Search Result
Results for "
Selective inhibitor of cell cycle
" in MedChemExpress (MCE) Product Catalog:
1
Biochemical Assay Reagents
3
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-50767
-
|
PD 0332991
|
CDK
|
Cancer
|
|
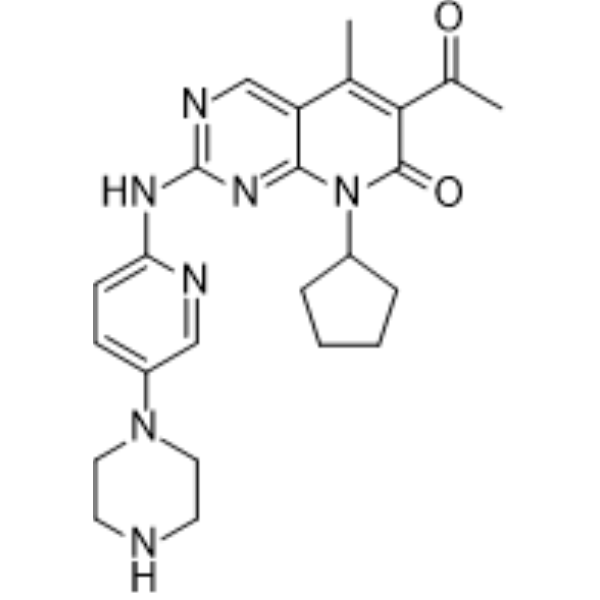
Palbociclib (PD 0332991) is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-13902
-
|
VE-822; VX-970; M6620
|
ATM/ATR
Apoptosis
STING
Caspase
|
Infection
Metabolic Disease
Cancer
|
|
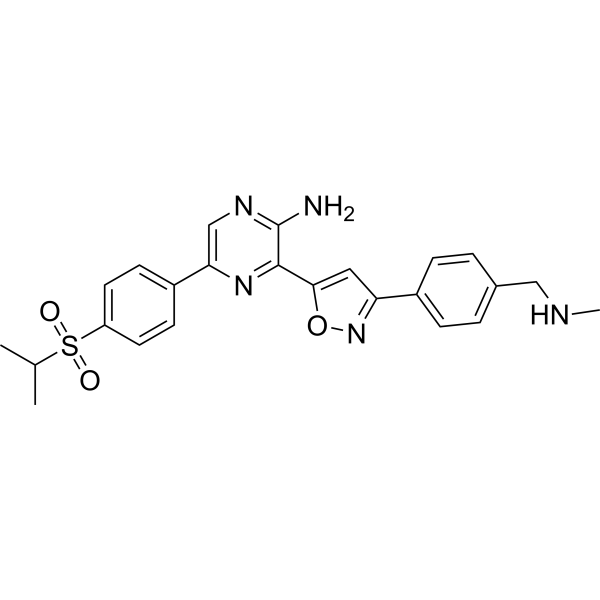
Berzosertib (VE-822) is an orally active, CNS-penetrant, and selective ATR kinase inhibitor. Berzosertib blocks ATR kinase activity, abrogates G2/M cell cycle checkpoint, impairs DNA damage repair. Berzosertib induces apoptosis, inhibnits conlony migration, inhibits cell proliferation, and activates cGAS-STING axes in cancer cells. Berzosertib can be used for the research of cancers, such as head and neck squamous cell carcinoma, and colorectal cancer .
|
-
-
- HY-10521
-
|
SB-480848
|
Phospholipase
Apoptosis
|
Cardiovascular Disease
Cancer
|
|
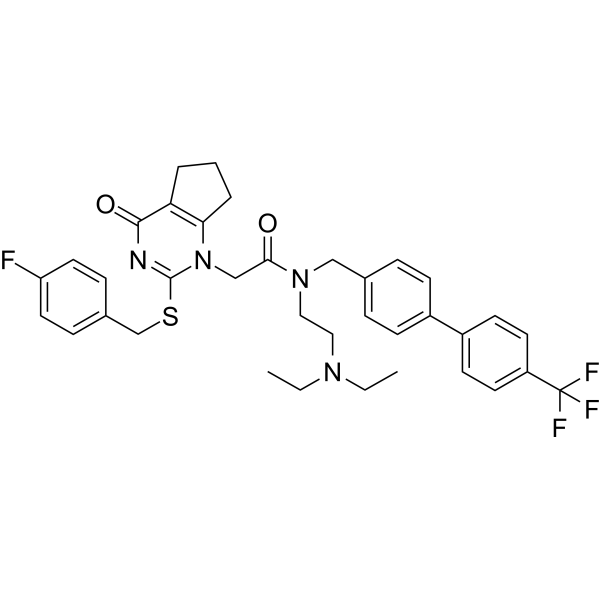
Darapladib (SB-480848) is an orally active, selective and reversible Lp-PLA2 inhibitor (IC50=0.25 nM). Darapladib can trigger irreversible actions on glioma cell apoptosis and induce cycle arrest. Darapladib can be used in the study of atherosclerosis and cancer .
|
-
-
- HY-16594
-
|
|
Proteasome
Cathepsin
Apoptosis
|
Cardiovascular Disease
Infection
Neurological Disease
Cancer
|
|
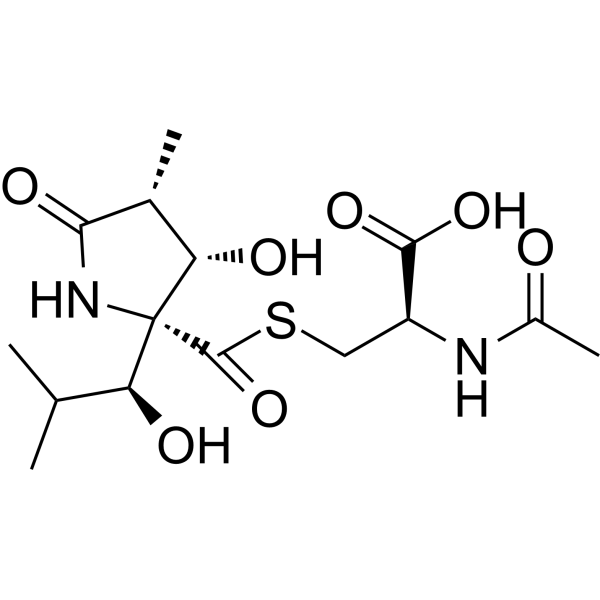
Lactacystin is a potent, orally active, irreversible, cell-permeable, selective 20S proteasome inhibitor (IC50 = 4.8 μM). Lactacystin also inhibits the lysosomal enzyme cathepsin A. Lactacystin inhibits cell growth and induces apoptosisand cell cycle arrest, and has antiviral and antioxidative activity. Lactacystin induces neurite outgrowth and hypertension. Lactacystin has the potential for the research of cancer, Neurological Disease, hypertension and Malaria, and so on [2] [6] .
|
-
-
- HY-50767C
-
|
PD-0332991 hydrochloride
|
CDK
|
Cancer
|
|
Palbociclib (PD 0332991) hydrochloride is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib hydrochloride has potent anti-proliferative activity and induces cell cycle arrest in cancer cells. Palbociclib hydrochloride can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-50767A
-
|
PD 0332991 monohydrochloride
|
CDK
|
Cancer
|
|
Palbociclib (PD 0332991) monohydrochloride is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib monohydrochloride has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-A0065
-
|
PD 0332991 isethionate
|
CDK
|
Infection
Neurological Disease
Cancer
|
|
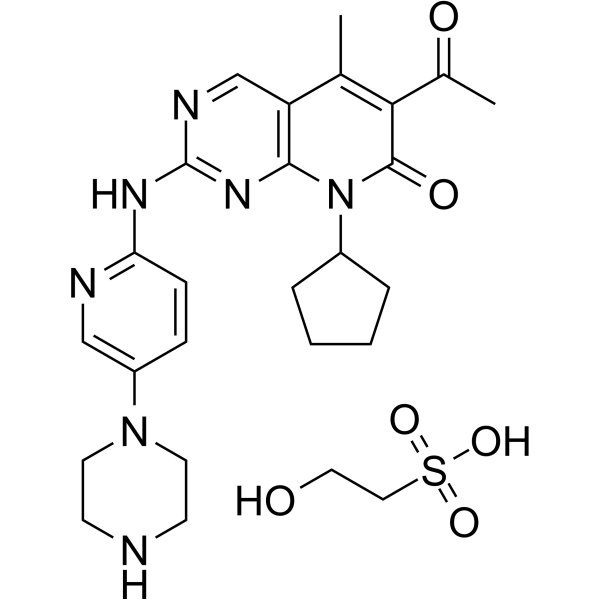
Palbociclib (PD 0332991) isethionate is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib isethionate has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-13630
-
|
BMY-40481
|
Topoisomerase
Bacterial
Autophagy
Apoptosis
|
Infection
Neurological Disease
Cancer
|
|
Etoposide phosphate (BMY-40481) is a potent anti-cancer chemotherapy agent and a selective topoisomerase II inhibitor to prevent re-ligation of DNA strands. Etoposide phosphate is the phosphate ester proagent of etoposide and is considered as active equivalent to Etoposide. Etoposide phosphate induces cell cycle arrest, apoptosis, and autophagy.
|
-
-
- HY-162001
-
|
|
CDK
|
Cancer
|
|
INX-315 is an orally active and selective CDK2 inhibitor that induces cell cycle arrest in the G1 phase. INX-315 reduces CDK2 substrate phosphorylation and inhibits tumor growth in a dose-dependent manner in xenograft mouse models. INX-315 may be used in cancer research .
|
-
-
- HY-B0990
-
|
|
Bacterial
Antibiotic
YAP
|
Infection
|
|
Thiostrepton is a thiazole antibiotic which selectively inhibits FOXM1. FOXM1 binds to YAP/TEAD complex. YAP/TEAD/FOXM1 complex binding at regulatory regions of genes governing cell cycle may impact cell proliferation .
|
-
-
- HY-12037A
-
|
ON-01910
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib (ON-01910) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3 kinase/Akt pathway, promots the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
-
- HY-158106
-
AZD8421
1 Publications Verification
|
CDK
|
Cancer
|
|
AZD8421 is a selective CDK2 inhibitor (IC50 = 9 nM) as well as achieving CDK family selectivity in cells versus key off-targets (CDK1, CDK4/6, CDK9), AZD8421 had no significant kinase inhibition outside the CDK family. AZD8421 inhibits cancer cell proliferation by inhibiting pRB phosphorylation, inducing cell cycle arrest in G1/S phase and senescence. AZD8421 can be studied in research for breast cancer and ovarian cancer .
|
-
-
- HY-100888
-
|
TAK-931
|
CDK
|
Cancer
|
|
Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM. Simurosertib has anti-cancer activity .
|
-
-
- HY-107407
-
|
|
Checkpoint Kinase (Chk)
CDK
PKC
Apoptosis
|
Cancer
|
|
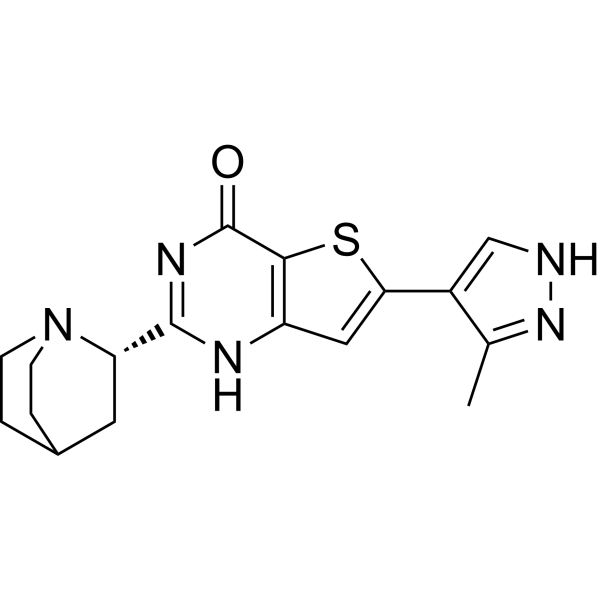
SB-218078 is a potent, selective, ATP-competitive and cell-permeable checkpoint kinase 1 (Chk1) inhibitor that inhibits Chk1 phosphorylation of cdc25C with an IC50 of 15 nM. SB-218078 is less potently inhibits Cdc2 (IC50 of 250 nM) and PKC (IC50 of 1000 nM). SB-218078 causes apoptosis by DNA damage and cell cycle arrest .
|
-
-
- HY-15728
-
|
IY-5511
|
Bcr-Abl
Apoptosis
STAT
JAK
Prion Protein
|
Infection
Neurological Disease
Cancer
|
|
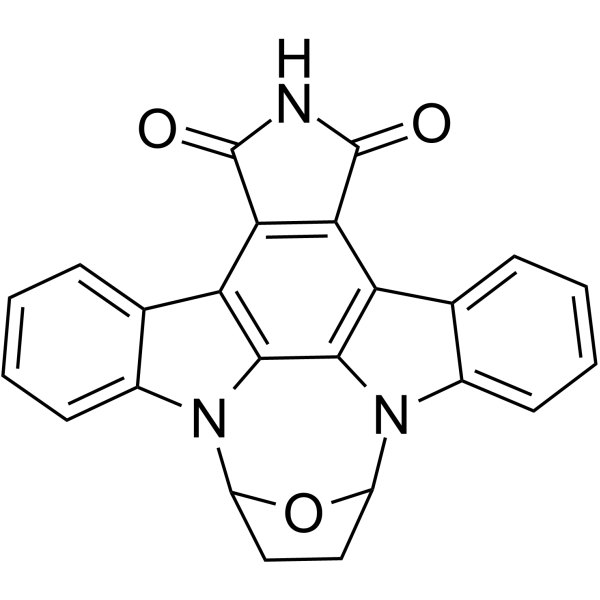
Radotinib (IY-5511) is an orally active and BBB-permeable selective tyrosine kinase Bcr-Abl1 inhibitor with an IC50 of 34 nM. Radotinib has anti-prion and anti-tumor activities. Radotinib can inhibit the proliferation, induce cell cycle arrest and apoptosis of tumor cells . Radotinib can be used in the research of cancer such as chronic myeloid leukemia and multiple myeloma, as well as neurodegenerative diseases such as prion diseases .
|
-
-
- HY-15611
-
|
|
CRM1
|
Cancer
|
|
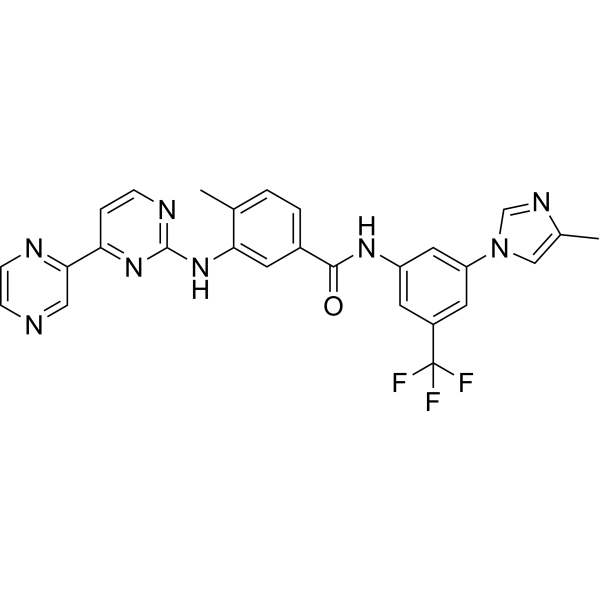
KPT-185 is an orally bioavailable and selective inhibitor of CRM1 and displays potent antiproliferative properties at submicromolar concentrations (IC50=100-500 nM), induces apoptosis, cell-cycle arrest, and myeloid differentiation in AML cell lines and patient blasts .
|
-
-
- HY-12246
-
XEN445
1 Publications Verification
|
Lipase
|
Cardiovascular Disease
Metabolic Disease
Cancer
|
|
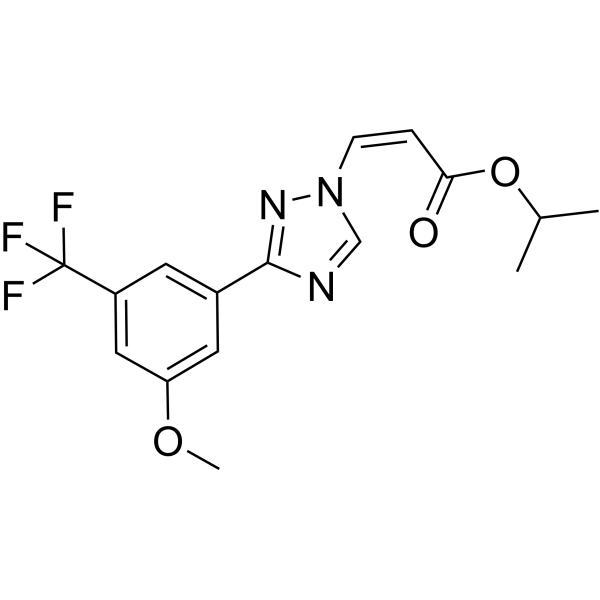
XEN445 is a potent, selective and orally active endothelial lipase (EL) inhibitor with an IC50 value of 0.237 μM. XEN445 selectively inhibits phospholipase enzymatic activity of LIPG. XEN445 raises plasma HDL and cholesterol levles. XEN445 induces G1 cell cycle arrest, reduces cell viability, suppresses cancer stem cell self-renewal, and inhibits tumor formation in LIPG-expressing triple-negative breast cancer cells, while showing no inhibitory effect on invasiveness or cancer stem cell stemness in these cells. XEN445 can be used for the research of cancer and metabolic disease, such as triple-negative breast cancer .
|
-
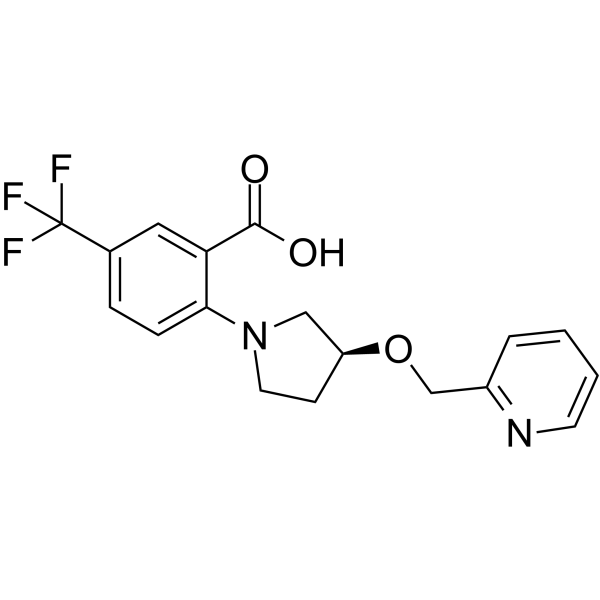
-
- HY-137497
-
|
|
Ras
Apoptosis
|
Cancer
|
|
KRAS inhibitor-9, a potent KRAS inhibitor (Kd=92 μM), blocks the formation of GTP-KRAS and downstream activation of KRAS. KRAS inhibitor-9 binds to KRAS G12D, KRAS G12C and KRAS Q61H protein with a moderate binding affinity. KRAS inhibitor-9 causes G2/M cell cycle arrest and induces apoptosis. KRAS inhibitor-9 selectively inhibits the proliferation of NSCLC cells with KRAS mutation but not normal lung cells .
|
-
-
- HY-112158
-
|
ERGi-USU
|
RIO Kinase
|
Cancer
|
|
NSC139021 (ERGi-USU) is a RIOK2 inhibitor with anticancer activity. RIOK2 can highly selectively inhibit the growth of ERG-positive cancer cells with IC50s of 30-400 nM against cell lines. RIOK2 also causes cell cycle arrest and apoptosis in glioblastoma via induction of Skp2 and Skp2-p27/p21-Cyclin E/CDK2-pRb signaling .
|
-
-
- HY-159480
-
|
|
Src
ATM/ATR
Apoptosis
mTOR
AMPK
|
Cancer
|
|
AD1058 is an orally active, selective, and BBB-permeable inhibitor of ATR (IC50: 1.6 nM). AD1058 exhibits anticancer activity by inhibiting tumor cell proliferation, inducing cell cycle arrest, and promoting apoptosis. AD1058 is suitable for research on advanced malignancies and brain metastases .
|
-

-
- HY-150109
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
Purinostat mesylate is a selective inhibitor of HDAC. Purinostat mesylate inhibits class I and class IIb HDACs with IC50s from 0.81 to 11.5 nM. Purinostat mesylate induces apoptosis and affects cell cycle of LAMA84 and 188 BL-2 cells, and shows potently anti-leukemia effects in vivo. Purinostat mesylate can be used for the research of lymphoblastic leukemia .
|
-
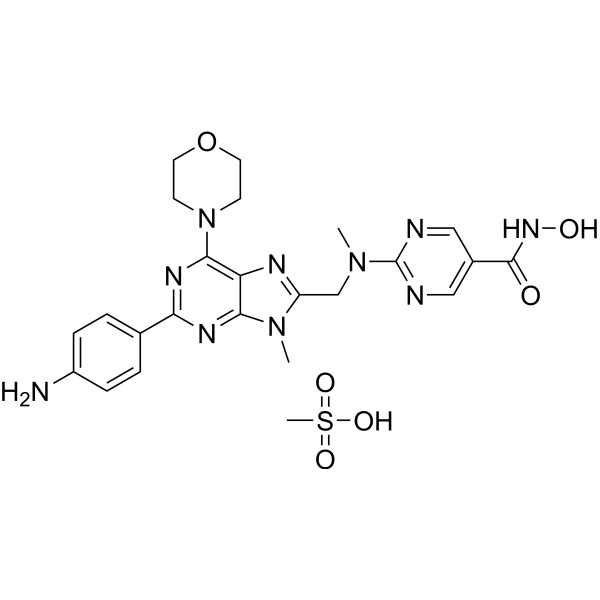
-
- HY-12037
-
|
ON-01910 sodium
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib sodium (ON-01910 sodium) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3K/Akt pathway, promotes the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib sodium is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
-
- HY-15485
-
|
|
Phosphodiesterase (PDE)
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
Zardaverine is an orally active and selective PDE3/4 inhibitor (IC50)=0.58 uM/0.17 uM) with potent bronchodilator activity. Zardaverine also selectively inhibits the proliferation of HCC cells and induces apoptosis and cycle arrest (G0/G1 phase). Zardaverine has good antitumor potential and is effective in both bronchial relaxation and reduction of inflammation in asthma .
|
-
-
- HY-132846
-
|
TAS0953/HM06
|
RET
Apoptosis
|
Cancer
|
|
Vepafestinib (TAS0953/HM06) is a next-generation brain-penetrant, selective and orally active RET inhibitor with an IC50 value of 0.33 nM. Vepafestinib inhibits the phosphorylation of RET and its downstream signaling pathways, thus blocking the growth and signal transduction of tumor cells and inducing cell cycle arrest and apoptosis. Vepafestinib can be used in the research of various RET-driven cancers, such as non-small cell lung cancer, thyroid cancer and other disease areas .
|
-
-
- HY-174979
-
|
|
Fat Mass and Obesity-associated Protein (FTO)
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
Dac590 is an orally active and selective obesity-associated protein (FTO) inhibitor with an IC50 of 6.06 nM. Dac590 shows highly selective over ALKBH5 and ALKBH3. Dac590 suppresses oncogenic FTO signaling, induces myeloid differentiation, G1-phase cell cycle arrest, and apoptosis in acute myeloid leukemia (AML) cells. Dac590 inhibits xenograft tumor growth and prolongs survival in acute myeloid leukemia mouse models with no observed toxicity. Dac590 can be used for the research of AML .
|
-

-
- HY-147868
-
|
|
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
DC-CPin711 is a potent and selective inhibitor of CREB-binding protein (CBP) bromodomain with an IC50 of 0.0626 μM. DC-CPin711 arrests cell cycle at G1 phase and induces apoptosis .
|
-
-
- HY-176142
-
|
|
CDK
c-Myc
Apoptosis
Bcl-2 Family
|
Cancer
|
|
YX0798 is a selective and orally active CDK9 inhibitor (Kd: 0.28 nM). YX0798 downregulates the oncoprotein c-MYC and pro-survival protein MCL-1. YX0798 disrupts the cell cycle and results in transcriptomic reprogramming, eventually leading to cell apoptosis. YX0798 has antitumor activity .
|
-

-
- HY-115565
-
|
|
CDK
Apoptosis
|
Cancer
|
|
CGP-74514 (Compound 13) is a highly selective cyclin-dependent kinase 1 (CDK1) inhibitor (IC50=25 nM). CGP-74514 inhibits CDK1/cyclin B complex activity, arrests the cell cycle at G2/M phase and induces tumor cell apoptosis. CGP-74514 is promising for research of bladder cancer .
|
-

-
- HY-157396
-
|
|
Aurora Kinase
Apoptosis
Caspase
|
Cancer
|
|
JAB-2485 is an orally active and selective Aurora kinase A (AURKA) inhibitor with an IC50 value of 0.327 nM. JAB-2485 exhibits inhibitory activity against various tumor cell lines such as neuroblastoma, triple-negative breast cancer, small cell lung cancer, and epithelial ovarian cancer. JAB-2485 can induce cell cycle arrest and apoptosis in tumor cells. JAB-2485 has antitumor activity .
|
-
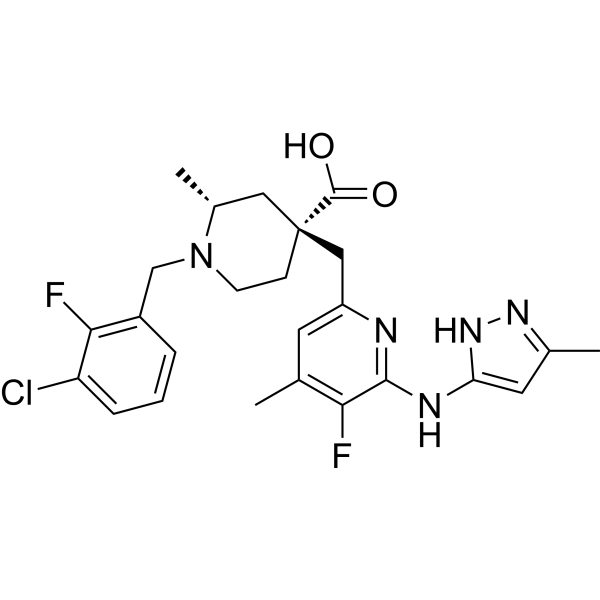
-
- HY-115452
-
|
|
JAK
Apoptosis
|
Cancer
|
|
G5-7, an orally active and allosteric JAK2 inhibitor, selectively inhibits JAK2 mediated phosphorylation and activation of EGFR (Tyr 1068) and STAT3 by binding to JAK2. G5-7 induces cell cycle arrest, apoptosis and possesses antiangiogenic effect. G5-7 has the potential for glioma study .
|
-
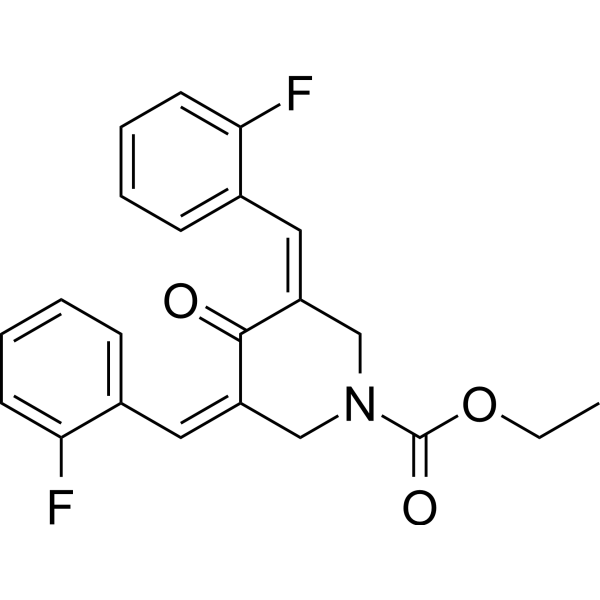
-
- HY-162471
-
|
|
DNA/RNA Synthesis
ATM/ATR
|
Cancer
|
|
GSK_WRN3 is selective WRN helicase inhibitor (pIC50 = 8.6). SK_WRN3 can selectively inhibit the growth of microsatellite unstable (MSI) cancer cells, induce DNA damage, and cause cell cycle arrest. GSK_WRN3 has anti-tumor activity .
|
-
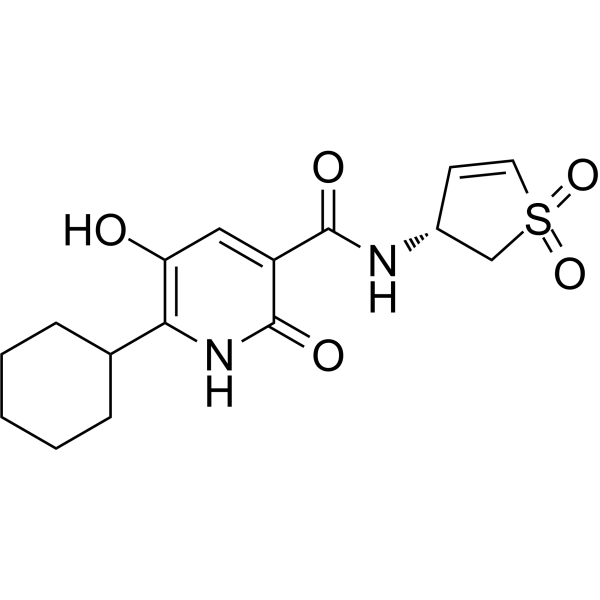
-
- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-
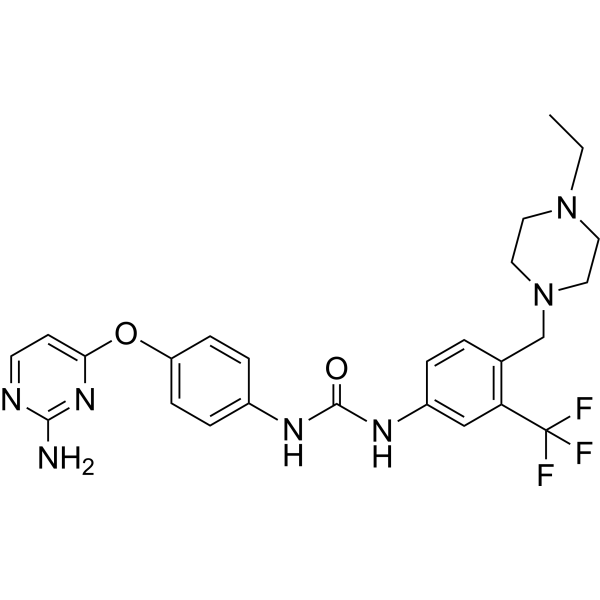
-
- HY-100888A
-
|
(R)-TAK-931
|
CDK
|
Others
|
|
(R)-Simurosertib ((R)-TAK-931) is the (R)-enantiomer of Simurosertib. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM .
|
-
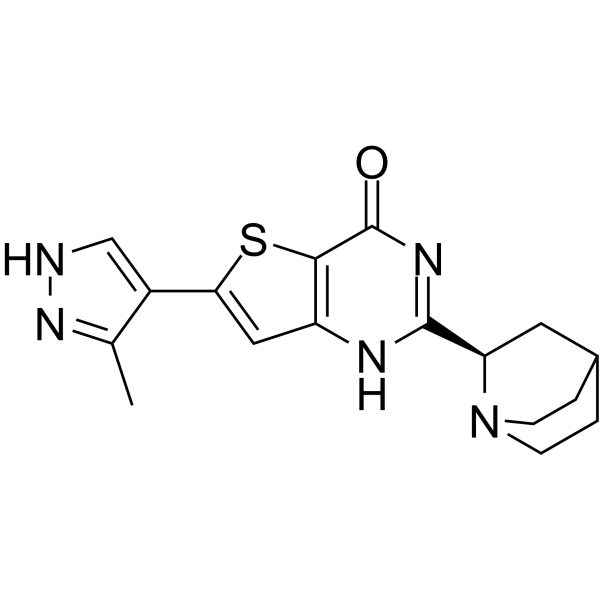
-
- HY-156278
-
|
|
Apoptosis
Autophagy
|
Cancer
|
|
FB49 is a highly selective inhibitor of Bcl-2-associated athanogene 3 (BAG3), with the Ki of 45 μM. FB49 inhibits the cell growth in human tumoral cells, but has no toxicity in human peripheral mononuclear cells. FB49 block cell cycle in G1 phase and to induce apoptosis as well as autophagy in medulloblastoma HD-MB03 treated cells .
|
-
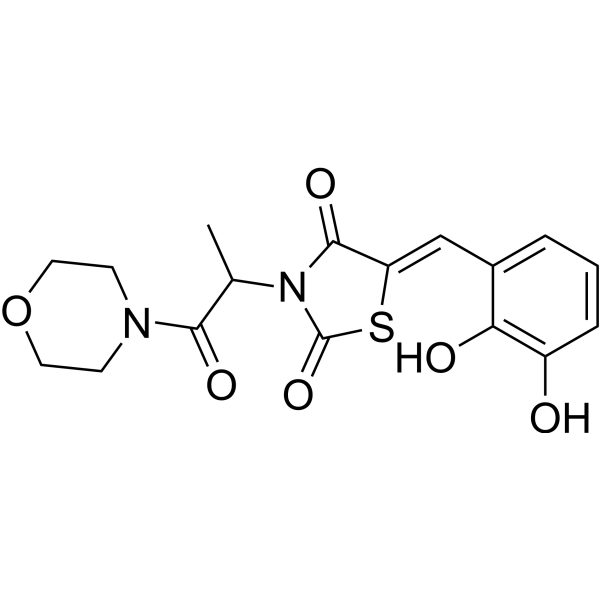
-
- HY-10815
-
|
|
Sigma Receptor
Apoptosis
|
Neurological Disease
Cancer
|
|
σ1 Receptor antagonist-1 is a highly potent and selective sigma 1 receptor antagonist (pKi=10.28). σ1 Receptor antagonist-1 inhibits cell growth, arrests cell cycle at G0/G1 phase and induces apoptosis of MCF-7/ADR cells .
|
-
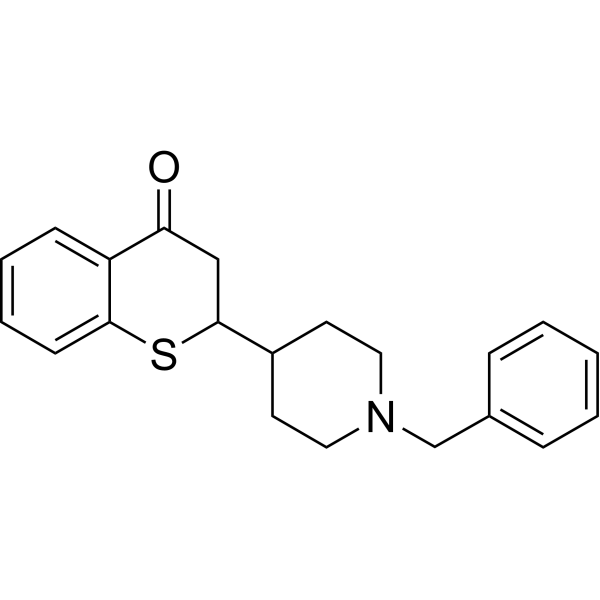
-
- HY-149912
-
|
|
Polo-like Kinase (PLK)
Trk Receptor
Apoptosis
|
Cancer
|
|
CZS-241 is an orally active and selective inhibitor of Polo-like Kinase (PLK) 4 (IC50=2.6 nM). CZS-241 inhibits TRKA with an IC50 value of 2.74 μM. CZS-241 induces apoptosis and arrests cell cycle at S/G2 phase. CZS-241 shows highly potent antiproliferative activity against leukemia cell lines, and exhibits safety against normal cell lines .
|
-
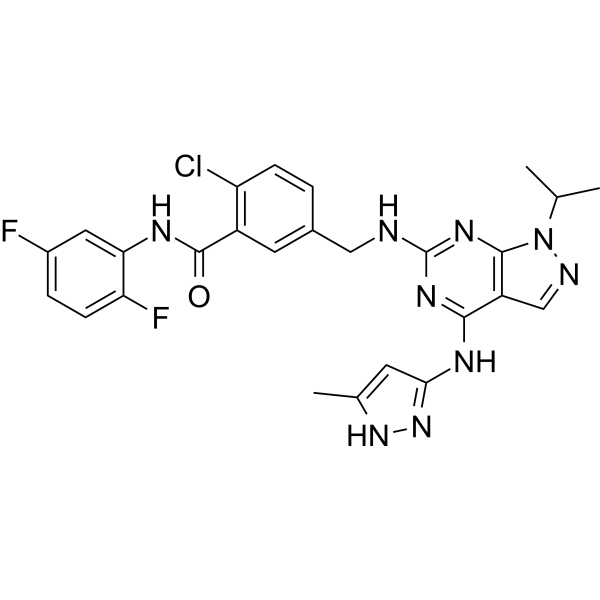
-
- HY-117102
-
|
|
Aryl Hydrocarbon Receptor
Checkpoint Kinase (Chk)
|
Cancer
|
|
ANI-7 is an activator of aryl hydrocarbon receptor (AhR) pathway. ANI-7 inhibits the growth of multiple cancer cells, and potently and selectively inhibits the growth of MCF-7 breast cancer cells with a GI50 of 0.56 μM. ANI-7 induces CYP1-metabolizing mono-oxygenases by activating AhR pathway, and also induces DNA damage, checkpoint Kinase 2 (Chk2) activation, S-phase cell cycle arrest, and cell death in sensitive breast cancer cell lines .
|
-
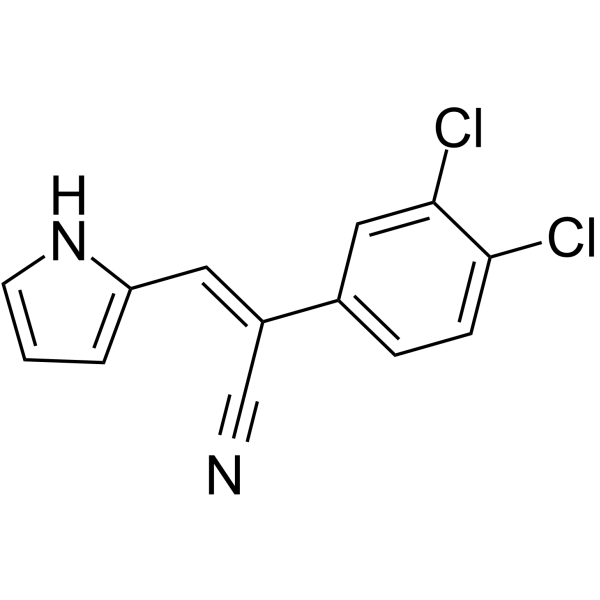
-
- HY-149075
-
|
|
Salt-inducible Kinase (SIK)
|
Cancer
|
|
MR22 is a potent pan-SIK (salt-inducible kinase) inhibitor. MR22 no longer exhibits activity on STE group kinases and displays excellent selectivity in a representative kinase panel. MR22-dependent SIK inhibition led to centrosome dissociation and subsequent cell-cycle arrest in ovarian cancer cells .
|
-
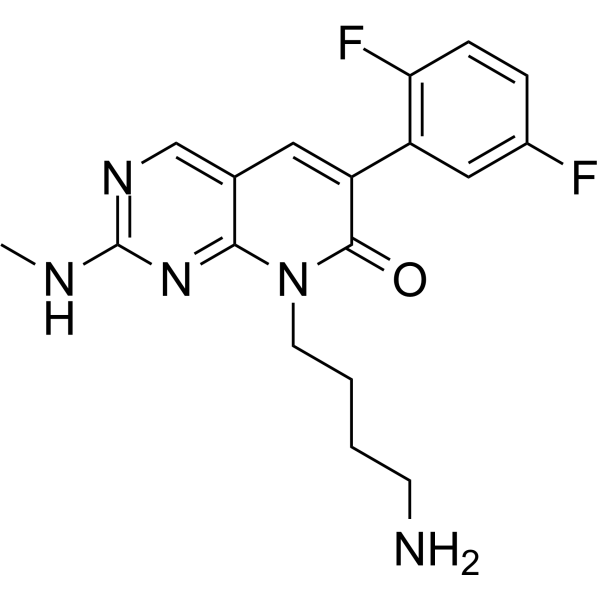
-
- HY-117884
-
|
|
Apoptosis
|
Cancer
|
|
(Rac)-CCT 250863 (compound rac-21) is a selective and reversible NEK 2 inhibitor with an IC50 of 0.073 µM. (Rac)-CCT 250863 shows good effects of inducing cell cycle arrest and also can antiproliferative in cells (Pomalidomide sensitive/resistant). (Rac)-CCT 250863 induces apoptosis when combines with Pomalidomide .
|
-
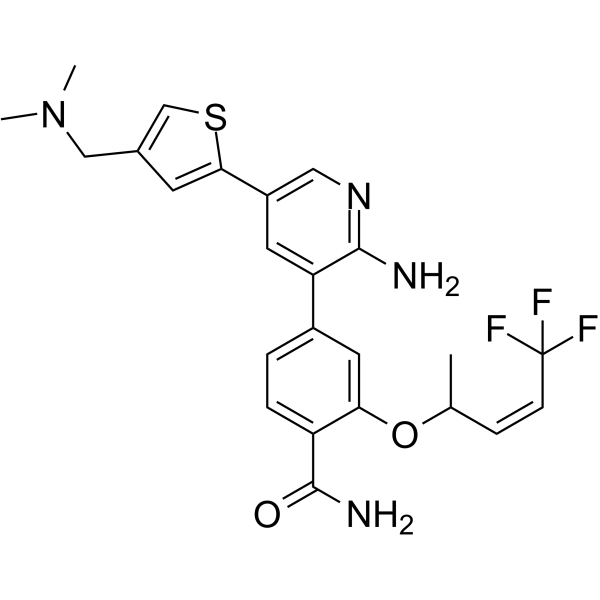
-
- HY-175785
-
|
|
Estrogen Receptor/ERR
Aryl Hydrocarbon Receptor
Apoptosis
MDM-2/p53
|
Cancer
|
|
X15695 is selective and orally active estrogen receptor (ERα) degrader. X15695 is an aryl hydrocarbon receptor (AHR) ligand. X15695 enables AHR to form a complex with the ERα, promoting its proteasomal degradation. X15695 inhibits the breast cancer cells proliferation, promotes cell cycle block and induces apoptosis. X15695 can be used for the study of breast cancer .
|
-

-
- HY-18969
-
|
|
CDK
|
Cancer
|
|
LDC3140 is a selective CDK7 inhibitor (IC50<5 nM). By inhibiting the activity of CDK7, LDC3140 affects the regulation of the cell cycle, leading to cell cycle arrest and thus inhibiting the proliferation of tumor cells. LDC3140 can be used in the research of cancer treatment .
|
-

-
- HY-18952A
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
(Z)-SU5614 is a potent FLT3 inhibitor and selectively induces growth arrest, apoptosis, and cell cycle arrest in Ba/F3 and AML cell lines expressing a constitutively activated FLT3 .
|
-
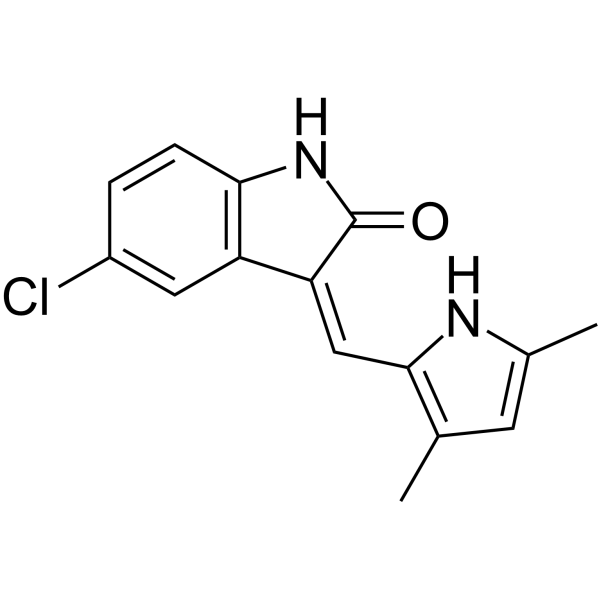
-
- HY-158143
-
|
|
Histone Methyltransferase
Apoptosis
|
Cancer
|
|
AZD3470 is an orally active MTA-cooperative PRMT5 inhibitor, selective for MTAP-deficient tumors. AZD3470 induces cell cycle G2/M phase alterations, DNA damage, apoptosis, and symmetric dimethylarginine reduction. AZD3470 alters alternative splicing, increases skipped exon events in DNA repair and cell cycle pathways, and inhibits cancer cell proliferation and tumor growth. AZD3470 can be used for the research of non-small cell lung cancer and MTAP-deleted solid tumors .
|
-

-
- HY-116861
-
|
|
MetAP
|
Cancer
|
|
A-357300 is a reversible and selective MetAP2 inhibitor with IC50s of 0.12 and 57 μM against MetAP2 and MetAP1. A-357300 induces cytostasis by cell cycle arrest at the G1 phase selectively in endothelial cells and in a subset of tumor cells. A-357300 inhibits angiogenesis both in vitro and in vivo and shows potent antitumor efficacy in carcinoma, sarcoma, and neuroblastoma murine models. A-357300 can be used for the studies of neuroblastoma, fibrosarcoma and breast cancer .
|
-

-
- HY-118976
-
|
CGP-74514A
|
CDK
Apoptosis
|
Cancer
|
|
CGP-74514 hydrochloride is a highly selective cyclin-dependent kinase 1 (CDK1) inhibitor (IC50: 25 nM). CGP-74514 hydrochloride inhibits CDK1/cyclin B complex activity, arrests the cell cycle at G2/M phase and induces tumor cell apoptosis. CGP-74514 hydrochloride is promising for research of bladder cancer .
|
-

-
- HY-13630A
-
|
BMY-40481 disodium
|
Topoisomerase
Autophagy
Apoptosis
|
Neurological Disease
Cancer
|
|
Etoposide phosphate disodium (BMY-40481 disodium) is a potent anti-cancer chemotherapy agent and a selective topoisomerase II inhibitor to prevent re-ligation of DNA strands. Etoposide phosphate disodium is the phosphate ester proagent of etoposide and is considered as active equivalent to Etoposide. Etoposide phosphate disodium induces cell cycle arrest, apoptosis, and autophagy.
|
-
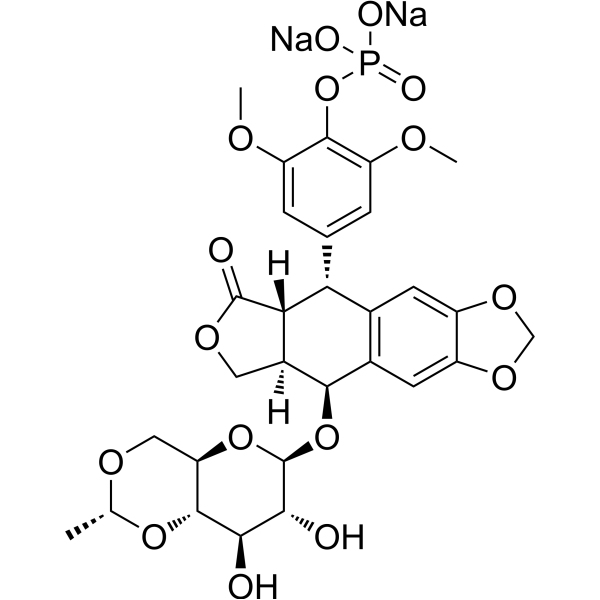
-
- HY-150636
-
|
|
Autophagy
Apoptosis
|
Cancer
|
|
Autophagy-IN-1 is a potent autophagy/mitophagy inhibitor, acts by selectively increasing the autophagic flux while blocking the autophagosome-lysosome fusion in cancer cells. Autophagy-IN-1 can induce apoptosis and cell cycle arrest. Autophagy-IN-1 significantly inhibits tumor growth in an HCT116 xenograft mouse model and with low toxicity. Autophagy-IN-1 can be used for researching colorectal cancer .
|
-
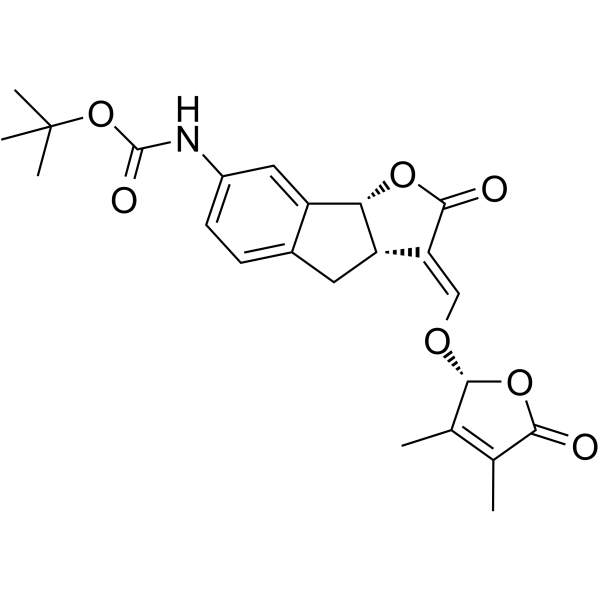
-
- HY-146812
-
|
|
G-quadruplex
|
Cancer
|
|
DIZ-3 is a selective multimeric G4 ligand based on a G4-ligand-dimerizing strategy. DIZ-3 intercalates into the G4-G4 interface, stabilizing the higher-order structure. DIZ-3 induces cell cycle arrest and apoptosis, and thus inhibits cell proliferation in alternative lengthening of telomere (ALT) cancer cells .
|
-
-
- HY-50767B
-
|
PD-0332991 dihydrochloride
|
CDK
|
Cancer
|
|
Palbociclib (PD 0332991) dihydrochloride is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib dihydrochloride has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-179406
-
|
|
PARP
DNA/RNA Synthesis
Reactive Oxygen Species (ROS)
Apoptosis
|
Cancer
|
|
PARP1-IN-49 is a selective PARP1 inhibitor with an IC50 of 23.56 nM and a Kd of 17.78 nM. PARP1-IN-49 shows a selectivity for PARP1 over PARP2. PARP1-IN-49 leads to the induction of DNA damage, cell cycle arrest, and apoptosis. PARP1-IN-49 also increases intracellular ROS levels and inhibits cell migration. PARP1-IN-49 can be used for the research of breast cancer and ovarian cancer .
|
-

- HY-14661
-
|
|
Kinesin
|
Cancer
|
|
SB-743921 free base is a potent selective inhibitor of the mitotic kinesin KSP (Eg5), with a Ki of 0.1 nM. SB-743921 free base can induce mitotic arrest, block cell cycle progression, induce apoptosis, and can be used in the research of myeloma, leukemia and other diseases .
|
-
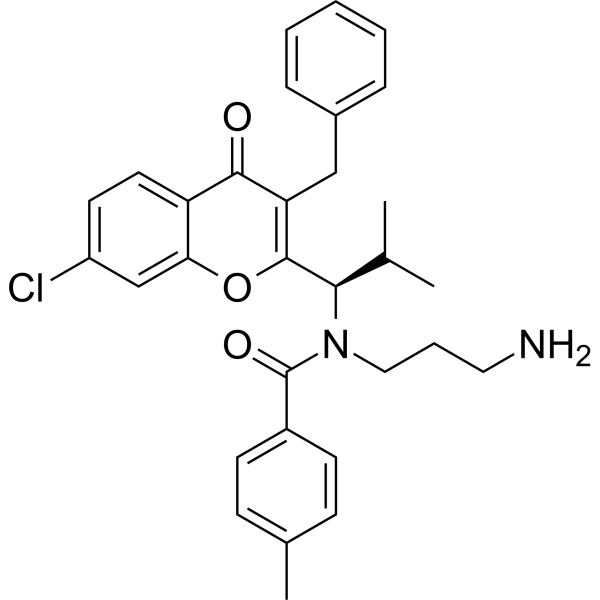
- HY-149024
-
|
|
VEGFR
Apoptosis
MDM-2/p53
|
Cancer
|
|
VEGFR-2-IN-23 (compound 11b) is a potent and selective VEGFR-2 inhibitor with an IC50 value of 0.34 nM. VEGFR-2-IN-23 shows antitumor activity. VEGFR-2-IN-23 induces apoptosis and cell cycle arrest at G1 phase .
|
-
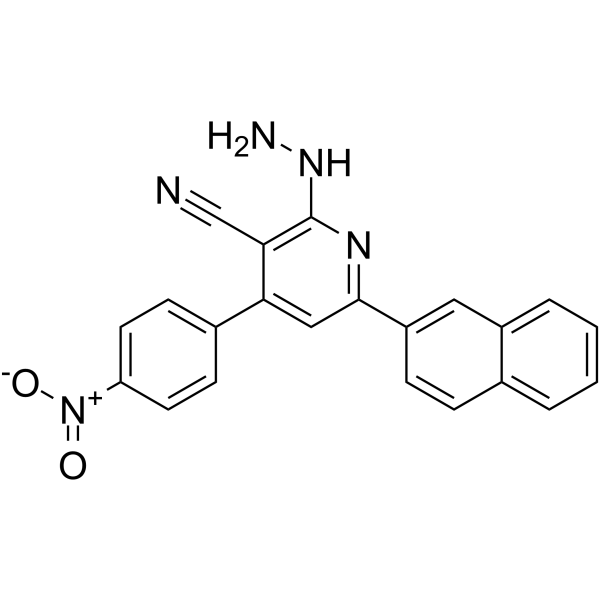
- HY-146729
-
|
|
ASK1
MAP3K
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
ASK1-IN-3 is a potent and selective ASK1 kinase inhibitor with IC50 of 33.8 nM, as well as inhibits several cell cycle regulating kinases. ASK1-IN-3 has strong HepG2 cancer cells apoptosis induction and potent cell cycle arrest activities .
|
-
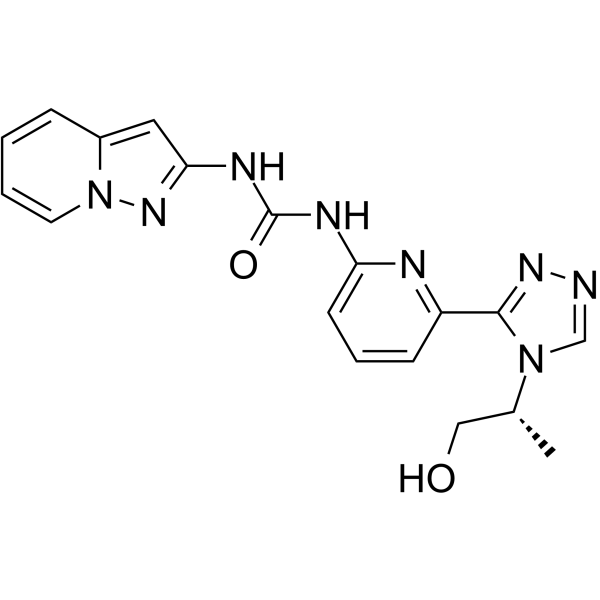
- HY-112314
-
|
|
Src
Bcr-Abl
Apoptosis
|
Cancer
|
|
AZD0424 is an orally active, and dual selective Src/Abl kinase inhibitor with potential antineoplastic activity . AZD0424 induces apoptosis and cell cycle arrest in lymphoma cells .
|
-
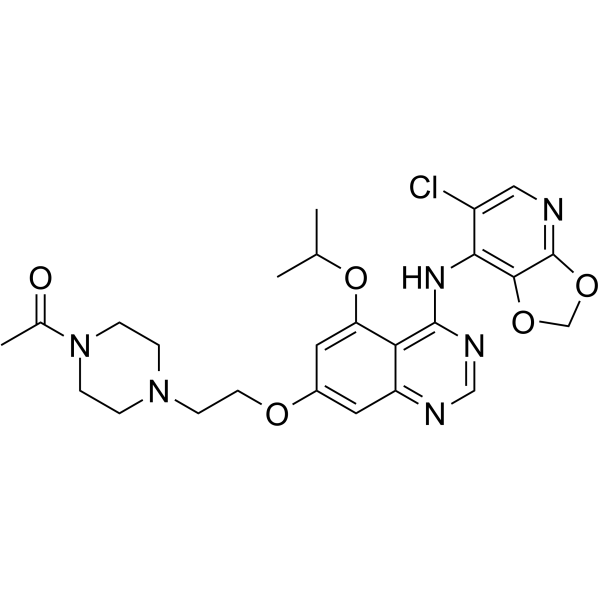
- HY-151905
-
|
|
c-Met/HGFR
|
Cancer
|
|
D6808 is a highly selective and potent c‑Met inhibitor with an IC50 value of 2.9 nM. D6808 induces cell apoptosis and cell cycle arrest. D6808 can be used for the research of NSCLC and gastric cancers .
|
-
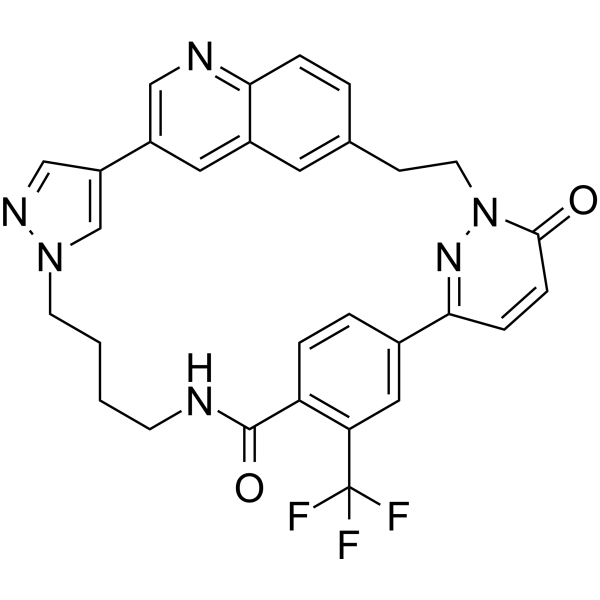
- HY-168263
-
|
|
Histone Methyltransferase
Apoptosis
|
Cancer
|
|
PRMT5-IN-45 (compound 36) is a potent and selective PRMT5 inhibitor with an IC50 of 3 nM. PRMT5-IN-45 potently reduces the level of symmetric dimethylarginines (sDMA) and inhibits the proliferation of MOLM-13 cell lines by inducing apoptosis and cell cycle arrest .
|
-

- HY-137135
-
|
|
Phosphatase
Apoptosis
ERK
p38 MAPK
JNK
|
Cancer
|
|
Cantharidic acid is a selective inhibitor for protein phosphatase 2 (PP2A) and protein phosphatase 1 (PP1). Cantharidic acid inhibits cell viability and arrest cell cycle at sub G1 phase, induces apoptosis in cells NPC-39 and HONE-1 through the upregulation of ERK1/2, p38, and JNK1/2 pathway .
|
-

- HY-125257
-
|
LY6
|
Phosphatase
SHP2
ERK
Akt
STAT
JAK
Apoptosis
|
Cancer
|
|
SHP2 inhibitor LY6 (LY6) is a selective SHP2 inhibitor with an IC50 of 9.8 μM, showing 7-fold selectivity over SHP1. SHP2 inhibitor LY6 inhibits SHP2-mediated cell signaling pathways and suppresses cell proliferation. SHP2 inhibitor LY6 elicits induces apoptosis and G2/M cell cycle arrest in lung cancer cells. SHP2 inhibitor LY6 can be used for the research of B cell acute lymphoblastic leukemia, acute myeloid leukemia, and lung cancer .
|
-
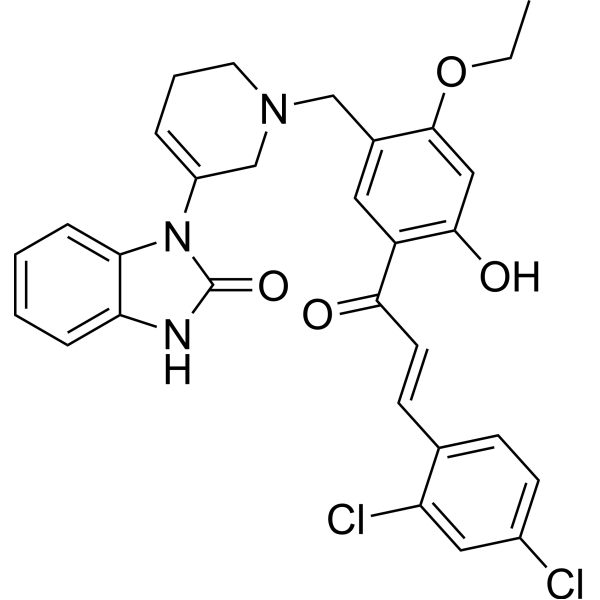
- HY-151407
-
|
|
CDK
|
Cancer
|
|
CDK1-IN-3 (8g) is a selective CDK1 inhibitor with IC50s of 36.8, 305.17 and 369.37 nM for CDK1, CDK2 and CDK5, respectively. CDK1-IN-3 inhibits the growth of cancer cells by affecting cell cycle. CDK1-IN-3 can be used for the research of cancer .
|
-
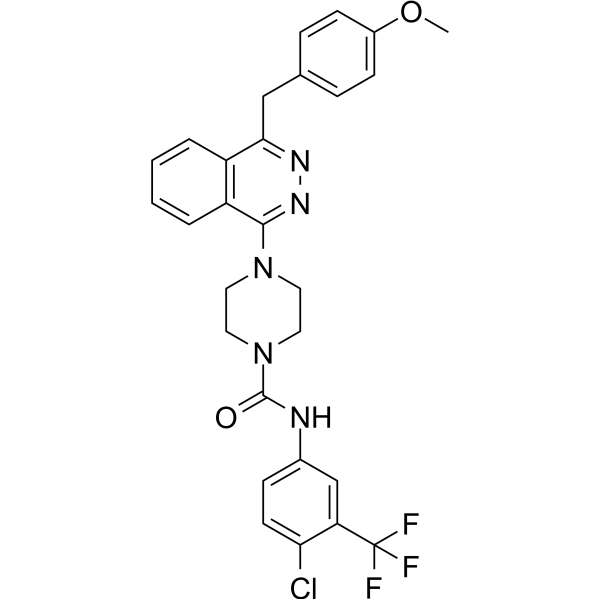
- HY-15882
-
|
|
Endogenous Metabolite
|
Cancer
|
|
GNE-783 is a selective CHK1 inhibitor that enhances the activity of gemcitabine. GNE-783 improves the efficacy of anti-metabolite DNA damage drugs by inactivating S-phase and G2-phase cell cycle checkpoints following DNA damage. GNE-783 selectively enhances the chemical synergy of certain drugs in different tumor types, for example, enhancing the activity of temozolomide only in melanoma cell lines .
|
-

- HY-110359
-
|
|
CDK
Apoptosis
|
Cancer
|
|
CGP-74514 dihydrochloride is a highly selective cyclin-dependent kinase 1 (CDK1) inhibitor (IC50=25 nM). CGP-74514 dihydrochloride inhibits CDK1/cyclin B complex activity, arrests the cell cycle at G2/M phase and induces tumor cell apoptosis. CGP-74514 dihydrochloride is promising for research of bladder cancer .
|
-

- HY-174404
-
|
|
Topoisomerase
Apoptosis
Bcl-2 Family
|
Cancer
|
|
Topoisomerase II inhibitor 23 is a potent topoisomerase II inhibitor (IC50 = 0.94 μM). Topoisomerase II inhibitor 23 shows high selectivity and exceptional cytotoxic activity in MCF-7, HepG2, and HCT116 cells. Topoisomerase II inhibitor 23 induces cell cycle arrest at the G1 phase, leading to inhibition of cell proliferation. Topoisomerase II inhibitor 2 induces apoptosis by up-regulating the pro-apoptotic Bax level and down-regulating the anti-apoptotic Bcl-2 level.
|
-

- HY-122359A
-
|
rel-L-Centchroman; Ormeloxifene
|
Estrogen Receptor/ERR
Apoptosis
Reactive Oxygen Species (ROS)
|
Cancer
|
|
rel-Levormeloxifene (rel-L-Centchroman) is the relative configuration of Levormeloxifene (HY-122359). rel-Levormeloxifene is a selective estrogen receptor modulator (SERM). rel-Levormeloxifene inhibits proliferation of leukemia cells with IC50 about 7 μM, arrests cell cycle at G0/G1 phase, and induces apoptosis. rel-Levormeloxifene induces differentation of myelogenesis leukemia, and enhances ROS production in K562 cells .
|
-

- HY-50767D
-
|
PD 0332991 orotate
|
CDK
|
Cancer
|
|
Palbociclib (PD 0332991) orotate is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib orotate has potent anti-proliferative activity and induces cell cycle arrest in cancer cells. Palbociclib orotate can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
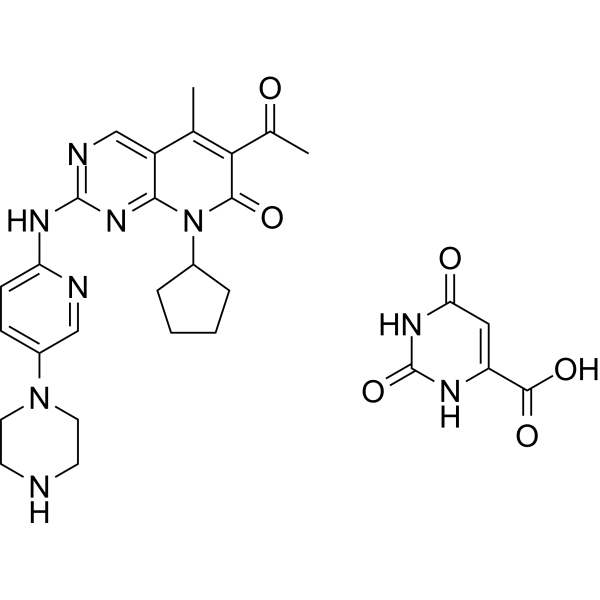
- HY-149912A
-
|
|
Polo-like Kinase (PLK)
Trk Receptor
Apoptosis
|
Cancer
|
|
CZS-241 hydrochloride is an orally active and selective inhibitor of Polo-like Kinase 4 (PLK4) (IC50=2.6 nM). CZS-241 hydrochloride inhibits TRKA with an IC50 value of 2.74 μM. CZS-241 hydrochloride induces apoptosis and arrests cell cycle at S/G2 phase. CZS-241 hydrochloride shows highly potent antiproliferative activity against leukemia cell lines, and exhibits safety against normal cell lines .
|
-

- HY-161339
-
|
|
Apoptosis
Sirtuin
|
Cancer
|
|
SIRT2-IN-13 (compound 7c) is an inhibitor of SIRT2 and induces apoptosis. SIRT2-IN-13 selectively inhibits colon cancer and leukemia cells and arrests the cell cycle in the G2 phase .
|
-

- HY-133838
-
|
BM41440
|
Parasite
PKC
|
Infection
Cancer
|
|
Ilmofosine (BM41440) is a potent and selective protein kinase C inhibitor. Ilmofosine induces cell cycle arrest at the G2 phase. Ilmofosine also is an anti-leishmanial agent .
|
-

- HY-164462
-
|
|
PKC
NF-κB
Apoptosis
|
Cancer
|
|
BHA536 is an orally active selective inhibitor for PKCα/β and NF-kB signaling pathway. BHA536 inhibits the proliferation of CD79-mutated ABC DLBCL cell, arrests cell cycle at G1 phase, and induces apoptosis in TMD8 cell. BHA536 exhibits antitumor efficacy in mice .
|
-

- HY-176727
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
WRN inhibitor 19 (Compound 40) is a WRN helicase inhibitor (IC50: 3.7 nM). WRN inhibitor 19 exhibits selective antiproliferative activity in WRN-dependent cancer cells. WRN inhibitor 19 inhibits WRN helicase function by competitively binding to the ATP site and induces DNA damage and cell cycle arrest. WRN inhibitor 19 can be used in the study of WRN-dependent cancers .
|
-

- HY-149451
-
|
|
RET
VEGFR
c-Myc
|
Cancer
|
|
SYHA1815 is an orally active RET inhibitor (IC50=0.9 nmol/L) with antitumor activity. SYHA1815 is more selective for RET than KDR (IC50=15.9 nmol/L). SYHA1815 arrests the G1 cell cycle and inhibits RET-driven cell proliferation by downregulating c-Myc .
|
-

- HY-146316
-
|
|
Topoisomerase
|
Cancer
|
|
Topoisomerase II inhibitor 6 (Compound 5), a tryptanthrin derivative, is a potent and selective inhibitor of topoisomerase II. Topoisomerase II inhibitor 6 exhibits antiproliferative activity on different tumor cell lines. Topoisomerase II inhibitor 6 blocks the cell cycle of CCRF-CEM in the G2 phase and induces DNA DSB. Topoisomerase II inhibitor 6 has the potential for the research of cancer diseases .
|
-
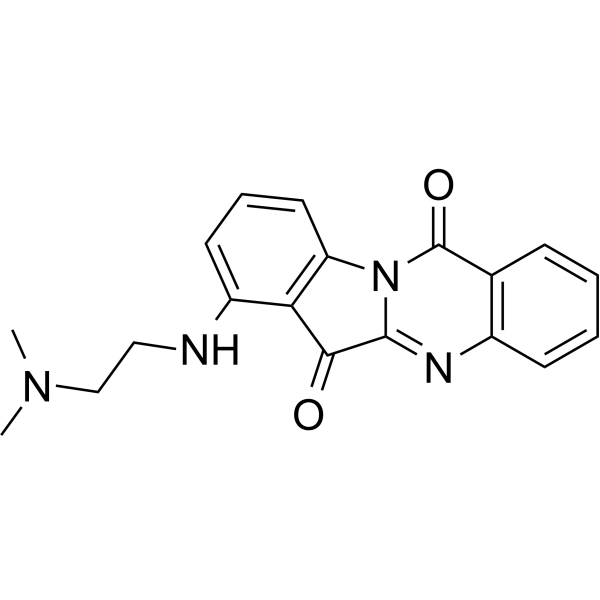
- HY-162416
-
|
|
CDK
|
Cancer
|
|
CDK7-IN-27 (Compound 37) is a selective inhibitor for cyclin-dependent kinase 7 (CDK7), with Ki of 3 nM. CDK7-IN-27 arrests the cell cycle at G0/G1 phase .
|
-
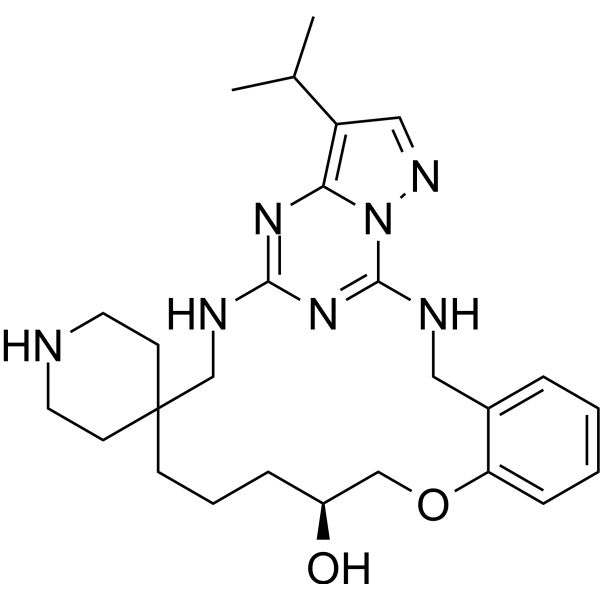
- HY-170954
-
|
|
Apoptosis
c-Met/HGFR
TAM Receptor
|
Cancer
|
|
C-Met/Axl-IN-1 (Compound 22a) is an orally active and selective type II c-Met/Axl inhibitor with IC50 values of 1 nM and 10 nM, respectively. C-Met/Axl-IN-1 can inhibit proliferation, induce cell cycle arrest and apoptosis of tumor cells. C-Met/Axl-IN-1 has strong anti-tumor activity .
|
-

- HY-P10226
-
|
|
Chloride Channel
CFTR
|
Endocrinology
|
|
PGD97 is a selective cyclic peptide inhibitor against CAL/CFTR interactions, with a KD value of 6 nM towards the CAL PDZ domain for its desulfide cyclized form. PGD97 (desulfide cyclized form) has selectivity ≥ 130-fold compared to NHERF1/2 PDZ domains. PGD97 is capable of stabilizing F508del-CFTR at the cell membrane and improving CFTR function required for proper fluid homeostasis in tne lung. PGD97 can be used for the research of cystic fibrosis .
|
-

- HY-159515
-
|
|
Apoptosis
DNA/RNA Synthesis
Reactive Oxygen Species (ROS)
|
Cancer
|
|
PBE-AMF is a prodrug that activates H2O2 with anticancer activity. PBE-AMF impedes tumor proliferation by inhibiting DNA synthesis, reducing ATP levels, inducing apoptosis, and arresting the cell cycle. PBE-AMF potently and selectively inhibits the proliferation of MDA-MB-231 cells (IC50=6.4 μM) while sparing non-cancerous MCF-10A cells .
|
-

- HY-163119
-
|
|
Pim
|
Cancer
|
|
Pim-1 kinase inhibitor 9 (compound 8b) is a selective inhibitor against Pim-1 kinase with IC50 value of 0.24 µM. Pim-1 kinase inhibitor 9 inhibits cell cycle of T47D at S phase. Pim-1 kinase inhibitor 9 reveals antitumor activity .
|
-
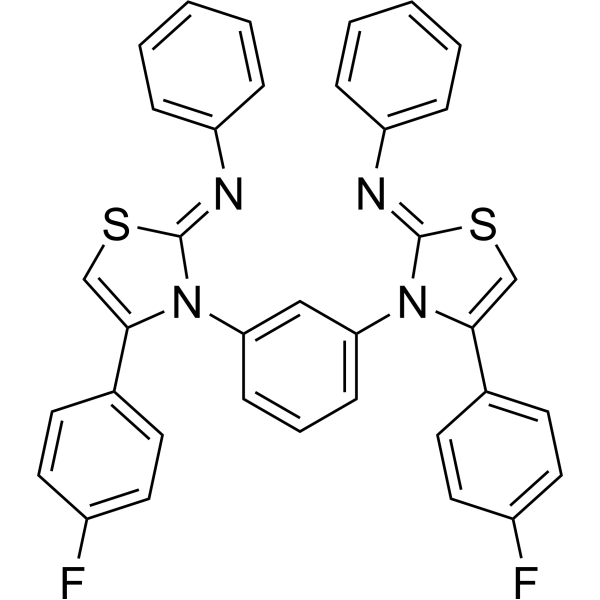
- HY-149376
-
|
|
Microtubule/Tubulin
|
Cancer
|
|
Tubulin inhibitor 38 (compound 14) is a tetrazole-based Tubulin inhibitor with antiproliferative potencies. Tubulin inhibitor 38 (100 nM,24 h) mediates mitotic arrest,blocks cell cycle at G2/M phase and induces apoptosis. Tubulin inhibitor 38 exhibits high cytotoxicity with high selectivity index among HeLa,MCF7,and U87 MG cells .
|
-
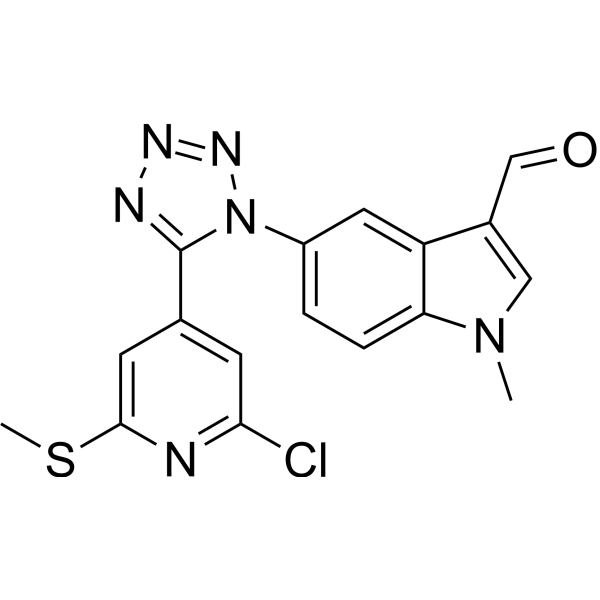
- HY-176066A
-
|
|
c-Myc
Apoptosis
|
Cancer
|
|
c-Myc inhibitor 16 iodide (Compound W11) is a selective c-Myc G-quadruplex (c-Myc G4) inhibitor. c-Myc inhibitor 16 iodide inhibits the transcription and translation of the c-Myc gene, disrupts the tumor cell cycle, arrests cell growth in the G0/G1 phase and activates the mitochondrial apoptosis pathway to induce early apoptosis of cancer cells. c-Myc inhibitor 16 iodide is promising for research of breast cancer .
|
-

- HY-151409
-
|
|
CDK
|
Cancer
|
|
CDK1-IN-5 (10h) is a selective CDK1 inhibitor with IC50s of 42.19, 188.71 and 354.15 nM for CDK1, CDK2 and CDK5, respectively. CDK1-IN-5 inhibits growth of cancer cells by affecting cell cycle. CDK1-IN-5 can be used for the research of cancer .
|
-
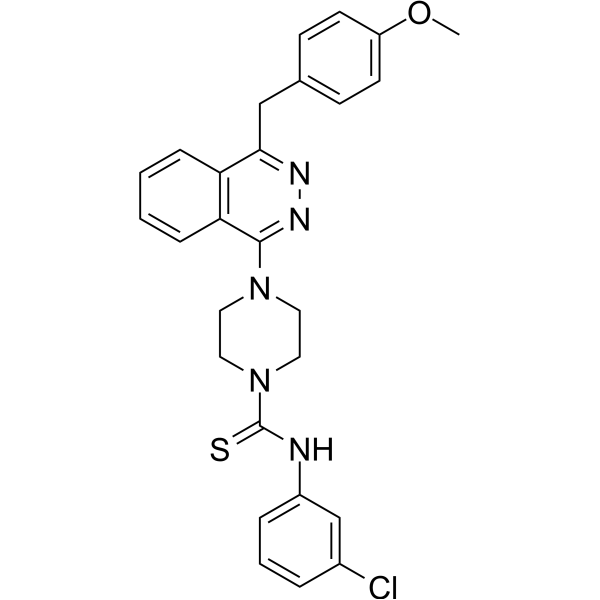
- HY-172255
-
|
|
PI4K
Apoptosis
Autophagy
PI3K
Akt
|
Cancer
|
|
PI4KIII beta inhibitor 4 (Compound 16) is a selective PI4KIIIβ inhibitor with an IC50 of 0.005 μM. PI4KIIIβ inhibitor 4 induces tumor cell apoptosis, cell cycle arrest, and autophagy by inhibiting the PI3K/AKT pathway. PI4KIIIβ inhibitor 4 can be used in the study of cancer .
|
-

- HY-151408
-
|
|
CDK
|
Cancer
|
|
CDK1-IN-4 (10d) is a selective CDK1 inhibitor with IC50s of 44.52, 624.93 and 135.22 nM for CDK1, CDK2 and CDK5, respectively. CDK1-IN4 inhibits the growth of cancer cells by affecting cell cycle. CDK1-IN-4 can be used for the research of cancer .
|
-
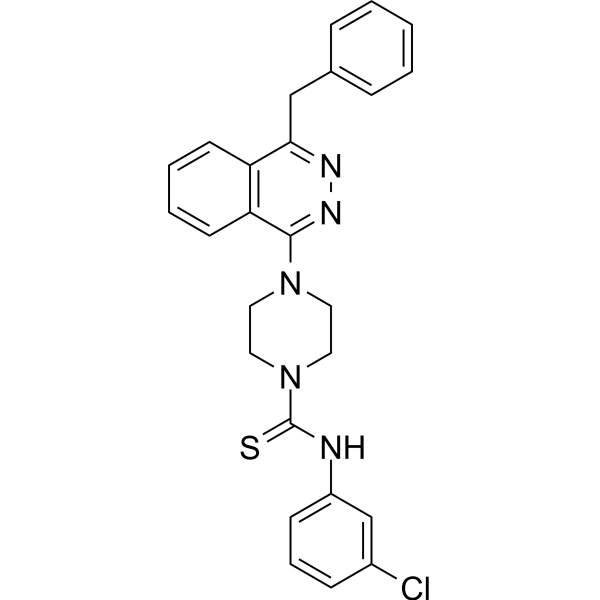
- HY-159560
-
|
|
Apoptosis
DNA/RNA Synthesis
|
Cancer
|
|
PBA-AMF can be activated by H2O2 to release Amonafide (AMF) (HY-10982). PBA-AMF selectively inhibited the proliferation of breast cancer cells, while sparing non-cancerous cells. PBA-AMF inhibits tumor proliferation by inhibiting DNA synthesis, reducing ATP levels, inducing apoptosis, and arresting the cell cycle. PBA-AMF can be used for research of tumors and other diseases associated with increased H2O2 levels .
|
-

- HY-125158
-
|
|
Aurora Kinase
Apoptosis
|
Cancer
|
|
HOI-07 is a selective Aurora B kinase inhibitor. HOI-07 blocks phosphorylation of histone H3 on Ser10 in
lung cancer cells. HOI-07 induces cell-cycle arrest, and apoptosis. HOI-07 has antitumor activity, and suppresses the tumor growth of A549, 143B and KHOS xenografts .
|
-

- HY-149291
-
|
|
ATM/ATR
Apoptosis
|
Cancer
|
|
ATM Inhibitor-7 is a potent and selective ataxia-telangiectasia mutated (ATM) inhibitor with an IC50 value of 1.0 nM. ATM Inhibitor-7 induces Apoptosis and cell cycle arrest at G2/M phase when combinanted with CPT-11 (HY-16562). ATM Inhibitor-7 combines with CPT-11 shows antitumor activity .
|
-
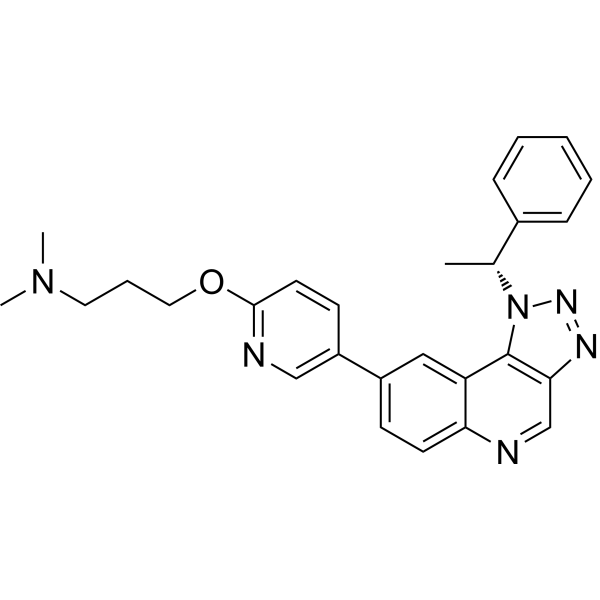
- HY-150695
-
|
|
Carbonic Anhydrase
Apoptosis
|
Cancer
|
|
hCAIX/XII-IN-5 (Coumarin 9a) a carbonic anhydrase (CA) inhibitor, and exhibits excellent hCA IX/XII selectivity (Ki=93.9 and 85.7 nM, respectively) over hCA I and hCA II. hCAIX/XII-IN-5 shows anti-proliferative activities to cancer cells. hCAIX/XII-IN-5 can delay cell cycle and induce apoptosis .
|
-
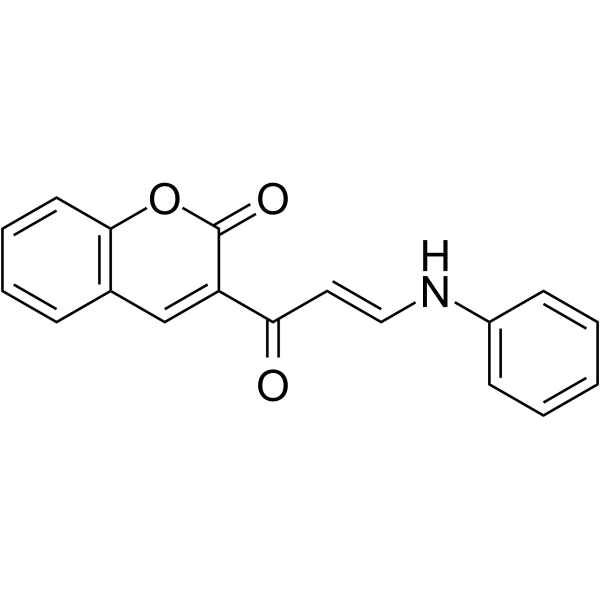
- HY-173057
-
|
|
BMX Kinase
Apoptosis
Autophagy
|
Cancer
|
|
BMX-IN-3 (B6a) is an irreversible and selective BMX inhibitor, with an IC50 of 12 nM. BMX-IN-3 (B6a) promots cell cycle arrest and apoptosis, triggers protective autophagy, and suppresses the BMX/AKT/mTOR pathway. BMX-IN-3 (B6a) can be used in the research for Gastric Carcinoma .
|
-

- HY-183146
-
|
|
CDK
|
Cancer
|
|
CDK2-IN-57 is a selective Cdk2/CycE inhibitor with an IC50 of 2.8 nM. CDK2-IN-57 inhibits Cdk1/2 kinase activity, blocks cell cycle progression, induces G0-phase cell cycle arrest, and prevents S-phase entry. CDK2-IN-57 can be used for the research of colon carcinoma .
|
-

- HY-114699
-
|
|
Endogenous Metabolite
|
Cancer
|
|
DC_YM21 is an inhibitor of menin-MLL interaction with potent and selective proliferation blocking activity. DC_YM21 can induce cell cycle arrest and differentiation of leukemia cells carrying MLL translocation. DC_YM21 shows potential application value in inhibiting MLL leukemia .
|
-

- HY-182436
-
|
|
Apoptosis
|
Cancer
|
|
MINA53-IN-1 is a selective MINA53 inhibitor with an IC50 of 1.5 μM. MINA53-IN-1 induces DNA damage, cell cycle arrest and apoptosis in tumor cells. MINA53-IN-1 can be used for cancer research .
|
-

- HY-179679
-
|
|
FAK
Apoptosis
|
Cancer
|
|
FAK-IN-29 is a selective FAK inhibitor with an IC50 of 0.5 nM. FAK-IN-29 can inhibit the proliferation and colony formation of MDA-MB-231 cells, and induce cell cycle arrest and apoptosis. FAK-IN-29 exhibits antitumor activity and can be used for the research of tumors such as triple-negative breast cancer .
|
-

- HY-182035
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
HDAC8-IN-16 is a selective histone deacetylase 8 (HDAC8) inhibitor with an IC50 of 0.16 μM. HDAC8-IN-16 induces cell apoptosis, triggers G2/M phase cell cycle arrest, and moderately inhibits cancer cell proliferation. HDAC8-IN-16 is applicable to relevant research on colorectal cancer .
|
-

- HY-181130
-
|
|
CDK
Apoptosis
|
Cancer
|
|
Anticancer agent 299 (compound P12) is a cell-cycle inhibitor, senescence inducer, apoptosis inducer, and antiproliferative agent. Anticancer agent 299 exhibits selective activity against cancer cells with minimal effects on non-tumoral chondrocyte cells at relevant concentrations. Anticancer agent 299 can be used for the research of ER+/HER2− breast cancer and BRAF-mutant melanoma .
|
-

- HY-181578
-
|
|
NAMPT
Apoptosis
|
Cancer
|
|
Nampt-IN-17 is an selective orally active NAMPT inhibitor with a human NAMPT IC50 of 17 nM and Ki of 25.9 nM. Nampt-IN-17 depletes intracellular NAD + and ATP, disrupts mitochondrial membrane potential, suppresses cell proliferation, self-renewal, invasion, and migration, induces cell-cycle arrest and apoptosis. Nampt-IN-17 exhibits selective activity against NAPRT-deficient gastric cancer cells. Nampt-IN-17 can be used for the research of NAPRT-deficient gastric cancer .
|
-

- HY-181639
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
HDAC6-IN-75 is a selective HDAC6 inhibitor with an IC50 of 0.17 nM against HDAC6. HDAC6-IN-75 induces the accumulation of acetylated α-tubulin in glioma cells. HDAC6-IN-75 triggers cell cycle changes, increases the SubG1 cell population, and promotes apoptosis in glioma cells and glioblastoma stem cells. HDAC6-IN-75 is applicable for glioma-related research .
|
-

- HY-13902A
-
|
VE-822 hydrochloride; VX-970 hydrochloride; M6620 hydrochloride
|
ATM/ATR
Apoptosis
STING
Caspase
|
Neurological Disease
Cancer
|
|
Berzosertib (VE-822) hydrochloride is an orally active, CNS-penetrant, and selective ATR kinase inhibitor. Berzosertib hydrochloride blocks ATR kinase activity, abrogates G2/M cell cycle checkpoint, impairs DNA damage repair. Berzosertib hydrochloride induces apoptosis, inhibnits conlony migration, inhibits cell proliferation, and activates cGAS-STING axes in cancer cells. Berzosertib hydrochloride can be used for the research of cancers, such as head and neck squamous cell carcinoma, and colorectal cancer .
|
-

- HY-180818
-
|
|
Anaplastic lymphoma kinase (ALK)
Apoptosis
|
Cancer
|
|
ALK-IN-33 (Compound 8q) is an orally active ALK inhibitor with an IC50 of 1.61 nM. ALK-IN-33 exhibits significant selective killing effect on ALK-positive cancer cells. ALK-IN-33 induces cell cycle arrest and apoptosis, effectively weakening the migration, invasion and long-term survival ability of cancer cells. ALK-IN-33 can be used for research on non-small cell lung cancer
|
-

- HY-179466
-
|
|
Microtubule/Tubulin
Apoptosis
Caspase
|
Cancer
|
|
BKT300 is a potent and selective protein regulator of cytokinesis 1 (PRC1) inhibitor. BKT300 inhibits PRC1 dephosphorylation at T481, disrupts actin and microtubule formation, induces G2/M cell cycle arrest, triggers mitotic catastrophe, and promotes apoptosis, thereby inhibiting proliferation and migration of acute myeloid leukemia (AML) cells while sparing normal cells. BKT300 inhibits tumor growth in mouse xenograft AML models. BKT300 can be used for the research of AML .
|
-

- HY-100888R
-
|
TAK-931 (Standard)
|
CDK
Reference Standards
|
Cancer
|
|
Simurosertib (Standard) is the analytical standard of Simurosertib (HY-100888). This product is intended for research and analytical applications. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM. Simurosertib has anti-cancer activity .
|
-

- HY-179460
-
|
IY-5511 dihydrochloride
|
Bcr-Abl
Apoptosis
STAT
JAK
Prion Protein
|
Neurological Disease
Cancer
|
|
Radotinib (IY-5511) dihydrochloride is an orally active and BBB-permeable selective tyrosine kinase Bcr-Abl1 inhibitor with an IC50 of 34 nM. Radotinib dihydrochloride has anti-prion and anti-tumor activities. Radotinib dihydrochloride can inhibit the proliferation, induce cell cycle arrest and apoptosis of tumor cells . Radotinib dihydrochloride can be used in the research of cancer such as chronic myeloid leukemia and multiple myeloma, as well as neurodegenerative diseases such as prion diseases .
|
-

- HY-183754
-
|
|
EGFR
PI3K
Akt
Apoptosis
|
Cancer
|
|
EGFR-IN-213 is a selective inhibitor of EGFR L858R/T790M/C797S with a human IC50 of 0.48 nM. EGFR-IN-213 acts as an antiproliferative agent, inducing apoptosis and cell cycle arrest, and inhibiting colony formation, cell migration, and tube formation. EGFR-IN-213 can be used for the research of non-small cell lung cancer, chronic myeloid leukemia, gastric cancer, prostate cancer .
|
-

- HY-174231
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-163 (Compound 13) is a competitive epidermal growth factor receptor (EGFR) inhibitor (IC50=0.079 μM, selective for HER-2 inhibition). EGFR-IN-163 induces tumor cell apoptosis and cell cycle arrest at G₂/M phase. EGFR-IN-163 is promising for research of estrogen receptor-positive (ER+) breast cancer .
|
-

- HY-177957
-
|
|
CDK
|
Cancer
|
|
CDK2-IN-52 (Compound Cpb No 39) is a selective cyclin-dependent kinase 2 (CDK2) inhibitor with a DC50 value of 1-10 nM. CDK2-IN-52 induces cell cycle arrest and suppresses tumor cell proliferation. CDK2-IN-52 is promising for research of CDK2-overexpressing malignancies such as breast and ovarian cancers .
|
-

- HY-172210
-
|
|
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
DDO-8958 is an orally active and selective BET BD1 inhibitor with a KD of 5.6 nM for BRD4 BD1. DDO-8958 exhibits low nanomolar inhibitory activity against all BET BD1 bromodomains except for BRDT BD1. DDO-8958 can inhibit the proliferation and migration, induce apoptosis and cell cycle arrest of tumor cells. DDO-8958 has anti-tumor activity .
|
-

- HY-142076A
-
|
|
CDK
|
Cancer
|
|
CDK4/6-IN-15 hydrochloride is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 hydrochloride potently inhibits cancer cells growth. CDK4/6-IN-15 hydrochloride arrests cell cycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
|
-

- HY-142076
-
|
|
CDK
|
Cancer
|
|
CDK4/6-IN-15 is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 potently inhibits cancer cells growth. CDK4/6-IN-15 arrests cell cycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
|
-
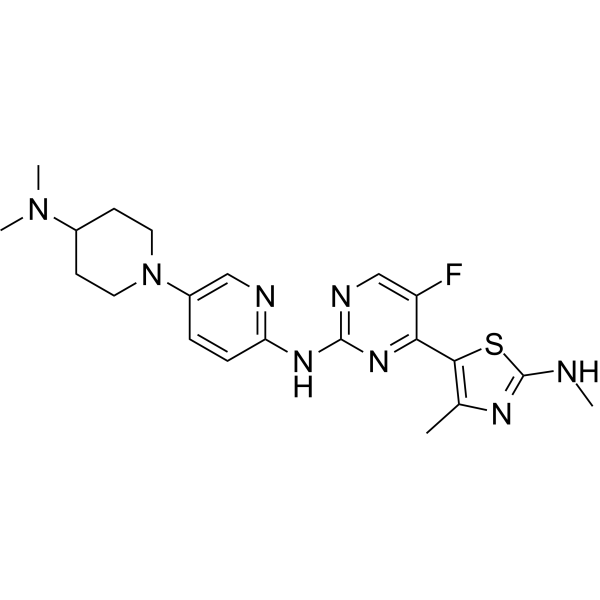
- HY-171180
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
AF615 is a small molecule inhibitor targeting the CDT1/Geminin protein complex (IC50 = 0.313 μM). AF615 selectively induces DNA damage, inhibits DNA synthesis, causes cell cycle arrest, and decreases the survival rate in cancer cell lines. AF615 displayed a Ki = 0.37 μM in case of Geminin-tCDT1 interaction and a Ki = 0.75 μM in case of Geminin-miniCDT1 interaction .
|
-

- HY-12286
-
|
|
Proteasome
Apoptosis
Autophagy
Caspase
Bcl-2 Family
NF-κB
PARP
|
Cancer
|
|
PI-1840 is a potent and selective chymotrypsin-like (CT-L) inhibitor for with an IC50 value of 27 nM. PI-1840 inhibits cell proliferation and arrest cell cycle at G2/M phase. PI-1840 induces apoptosis and induces autophagy. PI-1840 induces the accumulation of proteasome substrates p27, Bax, and IκB-α .
|
-
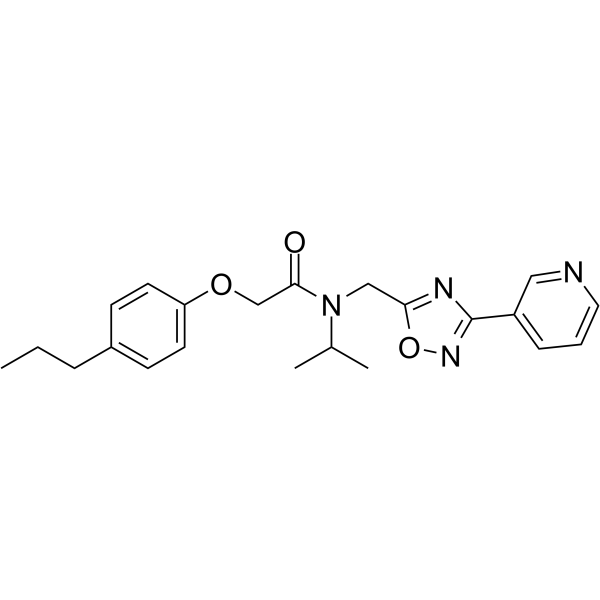
- HY-11107
-
|
|
c-Met/HGFR
Autophagy
Apoptosis
|
Cancer
|
|
PHA-665752 is a selective, ATP-competitive, and active-site inhibitor of the catalytic activity of c-Met kinase (Ki=4 nM; IC50=9 nM). PHA-665752 exhibits >50-fold selectivity for c-Met compared with a panel of diverse tyrosine and serine-threonine kinases. PHA-665752 induces apoptosis and cell cycle arrest, and exhibits cytoreductive antitumor activity .
|
-
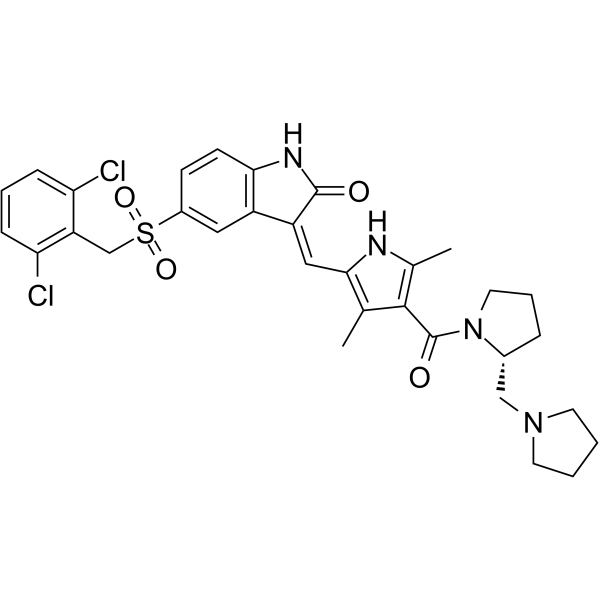
- HY-155285
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
YS-363 is a potent, selective, and orally active EGFR inhibitor, with IC50s of 0.96 nM and 0.67 nM for wild-type and L858R mutant forms of EGFR, respectively. YS-363 can induce G0/G1 cell cycle arrest and apoptosis .
|
-
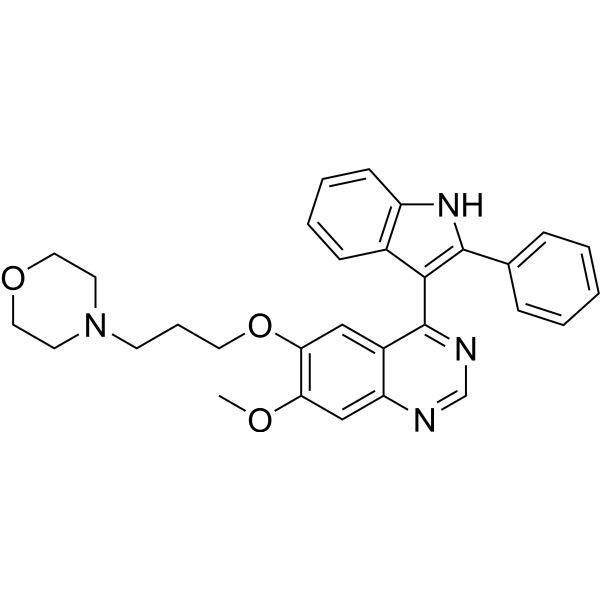
- HY-13630R
-
|
BMY-40481 (Standard)
|
Reference Standards
Topoisomerase
Bacterial
Autophagy
Apoptosis
|
Infection
Neurological Disease
Cancer
|
|
Etoposide phosphate (Standard) is the analytical standard of Etoposide phosphate. This product is intended for research and analytical applications. Etoposide phosphate (BMY-40481) is a potent anti-cancer chemotherapy agent and a selective topoisomerase II inhibitor to prevent re-ligation of DNA strands. Etoposide phosphate is the phosphate ester proagent of etoposide and is considered as active equivalent to Etoposide. Etoposide phosphate induces cell cycle arrest, apoptosis, and autophagy.
|
-

- HY-118798
-
|
NSC663285
|
Phosphatase
|
Cancer
|
|
DA 3003-2 is a potent and selectively Cdc25 inhibitor. DA 3003-2 shows antiproliferative activity. DA 3003-2 induces cell cycle arrest at the G2/M phase and increases the expression of P-tyr 15 Cdc2. DA 3003-2 has the potential for the research of prostate cancer .
|
-
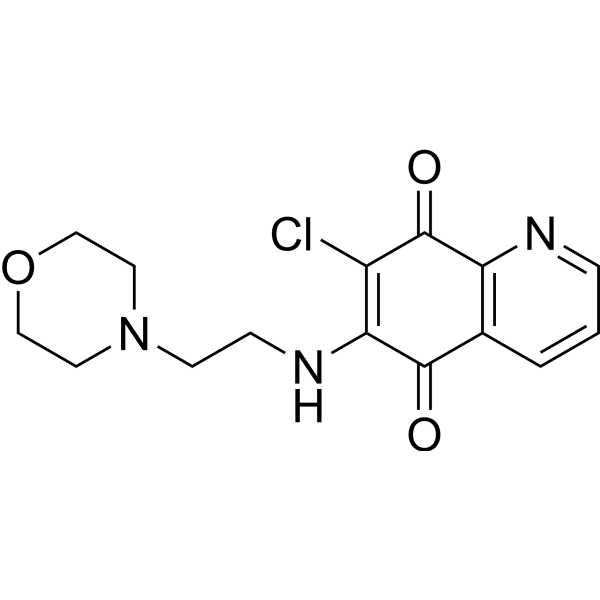
- HY-N7225
-
|
Gnidilatidin
|
DNA/RNA Synthesis
|
Cancer
|
|
Yuanhuacine (Gnidilatidin), a diterpene from Daphne genkwa, is an effective and highly selective inhibitor of the basal-like 2 (BL2) subtype of TNBC. Yuanhuacine can induce G2/M cell cycle arrest and has broad anti-tumor activity. Yuanhuacine is an orally active DNA damaging agent .
|
-
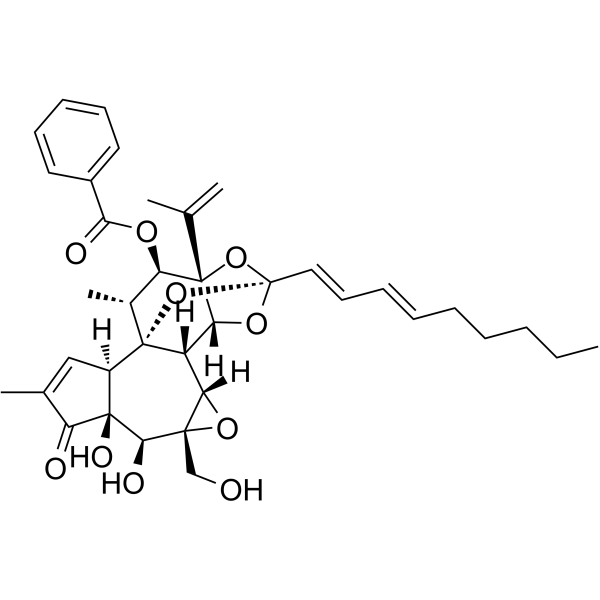
- HY-181030
-
|
|
Polo-like Kinase (PLK)
Apoptosis
CDK
Bcl-2 Family
|
Cancer
|
|
BDE30671203 is a highly selective PLK1 inhibitor (IC50 = 2.163 nM). BDE30671203 induces G2/M phase arrest and Apoptosis. BDE30671203 downregulates key cell cycle regulators (CDC20, CDK1) and the anti-apoptotic gene Bcl-2. BDE30671203 exhibits anticancer activity against non-small cell lung cancer, large cell lung cancer, and liver cancer .
|
-

- HY-181541
-
|
|
HDAC
Apoptosis
Wnt
β-catenin
MDM-2/p53
c-Myc
|
Cancer
|
|
HIT211504993 is a selective histone deacetylase 6 (HDAC6) inhibitor with an IC50 of 0.070 μM. HIT211504993 suppresses cancer cell proliferation, cause G1 phase cell cycle arrest and induces apoptosis. HIT211504993 inhibits Myc-driven tumorigenesis via nucleocytoplasmic acetylation, p53 modulation, and Wnt/β-catenin signaling modulation. HIT211504993 inhibits tumor growth in a colon cancer xenograft mouse model. HIT211504993 can be used for the research of colon cancer .
|
-

- HY-181579
-
|
|
HDAC
Apoptosis
PERK
Microtubule/Tubulin
|
Cancer
|
|
HDAC6-IN-74 is an orally active, selective histone deacetylase 6 (HDAC6) inhibitor with an IC50 of 0.036 μM. HDAC6-IN-74 induces tumor cell apoptosis, arrests cells at the S phase of the cell cycle, and impairs cell migration, invasion and colony-forming abilities. HDAC6-IN-74 exerts anticancer effects with no obvious toxicity. HDAC6-IN-74 can be used in the research of cancers such as liver cancer .
|
-

- HY-153361
-
|
|
PROTACs
Epigenetic Reader Domain
|
Inflammation/Immunology
Cancer
|
|
YD23 is a selective SMARCA2 PROTAC degrader with DC50 values of 64 nM and 297 nM in H1792 cells and H1975 cells. YD23 induces degradation of SMARCA2, which is synthetic lethal to SMARCA4. YD23 reduces chromatin accessibility only in SMARCA4 deficient cells, including cell cycle and cell growth regulatory genes. YD23 selectively inhibits growth of SMARCA4 mutant lung cancer cells. YD23 has potent tumor growth inhibitory activity in SMARCA4-mutant xenografts. YD23 can be used for the study of non-small cell lung cancer (NSCLC) .
|
-
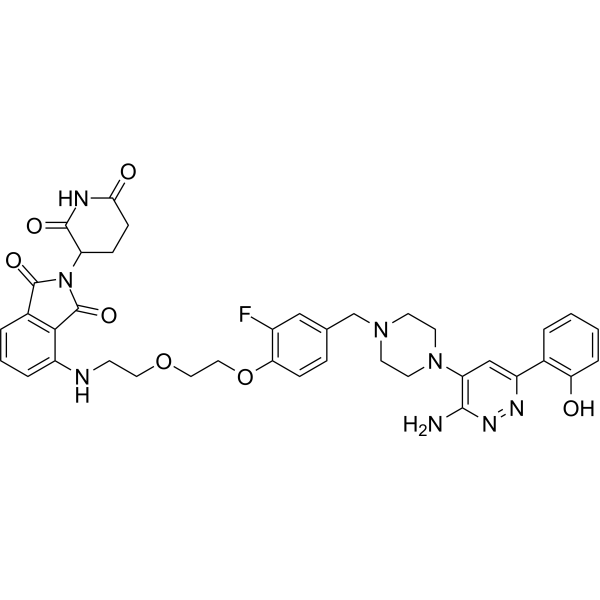
- HY-10227
-
|
PS-341; LDP-341; NSC 681239
|
Proteasome
NF-κB
Apoptosis
Autophagy
TREM receptor
|
Cancer
|
|
Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-
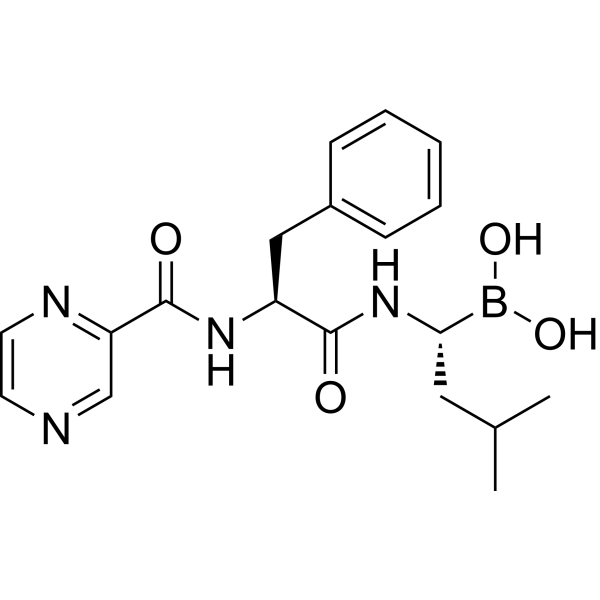
- HY-B1153
-
|
Glafenin
|
COX
CFTR
Apoptosis
Endoplasmic Reticulum Oxidoreductase 1 (ERO1)
|
Metabolic Disease
Inflammation/Immunology
|
|
Glafenine (Glafenin) is a non-selective, non-steroidal anti-inflammatory drug-based COX-1/COX-2 inhibitor. Glafenine exerts anti-inflammatory, anti-proliferative and anti-cell migration effects by inhibiting the arachidonic acid metabolic pathway and reducing prostaglandin synthesis. Glafenine can induce cell cycle arrest in vascular smooth muscle cells and endothelial cells and reduce the synthesis of the extracellular matrix protein Tenascin. Glafenine can be used in the research of inflammatory-related diseases, vascular restenosis and cystic fibrosis (CF) .
|
-
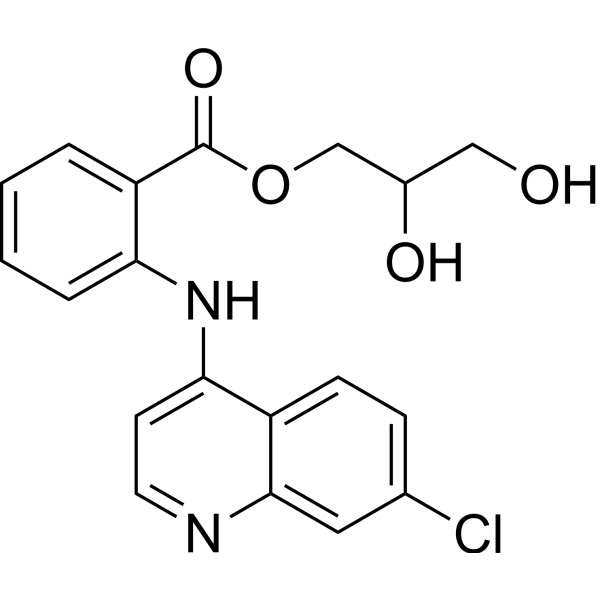
- HY-B1153A
-
|
Glafenin hydrochloride
|
COX
CFTR
Apoptosis
Endoplasmic Reticulum Oxidoreductase 1 (ERO1)
|
Metabolic Disease
Inflammation/Immunology
|
|
Glafenine (Glafenin) hydrochloride is a non-selective, non-steroidal anti-inflammatory drug-based COX-1/COX-2 inhibitor. Glafenine hydrochloride exerts anti-inflammatory, anti-proliferative and anti-cell migration effects by inhibiting the arachidonic acid metabolic pathway and reducing prostaglandin synthesis. Glafenine hydrochloride can induce cell cycle arrest in vascular smooth muscle cells and endothelial cells and reduce the synthesis of the extracellular matrix protein Tenascin. Glafenine hydrochloride can be used in the research of inflammatory-related diseases, vascular restenosis and cystic fibrosis (CF) .
|
-
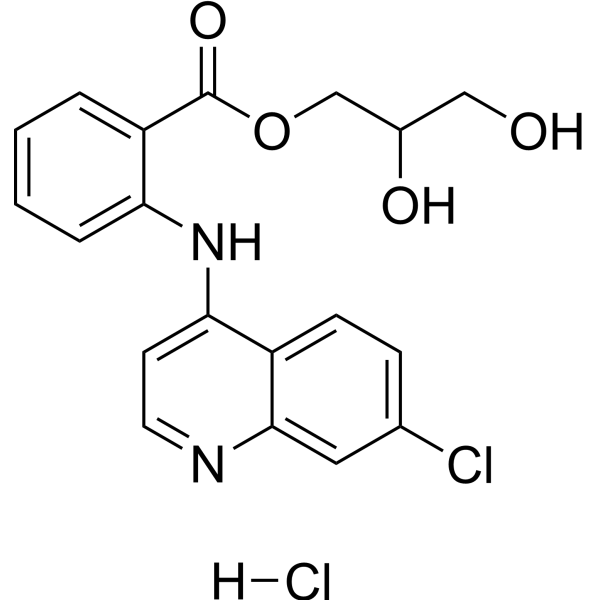
- HY-179045
-
|
|
Estrogen Receptor/ERR
Apoptosis
|
Cancer
|
ERα degrader 14 (Compound B7) is a potent ERα degrader. ERα degrader 14 exhibits significantly potent and selective anti-proliferative activity on two ERα-positive breast cancer cell lines (MCF-7 and T47D). ERα degrader 14 induces cell cycle arrest, inhibits cell migration, and induces cell apoptosis. ERα degrader 14 effectively inhibits tumor growth in mouse models. ERα degrader 14 can be used for the study of breast cancer .
|
-

- HY-161409
-
|
|
Androgen Receptor
Apoptosis
|
Cancer
|
|
SC912 is an AR-V7 inhibitor (IC50 = 0.36 μM). SC912 possesses safety, potency and selectivity. SC912 binds directly to AR-FL and AR-V7 proteins, inhibites nuclear localization and chromatin binding capabilities. SC912 exerts anticancer activity through inhibition of proliferation, induction of cell cycle arrest and apoptosis .
|
-
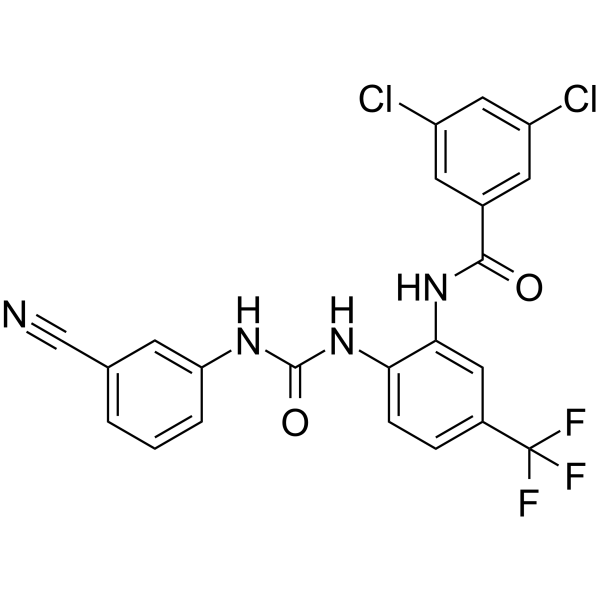
- HY-111033
-
|
|
MEK
ERK
Apoptosis
p38 MAPK
CDK
PARP
|
Inflammation/Immunology
Cancer
|
|
RO5068760 is a potent, orally active and selective non-ATP-competitive MEK1/2 inhibitor with an IC50 of 0.025 μM for MEK1. RO5068760 significantly inhibits MAPK pathway activity, thereby inducing G1 cell cycle arrest and apoptosis to inhibit cancer cell growth. RO5068760 exhibits significant efficacy in a broad spectrum of tumors with aberrant MAPK pathway activation. RO5068760 can be used for melanoma, colorectal cancer, non-small cell lung cancer (NSCLC), and pancreatic cancer research .
|
-

- HY-150772
-
|
|
Microtubule/Tubulin
HDAC
Apoptosis
Mitochondrial Metabolism
|
Cancer
|
|
Tubulin/HDAC-IN-1 is a dual tubulin and HDAC-IN-1 inhibitor through CH/π interaction with tubulin and hydrogen bond interaction with HDAC8. Tubulin/HDAC-IN-1 inhibits tubulin polymerization and selectively inhibits HDAC8 (IC50: 150 nM). Tubulin/HDAC-IN-1 has cytotoxicity against various human cancer cells, also arrests cell cycle in the G2/M phase and induces cell apoptosis. Tubulin/HDAC-IN-1 can be used in the research of hematologic and solid tumors such as neuroblastoma, leukemia .
|
-
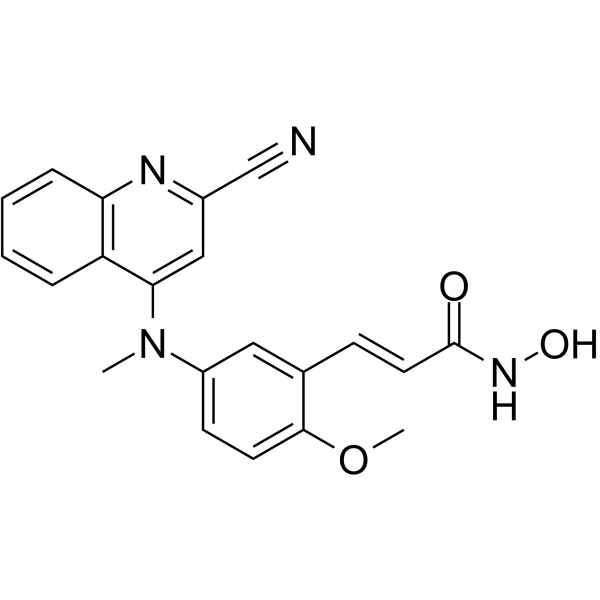
- HY-179032
-
|
|
VEGFR
PI3K
Akt
mTOR
Apoptosis
|
Cancer
|
|
VEGFR-2-IN-77 (Compound 10) is a VEGFR-2 inhibitor with an IC50 value of 139 nM. VEGFR-2-IN-77 inhibits the PI3K/AKT/mTOR pathway. VEGFR-2-IN-77 exhibits selective cytotoxicity, being particularly sensitive to leukemia and prostate cancer cells. VEGFR-2-IN-77 causes cell cycle arrest and apoptosis, inhibiting cell migration and invasion. VEGFR-2-IN-77 can be used for the study of leukemia and prostate cancer.
|
-

- HY-176734
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
CZL-S092 is a PLK4 inhibitor with an IC50 value of 0.9 nM and excellent selectivity over other PLK4 family members (PLK1, PLK2, and PLK3). CZL-S092 exhibits anti-neuroblastoma activity in vitro (IMR-32 cells, IC50 = 1.143 μM). CZL-S092 inhibits cell migration and halts the cell cycle and induces apoptosis. CZL-S092 can be used in studies of various cancers including neuroblastoma cancer .
|
-

- HY-179321
-
|
|
PROTACs
HDAC
|
Cancer
|
|
PROTAC HDAC4 Degrader-1 (compound SCT-1) is a potent and selective PROTAC HDAC4 degrader. PROTAC HDAC4 Degrader-1 reduces HDAC4 protein level, induces S phase cell cycle arrest, and inhibits cell colony formation, thereby inhibiting proliferation of the tumor cells. PROTAC HDAC4 Degrader-1 exhibits efficacy in a H460 mouse model. PROTAC HDAC4 Degrader-1 can be used for cancer research, such as lung cancer .
|
-

- HY-115906
-
|
|
FLT3
MNK
Apoptosis
|
Cancer
|
|
K783-0308 is a potent and selective dual inhibitor of FLT3 and MNK2 with IC50 values of 680 and 406 nM, respectively. K783-0308 inhibits the growth of MOLM-13 (IC50=10.5 µM) and MV-4-11 (IC50=10.4 µM) cells. K783-0308 promotes acute myeloid leukemia (AML) cell apoptosis and cell cycle arrests in the G0/G1 phase .
|
-
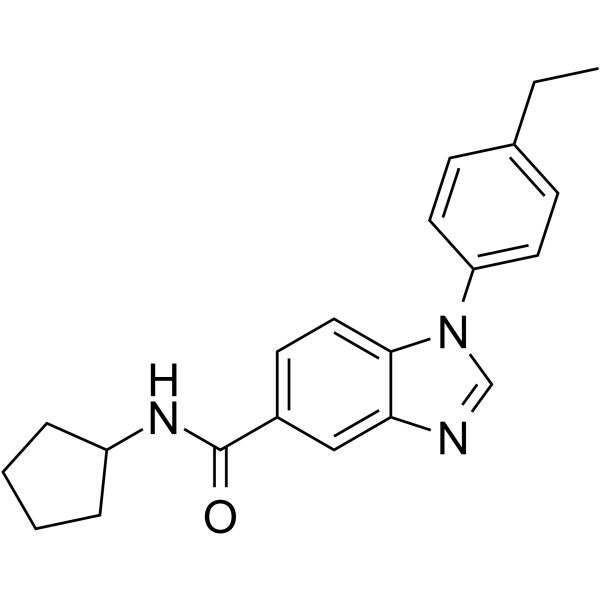
- HY-173557
-
|
|
Histone Methyltransferase
Apoptosis
|
Cancer
|
|
PRMT7-IN-2 (A33) is a selective PRMT7 inhibitor with an IC50 of 0.50 μM. PRMT7-IN-2 arrests cell cycle at G0/G1 phase, induces cell apoptosis, and inhibits cell growth in vivo and in vitro. PRMT7-IN-2 decreases the monomethylarginine level of PRMT7, increases expression of epithelial marker (E-cadherin, and reduces expression of mesenchymal markers such as N-cadherin, Vimentin, and ZEB2 .
|
-

- HY-178969
-
|
|
FAK
Pyk2
Apoptosis
|
Neurological Disease
Cancer
|
|
GZD-257 is a brain-penetrant, ATP-competitive FAK inhibitor (IC50 = 14.3 nM), performing 4.77-fold selectivity with FAK to Pyk2 (IC50 = 68.2 nM). GZD-257 can significantly induce apoptosis of U118MG cells and arrest the cell cycle at the G2/M phase. GZD-257 can be used for the study of Glioblastoma (GBM) .
|
-

- HY-W654305
-
|
PD 0332991-d4
|
Isotope-Labeled Compounds
CDK
|
Cancer
|
|
Palbociclib-d4 is deuterium labeled Palbociclib. Palbociclib (PD 0332991) is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-

- HY-N9684
-
|
|
EGFR
GSK-3
Hedgehog
Akt
ERK
Apoptosis
|
Cancer
|
|
Degalactotigonin is a saponin-selective inhibitor targeting the EGFR, GSK3β and Hedgehog/Gli1 pathways and can be isolated from Solanum nigrum (Solanum nigrum). Degalactotigonin inhibits EGFR phosphorylation and the downstream Akt/ERK signaling pathway, and at the same time inhibits the Hedgehog/Gli1 pathway through GSK3β inactivation, thereby inducing cancer cell apoptosis, arresting the cell cycle, and inhibiting migration and invasion. Degalactotigonin can be used in targeted research on malignant tumors such as pancreatic cancer and osteosarcoma .
|
-

- HY-183364
-
-

- HY-150615
-
|
|
SphK
Apoptosis
|
Cancer
|
|
SphK1-IN-2 is a potent, selective SphK1 inhibitor with IC50 values of 19.81 nM and >10 μM for SphK1 and SphK2, respectively. SphK1-IN-2 exhibits anti-proliferative activities and induces cell cycle arrest and apoptosis. SphK1-IN-2 can be used for cancer research .
|
-
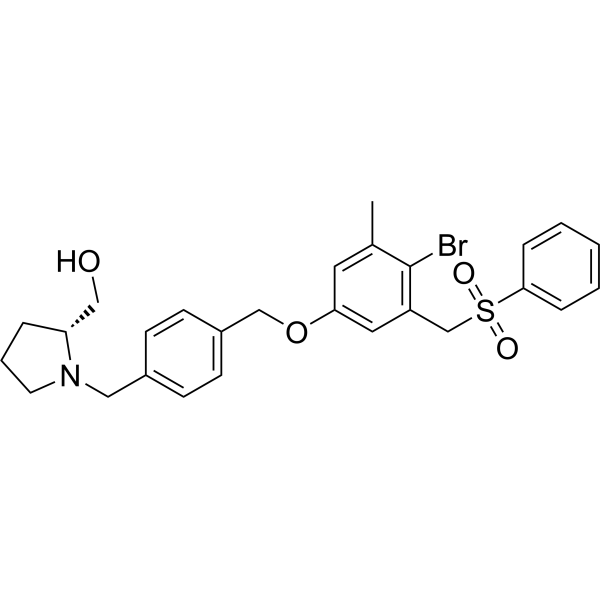
- HY-147892
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
HDAC-IN-42 (compound 14f) is a potent and selective HDAC inhibitor with IC50 values of 0.19 and 4.98 µM for HDAC1 and HDAC6, respectively. HDAC-IN-42 shows anticancer and anti-proliferative activity. HDAC-IN-42 induces apoptosis and cell cycle arrest at G2/M phase .
|
-
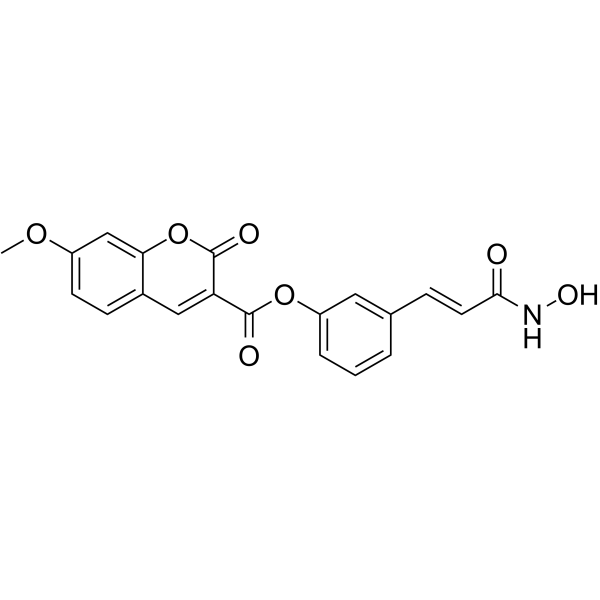
- HY-156182
-
|
|
JNK
Apoptosis
|
Cancer
|
|
JNK-IN-14 is a potent and selective JNK inhibitor with IC50 values of 1.81, 12.7 and 10.5 nM for JNK1, JNK2 and JNK3, respectively. JNK-IN-14 induces early-stage apoptosis and G2/M cell cycle arrest. JNK-IN-14 can be used for cancer research
|
-
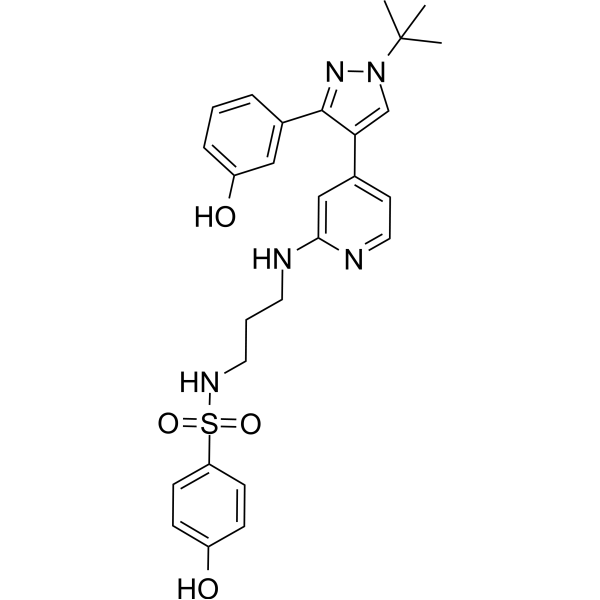
- HY-114302
-
CCB02
1 Publications Verification
|
Microtubule/Tubulin
|
Cancer
|
|
CCB02 is a selective CPAP-tubulin interaction inhibitor, binding to tubulin and competing for the CPAP binding site of β-tubulin, with an IC50 of 689 nM, and shows potent anti-tumor activity. CCB02 shows no inhibition on the cell cycle- and centrosome-related kinases, or the phosphorylation status of Aurora A, Plk1, Plk2, CDK2, and CHK1 .
|
-
- HY-100888AR
-
|
|
CDK
Reference Standards
|
Others
|
|
(R)-Simurosertib (Standard) is the analytical standard of (R)-Simurosertib (HY-100888A). This product is intended for research and analytical applications. (R)-Simurosertib ((R)-TAK-931) is the (R)-enantiomer of Simurosertib. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM .
|
-

- HY-101029
-
|
|
NEKs
Apoptosis
|
Cancer
|
|
MBM-55 is a potent NIMA-related kinase 2 (Nek2) inhibitor with an IC50 of 1 nM. MBM-55 shows a 20-fold or greater selectivity in most kinases with the exception of RSK1 (IC50=5.4 nM) and DYRK1a (IC50=6.5 nM). MBM-55 effectively inhibits the proliferation of cancer cells by inducing cell cycle arrest and apoptosis. MBM-55 shows antitumor activities, and no obvious toxicity to mice .
|
-
- HY-116217
-
|
FdCyd; NSC-48006
|
DNA Methyltransferase
|
Cancer
|
|
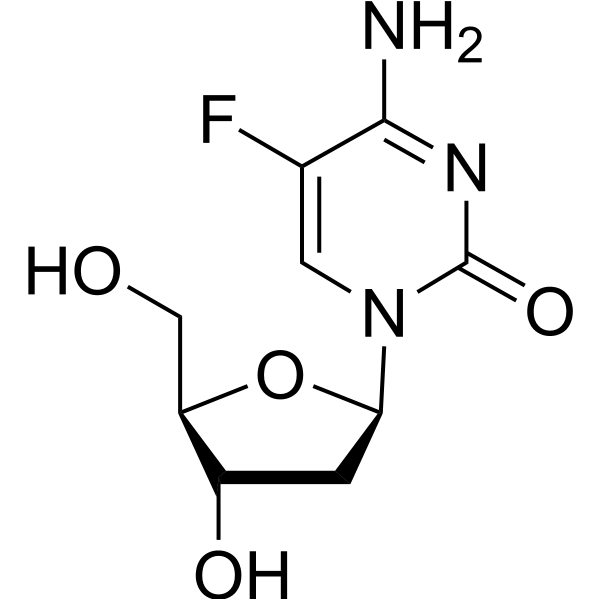
5-Fluoro-2'-deoxycytidine (FdCyd), a fluoropyrimidine nucleoside analogue, is a DNA methyltransferase (DNMT) inhibitor. 5-Fluoro-2'-deoxycytidine is a tumor-selective proagent of thymidylate synthase inhibitor 5-fluoro-2′-dUMP. 5-Fluoro-2'-deoxycytidine can induce cell cycle arrest in tumor cells through the DNA damage response, and it has anti-tumor activity .
|
-
- HY-176736
-
|
|
CDK
Apoptosis
|
Cancer
|
|
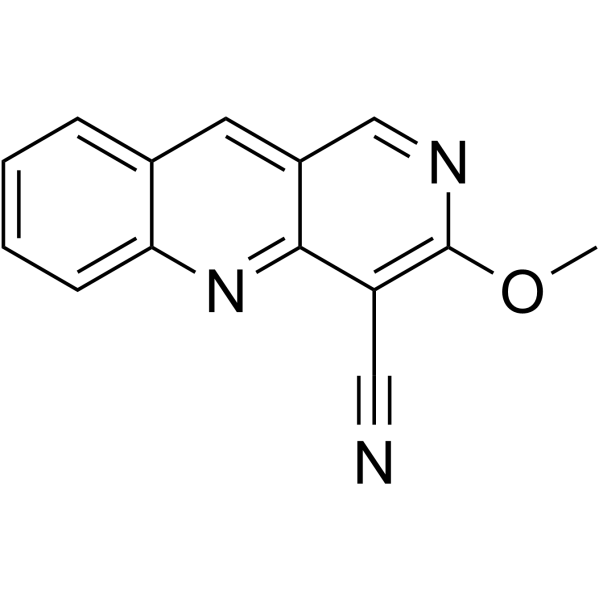
CDK9-IN-40 is a potent and orally active CDK9 inhibitor with an IC50 of 5.5 nM. CDK9-IN-40 shows high selectivity for CDK9 versus CDK1, CDK2, CDK4, and CDK6, respectively. CDK9-IN-40 can arrest cell cycle, induce cell apoptosis and inhibit tumor growth. CDK9-IN-40 exhibits strong anti-cancer activity .
|
-

- HY-169002
-
|
|
Phosphatase
|
Cancer
|
|
PP5-IN-2 is an orally active and selective protein phosphatase 5 (PP5) inhibitor with an IC50 value of 0.9 μM. PP5-IN-2 activates p53 and downregulates cyclin D1 and MGMT, which shows potency in cell cycle arrest and reverses Temozolomide (TMZ) (HY-17364) resistance in the U87 MG cell line. PP5-IN-2 effectively inhibits tumor growth in the xenograft mouse model .
|
-

- HY-15728S
-
|
IY-5511-d6
|
Isotope-Labeled Compounds
Bcr-Abl
Apoptosis
STAT
JAK
Prion Protein
|
Infection
Neurological Disease
Cancer
|
|
Radotinib-d6 is deuterium labeled Radotinib (HY-15728). Radotinib (IY-5511) is an orally active and BBB-permeable selective tyrosine kinase Bcr-Abl1 inhibitor with an IC50 of 34 nM. Radotinib has anti-prion and anti-tumor activities. Radotinib can inhibit the proliferation, induce cell cycle arrest and apoptosis of tumor cells . Radotinib can be used in the research of cancer such as chronic myeloid leukemia and multiple myeloma, as well as neurodegenerative diseases such as prion diseases .
|
-
- HY-150582
-
|
|
c-Met/HGFR
c-Kit
FLT3
Apoptosis
|
Cancer
|
|
c-Met-IN-14 (compound 26af) is a selective inhibitor of c-Met kinase from N-sulfonylamidine-based derivatives, with an IC50 value of 2.89 nM. c-Met-IN-14 shows anticancer activity by blocking phosphorylation of c-Met, and arrests cell cycle at G2/M phase. c-Met-IN-14 induces apoptosis of A549 cells in a dose-dependent manner .
|
-
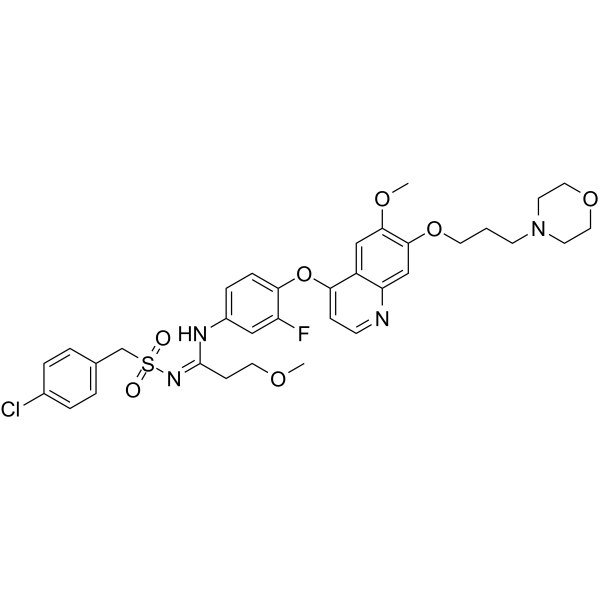
- HY-164129
-
|
|
CDK
Apoptosis
|
Cancer
|
|
CDK4-IN-3 (Compound 389) is a potent irreversible cyclin-dependent kinase 4 (CDK4) inhibitor (IC50=25 nM, >10-fold selective over CDK6). CDK4-IN-3 arrests the cell cycle at G₁ phase, and induces tumor cell apoptosis. CDK4-IN-3 is promising for research of solid tumors such as breast and lung cancers .
|
-

- HY-146980
-
|
|
Apoptosis
GLUT
|
Cancer
|
|
GLUT4-IN-2 is a potent and selective GLUT4 inhibitor with IC50s of 11.4 µM and 6.8 µM for GLUT1 and GLUT4, respectively. GLUT4-IN-2 induces cell apoptosis and cell cycle arrest at G0/G1phase. GLUT4-IN-2 shows potent antitumor activity .
|
-
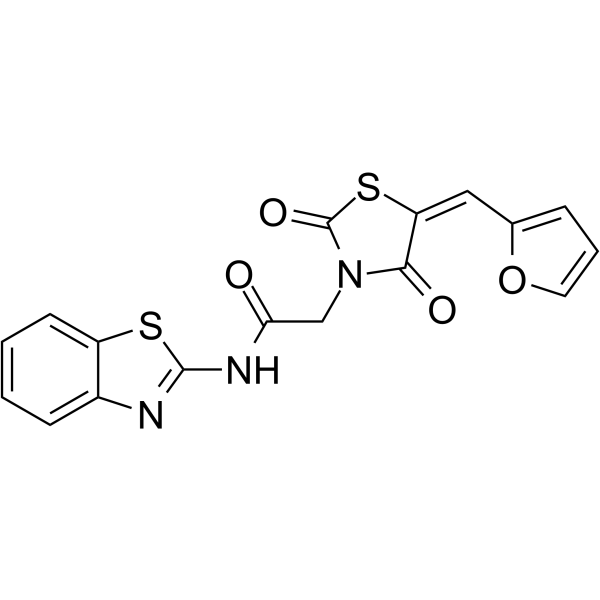
- HY-161372
-
|
|
PARP
c-Met/HGFR
Apoptosis
|
Cancer
|
|
PARP1/c-Met-IN-1 (Compound 16) is a selective dual inhibitor for PARP1 and c-Met, with IC50s of 3.3 and 32.2 nM, respectively. PARP1/c-Met-IN-1 induces cell apoptosis and cell cycle arrest in G2/M phase in MDA-MB-231 cells. PARP1/c-Met-IN-1 exhibits antitumor activity in mice .
|
-
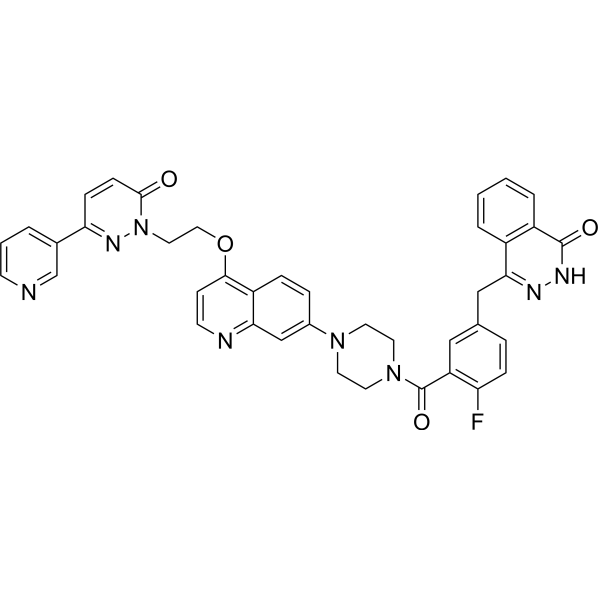
- HY-178150
-
|
|
Wee1
|
Cancer
|
|
WEE1-IN-14 (Compound 14) is a selective WEE1 inhibitor (IC50: 0.5 nM in L-RB-FEP calculations, 1.0 nM in ADP-Glo kinase assay). Inhibition of Wee1 in cancer cells disrupts the G2-M checkpoint, removes the regulatory controls on the cell cycle, and leads to early onset of mitotic failure followed by apoptosis of tumor cells. Therefore, WEE1-IN-14 is a useful tool for studying cancer biology .
|
-

- HY-147785
-
|
|
Pim
Caspase
Bcl-2 Family
Apoptosis
|
Cancer
|
|
Pim-1 kinase inhibitor 2 is a PIM-1 kinase inhibitor with a human IC50 of 0.63 μM. Pim-1 kinase inhibitor 2 exhibits high selectivity for cancer cells over normal cells. Pim-1 kinase inhibitor 2 induces apoptosis, increases active caspase-3 levels, upregulates BAX, downregulates Bcl-2, and elevates the Bax/Bcl-2 ratio. Pim-1 kinase inhibitor 2 suppresses cancer cell proliferation and induces cell cycle arrest. Pim-1 kinase inhibitor 2 can be used for the research of prostate carcinoma, hepatocellular carcinoma, and breast adenocarcinoma .
|
-
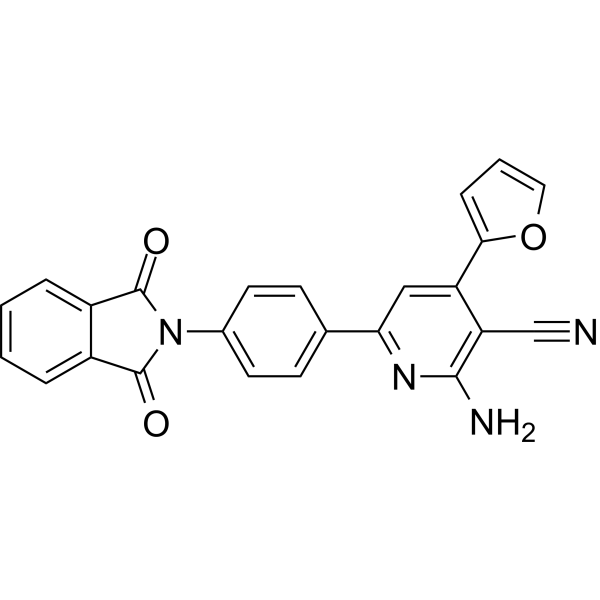
- HY-N14903
-
|
|
Antibiotic
Bacterial
|
Infection
Cancer
|
|
Oximidine III is an anti-tumor antibiotic. Oximidine III can selectively inhibit the growth of 3Y1 in rat fibroblasts with degeneration of various tumor genes. Oximidine III inhibits v-H-ras-3Y1, v-src-3Y1 cells and the normal 3Y1 cells with IC50s (nM) of 14, 4.5 and 140, respectively. Oximidine III stops RAS-or SRC-denatured cells at G1 phase of the cell cycle and increases p21WAF1 expression .
|
-

- HY-13260A
-
|
|
Akt
Autophagy
Apoptosis
|
Cancer
|
|
CCT128930 hydrochloride is a potent and selective inhibitor of AKT (IC50=6 nM). CCT128930 hydrochloride has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). CCT128930 hydrochloride induces cell cycle arrest, DNA damage, and autophagy. Antitumor activity .
|
-
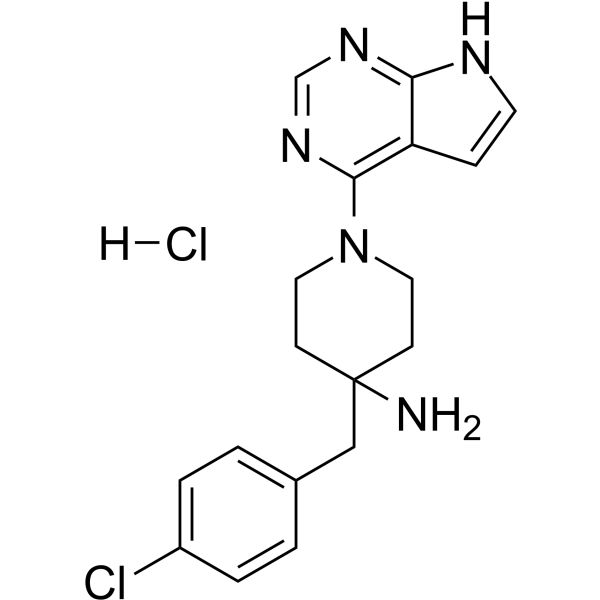
- HY-180200
-
|
|
Ras
ERK
|
Cancer
|
|
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1 cell cycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
|
-

- HY-131906
-
|
|
JAK
FLT3
Apoptosis
|
Cancer
|
|
JAK2-IN-7 is a selective JAK2 inhibitor with IC50s of 3, 11.7, and 41 nM for JAK2, SET-2, and Ba/F3 V617F cells, respectively. JAK2-IN-7 possesses >14-fold selectivity over JAK1, JAK3, FLT3. JAK2-IN-7 stimulates cell cycle arrest in the G0/G1 phase and induces tumor cellapoptosis. Antitumor activities .
|
-
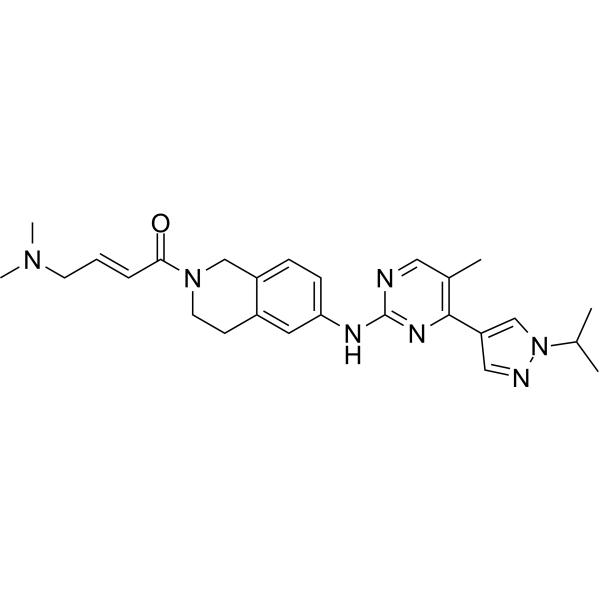
- HY-181979
-
|
|
HDAC
Apoptosis
|
Neurological Disease
Cancer
|
|
HDAC8-IN-15 is a selective HDAC8 inhibitor with an IC50 of 0.40 μM. HDAC8-IN-15 increases the acetylation level of the HDAC8 substrate SMC3 without altering the total protein level of SMC3. HDAC8-IN-15 reduces cancer cell viability, inhibits colony formation, slows cell migration, induces apoptosis, and causes cell cycle arrest at the SubG1 phase. HDAC8-IN-15 can be used in studies related to neuroblastoma .
|
-

- HY-150590
-
|
|
Proteasome
|
Cancer
|
|
20S Proteasome-IN-2 is a human 20S proteasome inhibitor. 20S Proteasome-IN-2 shows high selectivity to its β5 subunit with the IC50 of 0.18 μM. 20S Proteasome-IN-2 displays anti-proliferative effect in vitro and in vivo, and arrests cell cycle at G2/M .
|
-
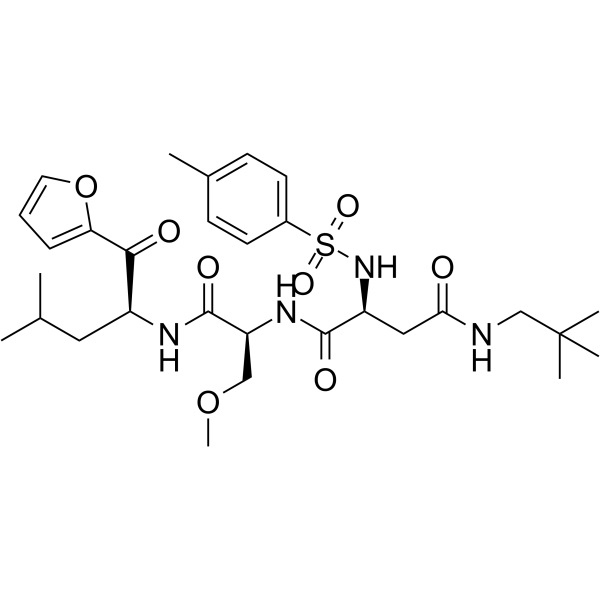
- HY-183297
-
|
|
CDK
|
Cancer
|
|
CDK4-IN-5 is a potent, orally active and selective CDK4 inhibitor. CDK4-IN-5 suppresses CDK4 expression and downregulates the CDK4/CyclinD1 complex. CDK4-IN-5 induces G0/G1 phase cell cycle arrest in bladder cancer cells via CyclinD1 expression suppression. CDK4-IN-5 selectively exerts activity against bladder cancer cells. CDK4-IN-5 can be used for the research of bladder cancer .
|
-

- HY-N2554
-
|
Ostenol
|
Monoamine Oxidase
PI3K
Akt
Apoptosis
|
Infection
Neurological Disease
Cancer
|
|
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cell cycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
|
-
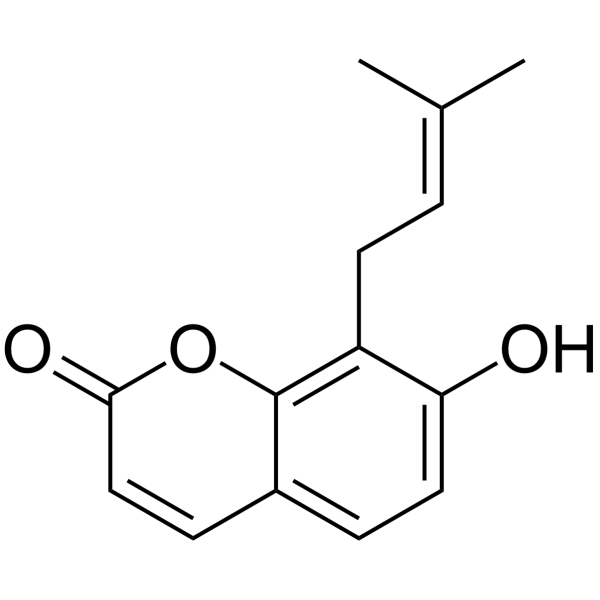
- HY-147646
-
|
|
CDK
Apoptosis
|
Cancer
|
|
CDK1/Cyc B-IN-1 (Compound 5) is a selective CDK1/Cyc B complex inhibitor with an IC50 of 97 nM. CDK1/Cyc B-IN-1 triggers apoptosis and G2/M cell cycle arrest. CDK1/Cyc B-IN-1 shows broad-spectrum cytotoxic action against cancer cell lines .
|
-
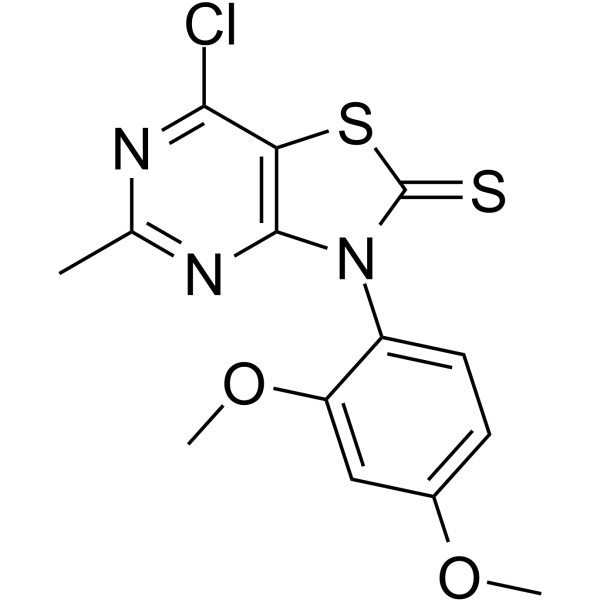
- HY-50767R
-
|
PD 0332991 (Standard)
|
Reference Standards
CDK
|
Cancer
|
|
Palbociclib (Standard) is the analytical standard of Palbociclib. This product is intended for research and analytical applications. Palbociclib (PD 0332991) is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-

- HY-148522
-
|
|
FLT3
|
Cancer
|
|
FLT3-IN-18 is a potent and selective FLT3 inhibitor with an IC50 value of 0.003 μM. FLT3-IN-18 induces apoptosis and cell cycle arrest at G1 phase. FLT3-IN-18 inhibits FLT3 and STAT5 phosphorylation. FLT3-IN-18 has the potential for the research of acute myeloid leukemia (AML) .
|
-
- HY-130841
-
|
|
APC
Ligands for Target Protein for PROTAC
|
Cancer
|
|
Apcin-A is a small molecule inhibitor that selectively targets the cell division cycle protein Cdc20 and is a derivative of Apcin (HY-110287). Apcin-A competitively binds to the D-box binding pocket of Cdc20 and inhibits substrate ubiquitination mediated by the anaphase promoting complex APC/C-Cdc20. Apcin-A also blocks the binding of Cdc20 to substrates (such as securin and cyclin B1), inhibiting anaphase initiation and cell cycle exit. Apcin-A can promote or prolong mitotic slippage in coordination with p31 comet under conditions of high spindle assembly checkpoint (SAC) activity. Apcin-A can be used to develop anti-mitotic drugs and overcome tumor chemotherapy resistance. Apcin-A can be used to synthesize PROTAC CP5V (HY-130257)[1][2][3].
|
-
- HY-124727
-
|
|
JAK
Apoptosis
|
Cancer
|
|
ZT55 is an orally active and highly-selective JAK2 inhibitor with an IC50 value of 0.031 μM. ZT55 inhibits the proliferation of JAK2 V617F-expressing HEL cell lines and induces apoptosis and cycle arrest. ZT-55 also effectively inhibits the growth of HEL xenograft tumours in a mice model. ZT-55 can be used in studies of myeloproliferative neoplasms, polycythemia vera and primary thrombocythemia .
|
-
- HY-10227S
-
|
PS-341-d8; LDP-341-d8; NSC 681239-d8
|
Isotope-Labeled Compounds
Proteasome
NF-κB
Apoptosis
Autophagy
TREM receptor
|
Cancer
|
|
Bortezomib-d8 is the deuterium labeled Bortezomib. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-
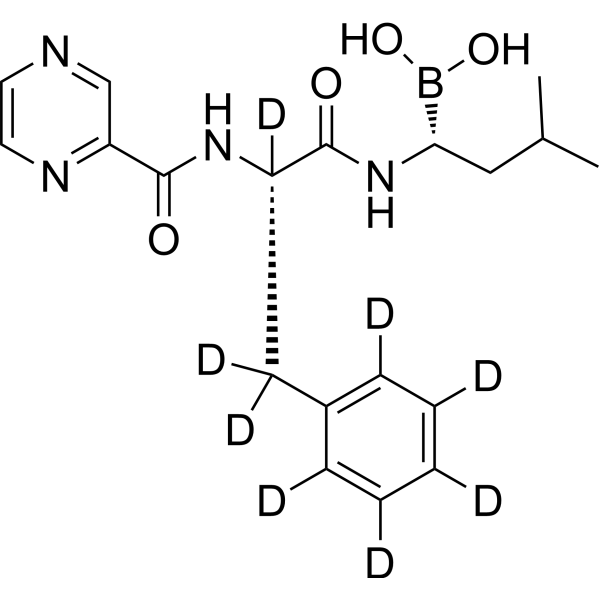
- HY-121382
-
|
|
Cholinesterase (ChE)
Apoptosis
Necroptosis
|
Neurological Disease
Cancer
|
|
Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
|
-
- HY-B1153R
-
|
Glafenin (Standard)
|
COX
CFTR
Apoptosis
Endoplasmic Reticulum Oxidoreductase 1 (ERO1)
Reference Standards
|
Metabolic Disease
Inflammation/Immunology
|
|
Glafenine (Standard) is the analytical standard of Glafenine. This product is intended for research and analytical applications. Glafenine (Glafenin) is a non-selective, non-steroidal anti-inflammatory drug-based COX-1/COX-2 inhibitor. Glafenine exerts anti-inflammatory, anti-proliferative and anti-cell migration effects by inhibiting the arachidonic acid metabolic pathway and reducing prostaglandin synthesis. Glafenine can induce cell cycle arrest in vascular smooth muscle cells and endothelial cells and reduce the synthesis of the extracellular matrix protein Tenascin. Glafenine can be used in the research of inflammatory-related diseases, vascular restenosis and cystic fibrosis (CF) .
|
-

- HY-B1153AR
-
|
Glafenin hydrochloride (Standard)
|
COX
CFTR
Apoptosis
Endoplasmic Reticulum Oxidoreductase 1 (ERO1)
Reference Standards
|
Metabolic Disease
Inflammation/Immunology
|
|
Glafenine (hydrochloride) (Standard) is the analytical standard of Glafenine (hydrochloride). This product is intended for research and analytical applications. Glafenine (Glafenin) hydrochloride is a non-selective, non-steroidal anti-inflammatory drug-based COX-1/COX-2 inhibitor. Glafenine hydrochloride exerts anti-inflammatory, anti-proliferative and anti-cell migration effects by inhibiting the arachidonic acid metabolic pathway and reducing prostaglandin synthesis. Glafenine hydrochloride can induce cell cycle arrest in vascular smooth muscle cells and endothelial cells and reduce the synthesis of the extracellular matrix protein Tenascin. Glafenine hydrochloride can be used in the research of inflammatory-related diseases, vascular restenosis and cystic fibrosis (CF) .
|
-

- HY-181491
-
|
|
Microtubule/Tubulin
EGFR
Akt
mTOR
Ras
Apoptosis
Autophagy
|
Cancer
|
|
Tubulin-IN-64 is a sulfonated styrylquinazoline derivative with high selectivity antitumor activity. Tubulin-IN-64 targets tubulin, inhibits the EGFR/Akt/mTOR and EGFR/Ras signaling pathways, induces cell cycle arrest, apoptosis and autophagy. Tubulin-IN-64 exhibits significant antitumor efficacy in the zebrafish GBM xenograft model. Tubulin-IN-64 can be used for the research on glioblastoma and leukemia .
|
-

- HY-10521R
-
|
SB-480848 (Standard)
|
Reference Standards
Phospholipase
Apoptosis
|
Cardiovascular Disease
Cancer
|
|
Darapladib (Standard) is the analytical standard of Darapladib. This product is intended for research and analytical applications. Darapladib (SB-480848) is an orally active, selective and reversible Lp-PLA2 inhibitor (IC50=0.25 nM). Darapladib can trigger irreversible actions on glioma cell apoptosis and induce cycle arrest. Darapladib can be used in the study of atherosclerosis and cancer .
|
-

- HY-169075
-
|
|
HDAC
CDK
Apoptosis
|
Cancer
|
|
CDK/HDAC-IN-4 is a high selective dual cyclin-dependent kinase (CDK)/histone deacetylase (HDAC) inhibitor with IC50 values of 88.4 and 168.9 nM, respectively. CDK/HDAC-IN-4 exhibits antiproliferative capacities against hematological and solid tumor cells. CDK/HDAC-IN-4 also induces MV-4-11 cell Apoptosis and S cell cycle arrests. CDK/HDAC-IN-4 possesses a significant antitumor potency in the MV-4-11 xenograft model .
|
-

- HY-182510
-
|
|
STAT
Interleukin Related
Apoptosis
|
Cancer
|
|
NTZ-24 is a selective STAT3 pathway inhibitor. NTZ-24 suppresses STAT3 phosphorylation at Tyr705, blocks STAT3-DNA interaction, and downregulates the levels of STAT3 downstream target proteins. NTZ-24 induces cell-cycle arrest and promotes apoptosis in cancer cells. NTZ-24 exerts significant antiproliferative activity against HeLa cells (IC50 = 3.3 μM). NTZ-24 can be used for the research of cervical cancer .
|
-

- HY-175553
-
|
|
VEGFR
Apoptosis
|
Cancer
|
|
VEGFR-2-IN-72 is a selective VEGFR-2 inhibitor with an IC50 of 22.2 nM. VEGFR-2-IN-72 exhibits anticancer activity against HepG2 and Hep3B human HCC cell lines with IC50s of 15.7 and 2.4 μM. VEGFR-2-IN-72 can cause a mild blockage of the cell cycle and induce cell aopoptosis. VEGFR-2-IN-72 can used for the study of hepatocellular carcinoma (HCC) .
|
-

- HY-184178A
-
|
|
Apoptosis
|
Cancer
|
|
HF-125 is an orally active, highly selective small-molecule inhibitor of Tribbles 2 (TRIB2). HF-125 promotes the destabilization and degradation of TRIB2 protein via the proteasome pathway. HF-125 downregulates neuroendocrine markers, induces cell cycle arrest and apoptosis, inhibits tumor cell colony formation and invasion, and reverses tumor cell resistance to Enzalutamide (HY-70002). HF-125 significantly inhibits tumor growth in SCID mouse xenograft models, and exerts synergistic inhibitory effects when combined with Enzalutamide. HF-125 can be used in research related to prostate cancer .
|
-

- HY-W181530
-
|
|
Molecular Glues
CDK
Apoptosis
Ligands for E3 Ligase
|
Cancer
|
|
NCT02 is a molecular glue degrader based on the E3 ubiquitin ligase DDB1 that targets CDK12 and its binding partner CCNK. NCT02 triggers the ubiquitination and proteasomal degradation of CCNK, thereby downregulating CDK12 protein levels and inhibiting its downstream signaling pathways. NCT02 can induce tumor cell apoptosis, arrest the cell cycle, and selectively inhibit the proliferation of colorectal cancer cells carrying TP53 defects or belonging to the consensus molecular subtype CMS4. NCT02 has the potential to inhibit tumor growth in in vitro and in vivo models .
|
-
- HY-132231
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
FD223 is a potent and selective phosphoinositide 3-kinase delta (PI3Kδ) inhibitor. FD223 displays high potency (IC50=1 nM) and good selectivity over other isoforms (IC50s of 51 nM, 29 nM and 37 nM, respectively for α, β and γ). FD223 exhibits efficient inhibition of the proliferation of acute myeloid leukemia (AML) cell lines by suppressing p-AKT Ser473 thus causing G1 phase arrest during the cell cycle. FD223 has potential for the research of leukemia such as AML .
|
-
- HY-101349
-
|
|
Dopamine Receptor
Apoptosis
Autophagy
PDGFR
ERK
mTOR
|
Neurological Disease
Cancer
|
|
L 741742 is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 induces G0/G1 cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cells in vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 can be used for the research of schizophrenia and glioblastoma .
|
-

- HY-101349A
-
|
|
Dopamine Receptor
Apoptosis
Autophagy
PDGFR
ERK
mTOR
|
Neurological Disease
Cancer
|
|
L 741742 hydrochloride is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 hydrochloride suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 hydrochloride induces G0/G1 cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cells in vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 hydrochloride can be used for the research of schizophrenia and glioblastoma .
|
-
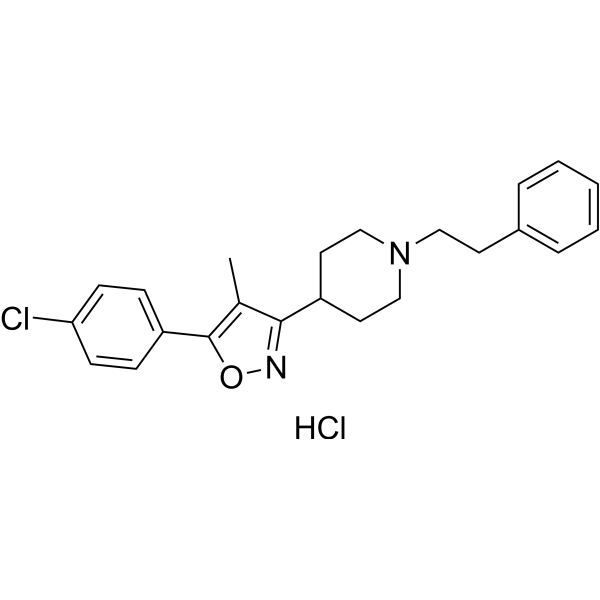
- HY-157941
-
|
|
ATM/ATR
Checkpoint Kinase (Chk)
DNA/RNA Synthesis
|
Cancer
|
|
ART0380 is a potent, selective and orally active ATR kinase inhibitor. ART0380 potently inhibits human ATR-ATRIP complex with an IC50 of 51.7 nM. ART0380 binds the ATP pocket of the ATR-ATRIP complex, blocks ATR-dependent Chk1 serine 345 phosphorylation, and induces cell cycle disorder and DNA damage. ART0380 demonstrates potent and selective antitumor activity in preclinical models with varying types of ataxia-telangiectasia mutated (ATM) gene aberrancy. ART0380 can be used for the research of cancer, such as colorectal cancer and prostate cancer .
|
-
- HY-169299
-
|
|
PARP
Topoisomerase
Apoptosis
|
Cancer
|
|
TOPOI/PARP-1-IN-2 (compound 6c) is a dual PARP-1 and topoisomerase 1 (TOPO-1) inhibitor with IC50s of 32.2 nM and 46.2 nM, respectively. TOPOI/PARP-1-IN-2 shows a selectivity for PARP-1 over PARP-2. TOPOI/PARP-1-IN-2 disrupts the cell cycle at the S phase and induces apoptosis in NCI-60 cancer cell lines .
|
-

- HY-168072
-
|
|
Interleukin Related
Apoptosis
Autophagy
|
Cancer
|
|
GP130-IN-1 (compound 49) is a potent GP130 inhibitor with significant in vitro antitumor activity and higher selectivity than Bazedoxifene (HY-A0031). GP130-IN-1 induces ultrastructural changes in cells, causing cell cycle arrest in the G0/G1 phase in a time-dependent manner and triggering apoptosis and autophagy. GP130-IN-1 can be used in the study of triple-negative breast cancer .
|
-

- HY-113914
-
|
Elraglusib
|
GSK-3
Apoptosis
Autophagy
|
Cancer
|
|
9-ING-41 (Elraglusib) is a maleimide-based ATP-competitive and selective glycogen synthase kinase-3β (GSK-3β) inhibitor with an IC50 of 0.71 μM. 9-ING-41 significantly leads to cell cycle arrest, autophagy and apoptosis in cancer cells. 9-ING-41 has anticancer activity and has the potential for enhancing the antitumor effects of chemotherapeutic agents .
|
-
- HY-50767AR
-
|
PD 0332991 monohydrochloride (Standard)
|
CDK
Reference Standards
|
Cancer
|
|
Palbociclib (monohydrochloride) (Standard) is the analytical standard of Palbociclib (monohydrochloride). This product is intended for research and analytical applications. Palbociclib (PD 0332991) monohydrochloride is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib monohydrochloride has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-

- HY-144293
-
|
|
Apoptosis
HDAC
|
Cancer
|
|
HDAC-IN-31 is a potent, selective and orally active HDAC inhibitor with IC50s of 84.90, 168.0, 442.7, >10000 nM for HDAC1, HDAC2, HDAC3, HDAC8, respectively. HDAC-IN-31 induces apoptosis and cell cycle arrests at G2/M phase. HDAC-IN-31 shows good antitumor efficacy. HDAC-IN-31 has the potential for the research of diffuse large B-cell lymphoma .
|
-
- HY-10227G
-
|
PS-341; LDP-341; NSC 681239
|
Proteasome
NF-κB
Apoptosis
Autophagy
TREM receptor
|
Cancer
|
|
Bortezomib (GMP) (PS-341 (GMP)) is Bortezomib (HY-10227) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-

- HY-175817
-
|
|
Wee1
Apoptosis
|
Cancer
|
|
PKMYT1-IN-10 is a selective PKMYT1 inhibitor with an IC50 of 3 nM. PKMYT1-IN-10 inhibits colony formation, induces apoptosis, and induces S-phase cancer cell cycle arrest. PKMYT1-IN-10 exhibits liver microsomal stability, favorable plasma stability, minimal CYPs inhibition. PKMYT1-IN-10 can be used for the studies of ovarian cancer and breast cancer .
|
-

- HY-28086
-
|
|
Phosphatase
|
Cancer
|
|
CDC25-IN-2 (Compound 1d) is a selective phosphatase Cdc25 inhibitor with IC50 values of 44.4, 55.2 and 53.9 μM for human Cdc25A, Cdc25B, Cdc25C. CDC25-IN-2 can block the cell cycle progression by inhibiting the activity of Cdc25 phosphatase. CDC25-IN-2 can be used for the research of cancer .
|
-

- HY-107592
-
|
|
IKK
STAT
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
ACHP (compound 4j) is a selective and orally active IκB kinase inhibitor with IC50 values of 8.5 nM and 250 nM for IKKβ and IKKα, respectively. ACHP can effectively inhibit the STAT3 signaling pathway and induce cancer cell cycle arrest and apoptosis. ACHP shows anti-inflammatory activity in a mouse ear edema model. ACHP can be used in anti-inflammatory and anti-cancer (such as multiple myeloma and leukemia) studies .
|
-
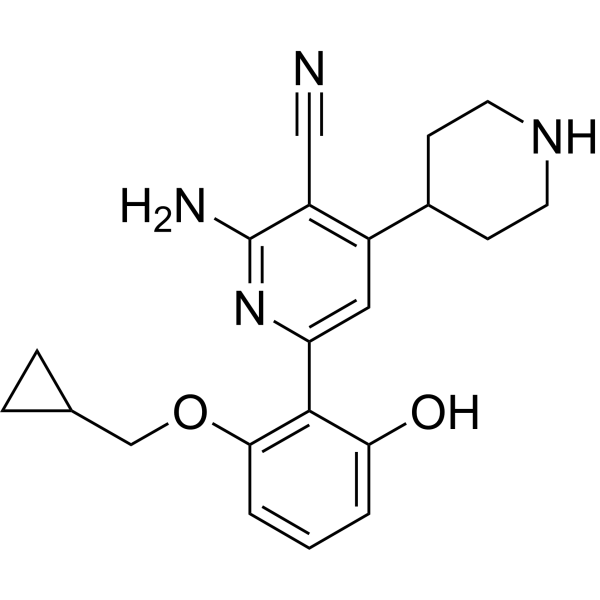
- HY-10325
-
|
EKI-785; WAY-EKI 785
|
EGFR
Apoptosis
|
Endocrinology
Cancer
|
|
CL-387785 (EKI-785; WAY-EKI 785) is an orally active EGFR inhibitor with an IC50 of 370 pM. CL-387785 inhibits EGF-stimulated EGFR autophosphorylation with an IC50 of approximately 5 nM. CL-387,785 exerts selective inhibition on cell lines overexpressing EGFR or c-erbB-2, whereas it shows weak inhibitory effects on cell lines with low expression of these two receptors. CL-387785 effectively induces cell cycle arrest and apoptosis. CL-387785 can be used for the research of non-small cell lung cancer and autosomal recessive polycystic kidney disease .
|
-
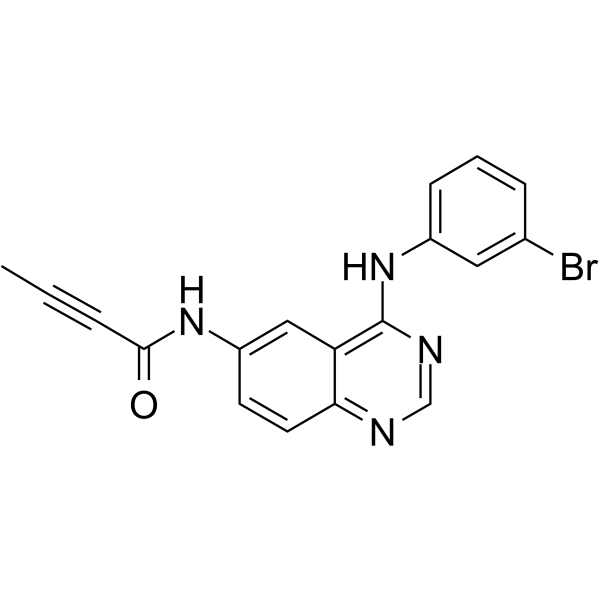
- HY-10227R
-
|
PS-341 (Standard); LDP-341 (Standard); NSC 681239 (Standard)
|
Reference Standards
Proteasome
NF-κB
Apoptosis
Autophagy
TREM receptor
|
Cancer
|
|
Bortezomib (Standard) is the analytical standard of Bortezomib. This product is intended for research and analytical applications. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-

- HY-183767
-
-

- HY-124012
-
|
|
DNA/RNA Synthesis
Apoptosis
|
Cancer
|
|
PCNA-I1 is a selective small molecule inhibitor targeting proliferating cell nuclear antigen (PCNA) with anticancer activity. PCNA-I1 can stabilize the PCNA trimer structure (Kd=0.14-0.41μM), reduce its binding to chromatin, induce tumor cell cycle arrest, inhibit DNA replication and repair, and enhance the anti-tumor effect of DNA damaging agents. PCNA-I1 can be used in the study of targeted therapy for prostate cancer, lung cancer and other tumors .
|
-
- HY-101029A
-
|
|
NEKs
Ribosomal S6 Kinase (RSK)
DYRK
Apoptosis
|
Cancer
|
|
MBM-55S is a potent NIMA-related kinase 2 (Nek2) inhibitor with an IC50 of 1 nM. MBM-55S shows a 20-fold or greater selectivity in most kinases with the exception of RSK1 (IC50=5.4 nM) and DYRK1a (IC50=6.5 nM). MBM-55S effectively inhibits the proliferation of cancer cells by inducing cell cycle arrest and apoptosis. MBM-55S shows antitumor activities, and no obvious toxicity to mice .
|
-
- HY-151154
-
|
|
EGFR
|
Cancer
|
|
EGFR/HER2/DHFR-IN-1 is a potent anticancer agent with high selectivity against MCF-7 breast cancer cells. EGFR/HER2/DHFR-IN-1 is a multiple inhibitor of EGFR/HER2 kinase and DHFR, with IC50s of 0.153 μM, 0.108 μM, 0.291 μM, respectively. EGFR/HER2/DHFR-IN-1 arrests cell cycle at G1/S and induces cells apoptosis .
|
-
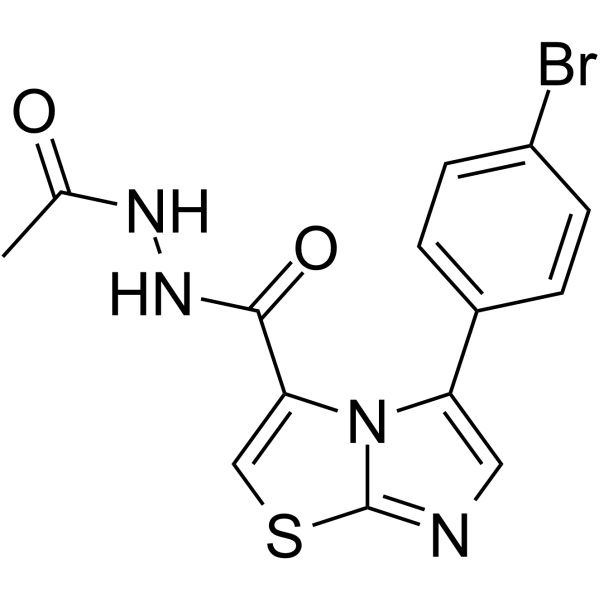
- HY-175208
-
|
|
Hippo (MST)
Caspase
Bcl-2 Family
Apoptosis
|
Cancer
|
|
MST3-IN-1 is a selective and orally active MST3 inhibitor, with an IC50 of 122.4 nM. MST3-IN-1 shows antiproliferative activity against HepG2 cell. MST3-IN-1 effectively induces apoptosis in HepG2 cells, and halts the cell cycle at the G2/M transition. MST3-IN-1 significantly suppressed tumor growth in HepG2 xenograft mice. MST3-IN-1 can be used for the study of liver cancer .
|
-

- HY-136910
-
|
USP7-IN-7
|
Deubiquitinase
MDM-2/p53
|
Cancer
|
|
USP7-797 (USP7-IN-7) is an orally available, selective USP7 inhibitor (IC50=0.5 nmol/L) with antitumor activity. USP7-797 reduces the level of MDM2, thereby increasing the stability and activity of p53, leading to cell cycle arrest and apoptosis. USP7-797 has low nanomolar cytotoxicity against p53 mutant cancer cell lines, p53 wild-type hematological tumors, and neuroblastoma cell lines .
|
-
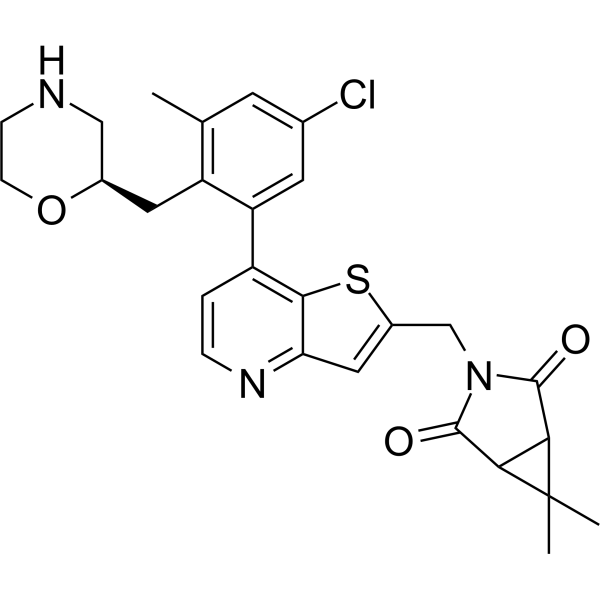
- HY-183098
-
|
|
PROTACs
IKK
Apoptosis
|
Cancer
|
|
UNC8209 is a selective PROTAC-based TANK-binding kinase 1 (TBK1) degrader. UNC8209 recruits cereblon (CRBN) to mediate ubiquitin-proteasome pathway-dependent TBK1 degradation and reduces AAK1, GAK, and AURKA abundance. UNC8209 suppresses tumor cell proliferation, impairs in vivo tumor growth, inhibits colony and clonogenic growth and enhances tumor cell sensitivity to TNFα or IFN-γ. UNC8209 modulates cell cycle and induces mild apoptosis. UNC8209 can be used for the research of clear cell renal cell carcinoma, non-small cell lung cancer, pancreatic ductal adenocarcinoma .
|
-

- HY-N12257
-
|
|
Cytochrome P450
Reactive Oxygen Species (ROS)
Apoptosis
|
Infection
Cancer
|
|
Antimycin A2 is a selective inhibitor of the cytochrome b-c1 complex in the mitochondrial electron transport chain. Antimycin A2 disrupts mitochondrial membrane potential and produces reactive oxygen species (ROS) by inhibiting electron transfer between cytochrome b and c. Antimycin A2 has bactericidal and piscicidal activity, as well as tumor cell growth inhibitory effects, and can induce S-phase cell cycle arrest and apoptosis in HeLa cells. Antimycin A2 is suitable for research of cervical cancer and fisheries management. Antimycin A2 can be naturally isolated from the fermentation products of Streptomyces sp. strains .
|
-

- HY-182005
-
|
|
EGFR
VEGFR
|
Cancer
|
|
EGFR/VEGFR2-IN-12 is a dual EGFR/VEGFR2 inhibitor, with an IC50 value of 64 nM against human EGFR and an IC50 value of 74 nM against human VEGFR2. EGFR/VEGFR2-IN-12 inhibits the phosphorylation of EGFR and VEGFR2, induces cell cycle arrest at the G1/S phase, activates apoptotic pathways, promotes PARP-1 cleavage, exhibits low micromolar antiproliferative activity, and shows much higher selectivity for cancer cells than normal cells. EGFR/VEGFR2-IN-12 is applicable for cancer-related research .
|
-

- HY-182914
-
|
|
EGFR
Akt
Bcl-2 Family
Apoptosis
|
Cancer
|
|
NGI‑189 is a selective OST‑A inhibitor. NGI‑189 inhibits the STT3A catalytic subunit of the OST complex and reduces N‑glycosylation of target glycoproteins. NGI‑189 blocks oncogenic and bypass signaling, reduces phosphorylation of EGFR, AKT, p70S6K and S6RP, and induces cell cycle arrest and apoptosis. NGI‑189 markedly suppresses tumor growth and induces tumor regression in non‑small cell lung cancer (NSCLC) xenograft models. NGI‑189 can be used for the research of EGFR‑mutant non‑small cell lung cancer .
|
-

- HY-156792
-
|
|
RIO Kinase
|
Cancer
|
|
RIOK2-IN-1 (com 4) is a potent and selective RIOK2 inhibitor (Kd=150 nM), but has low cellular activity (IC50=14,600 nM). RIOK2 is an atypical kinase associated with a variety of human cancers and is involved in ribosome maturation and cell cycle progression. The small molecule inhibitor CQ211 (HY-147655), an improvement of RIOK2-IN-1 as the lead compound, has good in vivo and in vitro activity, inhibits the proliferation of MKN-1 and HT-29 cancer cells, and can xenograft MKN in mice -1 model inhibits tumor progression .
|
-
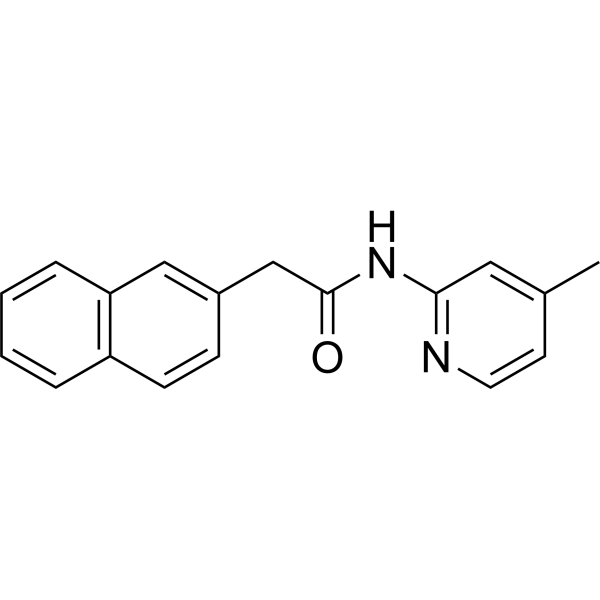
- HY-179019
-
|
|
HDAC
Microtubule/Tubulin
Apoptosis
Caspase
|
Neurological Disease
Cancer
|
|
HDAC-IN-94 is a potent, selective HDAC6 inhibitor (IC50 = 4.5 nM). HDAC-IN-94 shows >1000-fold selectivity over HDAC8 and shows minimal activity against other isoforms (HDAC1-3/10). HDAC-IN-94 induces α-tubulin hyperacetylation, apoptosis, and G2/M cell cycle arrest, exhibiting potent anti-tumor efficacy with low cytotoxicity. HDAC-IN-94 can be used for neuroblastoma and glioblastoma research .
|
-

- HY-100900
-
|
|
Deubiquitinase
|
Cancer
|
|
ML364 is a selective ubiquitin specific peptidase 2 (USP2) inhibitor (IC50=1.1 μM) with anti-proliferative activity, which direct binds to USP2 (Kd=5.2 μM), induces an increase in cellular cyclin D1 degradation and causes cell cycle arrest. ML364 increases the levels of mitochondrial ROS and decreases in the intracellular content of ATP .
|
-
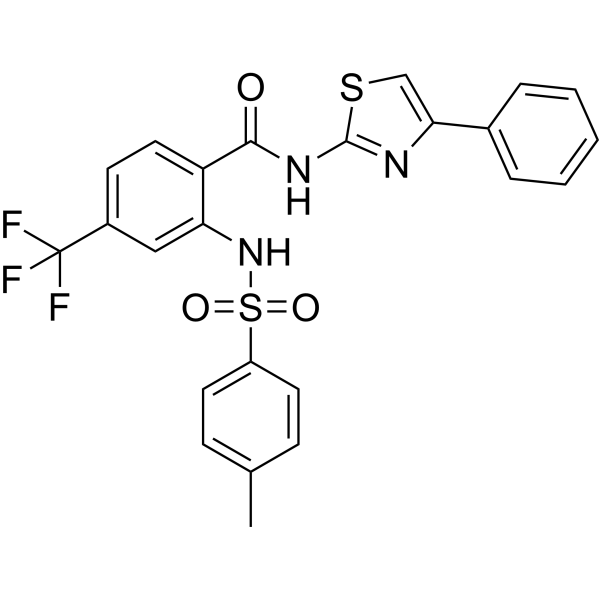
- HY-115909
-
|
|
CDK
|
Cancer
|
|
ZDLD20, a β-carboline, is orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 6.51 μM. ZDLD20 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cell cycle. ZDLD20 exhibits potent anticancer activity .
|
-
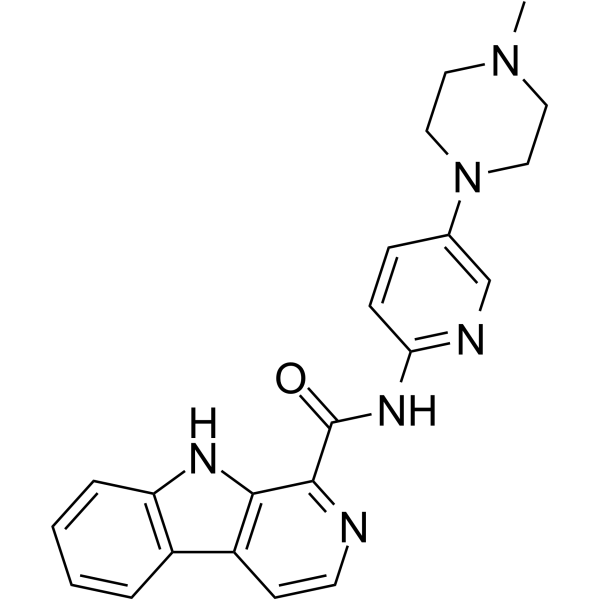
- HY-150511
-
|
|
Microtubule/Tubulin
Apoptosis
|
Cancer
|
|
3-(3-Phenoxybenzyl)amino-β-carboline is a potent tubulin inhibitor. 3-(3-Phenoxybenzyl)amino-β-carboline promotes selective degradation of αβ-tubulin heterodimers. 3-(3-Phenoxybenzyl)amino-β-carboline induces G2/M phase cell cycle arrest and apoptosis. 3-(3-Phenoxybenzyl)amino-β-carboline exhibits anticancer activity .
|
-
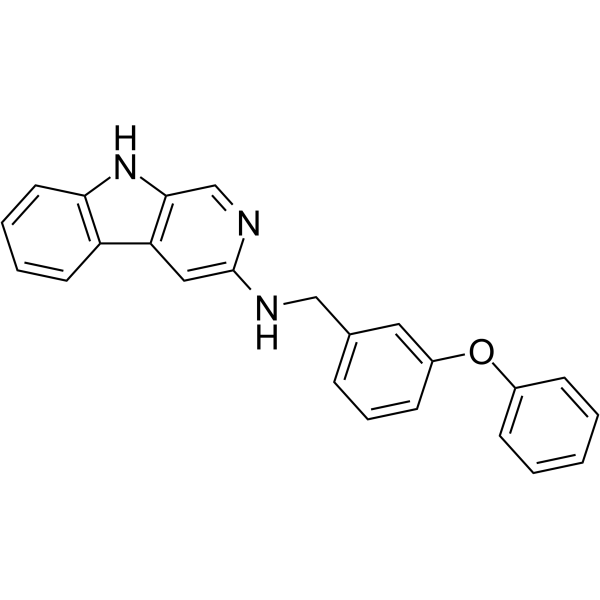
- HY-115589
-
|
|
Polo-like Kinase (PLK)
Apoptosis
|
Cancer
|
|
YLT-11 is a potent, selective and orally active PLK4 inhibitor with Kd values of >10000, 653, >10000, 5.2 nM for PLK1, PLK2, PLK3, PLK4, respectively. YLT-11 shows antiproliferative activity. YLT-11 induces Apoptosis and cell cycle arrest at G2/M phase. YLT-11 show anticancer activity .
|
-
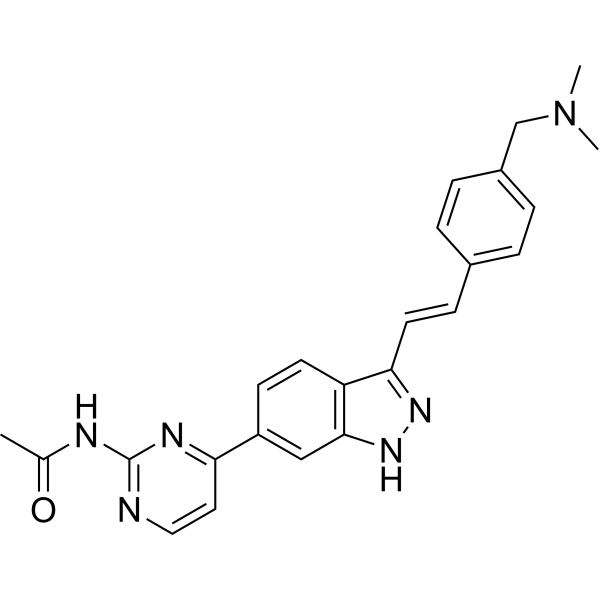
- HY-132844
-
|
HL-085
|
MEK
|
Cancer
|
|
Tunlametinib is a highly selective, orally active MEK1/2 inhibitor (IC50=1.9 nM, MEK1). Tunlametinib blocks the RAS-RAF-MEK-ERK signaling pathway, arrests tumor cell cycle and promotes apoptosis. Tunlametinib potently inhibits the proliferation of RAS/RAF mutant cancer cells (such as BRAF V600E, KRAS G12C mutant cells). Tunlametinib shows synergistic anti-tumor effects with BRAF/KRASG12C/SHP2 inhibitors, Docetaxel (HY-B0011). Tunlametinib can be used to study targeted therapy for RAS/RAF mutation-driven malignancies (such as melanoma, colorectal cancer, and non-small cell lung cancer) .
|
-
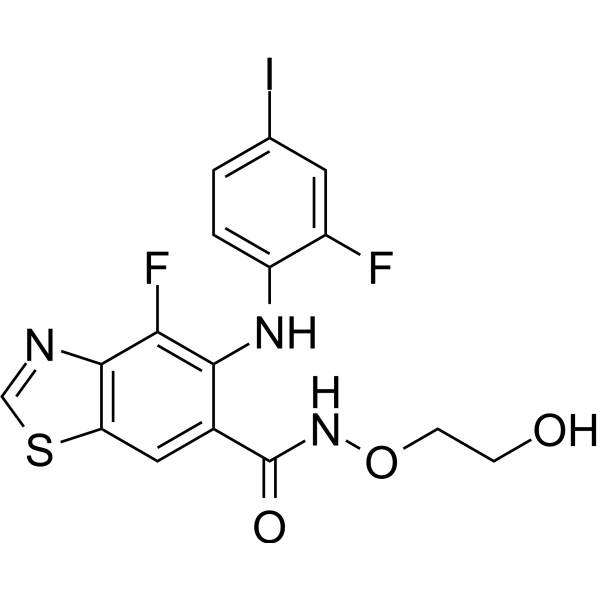
- HY-174302
-
|
|
Pim
HDAC
PARP
Apoptosis
|
Cancer
|
|
PIM-1/HDAC-IN-2 is a robust PIM/HDAC inhibitor (IC50 = 0.11 μM in MV4-11cells), which exerts a synergistic antiproliferative effect through a dual mechanism of inhibiting PIM1 kinase and selectively inhibiting HDAC6. PIM-1/HDAC-IN-2 induces cell apoptosis. PIM-1/HDAC-IN-2 remarkably induces the cleavage of PARP, thereby initiating the arrest of the cell cycle in G1 phase and a reduction in S phase. PIM-1/HDAC-IN-2 demonstrates significant anticancer efficacyin the MV4-11 xenograft model without notable toxicity[1].
|
-

- HY-180117
-
|
|
MMP
STAT
Apoptosis
|
Cancer
|
|
MMP-2/9-IN-2 (Compound 6k) is a MMP-2 and MMP-9 inhibitor, with IC50 values of 29.27 and 24.87 μM respectively. MMP-2/9-IN-2 exhibits good selective toxicity against multiple human hepatoma cell lines. MMP-2/9-IN-2 induces cell cycle arrest and apoptosis, significantly inhibits cell migration and invasion. MMP-2/9-IN-2 inhibits the phosphorylation of the STAT3 signaling pathway. MMP-2/9-IN-2 shows strong anti-tumor activity in a nude mouse xenograft model of HepG2 liver cancer cells .
|
-

- HY-159175
-
|
|
PROTACs
Deubiquitinase
Apoptosis
MDM-2/p53
|
Cancer
|
|
XM-U-14 is a selective PROTAC USP7 Degrader (DC50: 0.74 nM in inducing USP7 degradation in RS4;11 cell line). XM-U-14 upregulates the levels of p53 and p21. XM-U-14 also significantly inhibits acute lymphoblastic leukemia (ALL) cell growth (IC50: 0.5 nM and 8.3 nM for RS4;11 cells and Reh cells respectively). XM-U-14 induces apoptosis and cycle arrest. XM-U-14 inhibits tumor growth. (Blue: VHL ligand (HY-159465), Black: linker (HY-W539783); Pink: USP7 inhibitor (HY-159464)) .
|
-

- HY-180574
-
|
|
DYRK
|
Cancer
|
TSL2109 is an orally active and selective DYRK2 and CDK4/6 inhibitor with an IC50 value of 22 nM for DYRK2. TSL2109 exhibits high kinase selectivity over 93%. TSL2109 arrests cell cycle and induces apoptosis in virto. TSL2109 effectively overcomes Enzalutamide (HY-70002) resistance by suppressing tumor growth in vivo and virto. TSL2109 also shows CDK4/6 inhibitor resistance.TSL2109 demonstrates safety profile. TSL2109 can be used for prostate cancer research and breast cancer [1][2].
|
-

- HY-170921
-
|
|
Deubiquitinase
|
Cancer
|
|
USP1-IN-11 (compound 38-P2) is a selective, reversible, and noncompetitive USP1 (Ubiquitin-specific protease 1) inhibitor. USP1-IN-11 activates the DDR (DNA damage repair) pathway, induces cell cycle arrest and cell Apoptosis, and inhibits cell survival. USP1-IN-11 enhances the sensitivity of Olaparib (HY-10162)-resistant cells to Olaparib (HY-10162) and shows a synergetic effect with Andrographolide (HY-N0191) in BRCA-proficient cancer cells. USP1-IN-11 displays significant, dose-dependent antitumor efficacy in the MDA-MB-436 xenograft model .
|
-

- HY-182758
-
|
|
Topoisomerase
Apoptosis
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
Topoisomerase I-IN-21 is a promising topoisomerase I inhibitor with an IC50 of 18.79 μM. Topoisomerase I-IN-21 shows higher selectivity toward cancer cells over normal CD8 + cells. Topoisomerase I-IN-21 induces G0/G1 cell cycle arrest and apoptosis. Topoisomerase I-IN-21 activates the cGAS-STING pathway, leading to enhanced immune gene expression. Topoisomerase I-IN-21 can be used for research on leukemia, non-small-cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate, and breast cancers .
|
-

- HY-175002
-
|
|
PI3K
Akt
Apoptosis
|
Cancer
|
|
PI3Kδ-IN-24 is a selective PI3Kδ inhibitor (IC50 = 0.1 nM). PI3Kδ-IN-24 exhibits significant antiproliferative effects against PI3Kδ-overexpressing cancer cell lines. PI3Kδ-IN-24 reduces p-AKT levels and induces cell cycle arrest and apoptosis in tumor cells. PI3Kδ-IN-24 is useful in cancer research, such as diffuse large B-cell lymphoma (DLBCL) .
|
-

- HY-179375
-
|
|
CDK
Aurora Kinase
|
Cancer
|
|
LCI133 is afirst-in-class,nanomolar-potent, selective multikinase inhibitor targeting CDK4/6/9 and AURKA/B (IC50 = 4.7/10.2/4.1 nM and 2.8/10.6 nM). LCI133 induces S/G2 cell-cycle arrest and robust apoptosis in MYCN-amplified neuroblastoma BE(2)-C cells. LCI133 demonstrates significant antitumor efficacy in a BE(2)-C neuroblastoma xenograft model .
|
-

- HY-148712
-
|
|
Apoptosis
Sirtuin
|
Cancer
|
|
SIRT6 activator 12q is potent, selective and orally active SIRT6 activator with IC50 values of 171.20, >200, >200, >200, 0.58 μM for SIRT1, SIRT2, SIRT3, SIRT5, SIRT6, respectively. SIRT6 activator 12q inhibits cell growth and migration. SIRT6 activator 12q induces Apoptosis and cell cycle arrest at G2 phase. SIRT6 activator 12q shows anticancer activity .
|
-
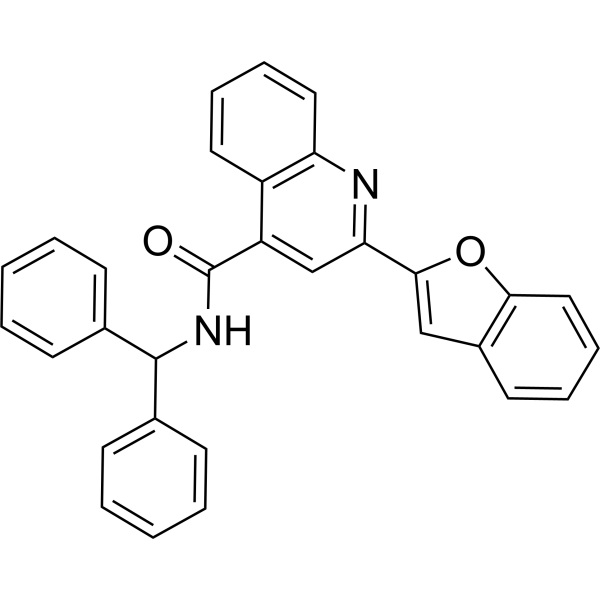
- HY-18200A
-
|
E5555 hydrochloride; ER-172594-00 hydrochloride
|
Protease Activated Receptor (PAR)
JAK
Apoptosis
|
Cardiovascular Disease
Cancer
|
|
Atopaxar hydrochloride (E5555 hydrochloride) is the hydrochloride salt form of Atopaxar (HY-18200). Atopaxar hydrochloride is an orally active, selective and reversible antagonist for thrombin receptor protease-activated receptor-1 (PAR-1). Atopaxar hydrochloride is the inhibitor for Janus kinase 1 (JAK1) and JAK2, which inhibits the JAK-STAT with EC50 of 5.90 μM in A549. Atopaxar hydrochloride inhibits the cell viability of A549 (IC50=7.02 μM), arrests the cell cycle at G1 phase and induces apoptosis. Atopaxar hydrochloride exhibits antiplatelet and antitumor activities. Atopaxar hydrochloride can be used for the research of atherothrombotic disease .
|
-

- HY-157486
-
|
|
Epigenetic Reader Domain
|
Cancer
|
|
KMI169 is a potent and selective inhibitor targeting lysine methyl-transferase (KMT9) (IC50 = 0.05 μM, Kd = 0.025 μM). KMI169 functions as a bi-substrate inhibitor targeting the cofactor S-5’-adenosyl-L-methionine (SAM) and substrate binding pockets of KMT9. KMI169 can downregulate target genes involved in cell cycle regulation and impair proliferation of tumor cells by inhibiting KMT9. KMI169 is a valuable tool to probe cellular KMT9 functions and can be research for combating diseases including prostate, lung, colon, and invasive bladder cancer .
|
-
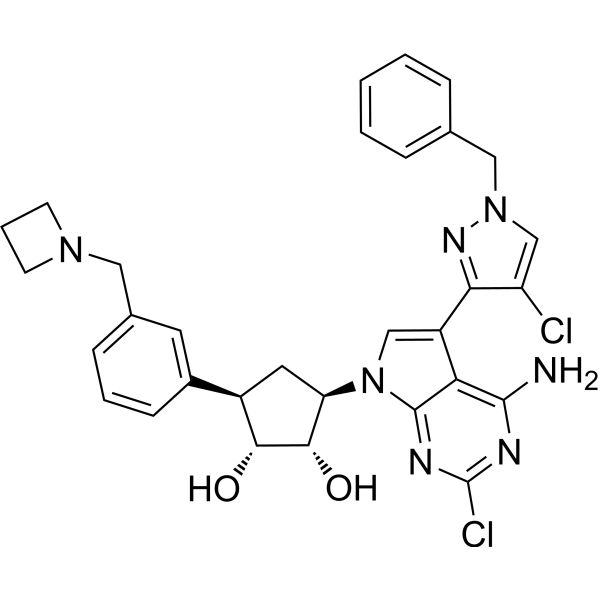
- HY-185011
-
|
|
Ceramidase
PPAR
TRP Channel
Interleukin Related
NEDD8-activating Enzyme
|
Inflammation/Immunology
Cancer
|
|
AM9053 is a selective, effective and slowly reversible inhibitor of N-acyl ethanolamine acid amidease (NAAA) (IC50 = 30 nM). The effect of AM9053 on FAAH activity is limited (IC50 > 100 nM). AM9053 inhibits the proliferation of colorectal cancer cells by activating the PPAR-α and TRPV1 dependent mechanisms and induces S-phase cell cycle arrest. AM9053 alleviates intestinal fibrosis by regulating macrophage activity and inhibiting the IL-23 signaling pathway. AM9053 leads to an increase in NAE levels, especially PEA and OEA. AM9053 can be used for the study of colorectal cancer and intestinal fibrosis .
|
-

- HY-155218
-
|
|
PROTACs
CDK
|
Cancer
|
|
TMX-2172 is a selective bivalent cereblon-recruiting PROTAC-based dual CDK2 and CDK5 degrader with IC50 values of 6.5 nM and 6.8 nM, respectively. TMX-2172 shows selectivity for CDK2 and CDK5 over other cell cycle CDKs (CDK1, CDK4, and CDK6) and transcriptional CDKs (CDK7 and CDK9). TMX-2172 inhibits CDK2/CDK5 enzymatic activity, induces their proteasomal degradation, reduces ASCL1 protein levels and half-life, induces cancer cell death, and exerts antiproliferative effects. TMX-2172 can be used for the research of ovarian cancer and small cell lung cancer .
|
-

- HY-100513
-
|
|
DNA/RNA Synthesis
Apoptosis
Antibiotic
|
Cancer
|
|
(±)-Dehydroaltenusin, an antibiotic, is a selective eukaryotic DNA polymerase α (pol α) inhibitor with an IC50 of 0.68 μM. (±)-Dehydroaltenusin can be isolated from fungus Alternaria tenuis. (±)-Dehydroaltenusin competitively inhibits the DNA template primer (Ki: 0.23 μM) and non-competitively suppresses the 2'-deoxyribonucleoside 5'-triphosphate substrate (Ki: 0.18 μM). (±)-Dehydroaltenusin induces the cancer cell S-phase cycle arrest and apoptosis. (±)-Dehydroaltenusin can be used for cancers like human adenocarcinoma research .
|
-

- HY-171785
-
|
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
CHK1-IN-12 (Compound example 1-5) is an orally active and highly selective checkpoint kinase 1 (CHK1) inhibitor with in vitro enzyme IC50≤10 nM and cellular IC50≤50 nM. CHK1-IN-12 inhibits the phosphorylation activity of CHK1 kinase to block the DNA damage response pathway, inducing tumor cell cycle arrest and apoptosis. CHK1-IN-12 is promising for research of cancers .
|
-

- HY-155489
-
|
|
Anaplastic lymphoma kinase (ALK)
Apoptosis
|
Cancer
|
|
DDO-2728 (compound 19) is a selective AlkB homologue 5 (ALKBH5) inhibitor with an IC50 of 2.97 μM. DDO-2728 increases the abundance of N 6 methyladenosine (m 6A) modifications, inducing cell apoptosis and cycle arrest. DDO-2728 suppresses tumor growth in the MV4 11 xenograft model with favorable safety profile, shows the potential of targeting ALKBH5 in cancer research .
|
-
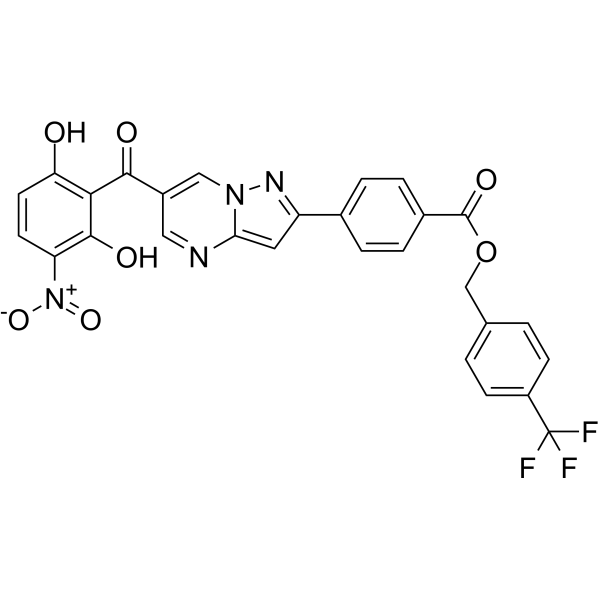
- HY-170402
-
|
|
Sirtuin
Apoptosis
|
Cancer
|
|
SIRT6-IN-4 (Compound 10d) is a selective inhibitor for SIRT6 with an IC50 of 5.68 μM. SIRT6-IN-4 inhibits the proliferation of MCF-7 with an IC50 of 8.30 μM. SIRT6-IN-4 arrests the cell cycle at G2/M phase, inhibits thecell migration and invasion of MCF-7, and induces apoptosis. SIRT6-IN-4 exhibits antitumor efficacy in mouse models .
|
-

- HY-153996
-
|
|
Deubiquitinase
Apoptosis
Bcr-Abl
STAT
c-Myc
|
Cancer
|
|
CT1113 is a selective, orally active USP25/USP28 inhibitor. CT1113 inhibits cancer cell proliferation, induces apoptosis, and causes G2/S phase cell cycle arrest. CT1113 reduces the levels of total BCR-ABL1, phosphorylated BCR-ABL1, total STAT5, and phosphorylated STAT5, but does not alter the mRNA level of BCR-ABL1. CT1113 is applicable to research related to pancreatic cancer, colon cancer, and acute leukemia .
|
-
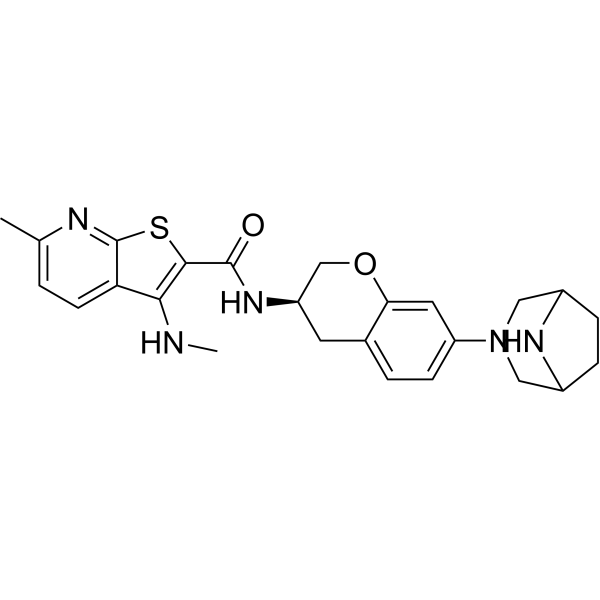
- HY-155982
-
|
|
STAT
|
Cancer
|
|
STAT3-IN-20 (Compound 40) is a selective STAT3 inhibitor (IC50: 0.65 μM). STAT3-IN-20 binds the SH2 domain to inhibit STAT3 phosphorylation, translocation, and downstream gene transcription. STAT3-IN-20 exhibits antiproliferative activities against STAT3-overactivated DU145 and MDA-MB-231 cancer cells (IC50: 2.97 μM and 3.26 μM respectively). STAT3-IN-20 induces cell cycle arrest and apoptosis .
|
-
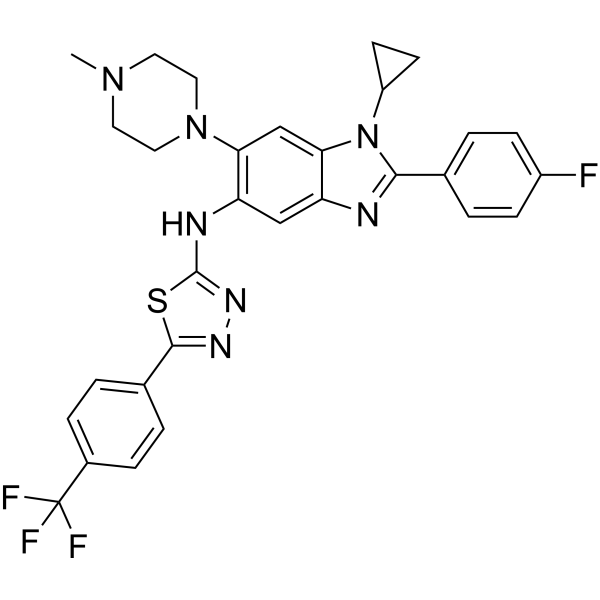
- HY-183771
-
|
|
CDK
Apoptosis
Reactive Oxygen Species (ROS)
Mitochondrial Metabolism
Bcl-2 Family
Caspase
|
Cancer
|
|
CDK4/6-IN-28 is a potent, orally active, and selective CDK4/6 inhibitor IC50 values of 14.02 and 10.03 nM, respectively. CDK4/6-IN-28 inhibits breast cancer cell colony formation, migration, and proliferation. CDK4/6-IN-28 induces G1-phase cell cycle arrest and apoptosis in breast cancer cells. CDK4/6-IN-28 exhibits tumor inhibitory activity in breast cancer xenograft mouse models. CDK4/6-IN-28 can be used for the research of breast cancer .
|
-

- HY-13224A
-
|
|
Endogenous Metabolite
|
Cancer
|
|
AZD4877 hydrochloride is a synthetic dynein inhibitor with potential anti-tumor activity. AZD4877 selectively inhibits the microtubule dynein KSP (also known as kinesin-5 or Eg5), which may lead to inhibition of mitotic spindle assembly. The action of AZD4877 may activate the spindle assembly checkpoint, leading to cell cycle arrest at the mitotic stage. AZD4877 may induce cell death in actively dividing tumor cells. AZD4877 may be less likely to cause peripheral neuropathy associated with microtubule-targeted compounds as it is not involved in post-mitotic processes. AZD4877 is essential for the formation of bipolar spindles and the proper segregation of sister chromosomes .
|
-

- HY-178391
-
|
|
ROS Kinase
p38 MAPK
ERK
Apoptosis
|
Cancer
|
|
SMU-037 is an orally active and selective ROS1 inhibitor that demonstrates potent activity (IC₅₀ = 6.8 nM) and possesses the ability to penetrate the blood-brain barrier. SMU-037 shows ~25-fold selectivity over ALK, and superior sensitivity against the G2032R mutation. SMU-037 attenuates phosphorylation of ROS1 and downstream MAPK-ERK signaling pathway, leading to cell cycle arrest and apoptosis. SMU-037 effectively suppresses tumor progression in both xenograft and intracranial mouse models. SMU-037 can be used for non-small cell lung cancer (NSCLC) research .
|
-

- HY-175261
-
|
|
CDK
Wee1
Checkpoint Kinase (Chk)
|
Cancer
|
|
DHI1 is an anti-leukemia agent with high selectivity for Jurkat (IC50 = 21.83 μM) and HL-60 (IC50 = 19.14 μM) leukemia cells and has low toxicity to non-cancerous cells. DHI1 can induce G2/M phase cell arrest in Jurkat and HL-60 leukemia cells, as well as S phase arrest in HL-60 cells, and has significant effects on cell cycle signaling molecules Wee1, cyclin B1, cdc2 on Tyr15, and Chk1. DHI1 inhibits the migration and invasion of Jurkat and HL-60 cells by disrupting cytoskeletal actin filaments. DHI1 can be used to study hematological malignancies .
|
-

- HY-101257
-
|
|
CDK
|
Cancer
|
|
YKL-5-124 is a potent, selective, irreversible and covalent CDK7 inhibitor with IC50s of 53.5 nM and 9.7 nM for CDK7 and CDK7/Mat1/CycH, respectively. YKL-5-124 is >100-fold greater selective for CDK7 than CDK9 and CDK2, and inactive against CDK12 and CDK13. YKL-5-124 induces a strong cell-cycle arrest, inhibits E2F-driven gene expression, and exhibits little effect on RNA polymerase II phosphorylation status .
|
-
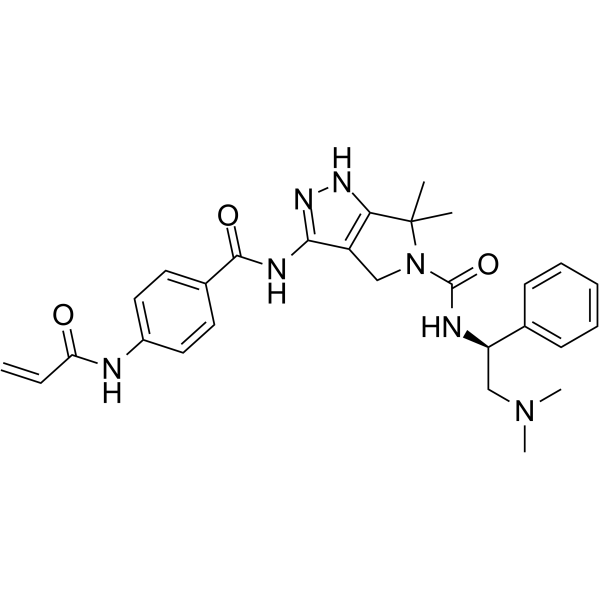
- HY-186045
-
|
|
Histone Methyltransferase
Apoptosis
DNA/RNA Synthesis
ATP-binding cassette (ABC) transporters
|
Cancer
|
|
SKLB06489 is a selective and orally active inhibitor of type I PRMT enzymes, with IC50 values of 64.55 nM (PRMT1), 4.21 nM (PRMT6), and 51.27 nM (PRMT8). SKLB06489 inhibits cell proliferation, colony formation, DNA replication, and DNA damage repair in cancer cells. SKLB06489 induces G0/G1-phase cell cycle arrest and apoptosis in cancer cells. SKLB06489 enhances intracellular cholesterol efflux via ABCA1 and ABCG1 upregulation, disrupts cholesterol metabolic homeostasis, and suppresses tumor growth in subcutaneous xenograft models. SKLB06489 can be used for the research of triple-negative breast cancer (TNBC) .
|
-

- HY-10325R
-
|
EKI-785 (Standard); WAY-EKI 785 (Standard)
|
Reference Standards
EGFR
Apoptosis
|
Endocrinology
Cancer
|
|
CL-387785 (EKI-785; WAY-EKI 785) (Standard) is the analytical standard of CL-387785 (HY-10325). This product is intended for research and analytical applications. CL-387785 is an orally active EGFR inhibitor with an IC50 of 370 pM. CL-387785 inhibits EGF-stimulated EGFR autophosphorylation with an IC50 of approximately 5 nM. CL-387,785 exerts selective inhibition on cell lines overexpressing EGFR or c-erbB-2, whereas it shows weak inhibitory effects on cell lines with low expression of these two receptors. CL-387785 effectively induces cell cycle arrest and apoptosis. CL-387785 can be used for the research of non-small cell lung cancer and autosomal recessive polycystic kidney disease.
|
-

- HY-163894
-
|
|
Apoptosis
HDAC
|
Cancer
|
|
HDAC6-IN-48 (compound 5i) is a potent and selective HDAC6 inhibitor with IC50 values of 5.16, 396.72, 638.08 nM for HDAC6, HDAC3, HDAC1, respectively. HDAC6-IN-48 induces apoptosis and cell cycle arrest at G0/G1 phase. HDAC6-IN-48 increases the protein expression of acetylated α-tubulin .
|
-

- HY-128769
-
|
|
CDK
|
Cancer
|
|
M2N12 is a potent and highly selective cell division cycle 25C protein phosphatase (Cdc25C) inhibitor with an IC50 value of 0.09 μM. M2N12 also has promising activity against Cdc25A and Cdc25B with IC50 values of 0.53 μM and 1.39 μM, respectively. M2N12 has anti-tumor activity and can be used for cancer research .
|
-
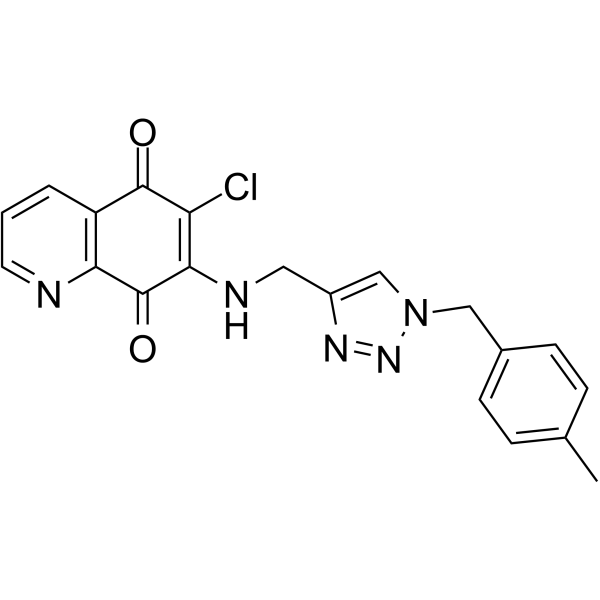
- HY-123486
-
|
|
Apoptosis
Bcr-Abl
|
Cancer
|
|
HS-438 is a potent and selective BCR-ABL inhibitor. HS-438 shows antiproliferative activity. HS-438 decreases the expression of phosphorylation of BCR-ABL (Tyr177). HS-438 induces apoptosis and cell cycle arrest at G0/G1 phase. HS-438 shows antitumor activity. HS-438 has the potential for the research of chronic myeloid leukemia (CML) .
|
-

- HY-118529
-
|
|
Phosphatase
|
Cancer
|
|
JUN-1111 is an irreversible and selective Cdc25 phosphatase inhibitor with IC50 values of 0.38, 1.8, 0.66, 28, 37 µM for Cdc25A, Cdc25B, Cdc25C, VHR, PTP1B, respectively. JUN-1111 induces cell cycle arrest at G1 and G2/M phases. JUN-1111 decreases the expression of phosphoCdk1 .
|
-
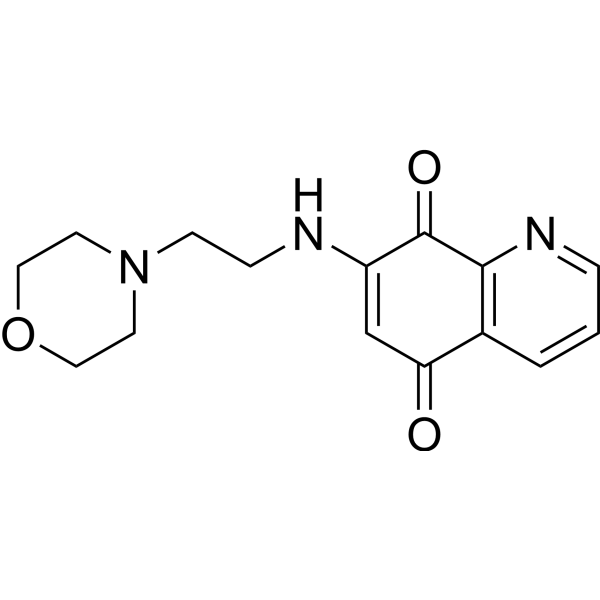
- HY-115908
-
|
|
CDK
Apoptosis
|
Cancer
|
|
ZDLD13, a β-carboline, is an orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 0.38 μM. ZDLD13 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cell cycle. ZDLD13 shows significant tumor growth inhibition in HCT116 tumor xenograft model .
|
-
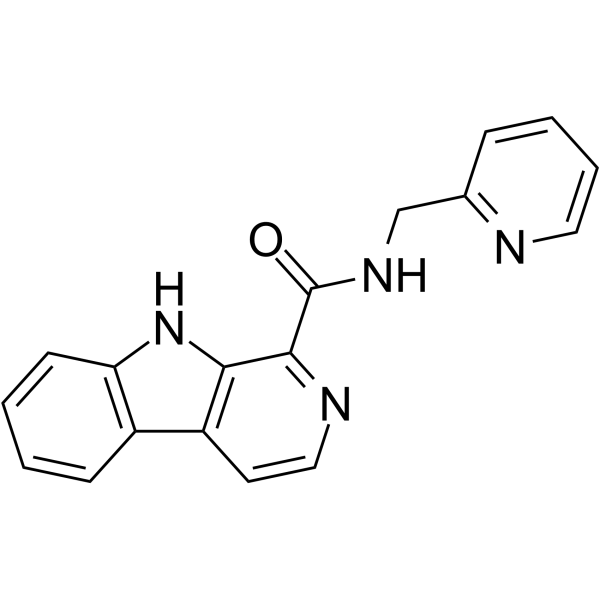
- HY-B0277
-
|
Ara-A; Adenine Arabinoside; 9-β-D-Arabinofuranosyladenine
|
Apoptosis
Fungal
Reactive Oxygen Species (ROS)
EBV
HSV
Antibiotic
DNA/RNA Synthesis
Drug Metabolite
|
Infection
Cancer
|
|
Vidarabine (Ara-A) is a nucleoside antibiotic isolated from Streptomyces, and a metabolite of Vidarabine phosphate (HY-B0277A). Vidarabine selectively inhibits viral DNA polymerase and cellular ribonucleotide reductase, thereby blocking viral replication. Vidarabine phosphate also exhibits antifungal activity, induces late-stage cellular apoptosis, and causes cell cycle arrest. Vidarabine phosphate can be used in research related to severe chronic active Epstein-Barr virus (EBV) infection, herpes infection, and candidiasis .
|
-
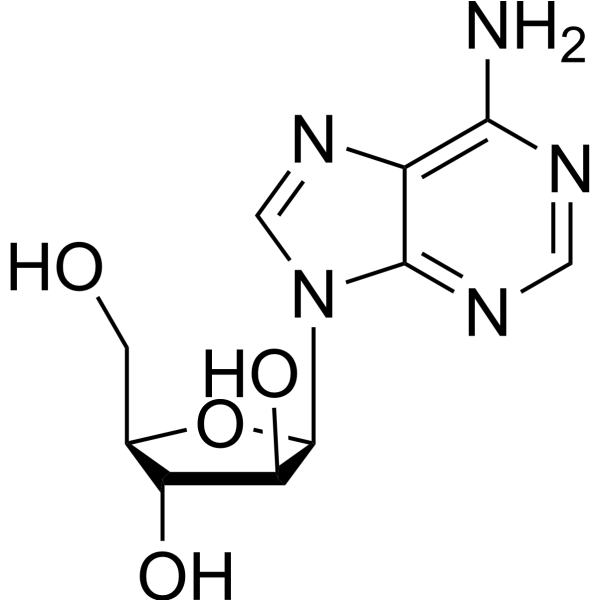
- HY-157932
-
|
|
Hippo (MST)
|
Cancer
|
|
MR24, a G-5555 (HY-19635) derivative, is a mammalian STE20-like (MST) kinase inhibitor. MR24 shows selectively to MST3/4 over MST1/2, with EC50 values of 57 and 583 nM, respectively. MR24 induces G1 phase cell cycle arrest. MR24 can be used for cancer research, such as breast, liver and lung cancers .
|
-
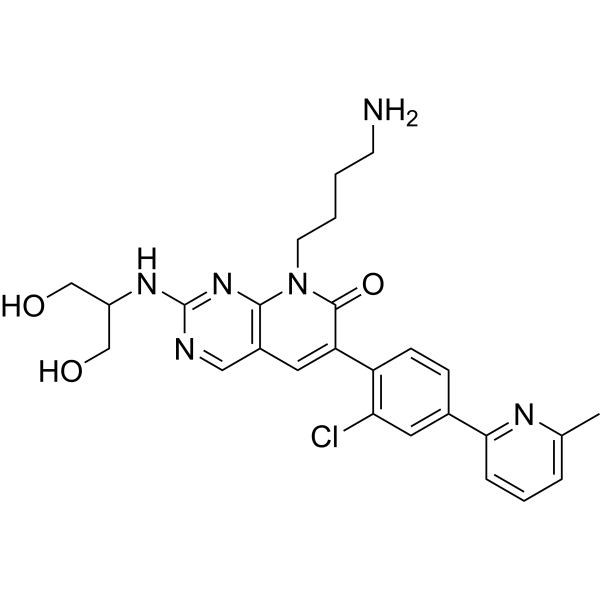
- HY-181954
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
ZW-49 is an orally active pan-EGFR inhibitor with IC50 values at 0.03-1.5 nM. ZW-49 inhibits all subgroups of EGFR mutations with selectivity over wild-type EGFR and other target families. ZW-49 blocks the ATP-binding pocket, occupies a conserved hydrophobic subpocket, avoids steric conflicts with PACC mutation P loops. ZW-49 inhibits cancer cells proliferation, induces G0/G1 phase cell-cycle arrest and apoptosis, and demonstrates anti-proliferative activity in xenograft mice models. ZW-49 can be used for the research of cancer, such as non-small cell lung cancer .
|
-

- HY-N2424
-
|
2-Phenyl-4-chromone
|
CDK
Apoptosis
Caspase
|
Cancer
|
|
Flavone is an anti-tumor compound that targets cell cycle regulatory proteins (such as cyclin B1) and apoptosis-related factors (such as p21waf1, PIG3). Flavone selectively induces mitochondrial-mediated apoptosis pathways in tumor cells, inhibits cyclin B1 protein expression, upregulates p21waf1, and activates p63/p73 proteins. Flavone has immunomodulatory functions that enhance natural killer cell (NK cell) activity and lymphocyte proliferation. Flavone is used in cancer research, especially for its inhibitory potential in solid tumor models such as esophageal cancer and liver cancer .
|
-
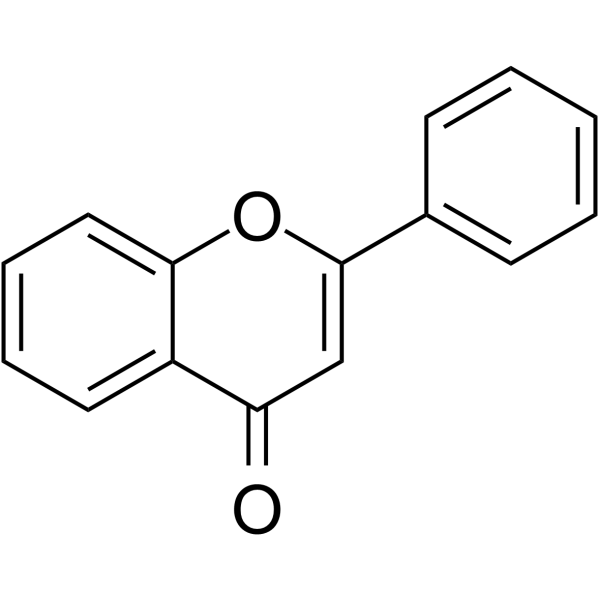
- HY-146751
-
|
|
PI3K
Akt
mTOR
Apoptosis
|
Cancer
|
|
PI3K/Akt/mTOR-IN-2 is a PI3K/AKT/mTOR pathway inhibitor. PI3K/Akt/mTOR-IN-2 possess anti-cancer effects and selectivity against MDA-MB-231 cells with IC50 value of 2.29 μM. PI3K/Akt/mTOR-IN-2 can induce cancer cell cycle arrest and apoptosis .
|
-
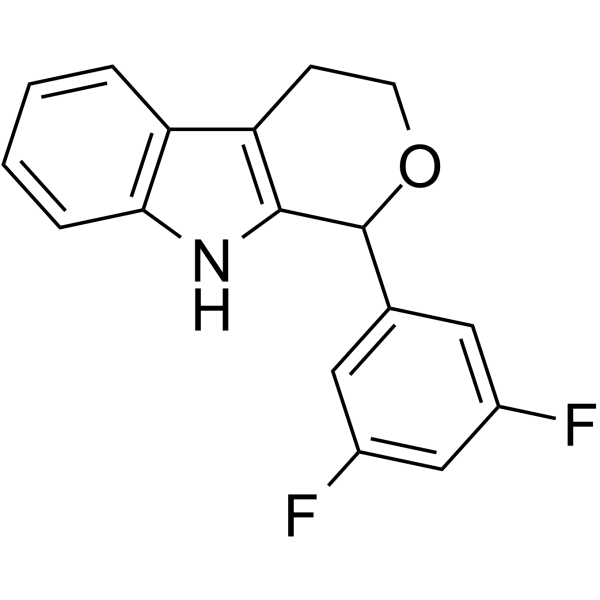
- HY-138008
-
|
|
Microtubule/Tubulin
Apoptosis
|
Cancer
|
|
WX-132-18B is a tubulin inhibitor with an IC50 of 0.45-0.99 nM. WX-132-18B selectively binds to the colchicine-binding site on tubulin, reduces microtubule content via depolymerization, and inhibits tubulin polymerization. WX-132-18B induces tumor cell cycle arrest, apoptosis and changes in nuclear membrane permeability, and decreases mitochondrial membrane potential. WX-132-18B exhibits antiproliferative activity against endothelial cells and human tumor cells, and inhibits the proliferation and growth of xenograft tumors in mice. WX-132-18B can be used in research related to sarcoma, non-small cell lung cancer, gastric cancer and breast cancer .
|
-

- HY-181996
-
|
|
Btk
Caspase
Apoptosis
PARP
|
Cancer
|
|
BTK-IN-47 (Compound 9e) is a covalent, selective BTK inhibitor with an IC50 of 5.15 nM against BTK. BTK-IN-47 inhibits the BTK signaling pathway, induces cell cycle arrest, and activates the canonical Caspase-dependent Apoptotic pathway (promoting the cleavage of Caspase-3, Caspase-7 and PARP), without inducing necroptosis, pyroptosis or ferroptosis. BTK-IN-47 exerts dose-dependent antiproliferative activity against hematologic tumor cell lines. BTK-IN-47 exhibits dose-dependent in vivo antitumor activity in a Ramos cell xenograft model in BALB/c nude mice. BTK-IN-47 can be used for the research of hematologic malignancies .
|
-

- HY-101029R
-
|
|
NEKs
Apoptosis
Reference Standards
|
Cancer
|
|
MBM-55 (Standard) is the analytical standard of MBM-55 (HY-101029). This product is intended for research and analytical applications. MBM-55 is a potent NIMA-related kinase 2 (Nek2) inhibitor with an IC50 of 1 nM. MBM-55 shows a 20-fold or greater selectivity in most kinases with the exception of RSK1 (IC50=5.4 nM) and DYRK1a (IC50=6.5 nM). MBM-55 effectively inhibits the proliferation of cancer cells by inducing cell cycle arrest and apoptosis. MBM-55 shows antitumor activities, and no obvious toxicity to mice .
|
-

- HY-183560
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
HDAC6-IN-82 is a selective HDAC6 inhibitor with an IC50 of 4.9 nM against HDAC6. HDAC6-IN-82 inhibits HDAC1 (112 nM), HDAC2 (737 nM), HDAC3 (623 nM), HDAC8 (1140 nM), HDAC10 (91.4 nM) and HDAC11 (219 nM). HDAC6-IN-82 reduces cancer cell viability, induces cell cycle arrest, triggers apoptosis, and increases the acetylation levels of H3K9 and α-tubulin. HDAC6-IN-82 can be used in cancer-related research such as leukemia .
|
-

- HY-121382R
-
|
|
Reference Standards
Apoptosis
Cholinesterase (ChE)
Necroptosis
|
Cardiovascular Disease
|
|
Cinosulfuron (Standard) is the analytical standard of Cinosulfuron. This product is intended for research and analytical applications. Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
|
-

- HY-168556
-
|
|
CDK
PROTACs
DNA/RNA Synthesis
|
Cancer
|
|
YJ9069 is a selective CDK12/CDK13 PROTAC degrader with an IC50 of 22.22 nM for in VCaP cells. CDK12/13 degradation rapidly triggers gene-length-dependent transcriptional elongation defects, leading to DNA damage and cell-cycle arrest. YJ9069 effectively inhibits proliferation in subsets of prostate cancer cells and significantly suppresses prostate tumor growth. (Pink: CDK12/CDK13 degradation agent (HY-168658); Black: Linker (HY-W015967); Blue: ligand for E3 ligase (HY-103596)) .
|
-

- HY-175473
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
HI042 is a FMS-like Tyrosine Kinase 3 (FLT3) inhibitor. HI042 shows IC50 values of 0.62 μM for MOLM-13, 0.33 μM for MV4-11, and 0.89 μM for OCI-AML3 cells. HI042 selectively reduces the viability of FLT3-internal tandem duplication
(FLT3-|TD) mutations-positive cell lines, induces apoptosis, disrupts cell cycle progression, and diminishes the clonogenic potential. HI042 can be used for the research of acute myeloid leukemia (AML) .
|
-

- HY-175208A
-
|
|
Hippo (MST)
Caspase
Bcl-2 Family
Apoptosis
|
Cancer
|
|
MST3-IN-1 TFA (Compound LD-1) is a selective and orally active MST3 inhibitor, with an IC50 of 122.4 nM. MST3-IN-1 TFA shows antiproliferative activity against HepG2 cell. MST3-IN-1 TFA effectively induces Apoptosis in HepG2 cells, and halts the cell cycle at the G2/M transition. MST3-IN-1 TFA significantly suppressed tumor growth in HepG2 xenograft mice. MST3-IN-1 TFA can be used for the study of liver cancer .
|
-

- HY-10447
-
|
EM-1421
|
Survivin
COX
TNF Receptor
Apoptosis
|
Infection
Inflammation/Immunology
Cancer
|
|
Terameprocol is an inhibitor targeting the Sp1 transcription factor, which can selectively inhibit the transcription of Sp1-dependent genes. Terameprocol exerts its effects by inhibiting Sp1-mediated gene transcription, such as reducing the expression of genes like CDC2, survivin and HMGB1, thereby arresting the cell cycle, inducing apoptosis, and suppressing the inflammatory response. Terameprocol exhibits anti-proliferative, pro-apoptotic, and anti-inflammatory activities and is currently mainly used in the research of diseases such as cancer and pulmonary arterial hypertension[1][2][3].
|
-
- HY-175684
-
|
|
JAK
STAT
Apoptosis
|
Cancer
|
|
JAK2-IN-14 is an orally active JAK2 inhibitor with an IC50 of 2 nM. JAK2-IN-14 demonstrates 89.5-, 80.5-, and 51-fold selectivity over JAK1, JAK3, and TYK2, respectively. JAK2-IN-14 inhibits STAT5 signaling pathway. JAK2-IN-14 causes tumor cell cycle arrest and apoptosis. JAK2-IN-14 can used for the study of myeloproliferative neoplasms (MPNs) .
|
-

- HY-179125
-
|
|
COX
Bcl-2 Family
Caspase
Apoptosis
|
Cancer
|
|
COX-2-IN-59 is a potent and selective COX-2 inhibitor with an IC50 of 0.052 μM. COX-2-IN-59 exhibits 200-fold selectivity over COX-1 (IC50 = 11.16 μM). COX-2-IN-59 reduces COX-2 levels, induces cell cycle arrest, and triggers apoptosis by increasing Bax expression, decreasing Bcl-2 levels, and activating caspase-3. COX-2-IN-59 can be used for the research of colon cancer .
|
-

- HY-101257B
-
|
|
CDK
|
Cancer
|
|
YKL-5-124 TFA is a potent, selective, irreversible and covalent CDK7 inhibitor with IC50s of 53.5 nM and 9.7 nM for CDK7 and CDK7/Mat1/CycH, respectively. YKL-5-124 TFA is >100-fold greater selective for CDK7 than CDK9 and CDK2, and inactive against CDK12 and CDK13. YKL-5-124 TFA induces a strong cell-cycle arrest, inhibits E2F-driven gene expression, and exhibits little effect on RNA polymerase II phosphorylation status .
|
-
- HY-173632
-
|
|
Ras
|
Cancer
|
|
AMG410 is a non-covalent and selective pan-KRAS inhibitor with IC50 values of 1-4 nM for KRAS G12D, KRAS G12V, and KRAS G13D. AMG410 shows greater than 100-fold selectivity against both HRAS and NRAS. AMG410 is a dual GTP(on)- and GDP(off)-state inhibitor (Kd(GDP-state) of 1 nM; Kd(GTP-state) of 22 nM). AMG410 blocks KRAS signaling in a cycling state-independent manner and also blocks proliferation in wildtype KRAS-amplified tumor cells. AMG410 can be used for the study of colorectal, pancreatic, and lung cancers .
|
-

- HY-178378
-
|
|
Histone Methyltransferase
GLP Receptor
Reactive Oxygen Species (ROS)
Autophagy
Apoptosis
|
Cancer
|
|
G9a-IN-4 is a G9a inhibitor with high selectivity (IC50 = 32 nM). G9a-IN-4 shows high selectivity against the other tested lysine/arginine methyltransferases. G9a-IN-4 exhibits high enzymatic activity against G9a and more potent antiproliferative effects against all tested cancer cells. G9a-IN-4 significantly suppresses the H3K9me2 level. G9a-IN-4 triggers autophagy by inducing the production of ROS, thus leading to cell apoptosis and cell cycle arrest at G0/G1 in CT26 colon cells. G9a-IN-4 can be used for the study of colon cancer .
|
-

- HY-180355
-
|
|
CDK
Ser/Thr Kinase
c-Myc
Apoptosis
|
Cancer
|
|
SY-5102 is a potent, selective and orally active cyclin-dependent kinase (CDK7) inhibitor with a Kd of 0.03 nM. SY-5102 shows anti-proliferative activity against HCC70 cells with an EC50 of 9 nM. SY-5102 can inhibit CDK7-mediated CAK function (downregulate CDK2 Thr160 phosphorylation) and TFIIH transcription function (downregulate RNA polymerase II Ser5 phosphorylation). SY-5102 can induce G2/M cell cycle arrest, downregulate the expression of the oncogene c-Myc, and ultimately trigger cancer cell apoptosis. SY-5102 can be used for the research of triple-negative breast cancer (TNBC) .
|
-

- HY-173481
-
|
|
CDK
|
Cancer
|
|
CDK9-IN-37 (Compound 24) is a CDK9 inhibitor (EC50: 5.5 nM) with weak inhibition on other CDK isoforms, showing high selectivity. CDK9-IN-37 has significant antiproliferative activity against acute myeloid leukemia MOLM-13 cells (IC50: 0.034 μM). CDK9-IN-37 inhibits the CDK9 signaling pathway, reduces the phosphorylation level of RNAP II CTD (Ser2), downregulates the anti-apoptotic protein McI-1, induces cell apoptosis, and arrests the cell cycle at the G2/M phase. CDK9-IN-37 can be used in the study of acute myeloid leukemia (AML) .
|
-

- HY-N7204
-
|
|
Monoamine Oxidase
Dopamine β-hydroxylase
Apoptosis
|
Neurological Disease
Inflammation/Immunology
|
|
4-Hydroxyderricin, the major active ingredients of Angelica keiskei Koidzumi, is an orally active, potent selective MAO-B (Monoamine oxidase inhibitors) inhibitor with an IC50 of 3.43 μM. 4-Hydroxyderricin also mildly inhibits dopamine β (DBH)-hydroxylase activity. 4-Hydroxyderricin has antidepressant activity, anti-allergic, anti-diabetic, anti-oxidant, and antitumor effects. 4-Hydroxyderricin promotes apoptosis and cell cycle arrest through regulating PI3K/AKT/mTOR pathway in hepatocellular cells. 4-Hydroxyderricin inhibits osteoclast formation and accelerates osteoblast differentiation . 4-Hydroxyderricin is promising for research of inflammatory diseases .
|
-
- HY-173008
-
|
|
Apoptosis
YTHDF
|
Cancer
|
|
YTHDF2-IN-1 (Compound CK-75) is a selective YTHDF2 inhibitor (Kd = 26.2 μM). YTHDF2-IN-1 binds to a small hydrophobic pocket on the YTH domain of YTHDF2, disrupting the interaction between YTHDF2 and m 6A-modified RNA. YTHDF2-IN-1 induces cell cycle arrest and Apoptosis. YTHDF2-IN-1 is applicable to research on chronic myeloid leukemia, colon cancer and choriocarcinoma .
|
-

- HY-147914
-
|
|
Apoptosis
|
Cancer
|
|
NSD2-IN-1 (compound 38) is a potent and high selective NSD2-PWWP1 (nuclear receptor-binding SET domain 2-PWWP1) inhibitor, with an IC50 of 0.11 μM. NSD2-IN-1 can bind to NSD2-PWWP1 and then affect the expression of genes regulated by NSD2. NSD2-IN-1 induces apoptosis and cell cycle arrest .
|
-
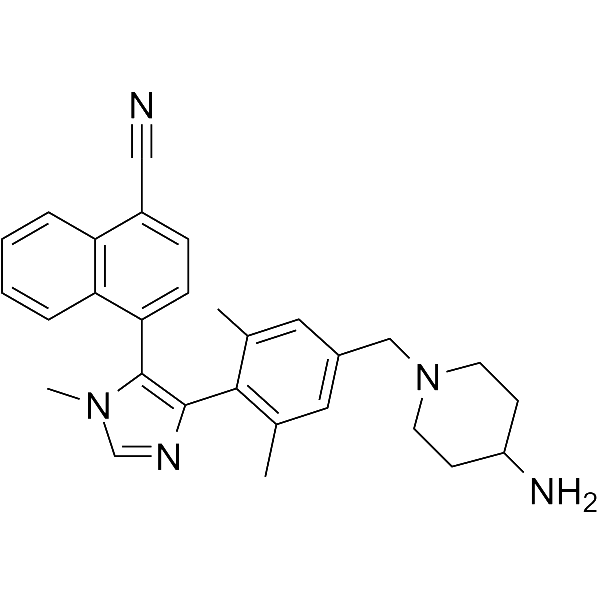
- HY-159936
-
|
|
HDAC
PPAR
Apoptosis
|
Cancer
|
|
CS4 is a selective HDAC inhibitor with the IC50 values of 38 nM, 12 nM, 5.8 μM, 19 μM and 61 μM against of HDAC1, HDAC6, HDAC8, HDAC4 and HDAC11, respectively. CS4 promotes α-tubulin and histone 3 acetylation. CS4 activates PPARγ and blocks glycolysis. CS4 induces cell cycle arrest at G2 phase and apoptosis, and shows anticancer effect both in vivo and in vitro .
|
-

- HY-157932A
-
|
|
Hippo (MST)
|
Cancer
|
|
MR24 TFA, a G-5555 (HY-19635) derivative, is a mammalian STE20-like (MST) kinase inhibitor. MR24 TFA shows selectively to MST3/4 over MST1/2, with EC50 values of 57 and 583 nM, respectively. MR24 TFA induces G1 phase cell cycle arrest. MR24 TFA can be used for cancer research, such as breast, liver and lung cancers .
|
-

- HY-N0863
-
|
NSC-698790; Smilax saponin B
|
Bcl-2 Family
Apoptosis
Akt
c-Myc
ERK
p38 MAPK
JNK
FOXO
|
Cancer
|
|
Methyl protodioscin (NSC-698790; Smilax saponin B) is a multi-target, selective, steroidal diglycoside inhibitor with antitumor activity that induces cell cycle arrest. The mechanism of action of Methyl protodioscin is complex, involving the induction of G2/M cell cycle arrest, regulation of the Bcl-2/Bax apoptotic pathway, inhibition of the Akt1/c-Myc axis and MAPK/ERK signaling, while simultaneously downregulating ADAM15 and inducing FOXO1 to reduce cholesterol synthesis. It also inhibits the JNK/c-Jun pathway, reducing the production of inflammatory factors (IL-6, TNF-α). Methyl protodioscin exhibits significant antitumor (inhibiting proliferation, migration, invasion, and inducing apoptosis), anti-inflammatory, and anti-restenosis activities. Methyl protodioscin can be used in research on lung cancer, prostate cancer, pancreatic cancer, and other tumors, as well as inflammatory diseases such as airway inflammation and enteritis .
|
-
- HY-181716
-
|
|
Ras
Apoptosis
|
Cancer
|
|
KRAS G12C-IN-74 is an orally active, selective KRAS G12C inhibitor with a target IC50 of 43.18 nM. KRAS G12C-IN-74 induces G0/G1 cell cycle arrest and apoptosis in KRAS G12C-mutant cancer cells. KRAS G12C-IN-74 is applicable for the research of KRAS G12C-mutant pancreatic cancer, colorectal cancer and lung cancer .
|
-

- HY-178114
-
|
|
Histone Methyltransferase
Apoptosis
|
Cancer
|
|
SKLB-0124 is a selective PRMT6 degrader with DC50s of 15.4 μM and a 16.4 μM in HCC827 and MDA-MB-435 cells. SKLB-0124 does not degrade PRMT1 or PRMT8. SKLB-0124 exhibits an IC50 on PRMT6 of 1.6 μM. SKLB-0124 induces proteasome dependent degradation of PRMT6 and significantly inhibits the proliferation. SKLB-0124 effectively induces apoptosis and cell cycle arrest. SKLB-0124 can be used for the studies of lung cancer and breast cancer .
|
-

- HY-182019
-
|
|
HDAC
SHMT
Apoptosis
Reactive Oxygen Species (ROS)
Ferroptosis
|
Cancer
|
|
HDAC11-IN-5 is a selective, potent and orally active HDAC11 inhibitor with an IC50 of 0.021 μM. HDAC11-IN-5 increases fatty acylation levels of substrate SHMT2 in AML cells. HDAC11-IN-5 induces apoptosis, G1 phase cell cycle arrest, ferroptosis, ROS production and terminal myeloid differentiation in AML cells. HDAC11-IN-5 demonstrates anti-tumor potency in an MLL-AF9-induced mouse AML model. HDAC11-IN-5 can be used for the research of cancer, such as acute myeloid leukemia .
|
-

- HY-116217R
-
|
FdCyd (Standard); NSC-48006 (Standard)
|
DNA Methyltransferase
Reference Standards
|
Cancer
|
|
5-Fluoro-2'-deoxycytidine (Standard) is the analytical standard of 5-Fluoro-2'-deoxycytidine. This product is intended for research and analytical applications. 5-Fluoro-2'-deoxycytidine (FdCyd), a fluoropyrimidine nucleoside analogue, is a DNA methyltransferase (DNMT) inhibitor. 5-Fluoro-2'-deoxycytidine is a tumor-selective proagent of thymidylate synthase inhibitor 5-fluoro-2′-dUMP. 5-Fluoro-2'-deoxycytidine can induce cell cycle arrest in tumor cells through the DNA damage response, and it has anti-tumor activity .
|
-

- HY-181627
-
|
|
MNK
DNA/RNA Synthesis
Interleukin Related
Eukaryotic Initiation Factor (eIF)
|
Cancer
|
|
ETC-501 is a blood-brain barrier-permeable, orally active, and selective MNK1/MNK2 inhibitor, with an IC50 of 0.033 μM against MNK1 and 0.111 μM against MNK2. ETC-501 inhibits glioblastoma cell proliferation, impairs DNA damage repair function, delays cell cycle progression, and suppresses ribosome biogenesis. ETC-501 enhances Temozolomide (HY-17364)-induced cellular senescence, attenuates the senescence-associated secretory phenotype, and increases cellular sensitivity to Navitoclax (HY-10087). ETC-501 is applicable to research related to glioblastoma .
|
-

- HY-173632A
-
|
|
Ras
|
Cancer
|
|
AMG410 diTFA is a non-covalent and selective pan-KRAS inhibitor with IC50 values of 1-4 nM for KRAS G12D, KRAS G12V, and KRAS G13D. AMG410 diTFA shows greater than 100-fold selectivity against both HRAS and NRAS. AMG410 diTFA is a dual GTP(on)- and GDP(off)-state inhibitor (Kd(GDP-state) of 1 nM; Kd(GTP-state) of 22 nM). AMG410 diTFA blocks KRAS signaling in a cycling state-independent manner and also blocks proliferation in wildtype KRAS-amplified tumor cells. AMG410 diTFA can be used for the study of colorectal, pancreatic, and lung cancers .
|
-

- HY-175252
-
|
|
PROTACs
EGFR
Apoptosis
|
Cancer
|
|
PROTAC EGFR degrader 14 is a potent and selective EGFR PROTACdegrader with a DC50 of about 2.9 nM and a Dmax of 93.1% for EGFR L858R/T790M/C797S. PROTAC EGFR degrader 14 selectively induces EGFR C797S degradation through a VHL and proteasome-dependent manner and downregulated EGFR-associated transcriptome and exhibits good selectivity over EGFR WT. PROTAC EGFR degrader 14 induces cell cycle arrest and apoptosis and significantly inhibits tumor growth. PROTAC EGFR degrader 14 can be used for the study of nonsmall cell lung cancer (NSCLC) (Pink: Target protein ligand: (HY-143337); Blue: E3 ligand (HY-125845); Black: Linker (HY-W004688)) .
|
-

- HY-10201
-
Sorafenib
Maximum Cited Publications
283 Publications Verification
Bay 43-9006
|
Raf
VEGFR
FLT3
Autophagy
Apoptosis
STAT
Akt
MMP
Cadherin
p38 MAPK
ERK
MEK
PI3K
PARP
Bcl-2 Family
|
Infection
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Sorafenib (Bay 43-9006) is a potent oral active multikinase inhibitor. Sorafenib blocks autophosphorylation and activity of receptor tyrosine kinases (VEGFR-2, VEGFR-3) and RAF family kinases, thereby suppressing the RAF/MEK/ERK and PI3K/Akt pathways, inhibiting STAT3 phosphorylation, and selectively inhibiting the MAPK pathway in cancer cells. Sorafenib induces cell cycle arrest, autophagy, apoptosis, and PARP cleavage, reduces Bcl-2, Bcl-XL, cyclin D1 levels, and activates Bak and Bax. Sorafenib inhibits tumor growth and metastasis in mouse and rat models. Sorafenib can be used for cancer research, such as colon, breast, non-small-cell lung cancer (NSCLC), ovarian, pancreatic, melanoma, colorectal and hepatocellular carcinoma .
|
-
- HY-10201A
-
|
Bay 43-9006 tosylate
|
Raf
VEGFR
FLT3
Autophagy
Apoptosis
STAT
Akt
MMP
Cadherin
p38 MAPK
ERK
MEK
PI3K
PARP
Bcl-2 Family
|
Infection
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Sorafenib (Bay 43-9006) tosylate is a potent oral active multikinase inhibitor. Sorafenib blocks autophosphorylation and activity of receptor tyrosine kinases (VEGFR-2, VEGFR-3) and RAF family kinases, thereby suppressing the RAF/MEK/ERK and PI3K/Akt pathways, inhibiting STAT3 phosphorylation, and selectively inhibiting the MAPK pathway in cancer cells. Sorafenib tosylate induces cell cycle arrest, autophagy, apoptosis, and PARP cleavage, reduces Bcl-2, Bcl-XL, cyclin D1 levels, and activates Bak and Bax. Sorafenib tosylate inhibits tumor growth and metastasis in mouse and rat models. Sorafenib tosylate can be used for cancer research, such as colon, breast, non-small-cell lung cancer (NSCLC), ovarian, pancreatic, melanoma, colorectal and hepatocellular carcinoma .
|
-
- HY-124309
-
|
|
Lactate Dehydrogenase
Apoptosis
|
Cancer
|
|
NHI-2 is a lactate dehydrogenase A (LDHA) inhibitor with an IC50 of 14.7 µM. NHI-2 shows selective for LDHA over LDHB (IC50 = 55.8 µM). NHI-2 is an efficient anti-glycolytic agent. NHI-2 enhances apoptosis, induces cell cycle arrest at S and G2 phases. NHI-2 has a broad spectrum anti-proliferative activity in cancer cells. NHI-2 affects extracellular acidification rate and ATP production. NHI-2 suppresses tumor growth in murine B78 melanoma tumor model .
|
-
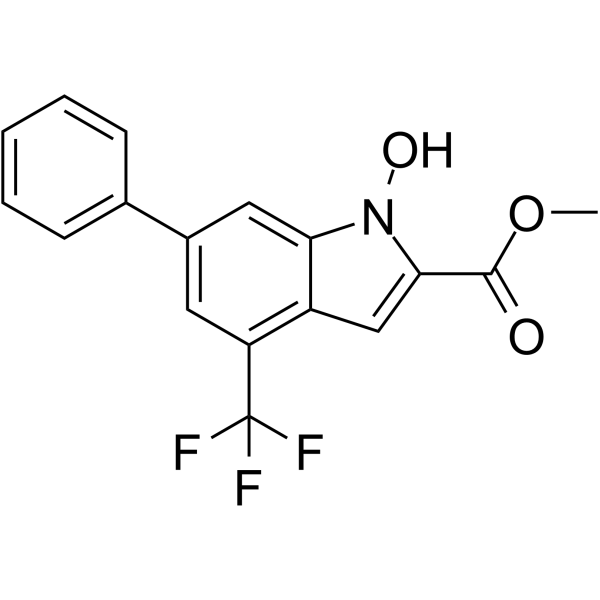
- HY-181051
-
|
|
Polo-like Kinase (PLK)
Apoptosis
|
Cancer
|
|
PLK1-IN-16 is a selective polo-like kinase 1 (PLK1) inhibitor with an IC50 of 0.25 nM. PLK1-IN-16 exhibits lower inhibitory potency against PLK2 and PLK3, induces G2 phase cell cycle arrest, induces apoptosis, and exhibits antiproliferative activity against tumor cells. PLK1-IN-16 can be stable under simulated gastric acid environmental conditions, and acceptable CYP 450 inhibition. PLK1-IN-16 can be used for the study of triple-negative breast cancer (TNBC), breast cancer, leukemia .
|
-

- HY-126251
-
|
|
CDK
Apoptosis
|
Cancer
|
|
CDK9-IN-7 (compound 21e) is a selective, highly potent, and orally active CDK9/cyclin T inhibitor (IC50=11 nM), which exhibits more potent over other CDKs (CDK4/cyclinD=148 nM; CDK6/cyclinD=145 nM). CDK9-IN-7 shows antitumor activity without obvious toxicity. CDK9-IN-7 induces NSCLC cell apoptosis, arrests the cell cycle in the G2 phase, and suppresses the stemness properties of NSCLC .
|
-
- HY-168953
-
|
|
P-glycoprotein
Apoptosis
Bcl-2 Family
Reactive Oxygen Species (ROS)
Caspase
|
Cancer
|
|
Lysosomal P-gp targeted agent 1 (Compound 14) is an anti-tumor agent targeting lysosomal P-glycoprotein (Pgp). Lysosomal P-gp targeted agent 1 is selectively transported into lysosomes by overexpressed Pgp, release nitric oxide (NO) to generate reactive oxygen species (ROS), resulting in lysosomal membrane permeabilization (LMP) and inducing apoptosis. Lysosomal P-gp targeted agent 1 can overcome P-glycoprotein-mediated drug resistance and lead to cell cycle arrest, but relatively low toxicity to normal cells. Lysosomal P-gp targeted agent 1 has antitumor activity, significantly inhibits tumor volume .
|
-
- HY-181850
-
|
|
G-quadruplex
DNA/RNA Synthesis
Cyclic GMP-AMP Synthase
STING
Autophagy
|
Cancer
|
|
Telomeric G4s ligand 2 is an orally active, selective ligand of telomeric G-quadruplex (G4), with an IC50 of 0.4 μM. Telomeric G4s ligand 2 binds to dimeric telomeric G4, inhibits the activities of DHX36 and BLM helicases. Telomeric G4s ligand 2 activates cGAS-STING and TERRA-ZBP1 pathways, inducing autophagy and G2/M cell cycle arrest, and exhibits antiproliferative effects across cancer cell lines. Telomeric G4s ligand 2 can be used for the study of colorectal cancer .
|
-

- HY-150688
-
|
|
JNK
|
Cancer
|
|
JAK3-IN-13 (compound 12n) is a potent, selective and orally active JAK3 inhibitor with IC50 values of 4728, 2039, 8, 365 nM for NK1, JNK2, JNK3, Tyk2, respectively. JAK3-IN-13 shows antiproliferative activity. JAK3-IN-13 induces cell cycle arrest at G0/G1 phase. JAK3-IN-13 shows antitumor activity .
|
-
- HY-112477A
-
|
(E/Z)-Hymenialdisine analogue-1
|
Checkpoint Kinase (Chk)
Casein Kinase
MEK
PKC
|
Cancer
|
|
(E/Z)-Chk2-IN-1 ((E/Z)-Hymenialdisine analogue-1) (Compound 1) is a potent and selective cell cycle kinase Chk2 inhibitor with an IC50 of 8 nM. (E/Z)-Chk2-IN-1 exhibits low inhibitory activity against other kinases (such as CK1δ, MEK1, and PKCα/βⅡ) with IC50 values both >89 nM. (E/Z)-Chk2-IN-1 can be used for the research of cancer .
|
-

- HY-100900R
-
|
|
Deubiquitinase
Reference Standards
|
Cancer
|
|
ML364 (Standard) is the analytical standard of ML364 (HY-100900). This product is intended for research and analytical applications. ML364 is a selective ubiquitin specific peptidase 2 (USP2) inhibitor (IC50=1.1 μM) with anti-proliferative activity, which direct binds to USP2 (Kd=5.2 μM), induces an increase in cellular cyclin D1 degradation and causes cell cycle arrest. ML364 increases the levels of mitochondrial ROS and decreases in the intracellular content of ATP .
|
-

- HY-164384
-
|
|
PI3K
Akt
mTOR
Apoptosis
|
Cancer
|
|
DFX117 is a selective, orally active inhibitor for PI3Kα and c-Met tyrosine kinase. DFX117 inhibits PI3K/Akt/mTOR pathway, inhibits the proliferation of NCI-H1975, NCI-H1993, and HCC827 with IC50s 0.02-0.08 µM. DFX117 arrests cell cycle at G0/G1 phase, induces apoptosis in A549 and NCI-H1975. DFX117 exhibits antitumor efficacy in mice .
|
-

- HY-123702
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
|
CAP-53194 is a selective Plk1 inhibitor with potential anticancer activity. CAP-53194 was identified by a high-throughput virtual screening approach using molecular docking, showing 100-fold selectivity for Plk1 over Plk2-4 and other cell cycle kinases. CAP-53194 is able to effectively exploit subtle differences between the binding sites of Plk1 and other Ser/Thr kinases, thereby enhancing their inhibitory effects. CAP-53194 meets the Lipinski compound analog criteria and passes other ADMET filters, indicating good compound compatibility. CAP-53194 belongs to a new class of potential Plk1 inhibitors suitable for subsequent compound development and testing .
|
-

- HY-179485
-
|
|
EGFR
VEGFR
COX
Caspase
Apoptosis
Bcl-2 Family
|
Cancer
|
|
EGFR/VEGFR2-IN-10 is a selective EGFR, VEGFR2 and COX2 inhibitor with IC50s of 8.5, 68 and 158 nM, respectively. EGFR/VEGFR2-IN-10 induces G1-phase cell cycle arrest in MCF-7 cells. EGFR/VEGFR2-IN-10 increases the Bax/Bcl-2 ratio, upregulates caspase-8, and elevates caspase-9 protein levels, confirming activation of the intrinsic apoptotic pathway. EGFR/VEGFR2-IN-10 demonstrates exceptional therapeutic potential by simultaneously inhibiting tumor proliferation, angiogenesis, and inflammation pathways while maintaining a favorable selectivity profile. EGFR/VEGFR2-IN-10 can be used as a research tool for cervical, liver, colon, and breast cancer studies .
|
-

- HY-183547
-
|
|
Wee1
CDK
|
Cancer
|
|
WEE1/PKMYT1-IN-4 is a selective WEE1/PKMYT1 inhibitor, with IC50 values of 0.185 μM and 2.2 nM for human WEE1 and PKMYT1, respectively. WEE1/PKMYT1-IN-4 inhibits phosphorylation of CDK1 at T14 and Y15, disrupting the G2/M cell cycle checkpoint. WEE1/PKMYT1-IN-4 can be used for the research of colorectal cancer and amplified breast cancer .
|
-

- HY-201330
-
|
RGT-419B
|
CDK
|
Cancer
|
|
GDC-4198 (RGT-419B) is an orally active CDK4/2 inhibitor with desired degrees of selectivity against kinases such as CDK6, CDK9 and GSK3β. GDC-4198 inhibits the activity of cyclin-CDK complexes, blocks phosphorylation of the retinoblastoma protein (pRb), arresting the cell cycle transition from G1 to S phase. GDC-4198 is promising for research of cancers, such as breast cancer (especially hormone receptor-positive, HER2-negative type), lung cancer, and colorectal cancer .
|
-

- HY-153278
-
|
CDK7-IN-21
|
CDK
DNA/RNA Synthesis
c-Myc
Early 2 Factor (E2F)
Topoisomerase
|
Cancer
|
|
Q901 (CDK7-IN-21) is a selective and potent CDK7 inhibitor with an IC50 of 10 nM. Q901 disrupts MYC and E2Fcell cycle control and DNA damage repair pathways. Q901 stabilizes TOP1-DPCs and sensitizes tumor to TOP1 inhibitors by suppressing RNAPII transition from initiation to elongation. Q901 can inhibit tumor growth and significantly enhances tumor growth inhibition combined with TOP1 inhibitors. Q901 can be used for the research of cancer, such as colon cancer and lung cancer .
|
-
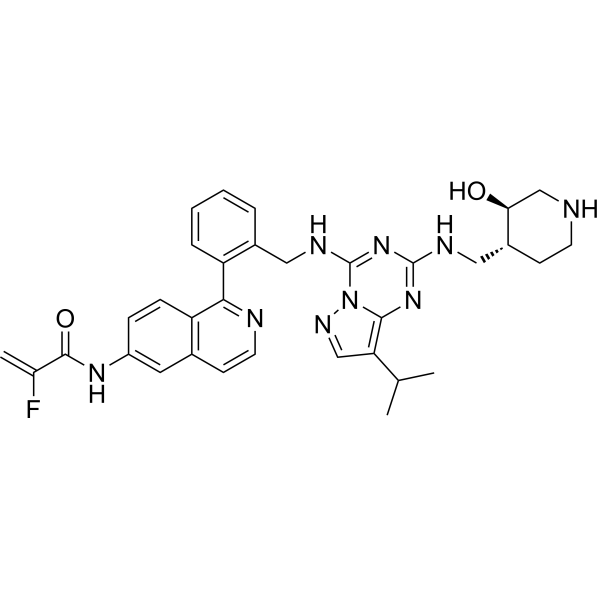
- HY-152226
-
|
|
HDAC
Apoptosis
|
Cancer
|
|
MC2590 is a potent pyridine-containing histone deacetylase (HDAC) inhibitor. MC2590 is a inhibitor of HDAC1-3, -6, -8, and -10 (class I/IIb-selective inhibitor) with IC50s of 0.015 μM-0.156 μM. MC2590 also inhibits HDAC isoforms HDAC4, HDAC5, HDAC7, HDAC9, HDAC11 with IC50s of 1.35 μM-3.98 μM. MC2625 induces G2/M cell cycle arrest and modulates pro- and anti-apoptotic microRNAs towards apoptosis induction .
|
-
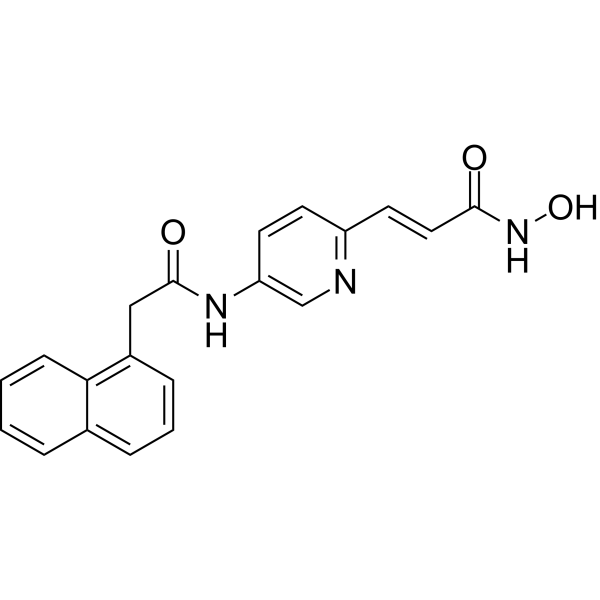
- HY-157125
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
PI3Kα-IN-14 (compound F8) is a selective PI3Kα inhibitor with an IC50 of 0.14 nM. PI3Kα-IN-14 induces a great decrease in mitochondrial membrane which caused cell cycle arrest at G1 phase and apoptosis in U87-MG cells. PI3Kα-IN-14 shows significant anti-proliferative activities against three tumor-derived cell lines (PC-3: IC50 of 0.28 μM; HCT-116: IC50 of 0.57 μM; and U87-MG: IC50 of 1.37 μM) .
|
-
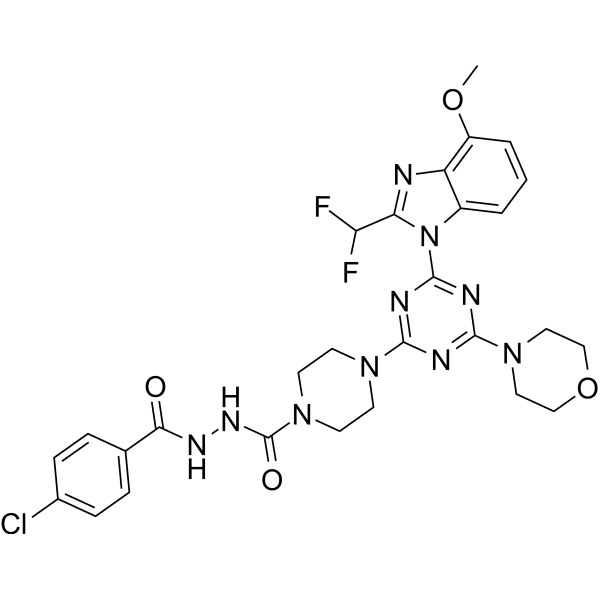
- HY-175834
-
|
|
Topoisomerase
Reactive Oxygen Species (ROS)
Apoptosis
Microtubule/Tubulin
|
Cancer
|
|
DNA/TOP2A-IN-1 is an inhibitor of DNA and TOP2A. DNA/TOP2A-IN-1 selectively binds to TOP2A, not TOP2B, and interacts with DNA and TOP2A to form a stable DM1-TOP2A-DNA ternary complex. DNA/TOP2A-IN-1 induces DNA damage, increases reactive oxygen species (ROS) and triggers apoptosis in triple-negative breast cancer (TNBC) cells. DNA/TOP2A-IN-1 disrupts microtubule distribution and induces cell cycle arrest. DNA/TOP2A-IN-1 shows strong antiproliferative activity and inhibits cell migration. DNA/TOP2A-IN-1 inhibits tumor growth and can be used for TNBC research .
|
-

- HY-181945
-
|
|
FLT3
Apoptosis
Reactive Oxygen Species (ROS)
|
Cancer
|
|
FLT3-IN-39 (Compound W4) is a selective FLT3 inhibitor, with an IC50 value of 16.0 nM against FLT3-ITD and an IC50 value of 20.4 nM against FLT3-D835Y. FLT3-IN-39 inhibits FLT3-ITD and FLT3-D835Y mutant kinases. FLT3-IN-39 induces G0/G1 cell cycle arrest and Apoptosis in cancer cells, and reduces intracellular ROS levels. FLT3-IN-39 exhibits anti-tumor activity against acute myeloid leukemia .
|
-

- HY-181766
-
|
|
EGFR
VEGFR
COX
Microtubule/Tubulin
Apoptosis
|
Neurological Disease
Cancer
|
|
EGFR/VEGFR2-IN-11 is an EGFR/VEGFR2 inhibitor with IC50 values of 0.62 μM (human EGFR), 2.26 μM (human VEGFR-2), 17.38 μM (human COX-2), and 19.31 μM (human tubulin). EGFR/VEGFR2-IN-11 inhibits COX-2 activity and tubulin polymerization. EGFR/VEGFR2-IN-11 induces cell cycle arrest and apoptosis. EGFR/VEGFR2-IN-11 exerts selective and antiproliferative activity against human cancer cells. EGFR/VEGFR2-IN-11 can be used for the research of colon carcinoma, breast carcinoma, leukemia, lymphoma, glioblastoma .
|
-

- HY-169093
-
|
|
PROTACs
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
MS41 is a selective eleven-nineteen leukemia (ENL) PROTAC degrader, with DC50s of 3.50 nM (MV4;11), 2.84 nM (SEMK2), 3.03 nM (Jurkat), and 26.58 nM (KASUMI1), respectively. MS41 effectively inhibits the growth of ENL-dependent leukemia cells, induces G1 cell cycle arrest and increases apoptosis. MS41 reduces the chromatin occupancy of ENL-associated transcription elongation machinery, and suppresses oncogenic gene expression and leukemia progression. Red: ENL ligand (HY-169094). Black: linker (HY-W105744). Blue: VHL ligand (HY-112078) .
|
-

- HY-143233
-
|
|
Pim
HDAC
Apoptosis
|
Cancer
|
|
PIM-1/HDAC-IN-1 (compound 4d) is a PIM-1 inhibitor, with an IC50 of 343.87 nM. PIM-1/HDAC-IN-1 has strong inhibitory activity and selectivity against HDAC 1 and HDAC 6, with IC50 values of 63.65 and 62.39 nM, respectively. PIM-1/HDAC-IN-1 exhibits apoptosis inducing potential in MCF-7 cell lines. PIM-1/HDAC-IN-1 shows pre-G1 apoptosis and cell cycle arrest at G2/M phase .
|
-
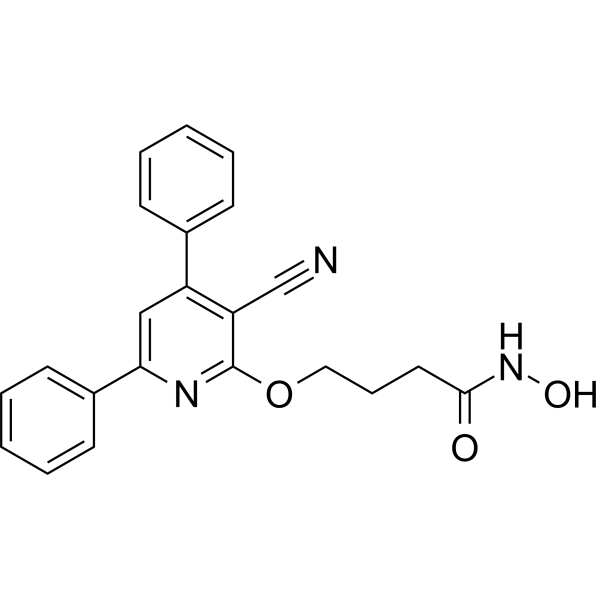
- HY-183366
-
|
|
HDAC
Caspase
Akt
|
Inflammation/Immunology
Cancer
|
|
HDAC1-IN-14 is an indole-based benzamide selective HDAC1 inhibitor with an IC50 of 77 nM. HDAC1-IN-14 acts as an antiproliferative agent, with GI50 values ranging from nanomolar to low micromolar levels in various cancer cells. HDAC1-IN-14 induces G0-G1 cell cycle arrest in colon cancer cells. HDAC1-IN-14 upregulates the expression of Caspase-3, Cyto-C and Bax, and downregulates the expression of AKT-1. HDAC1-IN-14 can be used in research related to leukemia, non-small cell lung cancer, colon cancer, central nervous system cancer, melanoma, ovarian cancer, renal cancer, prostate cancer and breast cancer .
|
-

- HY-182285
-
|
|
5-HT Receptor
DNA/RNA Synthesis
|
Cancer
|
|
FOXM1-IN-4 is a selective 5-HT7 receptor inhibitor with a Ki of 92 nM. FOXM1-IN-4 blocks 5-HT7 receptor signaling to reduce FOXM1, p-FOXM1, cyclin B1, and cdc25B levels. FOXM1-IN-4 acts as an antiproliferative, clonogenic inhibitor, and cell cycle inhibitor that induces G2/M arrest, reduces G0/G1 population. FOXM1-IN-4 can be used for the research of triple-negative breast cancer .
|
-

- HY-182285A
-
|
|
5-HT Receptor
DNA/RNA Synthesis
|
Cancer
|
|
FOXM1-IN-4 hydrochloride is a selective 5-HT7 receptor inhibitor with a Ki of 92 nM. FOXM1-IN-4 hydrochloride blocks 5-HT7 receptor signaling to reduce FOXM1, p-FOXM1, cyclin B1, and cdc25B levels. FOXM1-IN-4 hydrochloride acts as an antiproliferative, clonogenic inhibitor, and cell cycle inhibitor that induces G2/M arrest, reduces G0/G1 population. FOXM1-IN-4 hydrochloride can be used for the research of triple-negative breast cancer .
|
-

- HY-153278A
-
|
CDK7-IN-21 TFA
|
CDK
DNA/RNA Synthesis
c-Myc
Early 2 Factor (E2F)
Topoisomerase
|
Cancer
|
|
Q901 (CDK7-IN-21) TFA is a selective and potent CDK7 inhibitor with an IC50 of 10 nM. Q901 TFA disrupts MYC and E2Fcell cycle control and DNA damage repair pathways. Q901 TFA stabilizes TOP1-DPCs and sensitizes tumor to TOP1 inhibitors by suppressing RNAPII transition from initiation to elongation. Q901 TFA can inhibit tumor growth and significantly enhances tumor growth inhibition combined with TOP1 inhibitors. Q901 TFA can be used for the research of cancer, such as colon cancer and lung cancer .
|
-

- HY-B0277A
-
|
ara-AMP; ara-A 5'-monophosphate
|
EBV
HSV
Fungal
DNA/RNA Synthesis
Apoptosis
Drug Intermediate
Reactive Oxygen Species (ROS)
|
Infection
|
|
Vidarabine phosphate (ara-AMP; ara-A 5'-monophosphate) is a purine nucleoside antiviral agent and a prodrug of Vidarabine (HY-B0277). Vidarabine phosphate is rapidly converted into the antiviral active Vidarabine in vivo, which selectively inhibits viral DNA polymerase and cellular ribonucleotide reductase, thereby blocking viral replication. Vidarabine phosphate also exhibits antifungal activity, induces late-stage cellular apoptosis, and causes cell cycle arrest. Vidarabine phosphate can be used in research related to severe chronic active Epstein-Barr virus (EBV) infection, herpes infection, and candidiasis .
|
-
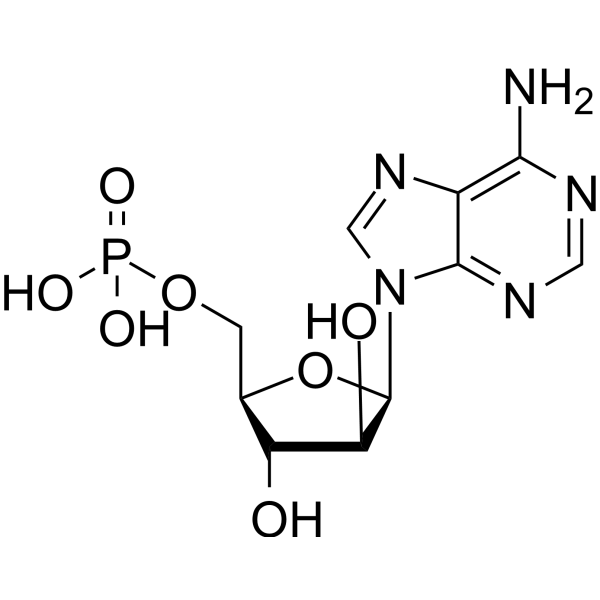
- HY-B0277R
-
|
Ara-A (Standard); Adenine Arabinoside (Standard); 9-β-D-Arabinofuranosyladenine (Standard)
|
Reference Standards
Apoptosis
Fungal
Reactive Oxygen Species (ROS)
EBV
HSV
Antibiotic
DNA/RNA Synthesis
Drug Metabolite
|
Infection
|
|
Vidarabine (Ara-A) (Standard) is the analytical standard of Vidarabine. This product is intended for research and analytical applications. Vidarabine is a nucleoside antibiotic isolated from Streptomyces, and a metabolite of Vidarabine phosphate (HY-B0277A). Vidarabine selectively inhibits viral DNA polymerase and cellular ribonucleotide reductase, thereby blocking viral replication. Vidarabine phosphate also exhibits antifungal activity, induces late-stage cellular apoptosis, and causes cell cycle arrest. Vidarabine phosphate can be used in research related to severe chronic active Epstein-Barr virus (EBV) infection, herpes infection, and candidiasis .
|
-

- HY-13614
-
|
|
Microtubule/Tubulin
Apoptosis
Caspase
|
Cardiovascular Disease
Cancer
|
|
E7974 is a selective inhibitor of α-tubulin (α-tubulin) with an IC50 of 3.9 μM. E7974 disrupts mitotic spindle formation, induces G2-M phase cell cycle arrest, initiates apoptosis, activates caspase-3, and induces poly (ADP-ribose) polymerase cleavage. E7974 reduces the area of choroidal neovascularization in mouse models, and exerts anti-angiogenic effects when loaded in modified micelles. E7974 can be used in research related to cancer and choroidal neovascularization .
|
-

- HY-119062
-
|
|
MetAP
CDK
|
Cancer
|
|
A-800141 is an orally active, selective, sulfonamide-based MetAP2 inhibitor (IC50=12 nM) that binds reversibly to MetAP2 and interacts with its manganese ions. A-800141 induces the production of N-terminal methionine-unprocessed GAPDH variants, which in turn triggers G1-phase cell cycle arrest, elevates p21 levels, and reduces the levels of phosphorylated Rb and total cyclin A. A-800141 exhibits anti-angiogenic and tumor growth inhibitory effects, and produces synergistic effects when combined with cytotoxic inhibitors or BCL-2 inhibitors. A-800141 has been widely used in scientific research related to B-cell lymphoma, neuroblastoma, prostate cancer, colon cancer, melanoma and other fields .
|
-

- HY-N16771
-
|
|
Caspase
Apoptosis
Bcl-2 Family
Bacterial
VEGFR
|
Cancer
|
|
Clausenidin is a selective inhibitor targeting apoptosis-related pathways, including the mitochondrial pathway and death receptor pathway, and vascular endothelial growth factor (VEGF). Clausenidin induces mitochondrial membrane depolarization by activating caspase-3, caspase-8 and caspase-9, upregulating the pro-apoptotic protein Bax and downregulating the anti-apoptotic protein Bcl-2. Clausenidin also inhibits VEGF expression and blocks angiogenesis, exerting anti-tumor activity. Clausenidin has inhibitory effects against Mycobacterium tuberculosis (MIC=200 μg/mL). Clausenidin can induce apoptosis in liver cancer cells, arrest the cell cycle in the G2/M phase, and inhibit tumor angiogenesis. Clausenidin can be used in the research of malignant tumors such as liver cancer .
|
-

- HY-170926
-
|
|
RET
Apoptosis
|
Cancer
|
CQ1373 is a potent RET inhibitor, demonstrating cellular potency with IC50 values of 13.0, 25.7 and 28.4 nM against BaF3 cells expressing CCDC6-RET, CCDC6-RET-G810C and CCDC6-RET-G810R, respectively. CQ1373 exhibits good selectivity toward wild-type RET and solvent front mutants G810C/R with IC50 values of 4.2, 7.1 and 32.4 nM, respectively. CQ1373 inhibits RET phosphorylation and downstream signaling through SHC. CQ1373 induces Apoptosis and cell cycle arrest in BaF3 cells. CQ1373 exhibits anti-tumor efficacy and can be used for cancer research .
|
-

- HY-181928
-
|
|
c-Met/HGFR
PARP
Apoptosis
|
Cancer
|
|
PARP1/c-Met-IN-3 (Compound L19) is a selective c-Met and PARP1 inhibitor, with an IC50 of 5.4 nM against c-Met and an IC50 of 3.7 nM against PARP1. PARP1/c-Met-IN-3 inhibits PARP2 enzymatic activity with an IC50 of 4.52 nM, and shows no specificity for PARP1 and PARP2. PARP1/c-Met-IN-3 induces cell cycle arrest and apoptosis. PARP1/c-Met-IN-3 exhibits anti-tumor activity against triple-negative breast cancer .
|
-

- HY-168477
-
|
|
HDAC
Autophagy
|
Cancer
|
|
HDAC1-IN-8 (compound 5c) is a potent and selective HDAC1 inhibitor with IC50 values of 11.94, 22.95, >500 µM for HDAC1, HDAC6, HDAC8, respectively. HDAC1-IN-8 shows antiproliferative activity. HDAC1-IN-8 induces cell cycle arrest at G1 and G2/M. HDAC1-IN-8 induces autophagy. HDAC1-IN-8 shows anticancer activity and has the potential for the research of lung cancer .
|
-

- HY-B0277AR
-
|
ara-AMP (Standard); ara-A 5'-monophosphate (Standard)
|
Reference Standards
Apoptosis
Drug Intermediate
Fungal
Reactive Oxygen Species (ROS)
EBV
HSV
DNA/RNA Synthesis
|
Infection
|
|
Vidarabine phosphate (ara-AMP; ara-A 5'-monophosphate) (Standard) is the analytical standard of Vidarabine phosphate. This product is intended for research and analytical applications. Vidarabine phosphate is a purine nucleoside antiviral agent and a prodrug of Vidarabine (HY-B0277). Vidarabine phosphate is rapidly converted into the antiviral active Vidarabine in vivo, which selectively inhibits viral DNA polymerase and cellular ribonucleotide reductase, thereby blocking viral replication. Vidarabine phosphate also exhibits antifungal activity, induces late-stage cellular apoptosis, and causes cell cycle arrest. Vidarabine phosphate can be used in research related to severe chronic active Epstein-Barr virus (EBV) infection, herpes infection, and candidiasis.
|
-

- HY-183013
-
|
|
PROTACs
PARP
Apoptosis
|
Cancer
|
|
PROTAC PARP2 degrader-1 is an orally active PARP2 PROTAC degrader with a DC50 of 2 μM. PROTAC PARP2 degrader-1 potently inhibits the enzymatic activities of PARP1 (IC50 = 2.74 nM) and PARP2 (IC50 = 0.32 nM), with approximately 10-fold higher selectivity for PARP2. PROTAC PARP2 degrader-1 induces cell cycle arrest and apoptosis, and exhibits significant anti-tumor efficacy in mouse models. PROTAC PARP2 degrader-1 can be used for the research of triple-negative breast cancer .
|
-

- HY-180119
-
|
|
IKK
NF-κB
Autophagy
|
Cancer
|
|
IKKβ-IN-5 is an orally active and selective IKKβ inhibitor with an IC50 of 7.5 nM. IKKβ-IN-5 directly inhibits IKKβ phosphorylation and attenuates NF κB mediated inflammatory and survival signals while promoting autophagy flux. IKKβ-IN-5 exhibits a 6-fold selectivity forIKKβ over the homologous kinase IKKα. IKKβ-IN-5 exerts robust antiproliferative effects through a dual mechanism involving G₂/M phase cell cycle arrest and autophagy activation, even under inflammatory stimulation in vitro. IKKβ-IN-5 demonstrates favorable pharmacokinetics and suppresses tumor growth in vivo. IKKβ-IN-5 can be used for colorectal cancer and potentially other inflammation driven malignancies research .
|
-

- HY-179020
-
|
|
PARP
VEGFR
DNA/RNA Synthesis
Apoptosis
Autophagy
|
Cancer
|
|
PARP/VEGFR3-IN-1 (Compound 18) is a dual PARP-VEGFR3 target inhibitor. Its IC50 values for PARP1, PARP2, and VEGFR3 are 0.0763, 0.0366, and 4.25 nM respectively. PARP/VEGFR3-IN-1 has no inhibitory effect on VEGFR1/2 and shows subtype selectivity. PARP/VEGFR3-IN-1 exhibits significant anti-proliferative activity in various cancer cells (leukemia, lung cancer, and triple-negative breast cancer). PARP/VEGFR3-IN-1 induces DNA damage and cell cycle arrest. PARP/VEGFR3-IN-1 triggers various forms of cell death by inducing apoptosis and autophagy. PARP/VEGFR3-IN-1 can be formulated into nanodelivery systems, significantly enhancing tumor targeting and therapeutic window .
|
-

- HY-173444
-
|
|
HDAC
Transferrin Receptor
Apoptosis
Ferroptosis
|
Cancer
|
|
HDAC11-IN-3 (Compound A9) is a selective HDAC11 inhibitor (IC50: 4.1 nM). HDAC11-IN-3 has inhibitory effects on U937 and OCI-AML2 acute myeloid leukemia (AML) cell lines (IC50: 10 μM). HDAC11-IN-3 has significant anti-AML activity, inducing apoptosis, cell cycle arrest, and differentiation. HDAC11-IN-3 upregulates the iron transporters transferrin (TF) and transferrin receptor (TFRC), and activates the p62-Keap1-Nrf2-HMOX1 pathway, which together lead to increased intracellular iron levels and induce ferroptosis in AML cells. HDAC11-IN-3 can be used alone or in combination with Cytarabine (HY-13605) for AML research .
|
-

- HY-179497
-
|
|
FLT3
VEGFR
Akt
STAT
ERK
Apoptosis
|
Cancer
|
FLT3-IN-37 (Compound 6z) is a potent inhibitor of FLT3-ITD, with IC50 values of 1.5 and 3.4 nM for FLT3-ITD and TEL-VEGFR2, respectively. FLT3-IN-37 exhibits high selectivity for wild-type FLT3 (WT) and c-Kit. FLT3-IN-37 inhibits FLT3 phosphorylation and downregulates the expression of p-Akt, p-STAT5, and p-ERK. FLT3-IN-37 exerts anti-leukemia effects by blocking the cell cycle and inducing apoptosis (apoptosis). FLT3-IN-37 can be used for research on acute myeloid leukemia (AML) .
|
-

- HY-W040129
-
|
|
DNA Alkylator/Crosslinker
DNA/RNA Synthesis
Apoptosis
Caspase
|
Infection
Neurological Disease
Cancer
|
|
Chromomycin A3 is an inhibitor that selectively binds to GC-rich DNA sequences. Chromomycin A3 targets the DNA minor groove after forming a dimer with Mg 2+. Chromomycin A3 inhibits DNA replication and transcription, blocks the binding of Sp1 transcription factor to target gene promoters, downregulates the expression of anti-apoptotic proteins such as FLIP, Mcl-1, and XIAP, and induces S-phase cycle arrest and caspase-dependent apoptosis in tumor cells. Chromomycin A3 can antagonize oxidative stress induced by glutathione depletion and neuronal apoptosis induced by Camptothecin (HY-15660). Chromomycin A3 can be used in basic research on malignant tumors such as cholangiocarcinoma, and is a potential chemosensitizer and GC-rich region probe .
|
-
- HY-174347
-
|
|
HSP
Casein Kinase
Apoptosis
|
Cancer
|
|
Hsp90-Cdc37-IN-4, a Celastrol (HY-13067) derivative, inhibits the Hsp90-Cdc37 protein-protein interaction (PPI). Hsp90-Cdc37-IN-4 selectively inhibits casein kinase 2 (CK2), reducing phosphorylation of its substrate Cdc37 at Serine 13. Hsp90-Cdc37-IN-4 induces G0/G1 cell cycle arrest and triggers apoptosis via the mitochondrial pathway. Hsp90-Cdc37-IN-4 demonstrates potent anti-breast cancer activity .
|
-

- HY-163985
-
|
|
PROTACs
FGFR
Apoptosis
|
Cancer
|
|
PROTAC FGFR2 degrader 1 (compound N5) is a PROTAC that effectively targets FGFR2 with DC50 of 6.46 nM, the FGFR2 IC50 is 0.08 nM. PROTAC FGFR2 degrader 1 has anti-proliferative activity and highly selective, induces G0/G1 arrest of KATOIII and SNU16 cell cycle and inhibits apoptosis by reducing the activation of p-ERK and p-PLCγ, the downstream proteins of FGFR2.
PROTAC FGFR2 degrader 1 inhibits gastric cancer cells remained above 50% at a concentration of 0.17 nM.
PROTAC FGFR2 degrader 1 potently inhibits the growth of SNU16 xenograft tumors in mouse model (Structure Note: Pink, FGFR2 activator: HY-18708; Blue, E3 ligase ligand: HY--10984; Black, linker: HY-163989; E3 ligase ligand + linker:HY-163986) .
|
-

- HY-170451
-
|
KT-253
|
PROTACs
MDM-2/p53
Apoptosis
|
Cancer
|
|
Seldegamadlin (KT-253) is a selective p53 stabilizer and a MDM2 PROTAC degrader (DC50 = 0.4 nM). Seldegamadlin inhibits the proliferation of cancer cell RS4;11 with an IC50 of 0.3 nM, arrests the cell cycle at G2/M phase, and induces apoptosis. Seldegamadlin upregulates p53 activity and overcomes the p53-MDM2 feedback loop. Seldegamadlin can be used for the study of hematologic and solid tumors, such as acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). (Pink: ligand for target protein MDM2 ligand 4 (HY-170452); Black: linker (HY-W001478); Blue: ligand for E3 ligase cereblon (HY-163927)) .
|
-

- HY-179034
-
|
|
Carbonic Anhydrase
c-Met/HGFR
Apoptosis
|
Cancer
|
|
CAIX/XII-IN-16 (Compound 5d) is a selective inhibitor of CA IX/XII and c-Met. CAIX/XII-IN-16‘s Ki values for hCA IX, hCA XII and CA I are 10.6, 31.8 and 2361 nM respectively, and its IC50 value for c-Met is 0.49 μM. CAIX/XII-IN-16 exhibits significantly enhanced cytotoxicity against colon cancer cells under hypoxic conditions. CAIX/XII-IN-16 causes cell cycle arrest and induces apoptosis (apoptosis). CAIX/XII-IN-16 significantly enhances the efficacy of Oxaliplatin (HY-17371) and 5-Fluorouracil (HY-90006). CAIX/XII-IN-16 can be used for studies on chemotherapy resistance related to hypoxia .
|
-

- HY-145601
-
|
TT 00420
|
Aurora Kinase
FGFR
VEGFR
|
Cancer
|
|
Tinengotinib (TT00420) is an orally active, spectrally selective small molecule kinase inhibitor targeting Aurora A/B (IC50=1.2-3.3 nM), FGFR1/2/3 (IC50=1.5-3.5 nM), VEGFRs, JAK1/2 and CSF1R. Tinengotinib blocks Aurora kinase-mediated cell cycle progression (inducing G2/M arrest), inhibits FGFR/JNK-JUN signaling pathway and activates MEK/ERK-dependent apoptotic pathway. Tinengotinib has the activity of anti-tumor proliferation, inducing apoptosis, inhibiting angiogenesis and regulating tumor microenvironment. Tinengotinib can be used in the study of triple-negative breast cancer (TNBC), gallbladder cancer and tumor immune microenvironment .
|
-
- HY-N12445
-
|
|
Topoisomerase
Caspase
Apoptosis
SOD
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Quercetin-3'-O-glucoside is an orally active flavonoid glycoside. Quercetin-3'-O-glucoside reduces liver glucose-6-phosphatase activity, alters serum insulin and glucose levels, and regulates the activities of antioxidant enzymes in the liver and kidney. Quercetin-3'-O-glucoside inhibits DNA topoisomerase II, induces S-phase cell cycle arrest and caspase-3-mediated apoptosis in hepatocellular carcinoma cells. Quercetin-3'-O-glucoside selectively inhibits EGFR-mediated signaling pathways targeting AKT, ERK1/2, FAK and MEK1/2. Quercetin-3'-O-glucoside inhibits growth factor-induced migration and invasion in pancreatic cancer cells. Quercetin-3'-O-glucoside exerts free radical scavenging effects. Quercetin-3'-O-glucoside is applicable to research related to pancreatic cancer, diabetes, hepatocellular carcinoma and malignant tumors .
|
-
- HY-183118
-
|
|
CDK
Apoptosis
|
Neurological Disease
Cancer
|
|
CID-078 is an orally active macrocyclic cyclin A and cyclin B inhibitor. CID-078 binds cyclin hydrophobic patches, disrupting interactions of cyclin A-Cdk2 with E2F1 and cyclin B-Cdk1 with Myt1, and selectively targets RxL binding motifs to block complex-substrate interactions. CID-078 induces DNA damage, G2/M cell cycle arrest, apoptosis, mitotic catastrophe, spindle assembly checkpoint activation, and neomorphic cyclin B-CDK2 complex formation, driving synthetic lethality in E2F-driven cancer cells. CID-078 can be used for the research of small cell lung cancer, non-small cell lung cancer, triple negative breast cancer, advanced solid tumors, luminal HR +/HER 2- breast cancer, RB1-altered solid tumors, and neuroblastoma .
|
-

- HY-147898
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
PI3K-IN-33 (Compound 6e) is a highly selective PI3K inhibitor with IC50 values of 11.73, 6.09 and 11.18 μM for PI3K-α、PI3K-β and PI3K-δ , respectively. PI3K-IN-33 arrests cell cycle at G2/M phase and induces apoptosis. PI3K-IN-33 can be used in leukemia research .
|
-
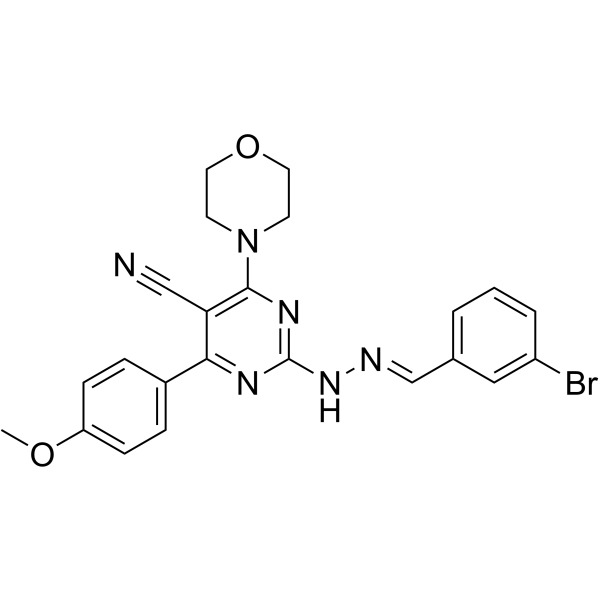
- HY-147900
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
PI3K-IN-35 (Compound 6l) is a highly selective PI3K inhibitor with IC50 values of 13.98, 7.22 and 10.94 μM for PI3K-α、PI3K-β and PI3K-δ, respectively. PI3K-IN-35 arrests cell cycle at G2/M phase and induces apoptosis. PI3K-IN-35 can be used in leukemia research .
|
-
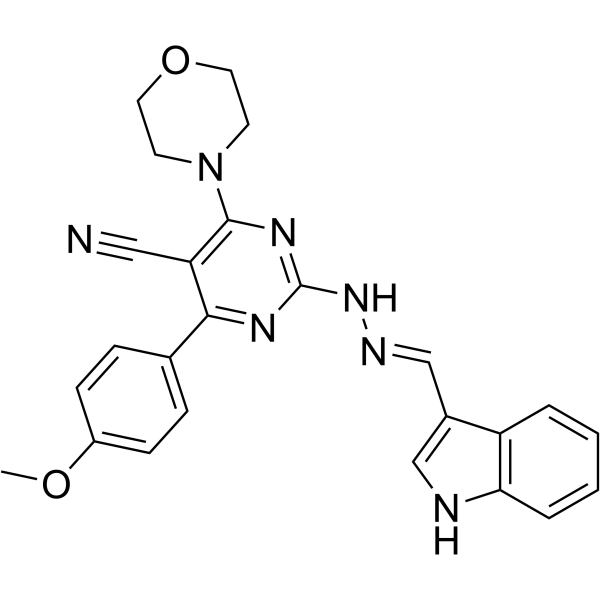
- HY-147899
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
PI3K-IN-34 (Compound 6g) is a highly selective PI3K inhibitor with IC50 values of 11.73, 6.09 and 11.18 μM for PI3K-α、PI3K-β and PI3K-δ , respectively. PI3K-IN-34 arrests cell cycle at G2/M phase and induces apoptosis. PI3K-IN-34 can be used in leukemia research .
|
-
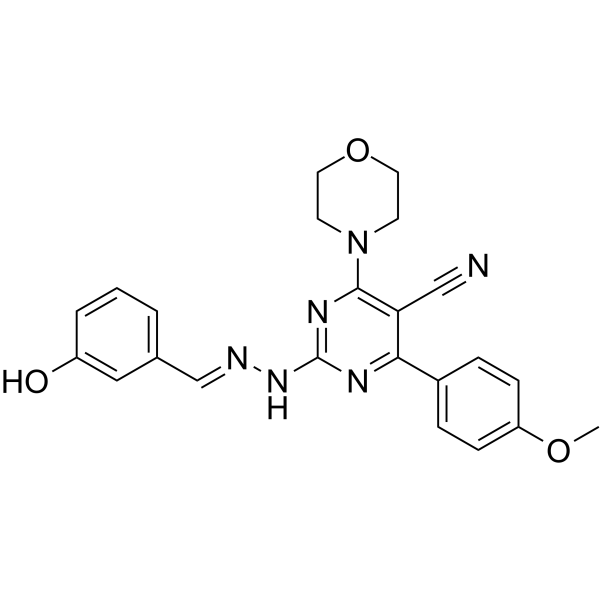
- HY-179499
-
|
|
PROTACs
Histone Methyltransferase
Apoptosis
|
Cancer
|
|
PROTAC EZH2 Degrader-9 is orally active EZH2 PROTAC degrader degrading EZH2 via the ubiquitin-proteasome pathway. PROTAC EZH2 Degrader-9 downregulates PRC2 core subunits and potent inhibition of H3K27me3 without affecting common CRBN neosubstrates while it was selective over GSp'T1 and ikZF1/3. PROTAC EZH2 Degrader-9 exhibits potent antiproliferative activity against multiple cancer cell lines by inducing cell cycle and apoptosis. PROTAC EZH2 Degrader-9 reverses PRC2-mediated gene silencing and inhibiting EZH2 non-catalytic target gene activation. PROTAC EZH2 Degrader-9 can be used for leukemia, lymphoma, and non-small cell lung cancer research .
|
-

- HY-164729
-
|
|
Antibody-Drug Conjugates (ADCs)
Topoisomerase
Apoptosis
|
Cancer
|
|
FZ-AD005 is a DLL3-targeting antibody-drug conjugate (ADC) with high selectivity, composed of the anti-DLL3 antibody FZ-A038 (HY-P990896), a dipeptide linker (Val-Ala), and DXd (HY-13631D). The Kd value of FZ-AD005 for human DLL3 ranges from 13.29 to 58.3 pmol/L. After binding to DLL3 on the cell surface, FZ-AD005 mediates endocytosis, and the payload DXd is released via cleavage by lysosomal cathepsins. DXd inhibits topoisomerase TopI to induce double-strand DNA breaks, cell cycle arrest and apoptosis, and FZ-AD005 exhibits bystander killing activity against adjacent DLL3-negative cells. FZ-AD005 shows stable circulation in vivo, has good tolerance and acceptable pharmacokinetic profiles in rats and cynomolgus monkeys, and effectively inhibits the growth of DLL3-expressing tumor cells. FZ-AD005 serves as a promising candidate molecule for research on small cell lung cancer and human neuroendocrine prostate cancer .
|
-

- HY-164184
-
|
|
Apoptosis
Caspase
HSP
Early 2 Factor (E2F)
DNA/RNA Synthesis
|
Cancer
|
|
Ly101-4B is an apoptosis inducer and multi-target inhibitor with antiproliferative, antitumor and cycytotoxic effects. Ly101-4B reduces HSF1 expression, inhibits microRNA-214 synthesis, downregulates HSP27, HSP70 and HSP90 expression, while suppressing E2F-dependent transcriptional activity and downregulating its target genes. Ly101-4B induces caspase 3/7-mediated apoptosis by reducing DNA synthesis, inhibiting the cell cycle and G1/S phase transition, without affecting RNA synthesis or inducing necrosis. Ly101-4B is selective for pancreatic ductal adenocarcinoma cells with different genotypes and varying degrees of E2F dependence. Ly101-4B can be used in research related to epithelial ovarian cancer and pancreatic ductal adenocarcinoma .
|
-

- HY-170576
-
|
|
FLT3
STAT
Apoptosis
|
Cancer
|
|
FLT3-IN-28 (Compound 12y) is an orally active FLT3 inhibitor with antitumor activity. FLT3-IN-28 selectively inhibits cancer cells harboring the FLT3 internal tandem duplication (ITD) mutation, with IC50 values of 85, 290, 130, 65, and 220 nM for BaF3-FLT3-ITD, BaF3-TEL-VEGFR2, MV4-11, MOLM-13, and MOLM-14 cell lines respectively (MV4-11 and MOLM-13/14 are acute myeloid leukemia (AML) cell lines carrying the FLT3-ITD mutation). Additionally, FLT3-IN-28 can downregulate the phosphorylation levels of FLT3 and STAT5 in MOLM-13 cells and induce cell cycle arrest and Apoptosis. FLT3-IN-28 has an oral bioavailability of 19.2% in SD rats and can prolong survival in a dose-dependent manner in NSG mice xenografted with MOLM-13 cells. FLT3-IN-28 holds promise for research in cancer fields related to FLT3-ITD .
|
-

- HY-180811
-
|
|
Anaplastic lymphoma kinase (ALK)
HDAC
Apoptosis
|
Neurological Disease
|
|
ALK/HDAC-IN-2 (Compound 19b) is an ALK/HDAC inhibitor with IC₅₀ values for ALK WT and total HDACs of 8 nM and 1.18 μM, respectively. ALK/HDAC-IN-2 exhibits inhibitory activity against ALK mutants G1202R, F1174L, and L1196M, with IC₅₀ values of 2.74, 9.23, and 34.28 nM, respectively. ALK/HDAC-IN-2 shows potent and selective inhibition against HDAC1 (IC₅₀ = 0.24 μM), while its inhibitory activity against HDAC7, HDAC6, and HDAC11 is weak (IC₅₀ > 10 μM). ALK/HDAC-IN-2 has broad-spectrum anti-proliferative activity against various cancer cells, inducing cell cycle arrest and apoptosis. ALK/HDAC-IN-2 can be used for the study of neuroblastoma .
|
-

- HY-176225
-
|
|
PROTACs
Src
Estrogen Receptor/ERR
Apoptosis
|
Cancer
|
|
BY13 is a SRC-3 PROTAC degrader with a DC50 of 0.031 μM. BY13 selectively blocks the ER signaling pathway over that of androgen receptor (AR)) through down-regulating ERα level. BY13 potently overcomes endocrine resistance in breast cancer by inducing cell cycle arrest in G1 phase and apoptosis, with superior effect over Fulvestrant (HY-13636). BY13 significantly inhibits the growth of drug-resistant breast tumors without obvious toxicity in LCC2 xenograft mice model . Pink: SRC-3 ligand (SI-2) (HY-101447); Blue: CRBN ligase ligand (HY-41547); Black: linker (HY-176226)
|
-

- HY-179155
-
|
|
PI3K
mTOR
Apoptosis
Bcl-2 Family
MDM-2/p53
Telomerase
Mitochondrial Metabolism
|
Inflammation/Immunology
Cancer
|
|
PI3K/mTOR-IN-19 is an orally active, potent, selective PI3K (IC50 = 4.23 nM) and mTOR (IC50 = 2.3 nM) inhibitor. PI3K/mTOR-IN-19 significantly inhibits Eca109 cell viability and induces apoptosis. PI3K/mTOR-IN-19 causes G0/G1 cell cycle arrest, decreased mitochondrial membrane potential, and demonstrates marked telomerase inhibitory activity. PI3K/mTOR-IN-19 modulates the expression of key apoptotic regulators (Bcl-2, Bax, and p53) and downregulates the PI3K/Akt/mTOR signaling pathway. PI3K/mTOR-IN-19 can be used for the study of esophageal cancer .
|
-

- HY-W040129R
-
|
|
Reference Standards
Bacterial
Fungal
Apoptosis
Antibiotic
|
Infection
Neurological Disease
Cancer
|
|
Chromomycin A3 (Standard) is the analytical standard of Chromomycin A3 (HY-W040129). This product is intended for research and analytical applications. Chromomycin A3 is an inhibitor that selectively binds to GC-rich DNA sequences. Chromomycin A3 targets the DNA minor groove after forming a dimer with Mg 2+. Chromomycin A3 inhibits DNA replication and transcription, blocks the binding of Sp1 transcription factor to target gene promoters, downregulates the expression of anti-apoptotic proteins such as FLIP, Mcl-1, and XIAP, and induces S-phase cycle arrest and caspase-dependent apoptosis in tumor cells. Chromomycin A3 can antagonize oxidative stress induced by glutathione depletion and neuronal apoptosis induced by Camptothecin (HY-15660). Chromomycin A3 can be used in basic research on malignant tumors such as cholangiocarcinoma, and is a potential chemosensitizer and GC-rich region probe .
|
-

- HY-155232
-
|
|
PI3K
Apoptosis
|
Cancer
|
|
PI3Kδ-IN-16 is a potent and selective PI3Kδ inhibitor with an IC50 value of 0.9 nM. PI3Kδ-IN-16 has a strong anti-proliferative effect on SU-DHL-6 cells, causing cell cycle arrest and inducing apoptosis. PI3Kδ-IN-16 tightly bins to PI3Kδ protein with a planar-shaped conformation. The kinase activity of PI3Kδ-IN-16 which is ~378-fold over PI3Kα, 412-fold over PI3Kβ, and 10-fold over PI3Kγ. PI3Kδ-IN-16 can be used for the study of hematologic malignancies .
|
-
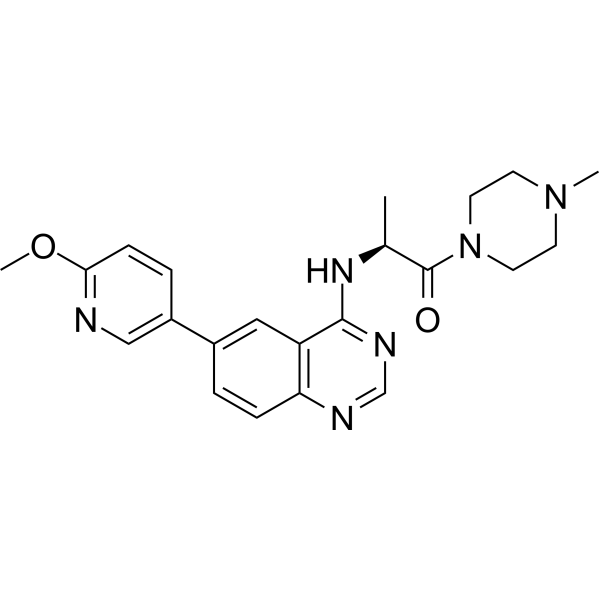
- HY-176239
-
|
|
PROTACs
PI3K
Akt
Apoptosis
Autophagy
|
Cancer
|
|
PROTAC PI3Kδ degrader-1 is a Lysine-targeted covalent PI3Kδ PROTAC degrader with a DC50 of 3.98 nM. PROTAC PI3Kδ degrader-1 has a potent antiproliferative activity and selective PI3Kδ inhibition (IC50: 8 nM). PROTAC PI3Kδ degrader-1 also significantly degrades p-AKT, induces cell cycle arrest in G1 phase and prompts cell apoptosis and autophagy. PROTAC PI3Kδ degrader-1 effectively inhibits the tumor growth in SU-DHL-6 xenograft mice model . Pink: PI3Kδ ligand (HY-169983); Blue: VHL ligase ligand (HY-112078); Black: linker (HY-W013381)
|
-

- HY-13817
-
|
|
Deubiquitinase
Autophagy
Apoptosis
|
Cardiovascular Disease
Neurological Disease
Endocrinology
Cancer
|
|
IU1 is a selective, reversible USP14 inhibitor with an IC50 of 4-5 μM. IU1 binds USP14’s catalytic cleft to block deubiquitinase activity. IU1 induces calpain-dependent Tau cleavage, causes ATP deficits, reduces E1~Ub thioester levels and 26S proteasome assembly. IU1 enhances 26S proteasome chymotrypsin-like activity, modulates LC3B-dependent autophagy flux, reduces cancer cell proliferation and migration, and blocks G0/G1 to S phase cell cycle transition in follicular thyroid cancer cells. IU1 activates autophagy-lysosomal and ubiquitin-proteasome pathways, triggers apoptosis, and reduces cervical cancer cell growth. IU1 enhances degradation of proteasome substrates linked to neurodegenerative disease, accelerates oxidized protein degradation, and increases oxidative stress resistance. IU1 can be used for the research of Alzheimer’s disease, follicular thyroid cancer, ischemic stroke, cervical cancer, and neurodegenerative disease .
|
-
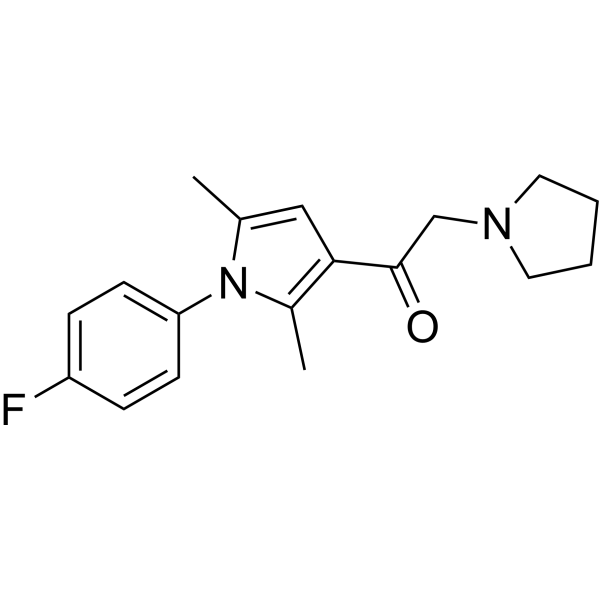
- HY-149824
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR T790M/L858R-IN-2 is a potent and selective EGFRT790M/L858R inhibitor with IC50 values of 3.5, 1290 nM for EGFRT790M/L858R, EGFR WT, respectively. EGFR T790M/L858R-IN-2 decreases the expression of p-EGFR, P-AKT, P-ERK1/2. EGFR T790M/L858R-IN-2 induces Apoptosis and cell cycle arrest in the G1 phase. EGFR T790M/L858R-IN-2 shows anti-cancer activity .
|
-
- HY-179388
-
|
|
PROTACs
Sirtuin
Apoptosis
Akt
mTOR
|
Cancer
|
|
PROTAC Sirt2 Degrader-2 is a highly efficient and selective PROTAC degrader targeting SIRT2. PROTAC Sirt2 Degrader-2 demonstrates the most potent anti-proliferative activity both in vitro and in vivo. PROTAC Sirt2 Degrader-2 leads to a marked increase in H4K16Ac levels. PROTAC Sirt2 Degrader-2 significantly suppresses clonogenic formation and migration, induces cell cycle arrest, and promotes apoptosis. PROTAC Sirt2 Degrader-2 inhibits the AKT/mTOR signaling pathway by indirectly degrading SIRT2 and blocking downstream protein phosphorylation, thereby disrupting the signaling cascade and suppressing tumor development. PROTAC Sirt2 Degrader-2 can be used for the study of ovarian cancer .
|
-

- HY-179498
-
|
|
FOXO
PTEN
ROCK
Epigenetic Reader Domain
PI3K
Akt
Apoptosis
|
Cancer
|
|
ROCK2-IN-13 is a selective ROCK2 inhibitor. ROCK2-IN-13 reduces nuclear expression by disrupting the interaction of ROCK2 with transcriptional co activators p300> and PGC 1α, repressing oncogenic transcription. ROCK2-IN-13 activates FOXO1 driven PTEN expression, leading to suppression of the PI3K/Akt pathway, induction of G2/M cell cycle arrest, and promotion of apoptosis. ROCK2-IN-13 ablates the nuclear transcriptional function of ROCK2 that sustains oncogenic signaling and restores the tumor suppressive PTEN/FOXO1 axis. ROCK2-IN-13 can be used for prostate cancer reseach .
|
-

- HY-181163
-
|
|
Caspase
COX
Cytochrome P450
Steroid Sulfatase
Apoptosis
|
Cancer
|
|
Caspase-3/7 activator 4 is a caspase-3 activator and caspase-7 activator. Caspase-3/7 activator 4 inhibits key enzymes in estrogen biosynthesis, including aromatase (IC50 = 38.3 nM) and steroid sulfatase (IC50 = 12.7 µM), and selectively suppresses COX-2 (IC50 = 5.38 µM). Caspase-3/7 activator 4 shows strong antioxidant activity (DPPH: IC50 = 16.26 µM). Caspase-3/7 activator 4 inhibits estrogen synthesis, suppresses estrogen availability, reduces prostaglandin production, increases caspase-3/7 expression, induces G0/G1 cell cycle arrest, induces apoptotic cell death, reduces circulating TNF-α and VEGFR-II levels, restores hepatorenal function markers and histoarchitecture, restores antioxidant defense enzyme activity, reduces lipid peroxidation, exerts antiproliferative activity against breast cancer cells, exerts antitumor activity in the Ehrlich ascites carcinoma models. Caspase-3/7 activator 4 can be used for the research of breast cancer, ehrlich ascites carcinoma .
|
-

- HY-P6292
-
|
|
PACAP Receptor
PKA
ERK
PI3K
Akt
GSK-3
|
Neurological Disease
Cancer
|
|
KS-133 is a bicyclic peptide with VIPR2 antagonistic activity that can cross the blood-brain barrier. KS-133 selectively blocks VIPR2-mediated Gq/Ca, Gs/cAMP, cAMP/PKA/ERK and PI3K/AKT/GSK3β signaling pathways. KS-133 inhibits VIPR2 agonist-induced CREB phosphorylation in the prefrontal cortex of mice. KS-133 shifts the polarization direction of macrophages toward M1. KS-133 attenuates cancer cell proliferation and reduces the cell cycle distribution level at the S-M phase. KS-133 exerts antitumor effects in a mouse model of colorectal cancer. KS-133 reverses cognitive decline in mouse models of psychiatric disorders. KS-133 can be used for research related to schizophrenia, colorectal cancer and breast cancer .
|
-

| Cat. No. |
Product Name |
Type |
-
- HY-164729
-
|
|
Fluorescent Dyes
|
|
FZ-AD005 is a DLL3-targeting antibody-drug conjugate (ADC) with high selectivity, composed of the anti-DLL3 antibody FZ-A038 (HY-P990896), a dipeptide linker (Val-Ala), and DXd (HY-13631D). The Kd value of FZ-AD005 for human DLL3 ranges from 13.29 to 58.3 pmol/L. After binding to DLL3 on the cell surface, FZ-AD005 mediates endocytosis, and the payload DXd is released via cleavage by lysosomal cathepsins. DXd inhibits topoisomerase TopI to induce double-strand DNA breaks, cell cycle arrest and apoptosis, and FZ-AD005 exhibits bystander killing activity against adjacent DLL3-negative cells. FZ-AD005 shows stable circulation in vivo, has good tolerance and acceptable pharmacokinetic profiles in rats and cynomolgus monkeys, and effectively inhibits the growth of DLL3-expressing tumor cells. FZ-AD005 serves as a promising candidate molecule for research on small cell lung cancer and human neuroendocrine prostate cancer .
|
-
- HY-10227G
-
|
PS-341; LDP-341; NSC 681239
|
Fluorescent Dyes
|
|
Bortezomib (GMP) (PS-341 (GMP)) is Bortezomib (HY-10227) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
| Cat. No. |
Product Name |
Type |
-
- HY-10227G
-
|
PS-341; LDP-341; NSC 681239
|
Biochemical Assay Reagents
|
|
Bortezomib (GMP) (PS-341 (GMP)) is Bortezomib (HY-10227) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-B0990
-
|
|
Bacterial
Antibiotic
YAP
|
Infection
|
|
Thiostrepton is a thiazole antibiotic which selectively inhibits FOXM1. FOXM1 binds to YAP/TEAD complex. YAP/TEAD/FOXM1 complex binding at regulatory regions of genes governing cell cycle may impact cell proliferation .
|
-
- HY-P6292
-
|
|
PACAP Receptor
PKA
ERK
PI3K
Akt
GSK-3
|
Neurological Disease
Cancer
|
|
KS-133 is a bicyclic peptide with VIPR2 antagonistic activity that can cross the blood-brain barrier. KS-133 selectively blocks VIPR2-mediated Gq/Ca, Gs/cAMP, cAMP/PKA/ERK and PI3K/AKT/GSK3β signaling pathways. KS-133 inhibits VIPR2 agonist-induced CREB phosphorylation in the prefrontal cortex of mice. KS-133 shifts the polarization direction of macrophages toward M1. KS-133 attenuates cancer cell proliferation and reduces the cell cycle distribution level at the S-M phase. KS-133 exerts antitumor effects in a mouse model of colorectal cancer. KS-133 reverses cognitive decline in mouse models of psychiatric disorders. KS-133 can be used for research related to schizophrenia, colorectal cancer and breast cancer .
|
-
- HY-129283A
-
|
|
Peptides
|
Cancer
|
|
Goralatide TFA is an inhibitor of cell cycle progression. Goralatide TFA enhances selectivity of hyperthermic purging of human progenitor cells. Goralatide TFA has the potential for the research of leukemia .
|
-
- HY-P10226
-
|
|
Chloride Channel
CFTR
|
Endocrinology
|
|
PGD97 is a selective cyclic peptide inhibitor against CAL/CFTR interactions, with a KD value of 6 nM towards the CAL PDZ domain for its desulfide cyclized form. PGD97 (desulfide cyclized form) has selectivity ≥ 130-fold compared to NHERF1/2 PDZ domains. PGD97 is capable of stabilizing F508del-CFTR at the cell membrane and improving CFTR function required for proper fluid homeostasis in tne lung. PGD97 can be used for the research of cystic fibrosis .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-16594
-
|
|
Structural Classification
Microorganisms
Antibiotics
Other Antibiotics
Source Classification
|
Proteasome
Cathepsin
Apoptosis
|
|
Lactacystin is a potent, orally active, irreversible, cell-permeable, selective 20S proteasome inhibitor (IC50 = 4.8 μM). Lactacystin also inhibits the lysosomal enzyme cathepsin A. Lactacystin inhibits cell growth and induces apoptosisand cell cycle arrest, and has antiviral and antioxidative activity. Lactacystin induces neurite outgrowth and hypertension. Lactacystin has the potential for the research of cancer, Neurological Disease, hypertension and Malaria, and so on [2] [6] .
|
-
-
- HY-B0990
-
-
-
- HY-N7204
-
|
|
Chalcones
Monophenols
Flavonoids
Phenols
Umbelliferae
Plants
Ondetia linearis Benth.
|
Monoamine Oxidase
Dopamine β-hydroxylase
Apoptosis
|
|
4-Hydroxyderricin, the major active ingredients of Angelica keiskei Koidzumi, is an orally active, potent selective MAO-B (Monoamine oxidase inhibitors) inhibitor with an IC50 of 3.43 μM. 4-Hydroxyderricin also mildly inhibits dopamine β (DBH)-hydroxylase activity. 4-Hydroxyderricin has antidepressant activity, anti-allergic, anti-diabetic, anti-oxidant, and antitumor effects. 4-Hydroxyderricin promotes apoptosis and cell cycle arrest through regulating PI3K/AKT/mTOR pathway in hepatocellular cells. 4-Hydroxyderricin inhibits osteoclast formation and accelerates osteoblast differentiation . 4-Hydroxyderricin is promising for research of inflammatory diseases .
|
-
-
- HY-W040129
-
|
|
Microorganisms
Source Classification
|
DNA Alkylator/Crosslinker
DNA/RNA Synthesis
Apoptosis
Caspase
|
|
Chromomycin A3 is an inhibitor that selectively binds to GC-rich DNA sequences. Chromomycin A3 targets the DNA minor groove after forming a dimer with Mg 2+. Chromomycin A3 inhibits DNA replication and transcription, blocks the binding of Sp1 transcription factor to target gene promoters, downregulates the expression of anti-apoptotic proteins such as FLIP, Mcl-1, and XIAP, and induces S-phase cycle arrest and caspase-dependent apoptosis in tumor cells. Chromomycin A3 can antagonize oxidative stress induced by glutathione depletion and neuronal apoptosis induced by Camptothecin (HY-15660). Chromomycin A3 can be used in basic research on malignant tumors such as cholangiocarcinoma, and is a potential chemosensitizer and GC-rich region probe .
|
-
-
- HY-B0277
-
-
-
- HY-N12257
-
|
|
Structural Classification
Monophenols
Microorganisms
Phenols
Source Classification
|
Cytochrome P450
Reactive Oxygen Species (ROS)
Apoptosis
|
|
Antimycin A2 is a selective inhibitor of the cytochrome b-c1 complex in the mitochondrial electron transport chain. Antimycin A2 disrupts mitochondrial membrane potential and produces reactive oxygen species (ROS) by inhibiting electron transfer between cytochrome b and c. Antimycin A2 has bactericidal and piscicidal activity, as well as tumor cell growth inhibitory effects, and can induce S-phase cell cycle arrest and apoptosis in HeLa cells. Antimycin A2 is suitable for research of cervical cancer and fisheries management. Antimycin A2 can be naturally isolated from the fermentation products of Streptomyces sp. strains .
|
-
-
- HY-N2424
-
|
2-Phenyl-4-chromone
|
Flavonoids
Classification of Application Fields
Flavones
Metabolic Disease
Endogenous metabolite
Disease Research Fields
Source Classification
|
CDK
Apoptosis
Caspase
|
|
Flavone is an anti-tumor compound that targets cell cycle regulatory proteins (such as cyclin B1) and apoptosis-related factors (such as p21waf1, PIG3). Flavone selectively induces mitochondrial-mediated apoptosis pathways in tumor cells, inhibits cyclin B1 protein expression, upregulates p21waf1, and activates p63/p73 proteins. Flavone has immunomodulatory functions that enhance natural killer cell (NK cell) activity and lymphocyte proliferation. Flavone is used in cancer research, especially for its inhibitory potential in solid tumor models such as esophageal cancer and liver cancer .
|
-
-
- HY-N0863
-
|
NSC-698790; Smilax saponin B
|
Structural Classification
other families
Classification of Application Fields
Plants
Disease Research Fields
Steroids
Source Classification
Cancer
|
Bcl-2 Family
Apoptosis
Akt
c-Myc
ERK
p38 MAPK
JNK
FOXO
|
|
Methyl protodioscin (NSC-698790; Smilax saponin B) is a multi-target, selective, steroidal diglycoside inhibitor with antitumor activity that induces cell cycle arrest. The mechanism of action of Methyl protodioscin is complex, involving the induction of G2/M cell cycle arrest, regulation of the Bcl-2/Bax apoptotic pathway, inhibition of the Akt1/c-Myc axis and MAPK/ERK signaling, while simultaneously downregulating ADAM15 and inducing FOXO1 to reduce cholesterol synthesis. It also inhibits the JNK/c-Jun pathway, reducing the production of inflammatory factors (IL-6, TNF-α). Methyl protodioscin exhibits significant antitumor (inhibiting proliferation, migration, invasion, and inducing apoptosis), anti-inflammatory, and anti-restenosis activities. Methyl protodioscin can be used in research on lung cancer, prostate cancer, pancreatic cancer, and other tumors, as well as inflammatory diseases such as airway inflammation and enteritis .
|
-
-
- HY-N2554
-
|
Ostenol
|
Coumarins
Phenols
Polyphenols
Phenylpropanoids
Kleinia odora (Forssk.) DC.
Umbelliferae
Plants
Source Classification
|
Monoamine Oxidase
PI3K
Akt
Apoptosis
|
|
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cell cycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
|
-
-
- HY-N7225
-
-
-
- HY-121382
-
-
-
- HY-N12445
-
|
|
Malvaceae
Structural Classification
Flavonols
Flavonoids
Abelmoschus manihot (Linn.) Medicus
Plants
Source Classification
|
Topoisomerase
Caspase
Apoptosis
SOD
|
|
Quercetin-3'-O-glucoside is an orally active flavonoid glycoside. Quercetin-3'-O-glucoside reduces liver glucose-6-phosphatase activity, alters serum insulin and glucose levels, and regulates the activities of antioxidant enzymes in the liver and kidney. Quercetin-3'-O-glucoside inhibits DNA topoisomerase II, induces S-phase cell cycle arrest and caspase-3-mediated apoptosis in hepatocellular carcinoma cells. Quercetin-3'-O-glucoside selectively inhibits EGFR-mediated signaling pathways targeting AKT, ERK1/2, FAK and MEK1/2. Quercetin-3'-O-glucoside inhibits growth factor-induced migration and invasion in pancreatic cancer cells. Quercetin-3'-O-glucoside exerts free radical scavenging effects. Quercetin-3'-O-glucoside is applicable to research related to pancreatic cancer, diabetes, hepatocellular carcinoma and malignant tumors .
|
-
-
- HY-100513
-
|
|
Structural Classification
Microorganisms
Antibiotics
Source Classification
|
DNA/RNA Synthesis
Apoptosis
Antibiotic
|
|
(±)-Dehydroaltenusin, an antibiotic, is a selective eukaryotic DNA polymerase α (pol α) inhibitor with an IC50 of 0.68 μM. (±)-Dehydroaltenusin can be isolated from fungus Alternaria tenuis. (±)-Dehydroaltenusin competitively inhibits the DNA template primer (Ki: 0.23 μM) and non-competitively suppresses the 2'-deoxyribonucleoside 5'-triphosphate substrate (Ki: 0.18 μM). (±)-Dehydroaltenusin induces the cancer cell S-phase cycle arrest and apoptosis. (±)-Dehydroaltenusin can be used for cancers like human adenocarcinoma research .
|
-
-
- HY-B0277R
-
-
-
- HY-N14903
-
|
|
Microorganisms
Antibiotics
Other Antibiotics
Source Classification
|
Antibiotic
Bacterial
|
|
Oximidine III is an anti-tumor antibiotic. Oximidine III can selectively inhibit the growth of 3Y1 in rat fibroblasts with degeneration of various tumor genes. Oximidine III inhibits v-H-ras-3Y1, v-src-3Y1 cells and the normal 3Y1 cells with IC50s (nM) of 14, 4.5 and 140, respectively. Oximidine III stops RAS-or SRC-denatured cells at G1 phase of the cell cycle and increases p21WAF1 expression .
|
-
-
- HY-121382R
-
|
|
Triterpenes
Structural Classification
Terpenoids
Plants
Caryophyllaceae
Source Classification
Gypsophila perfoliata Linn.
|
Reference Standards
Apoptosis
Cholinesterase (ChE)
Necroptosis
|
|
Cinosulfuron (Standard) is the analytical standard of Cinosulfuron. This product is intended for research and analytical applications. Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
|
-
-
- HY-W040129R
-
|
|
Microorganisms
Source Classification
|
Reference Standards
Bacterial
Fungal
Apoptosis
Antibiotic
|
|
Chromomycin A3 (Standard) is the analytical standard of Chromomycin A3 (HY-W040129). This product is intended for research and analytical applications. Chromomycin A3 is an inhibitor that selectively binds to GC-rich DNA sequences. Chromomycin A3 targets the DNA minor groove after forming a dimer with Mg 2+. Chromomycin A3 inhibits DNA replication and transcription, blocks the binding of Sp1 transcription factor to target gene promoters, downregulates the expression of anti-apoptotic proteins such as FLIP, Mcl-1, and XIAP, and induces S-phase cycle arrest and caspase-dependent apoptosis in tumor cells. Chromomycin A3 can antagonize oxidative stress induced by glutathione depletion and neuronal apoptosis induced by Camptothecin (HY-15660). Chromomycin A3 can be used in basic research on malignant tumors such as cholangiocarcinoma, and is a potential chemosensitizer and GC-rich region probe .
|
-
-
- HY-N9684
-
|
|
Plantaginaceae
Digitalis purpurea L.
Structural Classification
Natural Products
Plants
Source Classification
|
EGFR
GSK-3
Hedgehog
Akt
ERK
Apoptosis
|
|
Degalactotigonin is a saponin-selective inhibitor targeting the EGFR, GSK3β and Hedgehog/Gli1 pathways and can be isolated from Solanum nigrum (Solanum nigrum). Degalactotigonin inhibits EGFR phosphorylation and the downstream Akt/ERK signaling pathway, and at the same time inhibits the Hedgehog/Gli1 pathway through GSK3β inactivation, thereby inducing cancer cell apoptosis, arresting the cell cycle, and inhibiting migration and invasion. Degalactotigonin can be used in targeted research on malignant tumors such as pancreatic cancer and osteosarcoma .
|
-
-
- HY-N16771
-
|
|
Structural Classification
Rutaceae
Coumarins
Phenylpropanoids
Plants
Clausena excavata N. L. Burman
Source Classification
|
Caspase
Apoptosis
Bcl-2 Family
Bacterial
VEGFR
|
|
Clausenidin is a selective inhibitor targeting apoptosis-related pathways, including the mitochondrial pathway and death receptor pathway, and vascular endothelial growth factor (VEGF). Clausenidin induces mitochondrial membrane depolarization by activating caspase-3, caspase-8 and caspase-9, upregulating the pro-apoptotic protein Bax and downregulating the anti-apoptotic protein Bcl-2. Clausenidin also inhibits VEGF expression and blocks angiogenesis, exerting anti-tumor activity. Clausenidin has inhibitory effects against Mycobacterium tuberculosis (MIC=200 μg/mL). Clausenidin can induce apoptosis in liver cancer cells, arrest the cell cycle in the G2/M phase, and inhibit tumor angiogenesis. Clausenidin can be used in the research of malignant tumors such as liver cancer .
|
-
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-10227S
-
|
|
|
Bortezomib-d8 is the deuterium labeled Bortezomib. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-
-
- HY-W654305
-
|
|
|
Palbociclib-d4 is deuterium labeled Palbociclib. Palbociclib (PD 0332991) is an orally active selective CDK4 and CDK6 inhibitor with IC50 values of 11 and 16 nM, respectively. Palbociclib has potent anti-proliferative activity and induces cell cycle arrest in cancer cells, which can be used in the research of HR-positive and HER2-negative breast cancer and hepatocellular carcinoma .
|
-
-
- HY-15728S
-
|
|
|
Radotinib-d6 is deuterium labeled Radotinib (HY-15728). Radotinib (IY-5511) is an orally active and BBB-permeable selective tyrosine kinase Bcr-Abl1 inhibitor with an IC50 of 34 nM. Radotinib has anti-prion and anti-tumor activities. Radotinib can inhibit the proliferation, induce cell cycle arrest and apoptosis of tumor cells . Radotinib can be used in the research of cancer such as chronic myeloid leukemia and multiple myeloma, as well as neurodegenerative diseases such as prion diseases .
|
-
| Cat. No. |
Product Name |
|
Classification |
-
- HY-180200
-
|
|
|
Alkynes
|
|
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1 cell cycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
|
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-10227G
-
|
PS-341; LDP-341; NSC 681239
|
Proteasome
NF-κB
Apoptosis
Autophagy
TREM receptor
|
Cancer
|
|
Bortezomib (GMP) (PS-341 (GMP)) is Bortezomib (HY-10227) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Bortezomib (PS-341) is a reversible and selective proteasome inhibitor, and potently inhibits 20S proteasome (Ki=0.6 nM) by targeting a threonine residue. Bortezomib disrupts the cell cycle, induces apoptosis, and inhibits NF-κB. Bortezomib is the first proteasome inhibitor anticancer agent. Bortezomib can be used for the study of multiple myeloma (MM) . Bortezomib effectively inhibits TREM2 expression in tumor-associated macrophages (TAMs) .
|
-
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent:









































































































































































































































































































































































