitor of CYP2C19 human liver cytochrome. Ticlopidine inhibits CYP2C9 and CYP3A4 with IC50s of 26.0 and 32.3 μM, respectively.
Atazanavir-d6 is deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
Cedrol is a bioactive sesquiterpene, a potent competitive inhibitor of cytochrome P-450 (CYP) enzymes. Cedrol inhibits CYP2B6-mediated bupropion hydroxylase and CYP3A4-mediated midazolam hydroxylation with Ki of 0.9 μM and 3.4 μM, respectively. Cedrol also has weak inhibitory effect on CYP2C8, CYP2C9, and CYP2C19 enzymes . Cedrol is found in cedar essential oil and poetesses anti-septic, anti-inflammatory, anti-spasmodic, tonic, astringent, diuretic, insecticidal, and anti-fungal activities .
RG-12525 is a a specific, competitive and orally effective antagonist of the peptidoleukotrienes, LTC4, LTD4 and LTE4, inhibiting LTC4-, LTD4- and LTE4-inducd guinea pig parenchymal strips contractions, with IC50s of 2.6 nM, 2.5 nM and 7 nM, respectively; RG-12525 is also a peroxisome proliferator-activated receptor gamma (PPAR-gamma) agonist with IC50 of appr 60 nM and a potent inhibitor of CYP3A4, with a Ki value of 0.5 µM.
Mitotane (2,4′-DDD), an isomer of DDD and derivative of dichlorodiphenyltrichloroethane (DDT), is an antineoplastic agent, can be used to research adrenocortical carcinoma. Mitotane exert its adrenocorticolytic effect at least in part through lipotoxicity induced by intracellular free cholesterol (FC) accumulation. Mitotane can have direct pituitary effects on corticotroph cells. Mitotane can induce CYP3A4 gene expression via steroid and xenobiotic receptor (SXR) activation, and has agent-agent interactions .
Fipronil is a broad-spectrum insecticide effective against Lepidoptera species as well as thrips, locusts, ants, cockroaches, fleas and ticks. Fipronil selectively inhibits GABA receptor with IC50s of 30 nM and 1600 nM for cockroach and rat GABA receptors, respectively. Glutamate-gated chloride channels (GluCls), which are present in cockroaches but not in mammals, are sensitive to the blocking effect of Fipronil. Fipronil also induces apoptosis in HepG2 cells and promotes the expression of CYP1A1 and CYP3A4 mRNA in human hepatocytes .
ACP-5862-d4 is deuterium labeled ACP-5862. ACP-5862 is a major active, circulating, pyrrolidine ring-opened metabolite of Acalabrutinib with an IC50 of 5.0 nM for Bruton tyrosine kinase (BTK). ACP‐5862 is a weak time‐dependent inactivator of CYP3A4 and CYP2C8. Acalabrutinib is an orally active, irreversible, and highly selective BTK inhibitor, with an IC50 of 3 nM and EC50 of 8 nM[1][2].
GlcN-6-P Synthase-IN-1 (Compound 4d) is a Glucosamine-6-phosphate (GlcN-6-P) synthase inhibitor with an IC50 of 3.47 μM. GlcN-6-P Synthase-IN-1 exhibits significant antimicrobial activity. GlcN-6-P Synthase-IN-1 has good penetration in the CNS and is able to inhibit the cytochrome P450, CYP3A4 isoform .
Fenofibrate-d4 is the deuterium labeled Fenofibrate[1]. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively[2][3].
Fenofibrate (Standard) is the analytical standard of Fenofibrate. This product is intended for research and analytical applications. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
Nav1.7-IN-8 is a potent blockage of NaV1.7 with high selectivity for the inhibition of NaV1.7 over the subtypes hNaV1.1 and hNaV1.5. Nav1.7-IN-8 inhibits CYP2C9 and CYP3A4 with an IC50 of 0.17 μM and 0.077 μM, respectively. Nav1.7-IN-8 displays significant analgesic effects in rodent models of acute and inflammatory pain .
Dibenzylfluorescein (DBF) is a fluorogenic probe (Fluoresecent dye) that acts as a substrate for specific cytochrome P450 (CYP) isoforms, including CYP3A4, CYP2C8, CYP2C9, CYP2C19, and aromatase (CYP19). Dibenzylfluorescein is typically used near its Km value of 0.87-1.9 µM (Ex=485nm,Em=535nm). Dibenzylfluorescein is used to detect changes in CYP catalytic activity caused by drugs or disease .
Capmatinib (INC280; INCB28060) is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib is largely metabolized by CYP3A4 and aldehyde oxidase .
Ticlopidine-d4 is the deuterium labeled Ticlopidine. Ticlopidine (PCR 5332), an antithrombotic proagent, acts as an allosteric, noncompetitive inhibitor of CD39 with the IC50 of 81.7 μM. Ticlopidine blocks several NTPDase isoenzymes with IC50s of 170 μM and 149 μM for NTPDase2 and NTPDase3, respectively[1]. Ticlopidine is an inhibitor of CYP2C19 human liver cytochrome. Ticlopidine inhibits CYP2C9 and CYP3A4 with IC50s of 26.0 and 32.3 μM, respectively[2][3].
Capmatinib (INC280; INCB28060) hydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib hydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib hydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib hydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) dihydrochloride hydrate is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride hydrate can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride hydrate potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride hydrate is largely metabolized by CYP3A4 and aldehyde oxidase .
Capmatinib (INC280; INCB28060) dihydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
FGFR-IN-10 is an orally active inhibitor of FGFR and Cytochrome P450 (CYPs). FGFR-IN-10 inhibits wide type and V564F mutant FGFR2 with IC50s of 104.1 nM and 43.6 nM, respectively. FGFR-IN-10 also inhibits CYPs with IC50s of 3.33 μM (CYP2C9), 18.75 μM (CYP2C19), 4.34 μM (CYP2CD6), and 0.69 μM (CYP3A4), respectively .
Fenofibrate (GMP) is Fenofibrate (HY-17356) produced by using GMP guidelines. GMP small molecules work appropriately as an auxiliary reagent for cell therapy manufacture. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
BI 653048 phosphate is a selective and orally active nonsteroidal glucocorticoid (GC) agonist with an IC50 value of 55 nM . BI 653048 phosphate inhibits CP1A2, CYP2D6, CYP2C9, CYP2C19 and CYP3A4 isoforms’ activity and reduces affinity for the hERG ion channel (IC50>30 μM) . BI 653048 phosphate is extracted from patent WO2005028501A1 (Compound 103), is also a HCV NS3 protease inhibitor that can reduce viral loads infected with the hepatitis C virus .
AZD7325 is a potent and orally active partial selective PAM of GABAAα2 and Aα3 receptor (Ki=0.3 and 1.3 nM, respectively), and has less antagonistic efficacy at the Aα1 and Aα5 receptor subtypes . AZD7325 is a moderate CYP1A2 and a potent CYP3A4 inducer in vitro . AZD7325 has the potential for the investigation of anxiety and dravet syndrome . PAM: positive allosteric modulator.
BI 653048 is a selective and orally active nonsteroidal glucocorticoid (GC) agonist with an IC50 value of 55 nM . BI 653048 inhibits CP1A2, CYP2D6, CYP2C9, CYP2C19 and CYP3A4 isoforms’ activity and reduces affinity for the hERG ion channel (IC50>30 μM) . BI 653048 is extracted from patent WO2005028501A1 (Compound 103), is also a HCV NS3 protease inhibitor that can reduce viral loads infected with the hepatitis C virus .
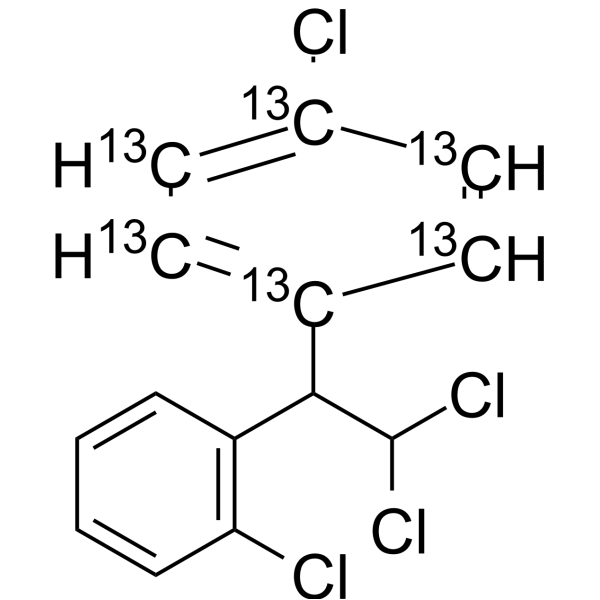
Mitotane- 13C6 is the 13C labeled Mitotane[1]. Mitotane (2,4′-DDD), an isomer of DDD and derivative of dichlorodiphenyltrichloroethane (DDT), is an antineoplastic agent, can be used to research adrenocortical carcinoma. Mitotane exert its adrenocorticolytic effect at least in part through lipotoxicity induced by intracellular free cholesterol (FC) accumulation. Mitotane can have direct pituitary effects on corticotroph cells. Mitotane can induce CYP3A4 gene expression via steroid and xenobiotic receptor (SXR) activation, and has agent-agent interactions[2][3][4][5].
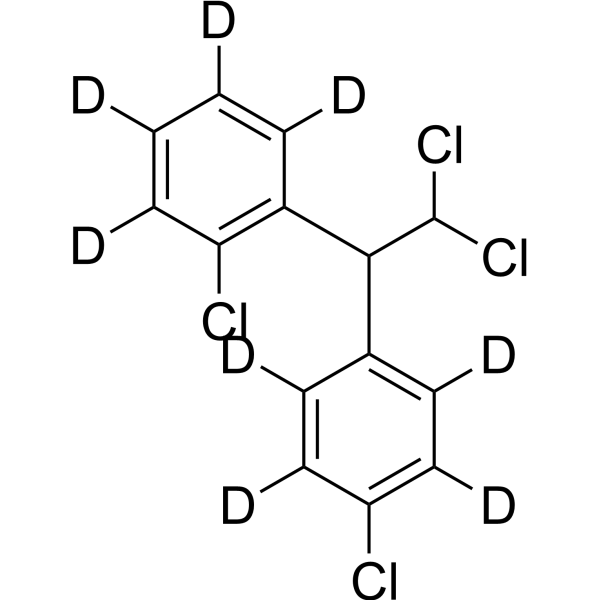
Mitotane-d8 is the deuterium labeled Mitotane[1]. Mitotane (2,4′-DDD), an isomer of DDD and derivative of dichlorodiphenyltrichloroethane (DDT), is an antineoplastic agent, can be used to research adrenocortical carcinoma. Mitotane exert its adrenocorticolytic effect at least in part through lipotoxicity induced by intracellular free cholesterol (FC) accumulation. Mitotane can have direct pituitary effects on corticotroph cells. Mitotane can induce CYP3A4 gene expression via steroid and xenobiotic receptor (SXR) activation, and has agent-agent interactions[2][3][4][5].
Fipronil (Standard) is the analytical standard of Fipronil. This product is intended for research and analytical applications. Fipronil is a broad-spectrum insecticide effective against Lepidoptera species as well as thrips, locusts, ants, cockroaches, fleas and ticks. Fipronil selectively inhibits GABA receptor with IC50s of 30 nM and 1600 nM for cockroach and rat GABA receptors, respectively. Glutamate-gated chloride channels (GluCls), which are present in cockroaches but not in mammals, are sensitive to the blocking effect of Fipronil. Fipronil also induces apoptosis in HepG2 cells and promotes the expression of CYP1A1 and CYP3A4 mRNA in human hepatocytes .
Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
GSK-25 is a potent, selective and orally bioavailable ROCK1 inhibitor (IC50=7 nM). GSK-25 maintains good selectivity against a panel of 31 kinases (>100 fold), as well as RSK1 and p70S6K (RSK1: IC50=398 nM, p70S6K: IC50=1 μM). GSK-25 inhibits P450 profile (IC50s of 2.5, 5.2, 2.5 µM for CYP2C9, CYP2D6, CYP3A4, respectively) .
Ginsenoside Rd (Standard) is the analytical standard of Ginsenoside Rd. This product is intended for research and analytical applications. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
Cepharanthine is a natural product that can be isolated from the plant Stephania cephalantha Hayata. Cepharanthine has anti-severe acute respiratory syndrome coronavirus 2 (anti-SARS-CoV-2) activities. Cepharanthine has good effective in suppressing viral proliferation (half maximal (50%) inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) values of 1.90 and 4.46 µM . Cepharanthine can also effectively reverses P-gp-mediated multidrug resistance in K562 cells and increase enhances the sensitivity of anticancer agents in xenograft mice model . Cepharanthine shows inhibitory effects of human liver cytochrome P450 enzymes CYP3A4, CYP2E1 and CYP2C9. Cepharanthine has antitumor, anti-inflammatory and antinociceptive effects .
Mirabegron impurity-1 is a potent and selective
β3- adrenergic receptor agonist. Mirabegron impurity-1
has the activity of inhibiting metabolism. Mirabegron impurity-1 can be used in
the study of the treatment of bladder impurity .
6β-Hydroxy Eplerenone is a metabolite of Eplerenone (HY-B0251). 6β-Hydroxy Eplerenone plays an important role in the research of hypertension, atherosclerosis, chronic systolic heart failure (HF) and cardiovascular (CV) .
GLPG-3221 is a potent, orally active corrector of CFTR (cystic fibrosis transmembrane conductance regulator), with an EC50 of 105 nM. GLPG-3221 can be uesd for the treatment of cystic fibrosis .
Glucocorticoid receptor-IN-2 (Compound WX019) is a selective glucocorticoid receptor (GR) modulator with anti-inflammatory effect. Glucocorticoid receptor-IN-2 exhibits very good transcriptional repressive activity with an IC50 of 0.171 nM against hMMP1, and comparable transcriptional activation activity with an EC50 of 0.94 nM against MMTV .
PI3K/mTOR Inhibitor-9 (Compound 1) is a potent mTOR and PI3K inhibitor with IC50 values of 38 nM, 6.6 μM, 6.6 μM and 0.8 μM against mTOR (phospho-S6 cellular assay), PI3Kα, PI3Kγ and PI3Kδ, respectively .
Sofpironium bromide (BBI 4000) is an anticholinergic agent used in the study of primary axillary hyperhidrosis (PAH). Sofpironium bromide reduces sweating by inhibiting M3 muscarinic receptors in eccrine glands at the application site. Sofpironium bromide also has a high afnity for the M1, M2, M4 and M5 subtypes .
Glucocorticoid receptor-IN-1 (Compound WX002) is a selective glucocorticoid receptor (GR) modulator with anti-inflammatory effect. Glucocorticoid receptor-IN-1 exhibits very good transcriptional repressive activity with an IC50 of 2.11 nM against hMMP1, and transcriptional activation activity with an EC50 of 5.59 nM against MMTV .
Esomeprazole sodium ((S)-Omeprazole sodium) is a potent and orally active proton pump inhibitor. Esomeprazole reduces acid secretion through inhibition of the H +, K +-ATPase in gastric parietal cells. Esomeprazole acts as an exosome inhibitor by blocking the exosome release via the inhibition of V-H +-ATPases . Esomeprazole has the potential for symptomatic gastroesophageal reflux disease research .
Esomeprazole magnesium salt ((S)-Omeprazole magnesium salt) is a potent and orally active proton pump inhibitor and reduces acid secretion through inhibition of the H +, K +-ATPase in gastric parietal cells. Esomeprazole magnesium salt has the potential for symptomatic gastroesophageal reflux disease research .
Esomeprazole potassium salt ((S)-Omeprazole potassium salt) is a potent and orally active proton pump inhibitor and reduces acid secretion through inhibition of the H +, K +-ATPase in gastric parietal cells. Esomeprazole potassium salt has the potential for symptomatic gastroesophageal reflux disease research .
Esomeprazole ((S)-Omeprazole) hemistrontium is a potent and orally active proton pump inhibitor and reduces acid secretion through inhibition of the H +, K +-ATPase in gastric parietal cells. Esomeprazole hemistrontium has the potential for symptomatic gastroesophageal reflux disease research .
Esomeprazole ((S)-Omeprazole) is a potent and orally active proton pump inhibitor and reduces acid secretion through inhibition of the H +, K +-ATPase in gastric parietal cells. Esomeprazole has the potential for symptomatic gastroesophageal reflux disease research .
Antipsychotic agent-2 (Compound 11) is a potent antipsychotic agent. Antipsychotic agent-2 shows affinities for 5-HT1A, 5-HT2A, 5-HT2C, D2 and H1 receptors with Kis of 56.6, 66.7, 552, 596 and 1140 nM, respectively. Antipsychotic agent-2 has BBB permeability .
Fluticasone furoate is a topical, intranasal, enhanced-affinity synthetic trifluorinated corticosteroid with a Kd of 0.3 nM. Fluticasone furoate has potent anti-inflamatory and anti-asthmatic activity, and low systemic exposure. Fluticasone furoate has the potential for allergic rhinitis treatment .
NSC-609249 hydrochloride is an impurity of Verapamil (HY-14275). Verapamil is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor .
Gemfibrozil 1-O-β-Glucuronide, a metabolite of Gemfibrozil (CI-719; HY-B0258), is a potent and competitive P450 (CYP) isoform CYP2C8 inhibitor with an IC50 of 4.07 μM .
TAK-715 is an orally active and potent p38 MAPK inhibitor with IC50s of 7.1 nM, 200 nM for p38α and p38β, respectively. TAK-715 inhibits casein kinase I (CK1δ/ε) to regulate activation of Wnt/β-catenin signaling. TAK-715 shows good significant efficacy in a rat arthritis model .
Resorufin benzyl ether (BzRes), a fluorogenic enzyme substrate, can be used to detect CYP3A4 enzyme activity. Resorufin benzyl ether modified with a recognizing moiety boronate, can be used for ONOO - detection via a self-immolation mechanism. Ex/Em=530-570 nm/590 nm .
Dibenzylfluorescein (DBF) is a fluorogenic probe (Fluoresecent dye) that acts as a substrate for specific cytochrome P450 (CYP) isoforms, including CYP3A4, CYP2C8, CYP2C9, CYP2C19, and aromatase (CYP19). Dibenzylfluorescein is typically used near its Km value of 0.87-1.9 µM (Ex=485nm,Em=535nm). Dibenzylfluorescein is used to detect changes in CYP catalytic activity caused by drugs or disease .
Fenofibrate (GMP) is Fenofibrate (HY-17356) produced by using GMP guidelines. GMP small molecules work appropriately as an auxiliary reagent for cell therapy manufacture. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
Fenofibrate (GMP) is Fenofibrate (HY-17356) produced by using GMP guidelines. GMP small molecules work appropriately as an auxiliary reagent for cell therapy manufacture. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
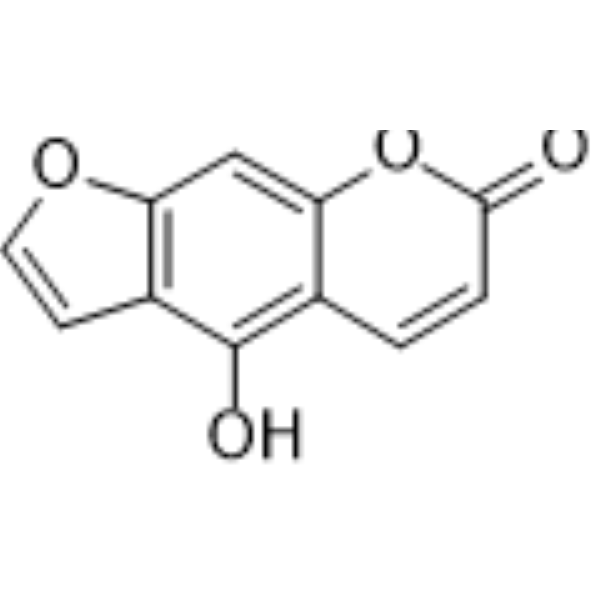
Licopyranocoumarin is an isoflavonoid that shows CYP3A4 inhibitory activity with an IC50 of 32 μM. Licopyranocoumarin has potent neuroprotective activities .
Peucedanol is a non-competitive inhibitor of CYP3A4 with a Ki value of 4.07 μM and a competitive inhibitor of CYP1A2 and CYP2D6 with Ki values of 3.39 μM and 6.77 μM, respectively .
1β-Hydroxydeoxycholic acid (1β-OH-DCA), a secondary bile acid, is a CYP3A biomarker. Deoxycholic acid is specifically metabolized into 1β-Hydroxydeoxycholic acid by CYP3A4 and CYP3A7 using recombinant human CYP450 enzymes .
Chalepensin, a furanocoumarin, is a competitive CYP2A6 inhibitor. Chalepensin also inhibits human CYP1A1, CYP1A2, CYP2A13, CYP2C9, CYP2D6, CYP2E1, and CYP3A4 to different extents .
Tetrahydrocurcumin is a Curcuminoid found in turmeric (Curcuma longa) that is produced by the reduction of Curcumin. Tetrahydrocurcumin inhibit CYP2C9 and CYP3A4.
Kushenol K, a flavonoid antioxidant isolated from the roots of Sophora flavescens. Kushenol K is a cytochrome P-450 3A4 (CYP3A4) inhibitor with a Ki value of 1.35 μM . Kushenol K shows weak antiviral activity against HSV-2 (EC50 of 147 μM) . Kushenol K also inhibits the activity of SGLT1 and SGLT2 .
Bergaptol is an inhibitor of debenzylation of the CYP3A4 enzyme with an IC50 of 24.92 μM. Recent studies have shown that it has anti-proliferative and anti-cancer properties .
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
Piperine, a natural alkaloid isolated from Piper nigrum L, inhibits P-glycoprotein and CYP3A4 activities with an IC50 value of 61.94±0.054 μg/mL in HeLa cell.
Kushenol M is a flavonoid from Sophora flavescens. Kushenol M is a cytochrome P450 (CYP) inhibitor, with IC50 values of 1.29 μM for CYP3A4 in human liver microsomes .
(±)14,15-Epoxyeicosatrienoic acid ((±)14(15)-EET) is the Cytochrome P450 metabolite of arachidonic acid. While CYP3A4 may be involved in breast cancer cell growth, (±)14,15-Epoxyeicosatrienoic acid may promote mitosis and anchorage-dependent cloning through STAT3 affected by CYP3A4. (±)14,15-Epoxyeicosatrienoic acid exhibits STAT3-dependent cell growth promotion and may also participate in the autocrine/paracrine pathway that drives cell growth .
Friedelin is isolated from isolated from the leaves of Maytenus ilicifolia(Mart). Friedelin is a noncompetitive inhibitor of CYP3A4 with IC50 and Kivalues of 10.79 μM and 6.16 μM, respectively. Friedelin is also a competitive inhibitor of CYP2E1 with IC50 and Ki values of 22.54 μM and 18.02 μM, respectively .
Thermopsoside is a flavone derivative isolated from Aspalathus linearis. Thermopsoside exhibits inhibitory effects on CYP450 isozymes with IC50 values of 6.0 μM, 9.5 μM, 12.0 μM, 32.0 μM, for CYP3A4, CYP2C19, CYP2D6 and CYP2C9, respectively .
Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration . Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp) . Atazanavir is also a SARS-CoV 3CL pro inhibitor with an IC50 of 3.49 μM .
Atazanavir (BMS-232632) sulfate, a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration . Atazanavir sulfate is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp) . Atazanavir sulfate is also a SARS-CoV 3CL pro inhibitor with an IC50 of
3.49 μM .
Xanthotoxol (8-Hydroxypsoralen) It is a kind of fragrant bean substance, and it is a CYP450 inhibitor. Xanthotoxol has anti-inflammatory, anti-inflammatory, and 5-HT antagonistic and protective effects. Xanthotoxol inhibited CYP3A4 sum CYP1A2 IC50s separation 7.43 μM sum 27.82 μM. Xanthotoxol can pass through MAPK and NF-κB, inhibiting inflammation.
Cedrol is a bioactive sesquiterpene, a potent competitive inhibitor of cytochrome P-450 (CYP) enzymes. Cedrol inhibits CYP2B6-mediated bupropion hydroxylase and CYP3A4-mediated midazolam hydroxylation with Ki of 0.9 μM and 3.4 μM, respectively. Cedrol also has weak inhibitory effect on CYP2C8, CYP2C9, and CYP2C19 enzymes . Cedrol is found in cedar essential oil and poetesses anti-septic, anti-inflammatory, anti-spasmodic, tonic, astringent, diuretic, insecticidal, and anti-fungal activities .
Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
Ginsenoside Rd (Standard) is the analytical standard of Ginsenoside Rd. This product is intended for research and analytical applications. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca 2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively.
Cepharanthine is a natural product that can be isolated from the plant Stephania cephalantha Hayata. Cepharanthine has anti-severe acute respiratory syndrome coronavirus 2 (anti-SARS-CoV-2) activities. Cepharanthine has good effective in suppressing viral proliferation (half maximal (50%) inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) values of 1.90 and 4.46 µM . Cepharanthine can also effectively reverses P-gp-mediated multidrug resistance in K562 cells and increase enhances the sensitivity of anticancer agents in xenograft mice model . Cepharanthine shows inhibitory effects of human liver cytochrome P450 enzymes CYP3A4, CYP2E1 and CYP2C9. Cepharanthine has antitumor, anti-inflammatory and antinociceptive effects .
The CYP3A4 protein, encoded by the CYP3A4 gene, is a key enzyme in drug metabolism and the synthesis of cholesterol, steroids, and lipids. It localizes to the endoplasmic reticulum and is induced by glucocorticoids and certain drugs. CYP3A4 Protein, Human (His) is the recombinant human-derived CYP3A4 protein, expressed by E. coli , with N-His labeled tag. The total length of CYP3A4 Protein, Human (His) is 502 a.a., with molecular weight of ~59.2 kDa.
CYP3A4 Protein, a cytochrome P450 monooxygenase, catalyzes diverse substrate metabolism, including sterols, hormones, and fatty acids. Mechanistically, it employs molecular oxygen for hydroxylation, impacting androgen metabolism, cholesterol, and retinoid pathways. Notably, it contributes to drug metabolism and regulates vitamin D catabolism, essential for calcium homeostasis. CYP3A4 Protein, Human (Cell-Free, His) is the recombinant human-derived CYP3A4 protein, expressed by E. coli Cell-free, with N-10*His labeled tag. The total length of CYP3A4 Protein, Human (Cell-Free, His) is 502 a.a., with molecular weight of 63.3 kDa.
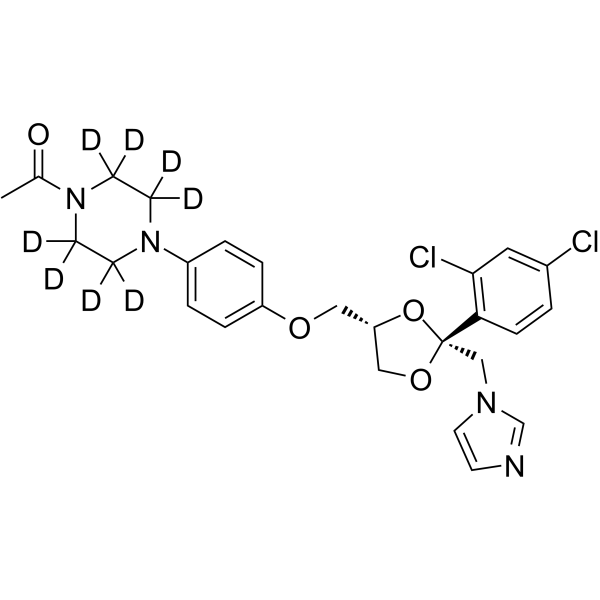
N-Desmethyl apalutamide-d4 is the deuterium-labeled N-Desmethyl-Apalutamide (HY-135331). N-Desmethyl Apalutamide is an active metabolite of Apalutamide. N-Desmethyl Apalutamide is a less potent antagonist of the androgen receptor and is responsible for one-third of the activity of Apalutamide. The formation of N-Desmethyl Apalutamide mediated predominantly by CYP2C8 and CYP3A4. N-Desmethyl Apalutamide is moderate to strong CYP3A4 and CYP2B6 inducer and has an excellent plasma-proteins bound concentration .
Tetrahydrocurcumin-d6 is a deuterium labeled Tetrahydrocurcumin. Tetrahydrocurcumin is a Curcuminoid which displays inhibitory activity for CYP2C9 and CYP3A4[1].
Carbosulfan-d18 is the deuterium labeled Carbosulfan. Carbosulfan inhibited relatively potently CYP3A4 and moderately CYP1A1/2 and CYP2C19 in pooled HLM (human livers). Carbosulfan activation is predominantly catalyzed in humans by CYP3A4.
(R)-Mirtazapine-d3 is a deuterium labeled (R)-Mirtazapine. (R)-Mirtazapine is a R(−)-enantiomer of Mirtazapine with antinociceptive properties in an animal model of acute thermal nociception. (R)-Mirtazapine is a 5-HT3 receptor antagonist. (R)-Mirtazapine is mainly metabolized by CYP3A4[1].
Verapamil-d7 is the deuterium labeled Verapamil (HY-14275). Verapamil ((±)-Verapamil) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil also inhibits CYP3A4. Verapamil has the potential for high blood pressure, heart arrhythmias and angina research .
Verapamil-d6 (CP-16533-1-d6) hydrochloride is the deuterium labled Verapamil (hydrochloride) (HY-A0064). Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research .
Brexpiprazole S-oxide-d8 is a deuterium labeled Brexpiprazole S-oxide. Brexpiprazole S-oxide is a main metabolite of Brexpiprazole and is metabolized by cytochrome P450 3A4 (CYP3A4). Brexpiprazole is an atypical antipsychotic agent and a partial agonist of human 5-HT1A and dopamine receptor with Kis of 0.12 nM and 0.3 nM, respectively. Brexpiprazole is also a 5-HT2A receptor antagonist with a Ki of 0.47 nM[1][2][3].
Topiroxostat-d4 is deuterium labeled Topiroxostat. Topiroxostat (FYX-051) is a potent and orally active xanthine oxidoreductase (XOR) inhibitor with an IC50 value of 5.3 nM and a Ki value of 5.7 nM. Topiroxostat exhibits weak CYP3A4-inhibitory activity (18.6%). Topiroxostat has the potential for hyperuricemia treatment[1][2].
Dehydroaripiprazole-d8 is deuterium labeled Dehydroaripiprazole. Dehydroaripiprazole (OPC-14857) is an active metabolite of Aripiprazole. Aripiprazole is an antipsychotic agent and is metabolized by CYP3A4 and CYP2D6 forming mainly Dehydroaripiprazole. Dehydroaripiprazole has with antipsychotic activity equivalent to Aripiprazole[1][2][3].
Clarithromycin-d3is the deuterium labeledClarithromycin(HY-17508) . Clarithromycin has a broad spectrum of antimicrobial activity. Clarithromycin inhibits the CYP3A4-catalyzed triazolam alpha-hydroxylation with the IC50 (Ki) value of 56 (43) μM . Clarithromycin significantly inhibits the HERG potassium current .Clarithromycin affects the autophagic flux by impairing the signaling pathway linking hERG1 and PI3K .
Verapamil-d3 (hydrochloride) is the deuterium labeled Verapamil hydrochloride. Verapamil hydrochloride ((±)-Verapamil hydrochloride) is a calcium channel blocker and a potent and orally active first-generation P-glycoprotein (P-gp) inhibitor. Verapamil hydrochloride also inhibits CYP3A4. Verapamil hydrochloride has the potential for high blood pressure, heart arrhythmias and angina research[1][2][3].
Stiripentol-d9 is the deuterium labeled Stiripentol. Stiripentol (STP) is an anticonvulsant agent, which can inhibit N-demethylation of CLB to NCLB mediatated by CYP3A4 (noncompetitively) and CYP2C19 (competitively) with Ki of 1.59±0.07 and 0.516±0.065 μM and IC50 of 1.58 and 3.29 μM, respectively[1][2].
Cletoquine-d4 is deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Cletoquine-d4-1 is the deuterium labeled Cletoquine. Cletoquine (Desethylhydroxychloroquine) is a major active metabolite of Hydroxychloroquine. Cletoquine is produced in the liver by CYP2D6, CYP3A4, CYP3A5, and CYP2C8 isoenzymes. Cletoquine is also a Chloroquine derivative and has the ability to against the chikungunya virus (CHIKV). Cletoquine has antimalarial effects and has the potential for autoimmune diseases treatment[1][2].
Atazanavir-d5 is the deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
Atazanavir-d9 is the deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
Fenofibrate-d6 is the deuterium labeled Fenofibrate. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively.
Dronedarone-d6 (hydrochloride) is the deuterium labeled Dronedarone. Dronedarone hydrochloride, a derivative of Amiodarone (HY-14187), is a class III antiarrhythmic agent for the study of atrial fibrillation (AF) and atrial flutter. Dronedarone hydrochloride is a potent blocker of multiple ion currents, including potassium current, sodium current, and L-type calcium current, and exhibits antiadrenergic effects by noncompetitive binding to β-adrenergic receptors. Dronedarone hydrochloride is a substrate for and a moderate inhibitor of CYP3A4[1][2][3][4].
Simvastatin acid-d6 (ammonium)mis the deuterium labeled Simvastatin acid ammonium. Simvastatin ammonium is an active metabolite of simvastatin lactone mediated by CYP3A4/5 in the intestinal wall and liver (pKa=5.5). Simvastatin ammonium reduces indoxyl sulfate-mediated reactive oxygen species and modulates OATP3A1 expression in cardiomyocytes and HEK293 cells transfected with the OATP3A1 gene[1].
Atazanavir-d6 is deuterium labeled Atazanavir. Atazanavir (BMS-232632), a highly selective HIV-1 protease inhibitor, is the first protease inhibitor approved for once-daily administration[1]. Atazanavir (BMS-232632) is a substrate and inhibitor of CYP3A4, and an inhibitor and inducer of P-glycoprotein (P-gp)[2]. Atazanavir is also a SARS-CoV 3CLpro inhibitor with an IC50 of 3.49 μM[3].
ACP-5862-d4 is deuterium labeled ACP-5862. ACP-5862 is a major active, circulating, pyrrolidine ring-opened metabolite of Acalabrutinib with an IC50 of 5.0 nM for Bruton tyrosine kinase (BTK). ACP‐5862 is a weak time‐dependent inactivator of CYP3A4 and CYP2C8. Acalabrutinib is an orally active, irreversible, and highly selective BTK inhibitor, with an IC50 of 3 nM and EC50 of 8 nM[1][2].
Fenofibrate-d4 is the deuterium labeled Fenofibrate[1]. Fenofibrate is a selective PPARα agonist with an EC50 of 30 μM. Fenofibrate also inhibits human cytochrome P450 isoforms, with IC50s of 0.2, 0.7, 9.7, 4.8 and 142.1 μM for CYP2C19, CYP2B6, CYP2C9, CYP2C8, and CYP3A4, respectively[2][3].
Ticlopidine-d4 is the deuterium labeled Ticlopidine. Ticlopidine (PCR 5332), an antithrombotic proagent, acts as an allosteric, noncompetitive inhibitor of CD39 with the IC50 of 81.7 μM. Ticlopidine blocks several NTPDase isoenzymes with IC50s of 170 μM and 149 μM for NTPDase2 and NTPDase3, respectively[1]. Ticlopidine is an inhibitor of CYP2C19 human liver cytochrome. Ticlopidine inhibits CYP2C9 and CYP3A4 with IC50s of 26.0 and 32.3 μM, respectively[2][3].
Mitotane- 13C6 is the 13C labeled Mitotane[1]. Mitotane (2,4′-DDD), an isomer of DDD and derivative of dichlorodiphenyltrichloroethane (DDT), is an antineoplastic agent, can be used to research adrenocortical carcinoma. Mitotane exert its adrenocorticolytic effect at least in part through lipotoxicity induced by intracellular free cholesterol (FC) accumulation. Mitotane can have direct pituitary effects on corticotroph cells. Mitotane can induce CYP3A4 gene expression via steroid and xenobiotic receptor (SXR) activation, and has agent-agent interactions[2][3][4][5].
Mitotane-d8 is the deuterium labeled Mitotane[1]. Mitotane (2,4′-DDD), an isomer of DDD and derivative of dichlorodiphenyltrichloroethane (DDT), is an antineoplastic agent, can be used to research adrenocortical carcinoma. Mitotane exert its adrenocorticolytic effect at least in part through lipotoxicity induced by intracellular free cholesterol (FC) accumulation. Mitotane can have direct pituitary effects on corticotroph cells. Mitotane can induce CYP3A4 gene expression via steroid and xenobiotic receptor (SXR) activation, and has agent-agent interactions[2][3][4][5].