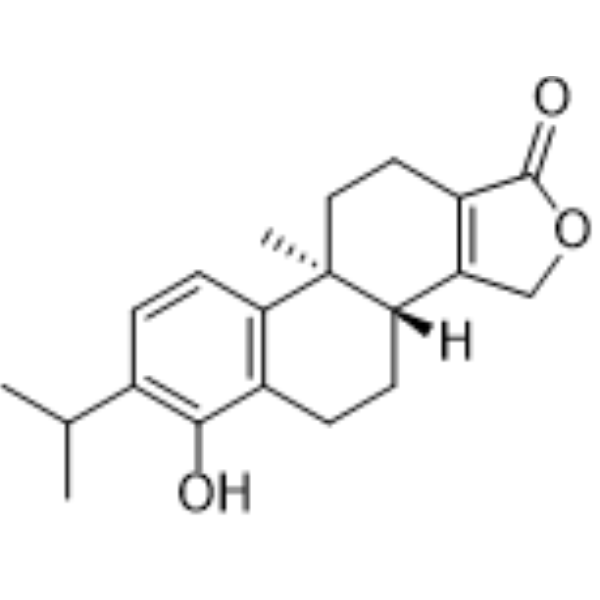
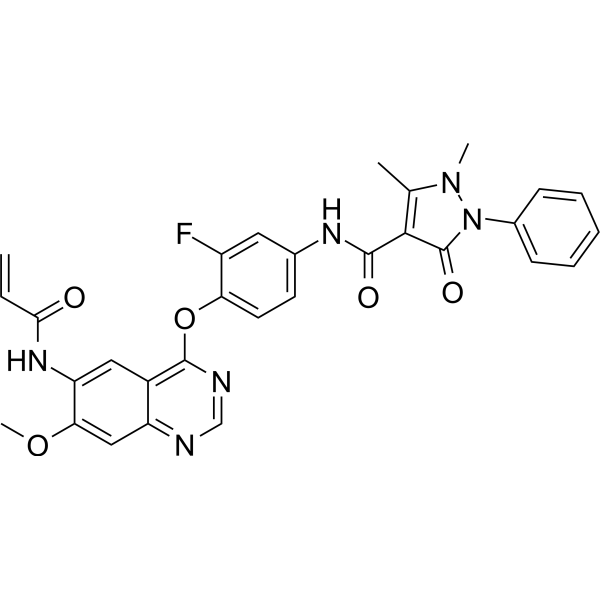
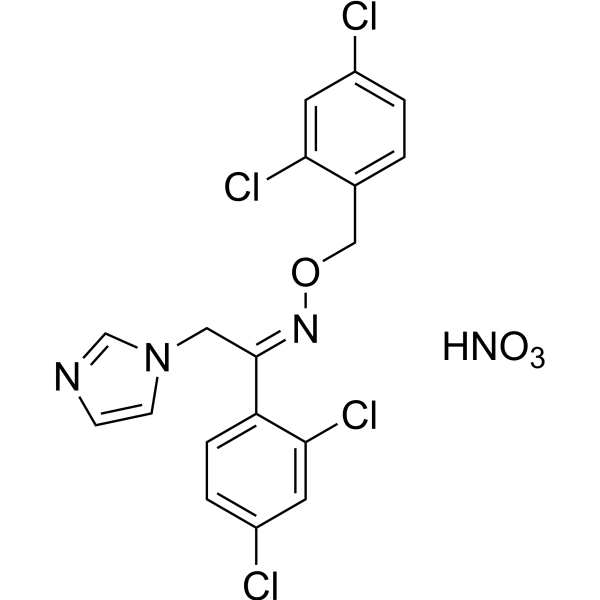
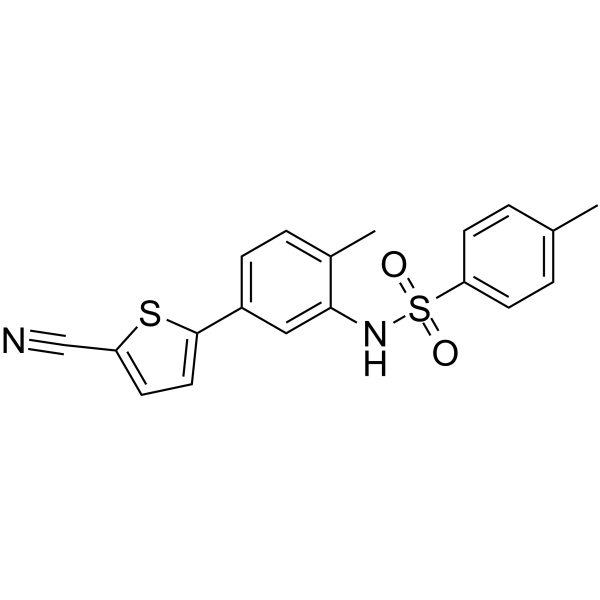
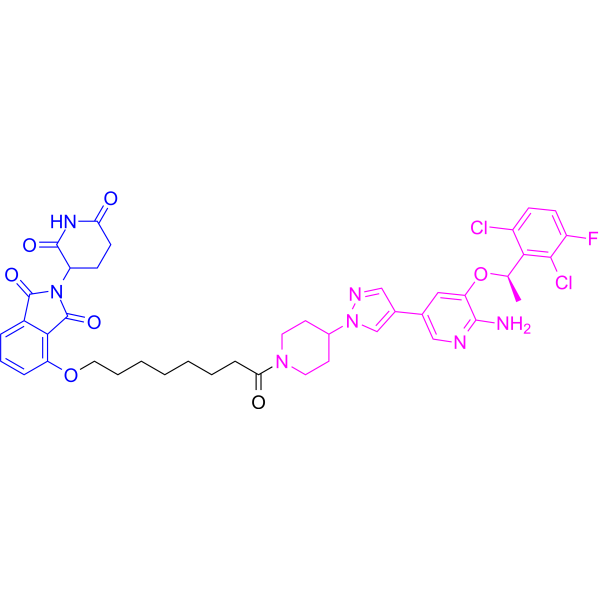
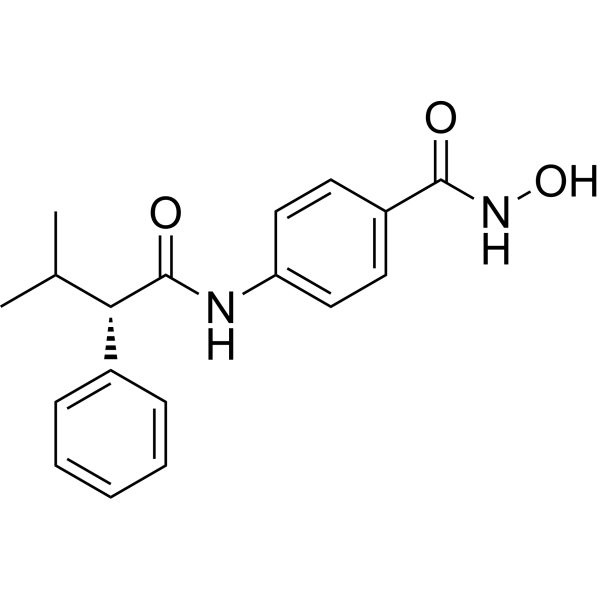
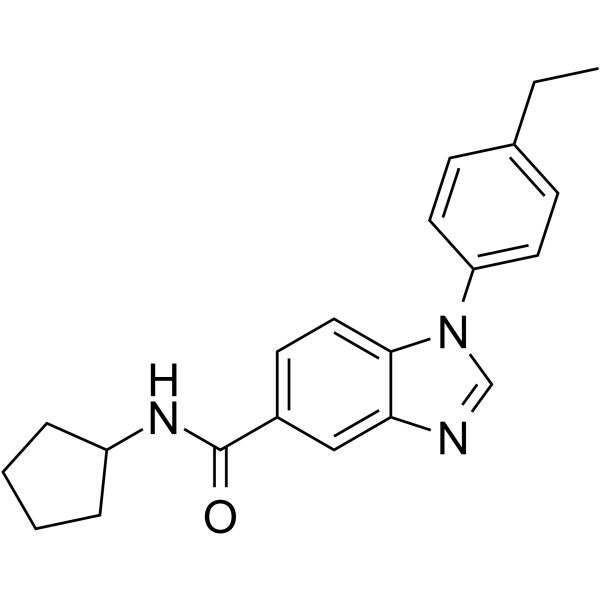
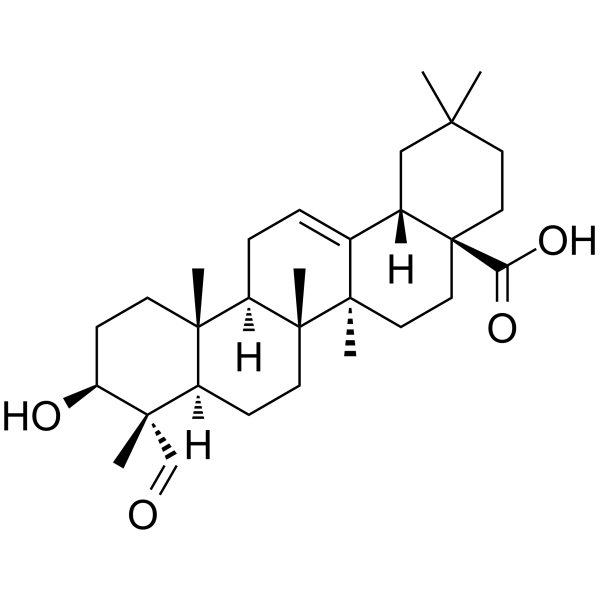
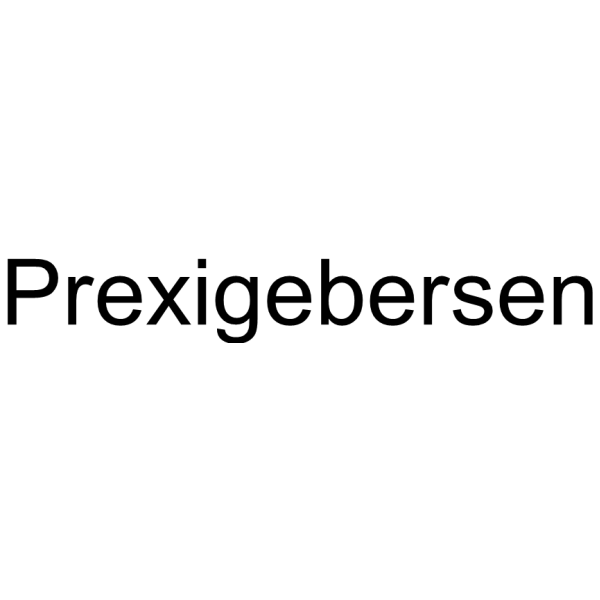From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
GY4137 is a sustained-release H2S donor possessing vasodilatory, antihypertensive, and anti-inflammatory activities. GY4137 can inhibit cell growth, induce apoptosis, and cause cell cycle arrest by blocking the STAT3 pathway, demonstrating potent anticancer activity .
INX-315 is an orally active and selective CDK2 inhibitor that induces cell cycle arrest in the G1 phase. INX-315 reduces CDK2 substrate phosphorylation and inhibits tumor growth in a dose-dependent manner in xenograft mouse models. INX-315 may be used in cancer research .
Barasertib (AZD1152), a pro-drug of Barasertib-hQPA, is a highly selective Aurora B inhibitor with an IC50 of 0.37 nM in a cell-free assay. Barasertib (AZD1152) induces growtharrest and apoptosis in cancer cells .
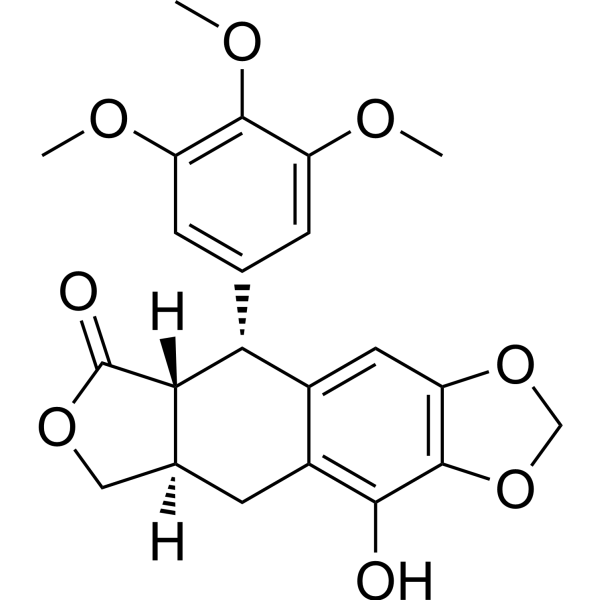
Costunolide ((+)-Costunolide) is a naturally occurring sesquiterpene lactone, with antioxidative, anti-inflammatory, antiallergic, bone remodeling, neuroprotective, hair growth promoting, anticancer, and antidiabetic properties. Costunolide can induce cell cycle arrest and apoptosis on breast cancer cells .
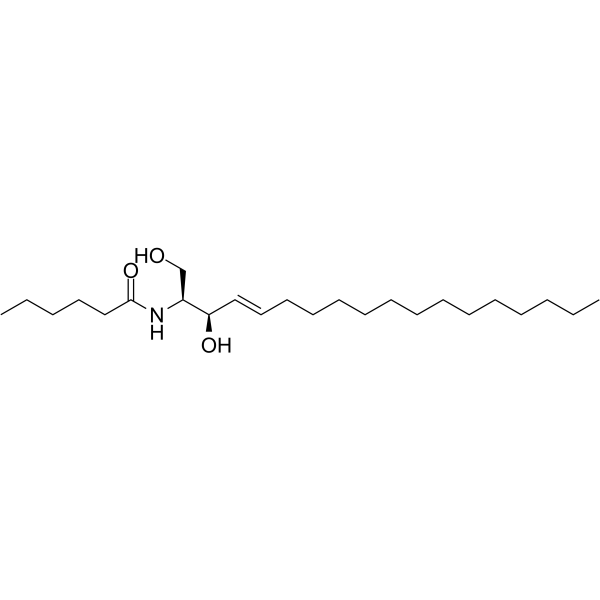
Ceramides Mixture is an endogenous ceramide and consists of hydroxy and non-hydroxy fatty acid-containing ceramides. Ceramides Mixture is a main lipid component of the permeability barrier in epidermis. Ceramides Mixture is involved in the regulation of growth inhibition, cell cycle arrest, and modulation of telomerase activity .
Barasertib-HQPA (AZD2811) is a highly selective Aurora B inhibitor with an IC50 of 0.37 nM in a cell-free assay. Barasertib-HQPA (AZD2811) induces growtharrest and apoptosis in cancer cells .
Cucurbitacin B belongs to a class of highly oxidized tetracyclic triterpenoids and is oral active. Cucurbitacin B inhibits tumor cell growth, migration and invasion and cycle arrest, but induces cell apoptosis. Cucurbitacin B has potent anti-inflammatory, antioxidant, antiviral, hypoglycemic, hepatoprotective, neuroprotective activity .
Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Triacetin (Glyceryl triacetate) is a synthetic compound that is a triester of glycerol and acetic acid, orally active. Triacetin increases acetate bioavailability in glioma cells. Triacetin induces glioma cell growtharrest and Apoptosis. Triacetin freely crosses the blood brain barrier/plasma membrane. Triacetin increases histone acetylation and enhances Temozolomide (HY-17364) (TMZ) chemotherapeutic efficacy .
WZB117 is a glucose transporter 1 (Glut1) inhibitor, which downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo.
AGX51 is the first-in-class pan-Id (inhibitors of DNA-binding/differentiation proteins) antagonist and degrader. AGX51 inhibits Id1-E47 interaction, leading to ubiquitin-mediated degradation of Ids, cell growtharrest, and viability reduction. AGX51 can inhibit TNBC and has an IC50 of about 25 nM. AGX51 can be used in cancer research.
Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
Terrestrosin D is an orally active apoptosis inducer. Terrestrosin D induces cell cycle arrest at the G1 and S phases, reduces mitochondrial membrane potential, and inhibits the growth of cancer cells and endothelial cells. Terrestrosin D is studied in castration-resistant prostate cancer and pulmonary fibrosis .
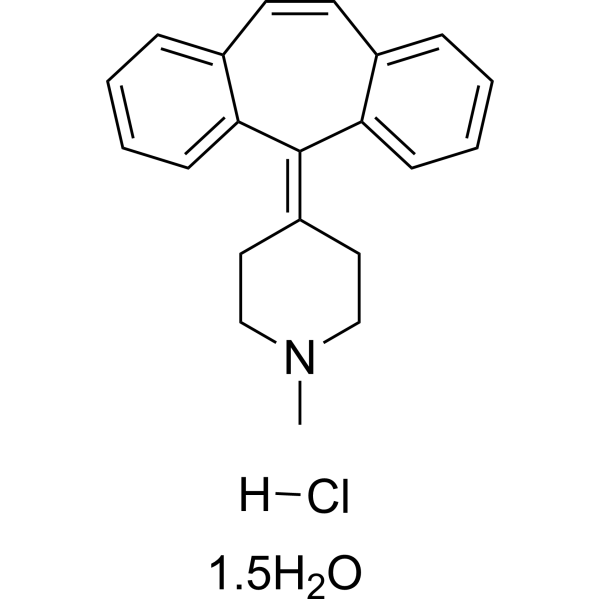
Cyproheptadine hydrochloride sesquihydrate acts as a p38 MAP kinase activator, CHK2 activator, histamine H1 receptor inhibitor and serotonin receptor inhibitor. Cyproheptadine hydrochloride sesquihydrate mediates cell cycle arrest via G1 phase arrest, G1/S transition arrest, G0/G1 phase arrest, reduced expression of cyclins D1/D2/D3, upregulated expression of HBP1, p16, p21, p27, and decreased phosphorylation of retinoblastoma protein. Cyproheptadine hydrochloride sesquihydrate induces Apoptosis by increasing PARP and cleaved PARP, as well as activating the mitochondrial caspase pathway. Cyproheptadine hydrochloride sesquihydrate inhibits tumor growth with extremely low toxicity to normal cells. Cyproheptadine hydrochloride sesquihydrate can be used in research related to hepatocellular carcinoma, multiple myeloma and acute myeloid leukemia .
OTS514 is a highly potent TOPK inhibitor with an IC50 of 2.6 nM. OTS514 strongly suppresses the growth of TOPK-positive cancer cells . OTS514 induces cell cycle arrest and apoptosis .
ER-076349 (Eribulin intermediate) is an inhibitor of tubulin polymerization, induces G2-M cell cycle arrest, and disrupts mitotic spindles. ER-076349 inhibits cancer cell growth, and inhibits tumor growth in several human tumor xenografts. ER-076349 is an analog of Halichondrin B .
SRT 2183 is a selective Sirtuin-1 (SIRT1) activator with an EC1.5 value of 0.36 μM . SRT 2183 induces growtharrest and apoptosis, concomitant with deacetylation of STAT3 and NF-κB, and reduction of c-Myc protein levels .
WJ460 is a potent inhibitor of myoferlin (MYOF) that interacts directly with MYOF. WJ460 inhibits the migration and growth, induces cell cycle arrest, mitochondrial autophagy, lipid peroxidation and ferroptosis in tumor cells. WJ460 has anti-tumor activity .
Legubicin (Doxorubicin-LNAA-Boc) is a novel conjugate of Doxorubicin (HY-15142A) and a Legumain-cleavable peptide linker. Legubicin is activated by Legumain to release leucine-doxorubicin while sparing normal tissues. Legubicin inhibits tumor cell growth and reduces DNA binding in non-legumain expressing cells. Legubicin completely arrests tumor growth in mice bearing CT26 tumors. Legubicin can be used for the study of colon carcinoma (CRC) .
HNHA is a potent HDAC inhibitor with an IC50 of 100 nM. HNHA arrests the cell cycle at the G1/S phase via p21 induction. HNHA inhibits tumor growth and tumor neovascularization. HNHA may be a potent anti-cancer agent against breast cancer .
TN-16 is a Microtubule polymerization inhibitor. TN-16 induces G2/M cell cycle arrest, metaphase mitotic arrest and Apoptotic cell death in cells, and blocks late Autophagic flux by inhibiting autophagosome-lysosome fusion. TN-16 suppresses tumor growth in syngeneic mouse breast cancer models. TN-16 can be used in research related to neuroblastoma, cervical cancer, breast cancer and other tumors .
MT 63-78 is a specific and potent direct AMPK activator with an EC50 of 25 μM. MT 63–78 also induces cell mitotic arrest and apoptosis. MT 63-78 blocks prostate cancer growth by inhibiting the lipogenesis and mTORC1 pathways. MT 63-78 has antitumor effects .
GSK_WRN3 is selective WRN helicase inhibitor (pIC50 = 8.6). SK_WRN3 can selectively inhibit the growth of microsatellite unstable (MSI) cancer cells, induce DNA damage, and cause cell cycle arrest. GSK_WRN3 has anti-tumor activity .
ANI-7 is an activator of aryl hydrocarbon receptor (AhR) pathway. ANI-7 inhibits the growth of multiple cancer cells, and potently and selectively inhibits the growth of MCF-7 breast cancer cells with a GI50 of 0.56 μM. ANI-7 induces CYP1-metabolizing mono-oxygenases by activating AhR pathway, and also induces DNA damage, checkpoint Kinase 2 (Chk2) activation, S-phase cell cycle arrest, and cell death in sensitive breast cancer cell lines .
2-Hydroxy-3-methylanthraquinone (compound 1) is a natural compound isolated from a water extract of Hedyotis diffusa WILLD. 2-Hydroxy-3-methylanthraquinone shows inhibitory activity against protein tyrosine kinases v-src and pp60src, and induces growtharrest and apoptosis in the HepG2 cancer cells .
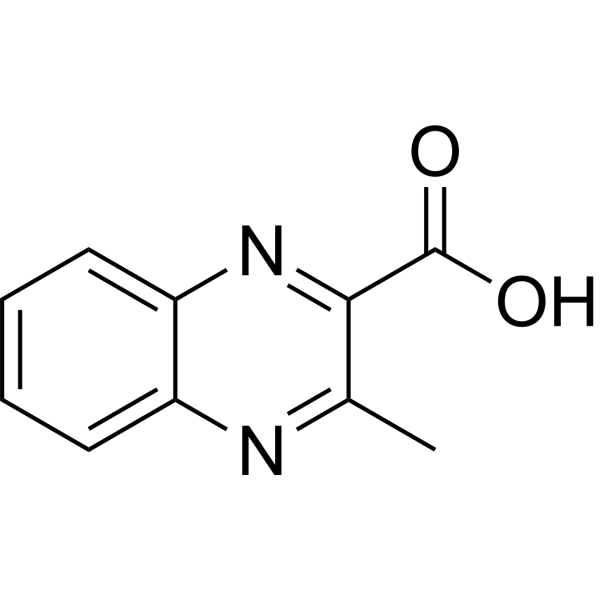
3-Methyl-2-quinoxalinecarboxylic acid (MQCA), an important N-oxide reductive metabolite of Quinocetone or Olaquindox, potently inhibits the growth of Chang liver cells through S phase arrest of the cell cycle .
8α-Tigloyloxyhirsutinolide 13-O-acetate (8αTGH) is a potent and orally active STAT3 inhibitor. 8α-Tigloyloxyhirsutinolide 13-O-acetate induces early oxidative stress and pyroptosis, and late DNA damage, cell cycle arrest, apoptosis in the TNBC cells. 8α-Tigloyloxyhirsutinolide 13-O-acetate suppresses tumor cell growth in vitro and tumor growth in vivo .
MC1742 is a potent HDAC inhibitor, with IC50s of 0.1 μM, 0.11 μM, 0.02 μM, 0.007 μM, 0.61 μM, 0.04 μM and 0.1 μM for HDAC1, HDAC2, HDAC3, HDAC6, HDAC8, HDAC10 and HDAC11, respectively. MC1742 can increase acetyl-H3 and acetyl-tubulin levels and inhibits cancer stem cells growth. MC1742 can induce growtharrest, apoptosis, and differentiation in sarcoma CSC .
Prexigebersen (BP1001) is an antisense oligonucleotide targeting Bcl-2 and Grb2. Prexigebersen exhibits antileukemic activity in cell models. Prexigebersen induces apoptosis (apoptosis), cell cycle arrest and ROS production in leukemia cells. Prexigebersen inhibits Grb2 expression, thereby suppressing tumor growth and survival. Prexigebersen can be used in studies related to acute myeloid leukemia .
Barasertib (AZD1152 dihydrochloride), a pro-drug of Barasertib-hQPA, is a highly selective Aurora B inhibitor with an IC50 of 0.37 nM in a cell-free assay. Barasertib (AZD1152 dihydrochloride) induces growtharrest and apoptosis in cancer cells .
FK-3000 is a potent anti-tumor agent that inhibits the growth of carcinoma cells through apoptosis and induction cell cycle arrest. FK-3000 also exhibit antiviral effects against HSV-1 and HIV-1 .
DTP3 TFA is a potent and selective GADD45β/MKK7 (growtharrest and DNA-damage-inducible β/mitogen-activated protein kinase kinase 7) inhibitor. DTP3 TFA targets an essential, cancer-selective cell-survival module downstream of the NF-κB pathway .
Costunolide (Standard) is the analytical standard of Costunolide. This product is intended for research and analytical applications. Costunolide ((+)-Costunolide) is a naturally occurring sesquiterpene lactone, with antioxidative, anti-inflammatory, antiallergic, bone remodeling, neuroprotective, hair growth promoting, anticancer, and antidiabetic properties. Costunolide can induce cell cycle arrest and apoptosis on breast cancer cells .
Phthalazinone pyrazole is a potent, selective, and orally active inhibitor of Aurora-A kinase with an IC50 of 0.031 μM. Phthalazinone pyrazole can arrests mitosis and subsequently inhibit tumor growth via apoptosis of proliferating cells. Phthalazinone pyrazole suppresses the epithelial-mesenchymal transition (EMT) during the differentiation of hepatocyte-like cells (HLCs) from human embryonic stem cells .
CHM-1, a microtubule-destabilizing agent, inhibits tubulin polymerization. CHM-1 is a potent and selective antimitotic antitumor activity against human hepatocellular carcinoma. CHM-1 induces growth inhibition and apoptosis via G2-M phase arrest in human hepatocellular carcinoma cells by activation of Cdc2 kinase activity .
3-Methyl-2-quinoxalinecarboxylic acid-d4 is the deuterium labeled 3-Methyl-2-quinoxalinecarboxylic acid. 3-Methyl-2-quinoxalinecarboxylic acid (MQCA), an important N-oxide reductive metabolite of Quinocetone or Olaquindox, potently inhibits the growth of Chang liver cells through S phase arrest of the cell cycle .
AHR agonist 3 is an aryl hydrocarbon receptor (AhR) agonist, that can induces cell cycle arrest or apoptosis via activation of tumor-suppressive transcriptional programs. AHR agonist 3 inhibits triple-negative breast cancer (TNBC) stem cell growth via AhR while exhibits minimal cytotoxicity against normal human primary cells and can be used for cancer research .
(Z)-SU5614 is a potent FLT3 inhibitor and selectively induces growtharrest, apoptosis, and cell cycle arrest in Ba/F3 and AML cell lines expressing a constitutively activated FLT3 .
Kazusamycin B is an antibiotic that could be isolated from the fermentation broth of Streptomyces sp. No. 81-484. Kazusamycin B inhibits cell growth and arrests cell cycle at G1 phase. Kazusamycin B can be used in research of cancer .
Oxcarbazepine (Standard) is the analytical standard of Oxcarbazepine. This product is intended for research and analytical applications. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
Aldehyde Dehydrogenase Isobutyramide is an orally active liver alcohol dehydrogenase (LADH2) inhibitor. Isobutyramide inhibits ethanol metabolism and enhances differentiation-related gene expression, inducing cell cycle arrest and significantly reducing the growth rate of prostate cancer cells (LNCaP). Isobutyramide is promising for research of advanced prostate cancer .
Autophagy inducer 7 (Compound SSA) is an Autophagy and Apoptosis inducer. Autophagy inducer 7 activates autophagy by inhibiting Akt/mTOR signaling and the expression of downstream proteins. Autophagy inducer 7 suppresses DNA synthesis and causes a G0-G1 cell-cycle arrest. Autophagy inducer 7 inhibits tumor cell growth .
DFPM activates plant resistance protein signaling in roots, and triggers root growtharrest. DFPM decreases root cell viability in accession Col-0. DFPM is light sensitive in aqueous solutions. DFPM becomes bioactive during light and oxygen-dependent modification .
Fluacrypyrim, a Miticide, is a STAT3 inhibitor. Fluacrypyrim significantly increases the protein tyrosine phosphatases(PTPs) activity. Fluacrypyrim inhibits the growth of leukemia cells by a predominant G1 arrest with significant decrease of the protein and mRNA levels of cyclin D1. Fluacrypyrim selectively inhibits STAT3 signaling, inducing growtharrest and apoptosis in STAT3-dependent cancer cells. Fluacrypyrim mitigates IR-induced hematopoietic system injury mainly by preventing apoptosis in the HSCs. Fluacrypyrim demonstrates significant analgesic and anti-inflammatory effects by inhibiting uterine smooth muscle contraction and inflammatory responses .
Asparanin A is an apoptosis inducer with anticancer activity. Asparanin A induces cell cycle arrest in the G0/G1 phase through mitochondria and PI3K/AKT signaling pathways, inhibiting cancer cell growth. Asparanin A also demonstrated in vivo efficacy in a mouse xenograft model of Ishikawa endometrial carcinoma, significantly inhibiting tumor growth .
OTS514 hydrochloride is a highly potent TOPK inhibitor, which inhibits TOPK kinase activity with a median inhibitory concentration (IC50) value of 2.6 nM. OTS514 hydrochloride strongly suppresses the growth of TOPK-positive cancer cells . OTS514 hydrochloride induces cell cycle arrest and apoptosis .
HBF-0079 is a selective anti-hepatocellular carcinoma agent. HBF-0079 induces cell cycle arrest and Apoptosis. HBF-0079 modulates cell-growth and anti-apoptotic signaling through AKT and mTOR. HBF-0079 exhibits antitumor activity against hepatocellular carcinoma .
SP1 is an α-peptide encoded by the mating pheromone MFα1 gene in Candida albicans, which can induce cell growtharrest at the mating type locus MTLa in Candida albicans. SP1 can be used in the study of the prevention and treatment of Candida albicans infection .
Oxcarbazepine-d4 (GP 47680-D4) is the deuterium labeled Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
PKCδ substrate acts as a nuclear transporter of ERK2 and is involved in ERK2 mediated gene activation. PKCδ is involved in the regulation of cell growth, proliferation, cell cycle arrest, and apoptosis by phosphorylating hBVR and other proteins. PKCδ substrate can be used to study the development of diseases, especially cancer biology .
Ocifisertib hydrochloride (CFI-400945 hydrochloride) is the hydrochloride salt form of Ocifisertib (HY-12300). Ocifisertib hydrochloride is an orally active PLK4 inhibitor with a Ki and an IC50 of 0.26 nM and 2.8 nM. Ocifisertib hydrochloride inhibits growth of various cancer cells, arrests cell cycles at G2/M phase, and induces apoptosis. Ocifisertib hydrochloride exhibits antitumor efficacy in mouse model .
PU3 is an Hsp90 inhibitor that competes with geldanamycin (GM) and others for the conserved ATP/ADP pocket of Hsp90. PU3 also induces degradation of proteins such as Her2 and inhibits breast cancer cell growth by causing retinoblastoma protein hypophosphorylation, G1 arrest, and cell differentiation. PU3 has the potential to be a cancer inhibitor. .
EGFR-IN-109 (compound 4) is an EGFR inhibitor, with the IC50 values of 25.8 and 182.3 nM for EGFR WT and EGFR T790M, respectively. EGFR-IN-109 arrests the cancer cells’ growth at the G2/M phase and induces both early and late apoptosis. EGFR-IN-109 can be used in cancer research .
CQ1373 is a potent RET inhibitor, demonstrating cellular potency with IC50 values of 13.0, 25.7 and 28.4 nM against BaF3 cells expressing CCDC6-RET, CCDC6-RET-G810C and CCDC6-RET-G810R, respectively. CQ1373 exhibits good selectivity toward wild-type RET and solvent front mutants G810C/R with IC50 values of 4.2, 7.1 and 32.4 nM, respectively. CQ1373 inhibits RET phosphorylation and downstream signaling through SHC. CQ1373 induces Apoptosis and cell cycle arrest in BaF3 cells. CQ1373 exhibits anti-tumor efficacy and can be used for cancer research .
8-Chloro-cAMP sodium is a cAMP analogue that induces growtharrest, and modulates cAMP-dependent PKA activity. 8-Chloro-cAMP sodium has anticancer activity .
EGFR-IN-135 (compound 3d) is a EGFR inhibitor with the IC50 of 0.086 μM. EGFR-IN-135 inhibitor cell growth and arrests the cell cycle at the S phase of reast cancer cell lines .
Azadirachtin D is an insect growth regulator targeting ecdysone receptor (EcR) in insects. Azadirachtin D blocks the 20-hydroxyecdysone signaling pathway, leading to developmental arrest or death in insects. Azadirachtin D is promising for research of agricultural pests .
EGFR-IN-96 (compound 7a) is a thieno[2,3-d]pyrimidine EGFR inhibitor that can induce apoptosis. EGFR-IN-96 arrests the growth of HepG2 cells in the S phase and G2/M phase, and inhibits the growth of cancer cells bearing EGFR wild-type and EGFR T790M .
Reticulol (K 251-1) is an inhibitor of cyclic adenosine 3', 5'-monophosphate phosphodiesterase. Reticulol shows antitumor activity independent with cell cycle arrest or apoptosis. Reticulol inhibits cell growth of murine melanoma cells and human lung tumor cells. Reticulol protects its lung metastasis via the bloodstream by inhibiting the growth of B16F10 melanoma .
DAT-230 is a microtubule inhibitor. DAT-230 induces cell apoptosis and results in microtubule de-polymerization and G2/M phase arrest. DAT-230 inhibits cell growthin vitro and in vivo .
CK0106023 is a potent and specific allosteric inhibitor of KSP with a Ki value of 12 nM, showing antitumor activity. CK0106023 causes mitotic arrest and growth inhibition in several tumor cell lines. CK0106023 exhibits antitumor activity in tumor-bearing mice .
GEM231 is an 18mer antisense oligonucleotide targeting the mRNA of the PKA-I (RIα regulatory subunit of cAMP dependent protein kinase type I ). GEM231 induces cell growtharrest, apoptosis, and differentiation in a variety of cancer cell lines in vitro and in tumors in vivo.
GEM231 sodium is an 18mer antisense oligonucleotide targeting the mRNA of the PKA-I (RIα regulatory subunit of cAMP dependent protein kinase type I ). GEM231 sodium induces cell growtharrest, apoptosis, and differentiation in a variety of cancer cell lines in vitro and in tumors in vivo.
Tubulin polymerization-IN-56 (compound 8l), an indazole derivative, is a potent tubulin polymerization inhibitor through interacting with the colchicine site, resulting in cell cycle arrest and cellular apoptosis. polymerization-IN-56 reduces cell migration and leads to more potent inhibition of tumor growth in vivo .
Oxcarbazepine-d4-1 is deuterium labeled Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
SK-7041 is a HDAC inhibitor with the IC50 of 172 nM. SK-7041 induces the hyperacetylation of histones H3 and H4 .SK-7041 inhibits tumor cell growthin vivo and in vitro, induces cell apoptosis, and arrests cell cycle at the G1 phase .
(S)-OTS514 is the S enantiomer of OTS514 (HY-18621). OTS514 is a highly potent TOPK inhibitor with an IC50 of 2.6 nM. OTS514 strongly suppresses the growth of TOPK-positive cancer cells . OTS514 induces cell cycle arrest and apoptosis .
IV-23 (Compound 20) is a potent Noxa mediated apoptosis inducer, and it is a promising anticancer agent with potential. IV-23 inhibits cell growths in vitro and in vivo, reduces colony formation, arrests cell cycle at M phase, and induces esophageal squamous cell carcinoma (ESCC) .
EGFR-IN-155 (compound 13a) is an EGFR inhibitor with IC50 values of 0.14 nM and 0.18 nM against EGFR TK and EGFR L858R, respectively. EGFR-IN-155 inhibits tumor growth, causes a cell cycle arrest at S phase, and and induces cell apoptosis .
PK7242 is an inducer of reactivation of mutant p53 in cancer cells. In cancer cells carrying the Y220C mutant, PK7242 binds to the p53-Y220C core domain and induces growth inhibition, cell-cycle arrest, and apoptosis.
Antitumor agent-168 (compound 21b) disrupts the microtubule network in tumor cells leading to G2/M cell cycle arrest and apoptosis induction. Antitumor agent-168 inhibits MCF-7 growth with an IC50 value of 1.4 nM .
4SC-207 is a potent, orally active microtubule inhibitor. 4SC-207 inhibits microtubule growth to inhibit tumor cell proliferation in vitro and in vivo, and promotes a mitotic delay/arrest, followed by apoptosis or aberrant divisions. 4SC-207 inhibits tumor growth in taxane resistant xenograft mouse models. 4SC-207 can be used for cancer research, such as colon adenocarcinoma and other malignancies .
YL93 is a dual inhibitors of MDM2/4 with Ki values of 0.64 μM and 1.1 nM for MDM4 and MDM2, respectively. YL93 induces cell-cycle arrest and apoptosis. YL93 shows p53-dependent cell growth inhibition .
Sulindac Sulfone-d6 is the deuterium labeled Sulindac sulfone (HY-B1787). Sulindac sulfone is an mTORC1 pathway inhibitor and a metabolite of Sulindac. Sulindac sulfone inhibits colon cancer cell growth and induces cell cycle arrest. Sulindac sulfone is used in cancer research .
Barasertib (Standard) is the analytical standard of Barasertib. This product is intended for research and analytical applications. Barasertib (AZD1152), a pro-drug of Barasertib-hQPA, is a highly selective Aurora B inhibitor with an IC50 of 0.37 nM in a cell-free assay. Barasertib (AZD1152) induces growtharrest and apoptosis in cancer cells .
LeuRS-IN-2 (Compound 9) is a Wolbachialeucyl-tRNA synthetase (LeuRS) inhibitor in the presence of adenosine monophosphate (AMP) with an EC50 value of 6 nM, efficiently arresting the growth of pathogenic host. LeuRS-IN-2 forms adenosine-based adducts inhibiting protein synthesis, which is promising for research of new antimicrobials with disrupting microbiota .
Oxcarbazepine-d8-1 is a deuterium of Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Oxcarbazepine-d8-1 has anti-cancer and anticonvulsant effects .
Apoptosis inducer 48 (5d) is an apoptotic agent. Apoptosis inducer 48 inhibits the growth of triple-negative breast cancer cells. Apoptosis inducer 48 attenuates proteasomal degradation via the ubiquitin-proteasome pathway, leading to G2/M phase cell cycle arrest and the induction of apoptotic .
c-Met degrader-1 (Compound H11) is an orally active c-Met degrader (through the ubiquitin proteasome system). c-Met degrader-1 has anti-hepatocellular carcinoma activity (HCC) and inhibits tumor growth in MHCC97H xenografts. c-Met degrader-1 inhibits HCC cell growth, arrests cell cycle, and induces apoptosis. c-Met degrader-1 may overcome resistance to type Ib inhibitors .
SSE1806 is a derivative of podophyllotoxin (a natural antimitotic agent) and a microtubule/tubulin inhibitor with significant anticancer and antiproliferative activities. The GI50 of SSE1806 on cancer cell growth ranges from 1.29-21.15 μM. SSE1806 causes mitotic abnormalities and G2/M phase arrest, increases p53 expression, and inhibits colon cancer organoid growth. SSE1806 is able to overcome multidrug resistance in cell lines overexpressing MDR-1 .
C2 L-Erythro ceramide (d18:1/2:0) is a cell-permeable sphingolipid. C2 L-Erythro ceramide (d18:1/2:0) induces cell cycle arrest in the G0/G1 phase and inhibits cell growth .
HR488B is an efficient HDAC1 inhibitor. HR488B specifically suppressed the growth of CRC cells by inducing cell cycle G0/G1 arrest and apoptosis. HR488B causes mitochondrial dysfunction, reactive oxygen species (ROS) generation, and DNA damage accumulation .
IMM-02 is a DID-DAD binding inhibitor with activity promoting actin assembly and microtubule stabilization. IMM-02 is able to trigger serum response factor-mediated gene expression and lead to cell cycle arrest and apoptosis. IMM-02 has shown the ability to slow tumor growth in a mouse colon cancer xenograft model .
(rac)-ZK-304709 is an isoform of ZK-304709 and is an orally active multi-targeted tumor growth inhibitor that inhibits multiple cell cycle-dependent kinases (CDKs), vascular endothelial growth factor receptor kinases (VEGF-RTKs), and platelet-derived growth factor receptor kinase β (PDGF-RTKβ). (rac)-ZK-304709 can dose-dependently inhibit the proliferation and colony formation of neuroendocrine tumor (NET) cells. (rac)-ZK-304709 directly acts on NET cells by inducing G2 cell cycle arrest and apoptosis, while reducing the expression of MCL1, survivin, and HIF1α. (rac)-ZK-304709 effectively controls tumor growth by inducing apoptosis and inhibiting tumor-induced angiogenesis, and may become a potential agent for inhibiting NET .
XSJ05 is a camptothecin (CPT) derivative that can inhibit topoisomerase I (Topo I) to exert anti-cancer activity. XSJ05 can trigger DNA double-strand breaks, leading to DNA damage. XSJ05 can inhibit the growth of colorectal cancer (CRC), arrest the cell cycle in G2/M phase, and induce apoptosis .
EGFR-IN-45 is a potent epidermal growth factor receptor (EGFR) pan inhibitor, with IC50s of 0.4 µM and 1.6 µM for EGFR and CDK2, respectively. EGFR-IN-45 also inhibit Topo I and Topo II. EGFR-IN-45 arrests cancer cells in the pre-G1 phase and induces apoptosis .
mTOR inhibitor-27 (Compound 7e) is a mammalian target of rapamycin (mTOR) inhibitor with an IC50 value of 5.47 μM. mTOR inhibitor-27 can induce tumor cell apoptosis and arrest the cell cycle in the S-phase, thereby inhibiting cancer cell growth. mTOR inhibitor-27 is promising for research of cancers, such as skin cancer .
HOI-07 is a selective Aurora B kinase inhibitor. HOI-07 blocks phosphorylation of histone H3 on Ser10 in
lung cancer cells. HOI-07 induces cell-cycle arrest, and apoptosis. HOI-07 has antitumor activity, and suppresses the tumor growth of A549, 143B and KHOS xenografts .
FGFR3-IN-11(compound B11) is a Fibroblast growth factor receptor 3 (FGFR3) inhibitor with a Ka value of 4.8 μM. FGFR3-IN-11 induces apoptosis, suppresses colony formation, and causes dose-dependent G0/G1 cell cycle arrest in cancer cells. FGFR3-IN-11 exerts anticancer activity against cancer cells with minimal toxicity toward normal hepatocytes and demonstrates tumor growth suppression in xenograft mouse models. FGFR3-IN-11 can be used for the research of hepatocellular carcinoma .
EGFR-IN-207 (Compound 5h) is an epidermal growth factor receptor (EGFR) kinase inhibitor with an IC50 of 0.21 μM. EGFR-IN-207 induces cell cycle arrest at the Sub-G1 phase and promotes Apoptosis. EGFR-IN-207 exhibits anticancer activity against lung cancer. EGFR-IN-207 shows extremely low toxicity in non-cancerous cell lines. EGFR-IN-207 can be used in lung cancer-related research .
N-Choloylphenylalanine (Compound 6273) (PheCA) is a VPR-induced growtharrest inhibitor. N-Choloylphenylalanine alleviates VPR-induced growtharrest. N-Choloylphenylalanine can be used in yeast and HIV-1 infection research .
PI3Kα-IN-29 is a potent, orally active and selective PI3Kα with an IC50 of 2.5 nM. PI3Kα-IN-29 exhibits >400-fold selectivity over PI3Kβ/δ/γ/mTOR. PI3Kα-IN-29 selectively degrades the H1047R mutant p110α protein and inhibits PI3Kα kinase activity. PI3Kα-IN-29 suppresses PI3K/AKT/mTOR signaling, induces G1 arrest, and inhibits migration. PI3Kα-IN-29 inhibits tumor growth in a T47 mouse model. PI3Kα-IN-29 can be used for the research of breast cancer .
Barasertib-HQPA (Standard) is the analytical standard of Barasertib-HQPA (HY-10126). This product is intended for research and analytical applications. Barasertib-HQPA (AZD2811) is a highly selective Aurora B inhibitor with an IC50 of 0.37 nM in a cell-free assay. Barasertib-HQPA (AZD2811) induces growtharrest and apoptosis in cancer cells .
Peligifatamab is a PSMA-targeted α-radioimmunoconjugate with an EC50 of 1.2 nM against human targets. Peligifatamab induces DNA damage, DNA double-strand breaks, cell cycle arrest and apoptosis (Apoptosis) in PSMA-positive prostate cancer cells. Peligifatamab reduces cell viability in a manner dependent on cellular PSMA expression levels. Peligifatamab inhibits tumor growth and tumor-induced abnormal bone growth in prostate cancer bone metastasis models. Peligifatamab exhibits antitumor efficacy in subcutaneous prostate cancer models and xenograft models. Peligifatamab can be used for the research of metastatic castration-resistant prostate cancer .
Tubulin/HDAC-IN-2 (Compound II-19k) is a dual inhibitor of Tubulin and HDAC, with an IC50 of 0.403 μM, 0.591μM, 3.552μM, 0.459μM for HDAC1/2/3/6. Tubulin/HDAC-IN-2 blocks cell cycle arrest at G2 phase, induces cell apoptosis. Tubulin/HDAC-IN-2 inhibits the growth of hematoma and solid tumor cells, reduces tumor metastasis, and also inhibits tumor growth in a liver tumor allograft mouse model .
3-Methyl-2-quinoxalinecarboxylic acid (Standard) is the analytical standard of 3-Methyl-2-quinoxalinecarboxylic acid. This product is intended for research and analytical applications. 3-Methyl-2-quinoxalinecarboxylic acid (MQCA), an important N-oxide reductive metabolite of Quinocetone or Olaquindox, potently inhibits the growth of Chang liver cells through S phase arrest of the cell cycle .
Autophagy-IN-1 is a potent autophagy/mitophagy inhibitor, acts by selectively increasing the autophagic flux while blocking the autophagosome-lysosome fusion in cancer cells. Autophagy-IN-1 can induce apoptosis and cell cycle arrest. Autophagy-IN-1 significantly inhibits tumor growth in an HCT116 xenograft mouse model and with low toxicity. Autophagy-IN-1 can be used for researching colorectal cancer .
V-11-0711 is a potent and selective Chk-α (IC50 = 20 nM) inhibitor. V-11-0711 can significantly reduce PCho levels. V-11-0711 causes a reversible growtharrest. V-11-0711 induces apoptosis at high concentrations. V-11-0711 can be used for the study of cervical cancer and triple-negative breast cancer .
Tubulin inhibitor 42 (Compound 14b) dose-dependently inhibited the activity of β-microtubulin (IC50 = 3.5 µM).Tubulin inhibitor 42 interferes with microtubule dynamic homeostasis, resulting in the arrest of the cancer cell cycle in the G2/M phase and inducing apoptosis. Tubulin inhibitor 42 significantly inhibits the angiogenic process in vitro and in vivo, preventing vascularization and tumor growth .
CAM2602 is an orally active Aurora A-TPX2 protein−protein interaction inhibitor with a human Kd of 19 nM for Aurora A. CAM2602 increases the proportion of PH3 positive cells while reducing P-T288 Aurora A levels. CAM2602 arrests tumor xenograft growth in mice. CAM2602 can be used for the research of cancer, such as acute T cell leukemia .
EGFR-IN-163 (Compound 13) is a competitive epidermal growth factor receptor (EGFR) inhibitor (IC50=0.079 μM, selective for HER-2 inhibition). EGFR-IN-163 induces tumor cell apoptosis and cell cycle arrest at G₂/M phase. EGFR-IN-163 is promising for research of estrogen receptor-positive (ER+) breast cancer .
Cinobufagin (Standard) is the analytical standard of Cinobufagin. This product is intended for research and analytical applications. Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
V-11-0711 hydrochloride is a potent and selective Chk-α (IC50 = 20 nM) inhibitor. V-11-0711 hydrochloride can significantly reduce PCho levels. V-11-0711 hydrochloride causes a reversible growtharrest. V-11-0711 induces apoptosis at high concentrations. V-11-0711 hydrochloride can be used for the study of cervical cancer and triple-negative breast cancer .
PROTAC SMARCA2 degrader-35 (Compound 43) is a selective SMARCA2PROTAC degrader with a DC50 < 0.1 μM for SMARCA2. PROTAC SMARCA2 degrader-35 has anticancer activity and regulates cancer cell proliferation and growth through cell cycle arrest and DNA replication inhibition in SMARCA4-deleted cancer cells .
Cinobufagine-d3 is the deuterium labeled Cinobufagin (HY-N0421). Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
EGFR/BRAFV600E-IN-4 (Compound 10f) is a dual EGFR and BRAF V600E inhibitor with IC50 values of 61 nM and 43 nM, respectively. EGFR/BRAFV600E-IN-4 arrests cell cycle, induces apoptosis in both early and late stages and inhibits cancer cells growth in vitro, and has a broad anticancer activity .
CFM-4 is a potent small molecular antagonist of CARP-1/APC-2 binding. CFM-4 prevents CARP-1 binding with APC-2, causes G2M cell cycle arrest, and induces apoptosis with an IC50 range of 10-15 μM. CFM-4 also suppresses growth of drug-resistant human breast cancer cells .
Pyridostatin (RR82) is a G-quadruplex DNA stabilizing agent (Kd=490 nM) and can target DNA and RNA G4s in cells. Pyridostatin promotes growtharrest in human cancer cells by inducing replication- and transcription-dependent DNA damage. Pyridostatin targets the proto-oncogene Src. Pyridostatin reduced SRC protein levels and SRC-dependent cellular motility in human breast cancer cells .
N-Desmethyldauricine is a NF-κB p65 inhibitor and apoptosis inducer. N-Desmethyldauricine reduces the protein expression level of p65, induces apoptosis, arrests the cell cycle at the G0/G1 phase, attenuates intercellular adhesion, and inhibits the growth of 3D spheroids of triple-negative breast cancer. N-Desmethyldauricine can be used in studies related to triple-negative breast cancer .
Ru (acac) 3 (Tris (acetylacetonato) ruthenium (III)) is a caspase-3 activator and Apoptosis inducer. Ru (acac) 3 exerts growth inhibitory effects on various cell lines in vitro by inhibiting DNA/RNA synthesis and inducing mild reversible S-phase cell cycle arrest. Ru (acac) 3 is commonly used in research related to ovarian cancer, osteosarcoma, cervical cancer, melanoma, and other fields .
Cucurbitacin B (Standard) is the analytical standard of Cucurbitacin B. This product is intended for research and analytical applications. Cucurbitacin B belongs to a class of highly oxidized tetracyclic triterpenoids and is oral active. Cucurbitacin B inhibits tumor cell growth, migration and invasion and cycle arrest, but induces cell apoptosis. Cucurbitacin B has potent anti-inflammatory, antioxidant, antiviral, hypoglycemic, hepatoprotective, neuroprotective activity .
Pyridostatin (RR82) hydrochloride is a G-quadruplex DNA stabilizing agent (Kd=490 nM) and can target DNA and RNA G4s in cells. Pyridostatin hydrochloride promotes growtharrest in human cancer cells by inducing replication- and transcription-dependent DNA damage. Pyridostatin hydrochloride targets the proto-oncogene Src. Pyridostatin hydrochloride reduced SRC protein levels and SRC-dependent cellular motility in human breast cancer cells .
Quinalizarin is a protein kinase CK2 inhibitor with a Ki of 0.052 μM. Quinalizarin exhibits antifungal and anticancer activities. Quinalizarin induces ROS production, apoptotic signaling, mitochondrial pathway activation, cell cycle arrest, and cytotoxicity in cancer cells. Quinalizarin inhibits hyphal growth, biofilm formation, and mature biofilm integrity of Candida albicans. Quinalizarin can be used in research related to cancer and fungal infections .
Cyclic-di-GMP sodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP sodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP sodium can be used in cancer research .
Cyclic-di-GMP is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP can be used in cancer research .
CCT128930 hydrochloride is a potent and selective inhibitor of AKT (IC50=6 nM). CCT128930 hydrochloride has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). CCT128930 hydrochloride induces cell cycle arrest, DNA damage, and autophagy. Antitumor activity .
CCT128930 is a ATP-competitive and selective inhibitor of AKT (IC50=6 nM for AKT2). CCT128930 has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). Antitumor activity.
Apoptosis inducer 46 is an apoptosis inducer. Apoptosis inducer 46 exhibits potent and selective growth inhibitory effects on metastatic triple-negative breast cancer (TNBC) cells. Apoptosis inducer 46 induces G2/M phase cell cycle arrest and apoptotic cell death in MDA-MB-231 cells, and blocks NF-κB nuclear translocation. Apoptosis inducer 46 can be used for the study of TNBC .
LSD1-IN-17 (compound 5b) is a potent LSD1 inhibitor. LSD1-IN-17 can inhibit LSD1-CoREST, MAO-A and MAO-B, with IC50 values of 0.005, 0.028, and 0.820 μM, respectively. LSD1-IN-17 displays cell growtharrest in prostate cancer LNCaP cells, with an IC50 of 17.2 μM .
DDO-2728 (compound 19) is a selective AlkB homologue 5 (ALKBH5) inhibitor with an IC50 of 2.97 μM. DDO-2728 increases the abundance of N 6 methyladenosine (m 6A) modifications, inducing cell apoptosis and cycle arrest. DDO-2728 suppresses tumor growth in the MV4 11 xenograft model with favorable safety profile, shows the potential of targeting ALKBH5 in cancer research .
Lestaurtinib (CEP-701) is an orally active and selective RPTKs (receptor protein tyrosine kinase) inhibitor, competitively inhibits ATP binding to the TrkA/B/C domain. Lestaurtinib inhibits RPTKs phosphorylation, with IC50s of 2, 25 and 0.9 nM for FLT3, TrkA and JAK2, respectively. Lestaurtinib induces apoptosis and cycle arrest, also can inhibit growth of tumor .
Cyclic-di-GMP diammonium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP diammonium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP diammonium can be used in cancer research .
STAT3-IN-12 is a potent STAT3 signal inhibitor that can inhibit IL-6 induced JAK/STAT3 signalling pathway activation. STAT3-IN-12 inhibits cancer cell growth, migration, and induce cell apoptosis as well as cycle arrest. STAT3-IN-12 can be used in cancer-related research, such as hepatocellular carcinoma (HCC) and oesophageal carcinoma .
ADAR1-IN-1 is a potent ADAR1 inhibitor. ADAR1-IN-1 significantly suppressed DU-145 cell proliferation (IC50 = 1.11 μM), clonogenicity, migration, and invasion, arrests cell cycle, and induces apoptosis. ADAR1-IN-1 safely and effectively inhibits tumor growth. ADAR1-IN-1 can be used for the study of prostate cancer (PCa) .
Anticancer agent 109 (compound 6-15) is an inhibitor of the Gas6-Axl axis with anti-cancer activity. Anticancer agent 109 inhibits the expression of Gas6 and Axl, and the expression p-PI3K and p-AKT in cancer cells, leads to G1 phase arrest and promotes cancer cells apoptosis, and inhibits tumor growth significantly in nude mouse tumor bearing models .
PQ32 is an antitumor agent that targets c-MYC Pu27 and KRAS G-quadruplexes. PQ32 inhibits tumor cell proliferation, arrests the cell cycle at the G2 phase, and induces apoptosis. PQ32 inhibits the expression of c-MYC and KRAS genes. PQ32 can inhibit tumor growth in mice and is used in the study of lymphoma, breast cancer, lung cancer, hepatocellular carcinoma, colorectal cancer, and other cancers .
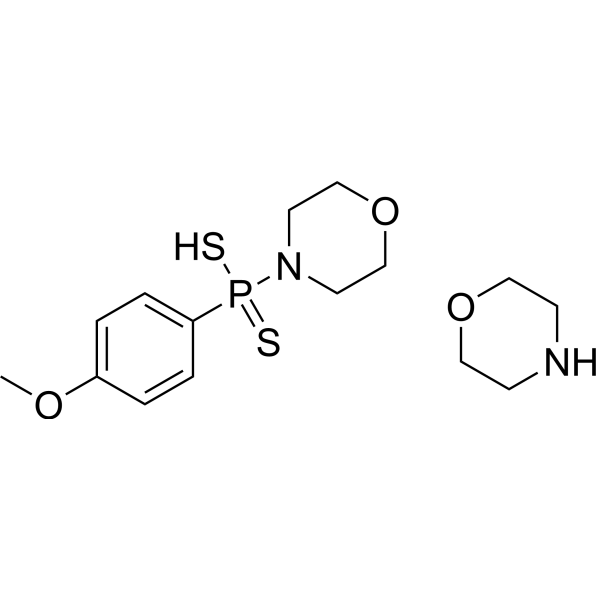
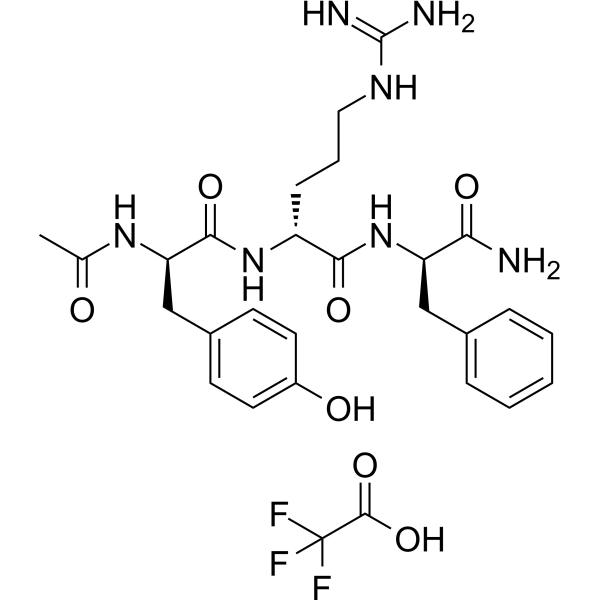
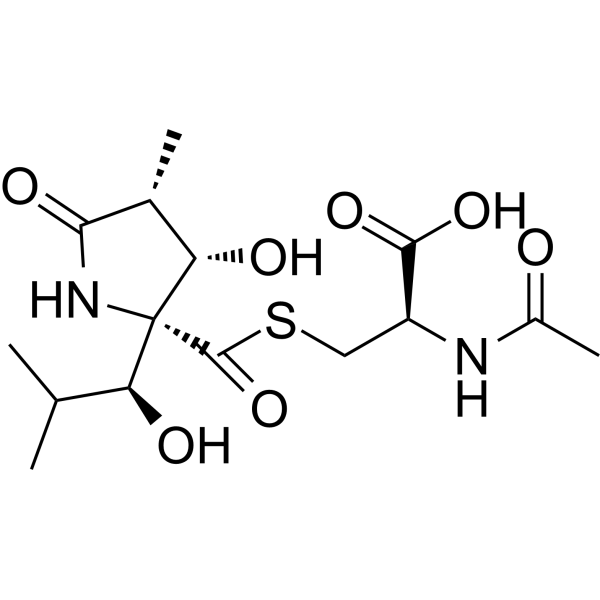
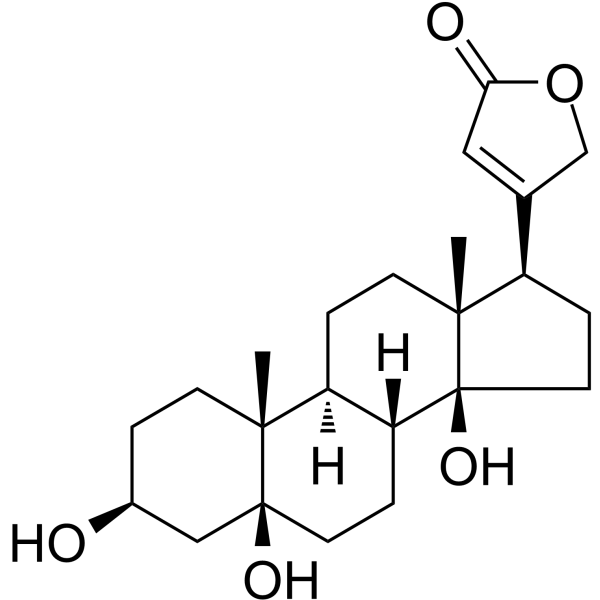
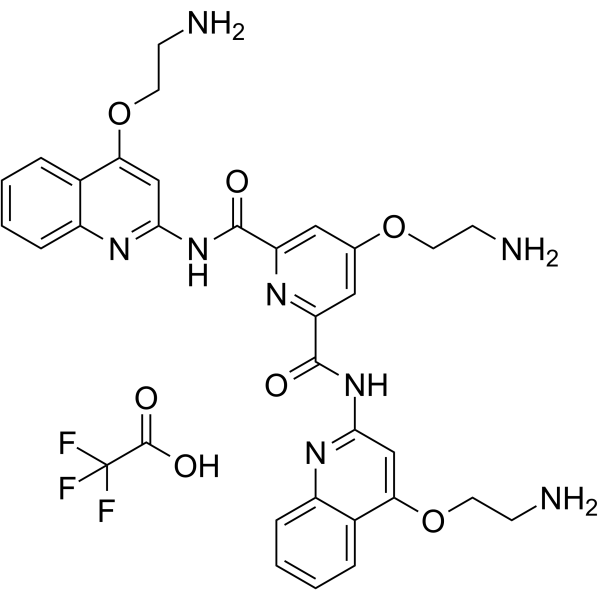
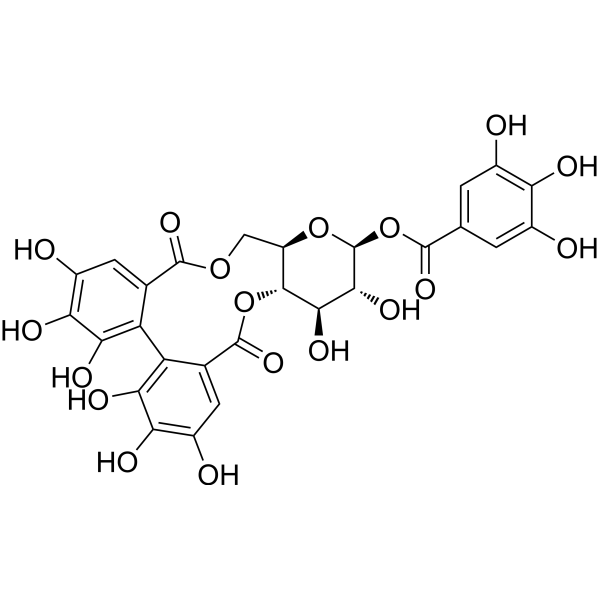
GYY4137 (Standard) is the analytical standard of GYY4137 (HY-107632). This product is intended for research and analytical applications. GY4137 is a sustained-release H2S donor possessing vasodilatory, antihypertensive, and anti-inflammatory activities. GY4137 can inhibit cell growth, induce apoptosis, and cause cell cycle arrest by blocKing the STAT3 pathway, demonstrating potent anticancer activity .
Discodermolide (NVP-XAA-296) is a potent microtubule-stabilizing agent with a Ki of 0.4 μM. Discodermolide stabilizes microtubules, induces G2 or M phase cell cycle arrest and apoptosis, leading to inhibition of cancer cell proliferation. Discodermolide competitively inhibits the binding of Paclitaxel (HY-B0015) to tubulin polymers, and inhibits the growth of Paclitaxel-resistant cells. Discodermolide can be used for breast and colon cancer research .
(-)-β-Peltatin is an aryltetrahydronaphthalene lignan. (-)-β-Peltatin exhibits antitumor activity and cytotoxicity against pancreatic cancer cells. (-)-β-Peltatin induces G2/M cell cycle arrest and apoptosis in pancreatic cancer cells. (-)-β-Peltatin inhibits the growth of subcutaneous xenografts of pancreatic cancer cells in nude mice. (-)-β-Peltatin can be used in pancreatic cancer-related research .
EGFR-IN-171 is an EGFR inhibitor with an IC50 value of 0.19 μM. EGFR-IN-171 also inhibits vascular endothelial growth factor 2 (VEGFR-2) with an IC50 value of 31.65 μM. EGFR-IN-171 can induce apoptosis and G2/M phase cell cycle arrest. EGFR-IN-171 can be used for cancer research, such as liver and breast cancer .
Cyclic-di-GMP disodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP disodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP disodium can be used in cancer research .
LSD1-IN-15 (compound 1b) is a potent LSD1 inhibitor. LSD1-IN-15 can inhibit LSD1-CoREST, MAO-A and MAO-B, with IC50 values of 0.149, 0.028, and 0.327 μM, respectively. LSD1-IN-15 displays cell growtharrest in prostate cancer LNCaP cells, with an IC50 of 9.9 μM .
LSD1-IN-16 (compound 4b) is a potent LSD1 inhibitor. LSD1-IN-16 can inhibit LSD1-CoREST, MAO-A and MAO-B, with IC50 values of 0.015, 0.024, and 0.366 μM, respectively. LSD1-IN-16 displays cell growtharrest in prostate cancer LNCaP cells, with an IC50 of 15.2 μM .
TM471-1 is an orally active and covalent Bruton's tyrosine kinase (BTK) inhibitor with IC50 values of 1.3 nM (BTK WT), >40,000 nM (BTK C481S), 7.9 nM (TEC) and 12.4 nM (TXK). TM471-1 inhibits cell growth in vivo and in vitro, arrests cell cycle at G0/G1 phase and induces cell apoptosis .
CDK2-IN-44 (Compound 46) is an inhibitor of cyclin-dependent kinase 2 (CDK2). CDK2-IN-44 can effectively inhibit the proliferation of cancer cells and exert its activity in inhibiting cancer cell growth by arresting the cell cycle, promoting Apoptosis, and inducing cellular senescence. CDK2-IN-44 holds promise for use in the research of ovarian cancer and breast cancer .
PARP-1/2-IN-2-IN-1 (Compound 12e) is a PARP1/2/CDK12 inhibitor (IC50: 34, 30 and 285 nM respectively). PARP-1/2-IN-2 inhibits DNA damage repair, promotes cell cycle arrest and apoptosis. PARP-1/2-IN-2 inhibits the growth of TNBC cells and TNBC xenograft tumor .
hIMB1636 is a humanized monoclonal antibody targeting Trop2. By binding to the conformational Trop2 epitope, hIMB1636 regulates related signaling pathways, triggers lysosomal endocytosis, and further induces apoptosis, cell cycle arrest, and antibody-dependent cellular cytotoxicity. hIMB1636 effectively inhibits tumor cell proliferation, migration and in vivo tumor growth, and also exerts bystander killing effect and mediates long-term retention. hIMB1636 can be conjugated with NOTA/DOTA for radiolabeling to enable immuno-PET imaging, or prepared as hIMB1636-LDP-AE to significantly inhibit the growth of breast cancer and lung cancer xenografts .
Antiestrogenic agent-1, an organophosphorus 13α-estrone derivative, is an antiestrogenic agent. Antiestrogenic agent-1 can disrupt estrogen signaling by inhibiting estrogen-mediated transcriptional activity. Antiestrogenic agent-1 can inhibit cancer cells proliferation, migration, invasion and induce G1-phase arrest. Antiestrogenic agent-1 mitigates estrogen-induced uterine growth in immature rats and inhibits tumor growth in a murine triple-negative breast cancer mice model. Antiestrogenic agent-1 can be used for the researches of cancer and endocrinology,such as breast cancer, oropharyngeal squamous cell carcinoma .
AZD3409 is a prenyl inhibitor that exhibits inhibitory activity against both farnesyl transferase and geranylgeranyl transferase I. AZD3409 inhibits the growth of breast cancer cells, with IC50s of 220 nM (MDA-MB-468), 180 nM (MDA-MB-361), and 290 nM (SK-Br-3). AZD3409 significantly reduces the activation level of AKT in breast cancer cell lines. AZD3409 induces G0/G1 phase arrest in MDA-MB-468 cells, causes G2/M phase arrest in MDA-MB-361 cells. AZD3409 can be used for the study of breast cancer .
PRMT5-IN-48 (compound D3) is an orally effective PRMT5 inhibitor (IC50=20.7 nM) with anticancer activity. PRMT5-IN-48 can inhibit the growth of various cancer cells, induce apoptosis (apoptosis), and arrest the cell cycle at the G0/G1 phase. PRMT5-IN-48 can be used for non-Hodgkin lymphoma (NHL) research .
Methoxyfenoterol is a β2-adrenergic receptor agonist. Methoxyfenoterol stimulates intracellular cAMP accumulation, inhibits tumor cell proliferation, induces G1 cell cycle arrest, upregulates cyclin-dependent kinase inhibitor p27, downregulates cyclin D1 and cyclin A, and inhibits Akt phosphorylation. Methoxyfenoterol crosses the blood-brain barrier and inhibits growth of astrocytoma xenografts. Methoxyfenoterol can be used for the research of astrocytoma, glioblastoma .
DH-18 is a matrix metalloproteinase-2 (MMP-2) inhibitor with the IC50 values of 139.45 nM, 518.11 nM and 833.34 nM for MMP-2, MMP-9 and MMP-8, respectively. DH-18 induces cell apoptosis and arrests cell cycle in the G0/G1 phase. DH-18 inhibits cell growth and can be used for study of chronic myeloid leukemia .
MS177 is an effective and fast-acting EZH2 degrader. MS177 is a PROTAC that consists of a CRBN ligand, linker, and a potent enzymatic EZH2 inhibitor C24 (C24 IC50): 12 nM). MS177 effectively depletes both canonical EZH2-PRC2 and noncanonical EZH2-cMyc complexes. MS177 induces leukaemia cell growth inhibition, apoptosis and cell cycle progression arrest .
CDK4/6-IN-15 hydrochloride is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 hydrochloride potently inhibits cancer cells growth. CDK4/6-IN-15 hydrochloride arrests cell cycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
SeSA-HCPT is an orally active dual-target inhibitor integrating Topo I and HDAC inhibition. SeSA-HCPT induces potent DNA damage, apoptosis, S-phase arrest in prostate cancer cells. SeSA-HCPT inhibits cancer cells proliferation and migration. SeSA-HCPT impairs homologous recombination by suppressing KIF4A-RAD51 signaling. SeSA-HCPT markedly inhibits CRPC tumor growth with minimal systemic toxicity .
NCP26 is an ATP-competitive ProRS inhibitor (with a KD of 271 nM in the absence of proline, and a KD of 0.35 nM in the presence of 100 μM proline). NCP26 activates AAR, induces G0/G1 phase arrest, Apoptosis, Caspase cleavage, and PARP cleavage. NCP26 downregulates MYC, TCF3, and CCND1. NCP26 inhibits the growth of multiple myeloma cells. NCP26 can be used for the research of multiple myeloma .
PU3 is a small molecule inhibitor of Hsp90 that competes with geldanamycin for Hsp90. PU3 induces degradation of proteins, including Her2, similar to geldanamycin. PU3 inhibits the growth of breast cancer cells, causing retinoblastoma protein hypophosphorylation, G1 arrest, and differentiation. PU3 represents a novel class of synthetic compounds that bind to Hsp90 and inhibit the proliferation of cancer cells. PU3 could provide a new strategy for the treatment of cancers .
CDK4/6-IN-15 is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 potently inhibits cancer cells growth. CDK4/6-IN-15 arrests cell cycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
LG308 is a novel synthetic compound with antimicrotubule activity. LG308 induces mitotic phase arrest and inhibits G2/M progression significantly which is associated with the upregulation of cyclin B1 and mitotic marker MPM-2 and the dephosphorylation of cdc2. LG308 also induces apoptosis and cell death. LG308 significantly suppresses tumor growth. LG308 with antimitotic activity has the potential for the research of prostate cancer .
ZDLD13, a β-carboline, is an orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 0.38 μM. ZDLD13 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cell cycle. ZDLD13 shows significant tumor growth inhibition in HCT116 tumor xenograft model .
EGFR/COX-2-IN-2 (Compound 10a) is a dual inhibitor targeting epidermal growth factor receptor (EGFR) (IC50= 6.0 μM) and cyclooxygenase-2 (COX-2) (IC50=50 μM). EGFR/COX-2-IN-2 induces S-phase cell cycle arrest and apoptosis. EGFR/COX-2-IN-2 is promising for research of cancers and inflammation-related diseases .
YVPGP is an oligopeptide exacted from Anthopleura anjunae. YVPGP has a significant antitumor activity by mediating PI3K/AKT/mTOR signaling pathway. YVPGP arrests DU-145 cells in the S phase and induces apoptosis via mitochondrial and death receptor pathways (caspase3, 7, 8, 9). YVPGP effectively inhibits tumor growth in DU-145 xenografts mice model, promising for prostate cancer research .
Human TGFBR2 mRNA encodes the human transforming growth factor beta receptor 2 (TGFBR2) protein, a transmembrane protein that has a protein kinase domain, forms a heterodimeric complex with TGF-beta receptor type-1, and binds TGF-beta. TGFBR2/ligand complex phosphorylates proteins, which then enter the nucleus and regulate the transcription of genes related to cell proliferation, cell cycle arrest, wound healing, immunosuppression, and tumorigenesis.
ZT55 is an orally active and highly-selective JAK2 inhibitor with an IC50 value of 0.031 μM. ZT55 inhibits the proliferation of JAK2 V617F-expressing HEL cell lines and induces apoptosis and cycle arrest. ZT-55 also effectively inhibits the growth of HEL xenograft tumours in a mice model. ZT-55 can be used in studies of myeloproliferative neoplasms, polycythemia vera and primary thrombocythemia .
Oxiconazole (Ro 13-8996) is a broad spectrum anti-fungal agent which can inhibit the growth of Candida, Aspergillus and Trichophyton. Oxiconazole is also a highly efficacious activator of CYP3A4 transactivation, which could be antagonized by Rifampicin (HY-B0272) in a competitive manner. Oxiconazole exhibits inhibitory effect against colorectal cancer (CRC) via peroxiredoxin-2 (PRDX2)-mediated autophagyarrest .
RAPTA-C (Ru(η6-p-cymene)Cl2(pta)) acts as an anti-cancer and anti-angiogenic agent. RAPTA-C exhibits anti-metastatic, anti-angiogenic, and anti-tumoral activities through protein and histone-deoxyribonucleic acid alterations. RAPTA-C exhibits cell growth inhibition by triggering G(2)/M phase arrest in cancer cells. RAPTA-C also enhances the levels of p53 and triggers the mitochondrial Apoptotic pathway, resulting in cytochrome C release and caspase-9 activation. RAPTA-C reduces the growth of tumors with the inhibition of angiogenesis in a ovarian carcinoma model .
EGFR-IN-169 is an epidermal growth factor (EGFR) (IC50 = 5.19 μM) inhibitor form panaxadiol. EGFR-IN-169 interferes with the migration and growth of colorectal cancer cells by inhibiting EGFR-mediated RalA/EMT pathway. EGFR-IN-169 shows an IC50 value of 4.46 μM and SI of 16.92 for HCT-116 cells. EGFR-IN-169 inhibits CDKs activity, induces G0/G1 cycle arrest and inhibits migration and invasion. EGFR-IN-169 reduces mitochondrial membrane potential and induces apoptosis and ROS production. EGFR-IN-169 can be used for the research of cancer, such as colorectal cancer .
BRD4/NAMPT-IN-1 (Compound A2) shows strong inhibitory effects on NAMPT and BRD4 (IC50=35 nM (NAMPT) and 58 nM (BRD4)). BRD4/NAMPT-IN-1 inhibits the growth and migration of hepatocellular carcinoma cells and promotes apoptosis. BRD4/NAMPT-IN-1 also shows potent anticancer effects in HCCLM3 xenograft mouse model, with no obvious toxic effects .
Triptophenolide (Hypolide) is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
EGFR/c-Met-IN-1 (compound TS-41) is a dual-target inhibitor of EGFR/c-Met. The IC50 for inhibiting EGFR L858R and c-Met is 68.1 nM and 0.26 nM respectively. . EGFR/c-Met-IN-1 induces apoptosis and cell cycle arrest in A549-P cells, downregulating the phosphorylation of EGFR, c-Met, and downstream AKT. EGFR/c-Met-IN-1 inhibits tumor growth in vitro and in vivo .
CFI-401870 is an orally active threonine tyrosine kinase (TTK (Mps1)) inhibitor with an IC50 of 3.1 nM. CFI-401870 exhibits IC50s against PLK4, KDR, AURKA and other kinases such as AURKB/INCENP were all greater than 1 μM.CFI-401870 inhibits the growth of various cancer cells, causing chromosome lag, an increase in aneuploidy and cell cycle arrest. CFI-401870 can be used for the study of cancers such as colon cancer .
PROTAC BRD9 Degrader-11 is a VHL-based BRD9PROTAC degraderwith an IC50 of 0.66 μM. PROTAC BRD9 Degrader-11 induces selective, proteasome-dependent degradation of BRD9 via the ubiquitin-proteasome system. PROTAC BRD9 Degrader-11 impairs cell viability, suppresses proliferation, and arrestsgrowth of acute myeloid leukemia cells. PROTAC BRD9 Degrader-11 can be used for the research of acute myeloid leukemia .
IKP-104 is a microtubule/tubulin inhibitor (IC50 = 1.31 μM). IKP-104 arrests cells in mitosis and the M phase by inhibiting microtubule polymerization and inducing cytoskeletal microtubule depolymerization. IKP-104 inhibits the growth of mouse and human tumor cell lines. IKP-104 exhibits anti-tumor effects in mouse ascites tumors and lung cancer models. IKP-104 is useful in the research of cancers such as leukemia, lung cancer and melanoma .
CDK9-IN-40 is a potent and orally active CDK9 inhibitor with an IC50 of 5.5 nM. CDK9-IN-40 shows high selectivity for CDK9 versus CDK1, CDK2, CDK4, and CDK6, respectively. CDK9-IN-40 can arrest cell cycle, induce cell apoptosis and inhibit tumor growth. CDK9-IN-40 exhibits strong anti-cancer activity .
Oxiconazole (Ro 13-8996) nitrate is a broad spectrum anti-fungal agent which can inhibit the growth of Candida, Aspergillus and Trichophyton. Oxiconazole nitrate is also a highly efficacious activator of CYP3A4 transactivation, which could be antagonized by Rifampicin (HY-B0272) in a competitive manner. Oxiconazole nitrate exhibits inhibitory effect against colorectal cancer (CRC) via peroxiredoxin-2 (PRDX2)-mediated autophagyarrest .
BR-cpd7 is a PROTAC degrader for fibroblast growth factor receptor FGFR1/2 with DC50 of 10 nM. BR-cpd7 arrests cell cycle, inhibits proliferations of FGFR1/2 aberrant activated tumor cells. (Pink: ligand for target protein FGFR-IN-12 (HY-160013); Black: linker; Blue: ligand for E3 ligase Thalidomide-NH-CH2-COOH (HY-131717))
all-trans-5,6-epoxy Retinoic acid (5,6-epoxy RA) is an agonist of all isoforms of the retinoic acid receptor (RAR; EC50s=77, 35, and 4 nM for RARα, RARβ, and RARγ, respectively). 5,6-epoxy RA (1 μM) also induces growtharrest of MCF-7 and NB4 cells in vitro. It is a natural metabolite of all-trans retinoic acid, which is a metabolite of vitamin A.
AKCI is a type of AURKC-IκBα interaction inhibitor, with an IC50 value of 24.9 μM. In MDA-MB-231 cells, AKCI can induce G2/M cell cycle arrest by regulating the p53/p21/CDC2/cyclin B1 pathway, inhibit cell migration and invasion, and reduce colony formation and tumor growth. AKCI can be used for research on breast cancer .
ELR510444 is an orally active tubulin inhibitor with an IC50 of 10 μM. ELR510444 binds to the colchicine-binding site on β-tubulin, inhibits tubulin assembly, depolymerizes microtubules and blocks HIF activity. ELR510444 induces cellular microtubule loss, abnormal mitotic spindle, mitotic arrest, apoptosis, morphological changes in tumor endothelial cells, and inhibits cancer cell proliferation, angiogenesis and tumor growth. ELR510444 can be used in research related to various cancers such as renal cell carcinoma .
Anticancer agent 57 (compound 14) potently inhibits MDA-MB-231, MDA-MB-468, and MCF-7 cell lines, with IC50s of 6.43 ~ 8.00 μM. Anticancer agent 57 induces cell cycle arrest and significantly promotes apoptosis. Anticancer agent 57 inhibits tumor growth in nude mice xenografted with MADMB-231 cells. Anticancer agent 57 can be used for researching triple negative breast cancer (TNBC) .
FLT3-IN-14 is a potent FLT3 inhibitor with IC50s of 5.6 nM and 1.4 nM for FLT3-WT and FLT3-ITD. FLT3-IN-14 reduces the phosphorylation of FLT3 (Y591), induces cell cycle arrest at G1 phase and apoptosis. FLT3-IN-14 significantly reduces the tumor growth in an MV4-11 xenograft mouse model .
CHNQD-01426 (Compound 4a) is an anticancer agent. CHNQD-01426 has cytotoxic activities against cancer cells. CHNQD-01426 significantly inhibits hepatocellular carcinoma cells proliferation via arresting S and G2/M phase cell cycle and induces apoptosis by inducing ROS production and elevating apoptosis-related proteins expression. CHNQD-01426 potently inhibits tumor growth in HepG2 xenograft mice model .
HIT211504993 is a selective histone deacetylase 6 (HDAC6) inhibitor with an IC50 of 0.070 μM. HIT211504993 suppresses cancer cell proliferation, cause G1 phase cell cycle arrest and induces apoptosis. HIT211504993 inhibits Myc-driven tumorigenesis via nucleocytoplasmic acetylation, p53 modulation, and Wnt/β-catenin signaling modulation. HIT211504993 inhibits tumor growth in a colon cancer xenograft mouse model. HIT211504993 can be used for the research of colon cancer .
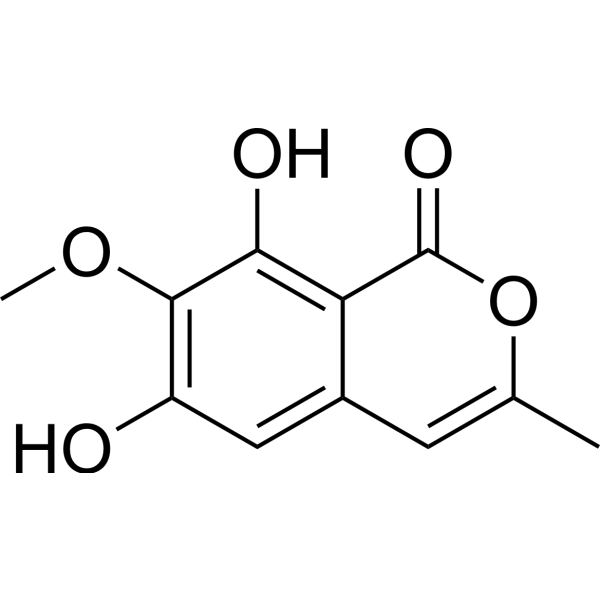
GYY4137 (GMP) is the GMP-grade version of GYY4137 (HY-107632). Small molecules of GMP grade can be used as adjuvant reagents in cell therapy. GYY4137 (GMP) is a sustained-release H2S donor possessing vasodilatory, antihypertensive, and anti-inflammatory activities. GYY4137 (GMP) can inhibit cell growth, induce apoptosis, and cause cell cycle arrest by blocking the STAT3 pathway, demonstrating potent anticancer activity .
Vepafestinib (TAS0953/HM06) is a next-generation brain-penetrant, selective and orally active RET inhibitor with an IC50 value of 0.33 nM. Vepafestinib inhibits the phosphorylation of RET and its downstream signaling pathways, thus blocking the growth and signal transduction of tumor cells and inducing cell cycle arrest and apoptosis. Vepafestinib can be used in the research of various RET-driven cancers, such as non-small cell lung cancer, thyroid cancer and other disease areas .
PP5-IN-2 is an orally active and selective protein phosphatase 5 (PP5) inhibitor with an IC50 value of 0.9 μM. PP5-IN-2 activates p53 and downregulates cyclin D1 and MGMT, which shows potency in cell cycle arrest and reverses Temozolomide (TMZ) (HY-17364) resistance in the U87 MG cell line. PP5-IN-2 effectively inhibits tumor growth in the xenograft mouse model .
NSC139021 (ERGi-USU) is a RIOK2 inhibitor with anticancer activity. RIOK2 can highly selectively inhibit the growth of ERG-positive cancer cells with IC50s of 30-400 nM against cell lines. RIOK2 also causes cell cycle arrest and apoptosis in glioblastoma via induction of Skp2 and Skp2-p27/p21-Cyclin E/CDK2-pRb signaling .
Lactacystin is a potent, orally active, irreversible, cell-permeable, selective 20S proteasome inhibitor (IC50 = 4.8 μM). Lactacystin also inhibits the lysosomal enzyme cathepsin A. Lactacystin inhibits cell growth and induces apoptosisand cell cycle arrest, and has antiviral and antioxidative activity. Lactacystin induces neurite outgrowth and hypertension. Lactacystin has the potential for the research of cancer, Neurological Disease, hypertension and Malaria, and so on [2] [6] .
Phoyunbene B is a similar substance to Resveratrol (HY-16561). Phoyunbene B exhibits stronger growth inhibitory activity against human liver cancer cells HepG2 compared to Resveratrol. Phoyunbene B induces G2/M phase cell cycle arrest and apoptosis. Phoyunbene B increases Bax/Bcl-2 and activates Caspase-3. Phoyunbene B inhibits the invasion and migration of cancer cells. Phoyunbene B can be used for research on liver cancer .
Mirvetuximab soravtansine (IMGN853) solution is an anti-folate receptor α (FRα) antibody drug-conjugate (ADC) consisting of the cytotoxic maytansinoid, DM4, covalently linked to the humanized monoclonal antibody M9346A, and the drug-linker conjugate for ADC is sulfo-SPDB-DM4 (HY-101141). Mirvetuximab soravtansine selectively binds to folate receptor 1 (FOLR1). Mirvetuximab soravtansine has an anti-proliferative effect via growtharrest and augmented DNA damage .
c-Myc inhibitor 16 iodide (Compound W11) is a selective c-Myc G-quadruplex (c-Myc G4) inhibitor. c-Myc inhibitor 16 iodide inhibits the transcription and translation of the c-Myc gene, disrupts the tumor cell cycle, arrests cell growth in the G0/G1 phase and activates the mitochondrial apoptosis pathway to induce early apoptosis of cancer cells. c-Myc inhibitor 16 iodide is promising for research of breast cancer .
5,8-Epidioxyergosta-6,9(11),22-trien-3-ol (9,11-Dehydroergosterol peroxide), an important steroid from medicinal mushroom, exerts antitumor activity in several tumor types. 5,8-Epidioxyergosta-6,9(11),22-trien-3-ol inhibits HT29 cell growth by inducing CDKN1A expression, thus causing cell cycle arrest and apoptosis .
cis-V-11-0711 is the cis form of V-11-0711 (HY-120337). V-11-0711 is a potent and selective Chk-α (IC50 = 20 nM) inhibitor. V-11-0711 can significantly reduce PCho levels. V-11-0711 causes a reversible growtharrest. V-11-0711 induces apoptosis at high concentrations. V-11-0711 can be used for the study of cervical cancer and triple-negative breast cancer .
Tubulin inhibitor 26 (compound 3c) is a potent inhibitor of tubulin. Tubulin inhibitor 26 is an indazole derivative compound. Tubulin inhibitor 26 shows noteworthy low nanomolar potency against HepG2, HCT116, SW620, HT29 and A549 cancer cell lines. Tubulin inhibitor 26 arrests tumor cell in G2/M phase and induced cell apoptosis. Tubulin inhibitor 26 suppresses tumor growth in vivo without affecting the mice body weight .
PLK1-IN-13 is a selective and orally active PLK1 inhibitor (IC50: 0.27 nM). PLK1-IN-13 also inhibits PLK2 (IC50: 12.72 nM) and PLK3 (IC50: 4.12 nM). PLK1-IN-13 arrests cell at G2 phase, induces apoptosis and down-regulates the transcription of the proliferation-related oncogene c-MYC. PLK1-IN-13 inhibits tumor growth, and can be used for research of acute myeloid leukemia (AML) .
2α-Ferrocenylmethyl-DHT is a dihydrotestosterone-derived ferrocene-steroid conjugate. 2α-Ferrocenylmethyl-DHT inhibits the growth of various cancer cells. 2α-Ferrocenylmethyl-DHT induces S-phase cell cycle arrest in ovarian cancer cells, elevates intracellular iron levels, triggers ROS-dependent cell death, and disrupts the integrity of multicellular tumor spheroids of ovarian cancer cells. 2α-Ferrocenylmethyl-DHT can be used in the research of prostate cancer and ovarian cancer .
Apoptosis inducer 43 is an apoptosis inducer. Apoptosis inducer 43 can induce apoptosis, SubG0-G1 cell cycle arrest, secondary necrosis, and upregulate caspase-3, p53, and Bax/Bcl-2 expression in HCT116 cells. Apoptosis inducer 43 can inhibit tumor growth in a solid Ehrlich carcinoma (SEC) mouse model. Apoptosis inducer 43 can be used to study cancers such as colon cancer, leukemia, and non-small cell lung cancer .
BRM/BRG1 ATP-IN-8 (Compound Cpd14) is an orally active BRG1/BRM ATPase domain inhibitor. BRM/BRG1 ATP-IN-8 exerts antitumor effects by disrupting aberrant chromatin remodeling in cancer cells, inducing apoptosis and growtharrest. BRM/BRG1 ATP-IN-8 is promising for research of BRG1/BRM-dependent malignancies, including hematological cancers, prostate cancer, breast cancer, and Ewing's sarcoma .
Pro-GA is a γ-glutamyl cyclotransferase (GGCT) inhibitor. Pro-GA inhibits the enzymatic activity of GGCT, disrupts glutathione homeostasis, induces the production of mitochondrial ROS, and upregulates the expression of p21, p27 and p16 in cells. Pro-GA inhibits the growth of cancer cells, induces cell cycle arrest and cellular senescence. Pro-GA exerts anti-tumor effects in breast cancer xenograft mouse models. Pro-GA can be used in research related to bladder cancer and breast cancer .
Cernuumolide J (TMJ-105) is an JAK2/STAT3 inhibitor. Cernuumolide J induces G2/M phase arrest and apoptosis in HEL leukemia cells by downregulating the phosphorylation of JAK2, STAT3, and Erk, and activating the phosphorylation of JNK and p38 MAPK. Cernuumolide J inhibits HEL leukemia cell growth in a time- and concentration-dependent manner, with an IC50 value of 1.79 μM. Cernuumolide J can be used for research in the field of anti-cancer therapy .
Tamarixetin (4'-O-Methyl Quercetin) is an orally active natural flavonoid derivative of quercetin and caseinolytic protease p (ClpP) inhibitor with anti-inflammatory, antioxidant and antitumor effects. Tamarixetin inhibits the hydrolytic activity of ClpP to the fluorescent substrate Suc-LY-AMC with an IC50 of 49.73 μM, which can be used for the study of Staphylococcus aureus infection. Tamarixetin inhibits tumor cell growth, induces apoptosis, and cell cycle arrest. Tamarixetin prevents cardiac hypertrophy by inhibiting the NFAT and AKT pathways .
I3IN-002 is a small-molecule RNA-binding protein IGF2BP3 inhibitor with an IC50 value of approximately 2 μM in SEM cells. I3IN-002 interferes with interaction with m6 A-modified mRNAs, disrupting the stabilization of target genes (such as CDK6, MYC, and BCL2) to inhibit leukemic cell growth, induce cell cycle arrest, and promote apoptosis. I3IN-002 is promising for research of B-cell acute lymphoblastic leukemia .
PRO-6E is an oral active PROTAC based on Cereblon ligand, and induces the degradation of MET with maximum degradation of 81.9% at 1 μM in MKN-45 cells. PRO-6E inhibits tumor growthin vivo and in vitro. PRO-6E induces cell apoptosis and induces cell arrest (Sturcture Note:(Blue: Cereblon ligand (HY-103596), Black: linker;Pink: ALK/c-Met inhibitor Crizotinib (HY-50878)) .
AR-42 (HDAC-42; OSU-HDAC42) is a potent, orally bioavailable pan-HDAC inhibitor (IC50=16 nM). AR-42 induces growth inhibition, cell-cycle arrest, apoptosis, and activation of caspases-3/7. AR-42 promotes hyperacetylation of H3, H4, and alpha-tubulin, and up-regulation of p21. AR-42 shows cytotoxicity against various human cancer cell lines .
K783-0308 is a potent and selective dual inhibitor of FLT3 and MNK2 with IC50 values of 680 and 406 nM, respectively. K783-0308 inhibits the growth of MOLM-13 (IC50=10.5 µM) and MV-4-11 (IC50=10.4 µM) cells. K783-0308 promotes acute myeloid leukemia (AML) cell apoptosis and cell cycle arrests in the G0/G1 phase .
BKT300 is a potent and selective protein regulator of cytokinesis 1 (PRC1) inhibitor. BKT300 inhibits PRC1 dephosphorylation at T481, disrupts actin and microtubule formation, induces G2/M cell cycle arrest, triggers mitotic catastrophe, and promotes apoptosis, thereby inhibiting proliferation and migration of acute myeloid leukemia (AML) cells while sparing normal cells. BKT300 inhibits tumor growth in mouse xenograft AML models. BKT300 can be used for the research of AML .
Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
PRMT7-IN-2 (A33) is a selective PRMT7 inhibitor with an IC50 of 0.50 μM. PRMT7-IN-2 arrests cell cycle at G0/G1 phase, induces cell apoptosis, and inhibits cell growthin vivo and in vitro. PRMT7-IN-2 decreases the monomethylarginine level of PRMT7, increases expression of epithelial marker (E-cadherin, and reduces expression of mesenchymal markers such as N-cadherin, Vimentin, and ZEB2 .
MDL-27048, a tubulin inhibitor, binds competitively, reversibly to the Colchicine (HY-16569)-binding site on tubulin heterodimers. MDL-27048 inhibits microtubule assembly, induces slow depolymerization of preassembled microtubules, disrupts microtubule polymerization-depolymerization dynamics, and disrupts cytoplasmic microtubule networks. MDL-27048 exerts growth inhibitory effects on human cancer cells, induces mitotic arrest, and does not disrupt actin filaments at microtubule-depolymerizing concentrations. MDL-27048 can be used for the research of malignant tumors .
CHM-1 (Standard) is the analytical standard of CHM-1 (HY-103257). This product is intended for research and analytical applications. CHM-1, a microtubule-destabilizing agent, inhibits tubulin polymerization. CHM-1 is a potent and selective antimitotic antitumor activity against human hepatocellular carcinoma. CHM-1 induces growth inhibition and apoptosis via G2-M phase arrest in human hepatocellular carcinoma cells by activation of Cdc2 kinase activity .
8-Gingerol can be found in the rhizome of ginger (Z. officinale) and has oral bioactivity. It activates TRPV1, with an EC50 value of 5.0 µM. 8-Gingerol inhibits COX-2 and also suppresses the growth of H. pylori in vitro. Additionally, 8-Gingerol exhibits anticancer, antioxidant, and anti-inflammatory properties by inhibiting the epidermal growth factor receptor (EGFR) and modulating its downstream STAT3/ERK pathway to suppress the proliferation, migration, and invasion of colorectal cancer cells. 8-Gingerol also exerts immunosuppressive effects by inhibiting oxidative stress, inducing cell cycle arrest, promoting apoptosis, and regulating autophagy. Furthermore, 8-Gingerol has cardioprotective effects. 8-Gingerol is promising for research in the fields of cancer, infection, immunosuppression, and cardiovascular diseases.
RO5068760 is a potent, orally active and selective non-ATP-competitive MEK1/2 inhibitor with an IC50 of 0.025 μM for MEK1. RO5068760 significantly inhibits MAPK pathway activity, thereby inducing G1 cell cycle arrest and apoptosis to inhibit cancer cell growth. RO5068760 exhibits significant efficacy in a broad spectrum of tumors with aberrant MAPK pathway activation. RO5068760 can be used for melanoma, colorectal cancer, non-small cell lung cancer (NSCLC), and pancreatic cancer research .
APD-94 is a dual inhibitor targeting tubulin and Bmi-1. APD-94 interfers tubulin normal polymerization. APD-94 suppresses the expression of Bmi-1. APD-94 causes cell cycle arrest at the G2/M phase in cancer cells and induces apoptosis, thus inhibiting cancer cell proliferation. APD-94 represses the growth of HT29 cell xenografts in NOD/SCID mice. APD-94 can be used for colorectal cancer study .
RGN6024 is a brain-penetrant, orally active and reversible small molecule tubulin destabilizer. RGN6024 inhibits microtubule polymerization both in biochemical and cellular assays, binds to the colchicine binding pocket of β-tubulin (SPR: Kd = 6.7 μM; tryptophan assay: Kd = 7.4 μM), and triggers G2/M arrest in glioblastoma (GB) cells. RGN6024 retains activity in βIII-tubulin overexpressing cells. RGN6024 inhibits tumor growth in a GB xenograft mouse model. RGN6024 can be used for the study of glioblastoma (GB) .
FLT3-IN-21 (compound LC-3) is a potent FLT3 inhibitor (IC50: 8.4 nM) and induces apoptosis. FLT3-IN-21 can arrest the cell cycle in the G1 phase and inhibit the proliferation of FLT3-ITD-positive AML cells MV-4-11 (IC50: 5.3 nM). In mice, FLT3-IN-21 (10 mg/kg/d) inhibited tumor growth in the MV-4-11 xenograft model (TGI=92.16%) .
Lepadin H is a ferroptosis inducer and apoptosis inducer with in vitro cytotoxicity and in vivo antitumor efficacy against cancer cells. Lepadin H reduces GPX4 and SLC7A11 levels, increases p53 and ACSL4 expression, drives lipid hydroperoxide production, elevates reactive oxygen species (ROS) levels, reduces cellular glutathione (GSH) levels, induces lipid peroxidation and G2/M phase cell cycle arrest, and suppresses clonogenic growth and migration of cancer cells.Lepadin H can be used for the research of melanoma .
Dac590 is an orally active and selective obesity-associated protein (FTO) inhibitor with an IC50 of 6.06 nM. Dac590 shows highly selective over ALKBH5 and ALKBH3. Dac590 suppresses oncogenic FTO signaling, induces myeloid differentiation, G1-phase cell cycle arrest, and apoptosis in acute myeloid leukemia (AML) cells. Dac590 inhibits xenograft tumor growth and prolongs survival in acute myeloid leukemia mouse models with no observed toxicity. Dac590 can be used for the research of AML .
MG-3C is a potent matrix metalloproteinase 9 (MMP-9) inhibitor. MG-3C can selectively kill non-small-cell lung cancer (NSCLC) cells harboring the EGFR T790M mutation. MG-3C blocks the EGFR/STAT3 signaling pathway, inducing G2/M phase arrest, growth inhibition, and apoptosis of cancer cells. MG-3C is promising for research of lung cancer .
Pyridostatin (RR82) trihydrochloride is a G-quadruplex (G4) inducer/stabilizer with a Kd of 490 nM. Pyridostatin trihydrochloride also acts as an inhibitor of Zika virus (ZIKV) NS2B-NS3 protease, with an IC50 of 11.0 μM. Pyridostatin trihydrochloride interacts with G-quadruplex structures, regulates the expression of SRC and SUB1, and induces replication- and transcription-dependent DNA damage, growtharrest, and genomic instability. Pyridostatin trihydrochloride exhibits antiproliferative and antiviral activities. Pyridostatin trihydrochloride can be used in studies related to breast cancer, cervical cancer, and Zika virus infection .
SL43 is an orally active and potent SOS1 inhibitor with a Kd of 0.16 μM. SL43 disrupts SOS1-KRAS interaction, inhibits SOS1-mediated nucleotide exchange on KRAS mutants, and suppresses RAS-MAPK signaling. SL43 exerts antiproliferative activity against KRAS-mutant cancer cells, induces early apoptosis and G1 phase cell cycle arrest, and reduces phosphorylated MEK and ERK levels. SL43 suppresses tumor growth in a colorectal cancer xenograft model .
Periplogenin is an orally active cardiac glycoside found in Cortex periplocae. Periplogenin can induce ROS production and necroptosis and cause G0/G1 phase arrest. Periplogenin can inhibit pyroptosis by regulating the NLRP3/Caspase-1/GSDMD signaling. Periplogenin suppresses growth of prostate carcinoma cells by docking to an ATP1A1 protein pocket and forming a hydrogen bond with T804. Periplogenin can be used for the researches of cancer, inflammation and immunology, such as prostate carcinoma, rheumatoid arthritis and psoriasis .
HS-36 is a highly selective and orally active dual inhibitor of CDK4 and CDK9 with IC50 values of 18.9 and 4.2 nM respectively. HS-36 exhibits nanomolar-level potent activity against various cancer cells, inducing G0/G1 phase arrest and promoting cell apoptosis. HS-36 efficiently inhibits tumor growth in a mouse model of MV-4-11 tumors. HS-36 can be used for the study of acute myeloid leukemia .
Anticancer agent 304 is an anticancer agent. Anticancer agent 304 binds to CDC45 with a Kd value of 83.0 μM. Anticancer agent 304 arrests the cell cycle of liver cancer cells at the G2/M phase, induces Apoptosis by upregulating C-PARP-1 and downregulating PARP-1 and BCL-2, and inhibits the migration, invasion and proliferation of liver cancer cells. Anticancer agent 304 suppresses tumor growth in animal models of hepatocellular carcinoma. Anticancer agent 304 is applicable to research related to liver cancer .
ERα degrader 14 (Compound B7) is a potent ERα degrader. ERα degrader 14 exhibits significantly potent and selective anti-proliferative activity on two ERα-positive breast cancer cell lines (MCF-7 and T47D). ERα degrader 14 induces cell cycle arrest, inhibits cell migration, and induces cell apoptosis. ERα degrader 14 effectively inhibits tumor growth in mouse models. ERα degrader 14 can be used for the study of breast cancer .
Cystatin B agonist 1 is an orally active MMP-2/9 inhibitor. Cystatin B agonist 1 exhibits inhibitory effects with IC50 values of 3.95 and 3.43 μM against U87 and T98G cells, respectively. Cystatin B agonist 1 induces the cell-cycle arrest at S phase, inhibits angiogenesis, and suppresses migration and invasion of MG cells. Cystatin B agonist 1 inhibits tumor growth in U87 MG xenograft model. Cystatin B agonist 1 can be used for the study of malignant glioma (MG) .
VEGFR-2-IN-58 (Compound 7b) inhibits VEGFR-2 with an IC50 of 42.5 nM. VEGFR-2-IN-58 displays selective cytotoxicity against cancer cells. VEGFR-2-IN-58 shows cellular growtharrest at the G2/M phase in cancer cells. VEGFR-2-IN-58 induces cancer cells Apoptosis, increasing BAX expression and reducing Bcl2 expression. VEGFR-2-IN-58 inhibits wound closure in cancer cells .
Triptophenolide (Standard) (Hypolide) is the analytical standard of Triptophenolide (HY-N0475). This product is intended for research and analytical applications. Triptophenolide is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
L 741742 is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 induces G0/G1 cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cells in vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 can be used for the research of schizophrenia and glioblastoma .
L 741742 hydrochloride is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 hydrochloride suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 hydrochloride induces G0/G1 cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cells in vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 hydrochloride can be used for the research of schizophrenia and glioblastoma .
Globulol is a terpenoid metabolite and Antimicrobial agent. Globulol can be isolated from Alpinia oxyphylla Miq. Globulol binds to PAK4, reduces the expression level of PAK4 in cancer cells, decreases the phosphorylation of AKT, and downregulates the expressions of STAT3, phosphorylated STAT3, and PD-L1. Globulol promotes the secretion of CCL4 by cancer cells. Globulol reduces the viability and proliferation ability of cancer cells, induces G0/G1 cell cycle arrest and Apoptosis in cancer cells, and inhibits cancer cell migration and the integrity of 3D tumor spheres. Globulol enhances the relevant effects of anti-PD-1 agents in the cancer cell microenvironment. Globulol exhibits anticancer activity against liver cancer. Globulol inhibits the mycelial growth of phytopathogenic fungi and the growth of phytopathogenic bacteria. Globulol can be used in studies related to hepatocellular carcinoma .
Skp2 inhibitor 3 is an orally active inhibitor of the Skp2-Cks1 complex, with an IC50 of 4.86 μM. Skp2 inhibitor 3 reduces Skp2 protein expression while upregulating the expression of p21 and p27. Skp2 inhibitor 3 inhibits colony formation and migration of cancer cells, and induces cell cycle arrest at the S phase. Skp2 inhibitor 3 suppresses tumor growth in xenograft mice. Skp2 inhibitor 3 can be used in the research of gastric cancer and prostate cancer .
TSL2109 is an orally active and selective DYRK2 and CDK4/6 inhibitor with an IC50 value of 22 nM for DYRK2. TSL2109 exhibits high kinase selectivity over 93%. TSL2109 arrests cell cycle and induces apoptosis in virto. TSL2109 effectively overcomes Enzalutamide (HY-70002) resistance by suppressing tumor growth in vivo and virto. TSL2109 also shows CDK4/6 inhibitor resistance.TSL2109 demonstrates safety profile. TSL2109 can be used for prostate cancer research and breast cancer [1][2].
DNMT-IN-6 is a DNA methyltransferase inhibitor with activity against DNMT1, DNMT3A, and DNMT3B. DNMT-IN-6 drives demethylation, and restores TMS1 tumor suppressor gene expression. DNMT-IN-6 induces apoptosis, causes G2/M phase arrest, disrupts mitochondrial integrity, and activates the intrinsic caspase cascade (3/7/9). DNMT-IN-6 inhibits tumor growth, and improves survival in xenograft models. DNMT-IN-6 can be used for the research of cancer, such as diffuse large B-cell lymphoma .
Oxiconazole nitrate (Standard) is the analytical standard of Oxiconazole nitrate. This product is intended for research and analytical applications. Oxiconazole (Ro 13-8996) nitrate is a broad spectrum anti-fungal agent which can inhibit the growth of Candida, Aspergillus and Trichophyton. Oxiconazole nitrate is also a highly efficacious activator of CYP3A4 transactivation, which could be antagonized by Rifampicin (HY-B0272) in a competitive manner. Oxiconazole nitrate exhibits inhibitory effect against colorectal cancer (CRC) via peroxiredoxin-2 (PRDX2)-mediated autophagy arrest .
WNY0824 (PLK1/BRD4-IN-1) is an orally active dual inhibitor of PLK1 and BET protein families, with IC50 values of 22, 402.5, 150.7, 103.9, and 311.9 nmol/L for PLK1, BRD2, BRD3, BRD4, and BRDT, respectively. WNY0824 induces cell cycle arrest and apoptosis by inhibiting AR- and MYC-mediated transcriptional processes. In addition, WNY0824 also inhibits tumor growth in Enzalutamide (HY-70002) resistant CRPC xenograft tumor models .
Tubulin polymerization-IN-86 (Compound B6) is an effective inhibitor of tubulin polymerization. Tubulin polymerization-IN-86 effectively inhibits microtubulin polymerization by binding to the colchicine binding sites on microtubulin, thereby disrupting the microtubule cytoskeleton within the cell. Tubulin polymerization-IN-86 exhibits potent anti-proliferative activity against a variety of cancer cell lines. Tubulin polymerization-IN-86 induces cell cycle arrest, apoptosis, inhibits cell migration, invasion, and long-term survival ability. Tubulin polymerization-IN-86 inhibits tumor growth in mice and can be used for the study of melanoma .
MTHFD2-IN-8 is a selective MTHFD2 inhibitor with an IC50 of 0.066 μM. MTHFD2-IN-8 directly binds to intracellular mitochondrially localized protein MTHFD2 and accumulates selectively in tumor mitochondria. MTHFD2-IN-8 increases intracellular ROS levels, induces mitochondrial membrane potential depolarization, arrests cell cycle at G0/G1 phase, promotes apoptosis in cancer cells. MTHFD2-IN-8 inhibits tumor growth in a mouse colon cancer graft model .
HA-9104 is a potent and selective inhibitor of cullin-5 neddylation via virtually targeting the V30 pocket of UBE2F. HA-9104 binds to UBE2F, reduces its protein levels, and consequently inhibits cullin-5 neddylation. HA-9104 has potent growth suppression and radiosensitizing activities via targeting the UBE2F-CRL5 axis and causing DNA damage, leading to induction of apoptosis and G2/M arrest in lung and pancreatic cancer cells .
PROTAC ATR degrader-3 is a potent CRBN-based ATRPROTAC degrader with a DC50 of 127 nM. PROTAC ATR degrader-3 also degrades CHK1 with an DC50 of 135 nM. PROTAC ATR degrader-3 inhibits cancer cells proliferation, migration and invasion, triggers apoptosis and induces S phase arrest and DNA damage. PROTAC ATR degrader-3 achieves tumor growth inhibition in LoVo xenograft mouse model without apparent toxicity. PROTAC ATR degrader-3 can be used for the research of colorectal cancer .
Dehydrozingerone is a ginger-derived component and cyclin D1 inhibitor that downregulates cyclin D1 expression and induces cell cycle G1 phase arrest. Dehydrozingerone reduces the proliferative capacity of castration-resistant prostate cancer cells under in vitro conditions. Dehydrozingerone reduces subcutaneous tumor growth by inhibiting cell proliferation and angiogenesis. Dehydrozingerone exerts antibacterial and antifungal activities via its α,β-unsaturated carbonyl conjugated system. Dehydrozingerone can be used in studies related to castration-resistant prostate cancer, bacterial infections, and food spoilage fungal infections .
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1 cell cycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
Tamarixetin (Standard) is the analytical standard of Tamarixetin. This product is intended for research and analytical applications. Tamarixetin (4'-O-Methyl Quercetin) is an orally active natural flavonoid derivative of quercetin and caseinolytic protease p (ClpP) inhibitor with anti-inflammatory, antioxidant and antitumor effects. Tamarixetin inhibits the hydrolytic activity of ClpP to the fluorescent substrate Suc-LY-AMC with an IC50 of 49.73 μM, which can be used for the study of Staphylococcus aureus infection. Tamarixetin inhibits tumor cell growth, induces apoptosis, and cell cycle arrest. Tamarixetin prevents cardiac hypertrophy by inhibiting the NFAT and AKT pathways .
TOPOI/PARP-1-IN-1 (Compound B6) is an orally active, low cytotoxic TOPOI/PARP dual inhibitor with an IC50 value of 0.09 μM for PARP1. TOPOI/PARP-1-IN-1 can effectively inhibit the proliferation and migration of cancer cells. TOPOI/PARP-1-IN-1 also causes cell cycle arrest in the G0/G1 phase and induces apoptosis. The tumor growth inhibition rate (TGI) of TOPOI/PARP-1-IN-1 in mice is 75.4% .
VEGFR-2-IN-71 is a dual VEGFR2/tubulin inhibitor. VEGFR-2-IN-71 inhibits tumor cell proliferation and induces apoptosis and cell cycle arrest. VEGFR-2-IN-71 inhibits angiogenesis in the chick chorioallantoic membrane (CAM) model. VEGFR-2-IN-71 inhibits tumor growth in the HGC-27 xenograft model by inhibiting VEGFR2 and tubulin. VEGFR-2-IN-71 has low oral bioavailability in rats. VEGFR-2-IN-71 can be used in cancer research .
ASX-173 is an orally active inhibitor of asparagine synthetase (ASNS) (IC50 = 0.113 μM, Ki = 0.4 nM). ASX-173 enhances the anticancer activity of L-asparaginase (ASNase) (HY-P1923). ASX-173 disrupts nucleotide synthesis and induces leukemia cell cycle arrest, apoptosis and autophagy in leukemia cells in combination with ASNase. ASX-173 slows the growth of OCI-AML2 xenografts in combination with ASNase. ASX-173 is indicated for the study of acute lymphoblastic leukemia, acute myeloid leukemia, colorectal cancer, and other cancers .
Antimycin A2 is a selective inhibitor of the cytochrome b-c1 complex in the mitochondrial electron transport chain. Antimycin A2 disrupts mitochondrial membrane potential and produces reactive oxygen species (ROS) by inhibiting electron transfer between cytochrome b and c. Antimycin A2 has bactericidal and piscicidal activity, as well as tumor cell growth inhibitory effects, and can induce S-phase cell cycle arrest and apoptosis in HeLa cells. Antimycin A2 is suitable for research of cervical cancer and fisheries management. Antimycin A2 can be naturally isolated from the fermentation products of Streptomyces sp. strains .
NHI-2 is a lactate dehydrogenase A (LDHA) inhibitor with an IC50 of 14.7 µM. NHI-2 shows selective for LDHA over LDHB (IC50 = 55.8 µM). NHI-2 is an efficient anti-glycolytic agent. NHI-2 enhances apoptosis, induces cell cycle arrest at S and G2 phases. NHI-2 has a broad spectrum anti-proliferative activity in cancer cells. NHI-2 affects extracellular acidification rate and ATP production. NHI-2 suppresses tumor growth in murine B78 melanoma tumor model .
pan-KRAS-IN-5 is a pan-KRAS translation inhibitor by targeting 5′-UTR RNA G-quadruplexes (rG4s). pan-KRAS-IN-5 strongly binds to and stabilizes KRAS rG4s, inhibits KRAS translation, and blocks the MAPK and PI3K-AKT pathways. pan-KRAS-IN-5 induces cell cycle arrest, prompts apoptosis in KRAS-driven cancer cells. pan-KRAS-IN-5 inhibits tumor growth and KRAS expression in KRAS-mutant xenograft. KRAS-IN-5 can be used for KRAS-driven cancer research .
HDAC1/CDK7-IN-1 (compound 8e) is a dual CDK7 and HDAC1 inhibitor with IC50s of 893 nM and 248 nM, respectively. HDAC1/CDK7-IN-1 inhibits the growth cells of MDA-MB-231, MCF-7, A549, and HCT-116 cancer cells. HDAC1/CDK7-IN-1 induces cell cycle arrest and apoptosis in HCT-116 cells, as well as hindered the migration of HCT-116 cells .
YJ9069 is a selective CDK12/CDK13 PROTAC degrader with an IC50 of 22.22 nM for in VCaP cells. CDK12/13 degradation rapidly triggers gene-length-dependent transcriptional elongation defects, leading to DNA damage and cell-cycle arrest. YJ9069 effectively inhibits proliferation in subsets of prostate cancer cells and significantly suppresses prostate tumor growth. (Pink: CDK12/CDK13 degradation agent (HY-168658); Black: Linker (HY-W015967); Blue: ligand for E3 ligase (HY-103596)) .
DDO-6600 is a covalent Hsp90 inhibitor. DDO-6600 disrupts the interaction between Hsp90 and its co-chaperone protein Cdc37, thereby inducing the degradation of kinase client proteins (such as AKT, CDK4, c-Raf). DDO-6600 has inhibitory activity against various cancer cells. DDO-6600 inhibits the migration and invasion of HCT-116 cells, and induces cell cycle arrest and apoptosis. DDO-6600 significantly inhibits tumor growth in the HCT-116 xenograft tumor model. DDO-6600 can be used for research on colorectal cancer .
Pyridostatin (RR82) TFA is a G-quadruplex (G4) inducer/stabilizer with a Kd of 490 nM. Pyridostatin TFA also acts as an inhibitor of Zika virus (ZIKV) NS2B-NS3 protease, with an IC50 of 11.0 μM. Pyridostatin TFA interacts with G-quadruplex structures, regulates the expression of SRC and SUB1, and induces replication- and transcription-dependent DNA damage, growtharrest, and genomic instability. Pyridostatin TFA exhibits antiproliferative and antiviral activities. Pyridostatin TFA can be used in studies related to breast cancer, cervical cancer, and Zika virus infection .
Isocryptotanshinone is a dual STAT3 and PTP1B (IC50 = 56.1 μM) inhibitor. Isocryptotanshinone inhibits STAT3 by binding to the STAT3 SH2 domain to block phosphorylation and nuclear translocation [1][2]. Isocryptotanshinone exerts its anti-proliferative effect via the induction of cell cycle arrest, apoptosis, and pro-death autophagy, through the regulation of STAT3, AKT/mTOR and MAPK signaling pathways. Isocryptotanshinone suppresses the xenograft gastric cancer (GC) tumor growth in BALB/c nude mice. Isocryptotanshinone can be used for cancer research, such as lung cancer, breast cancer and GC .
TMLB-C16 is a potent and orally active B3GAT3 inhibitor with a KD of 3.962 μM. TMLB-C16 suppresses proliferation and migration, and induces cell cycle arrest and apoptosis in MHCC-97H (IC50 = 6.53 μM) and HCCLM3 cells (IC50 = 6.22 μM). TMLB-C16 inhibits tumor growth in both MHCC-97H and HCCLM3 xenograft tumor mouse models without causing obvious toxicity. TMLB-C16 can be used for hepatocellular carcinoma research .
Tubulin polymerization-IN-62 (Compound 14b) is an inhibitor for microtubule polymerization (IC50 is 7.5 μM) and a degrader for α- and β-tubulin. Tubulin polymerization-IN-62 inhibits proliferation of cancer cells MCF-7, A549 and HCT-116, with IC50 of 32, 60 and 29 nM, respectively. Tubulin polymerization-IN-62 arrests the cell cycle at G2/M phase, inhibits the migration of MCF-7. Tubulin polymerization-IN-62 exhibits antitumor efficacy with a tumor growth inhibition rate (TGI) of 74.27% in 4T1 homograft mouse model .
USP1-IN-15 is an orally active and selective USP1 inhibitor with an IC50 of 12.3 nM. USP1-IN-15 has a high specificity for USP1 with negligible inhibition against all off-target DUBs. USP1-IN-15 suppresses colony formation, induces S-phase arrest, and stabilizes ubiquitinated PCNA. USP1-IN-15 also shows synergistic antiproliferative activity. USP1-IN-15 achieves significant tumor growth inhibition in vivo. USP1-IN-15 can be used for BRCA-mutated breast cancer .
MEK-IN-10 is an orally active pan-MEK/RAF non-degrading molecular glue with an IC50 of 782 nM against human MEK1. MEK-IN-10 blocks the phosphorylation of MEK and ERK, induces and stabilizes the MEK1-RAF complex, and disrupts the RAS-MAPK signaling pathway. MEK-IN-10 induces apoptosis in cancer cells and arrests cells at the G0/G1 phase. MEK-IN-10 induces tumor growth inhibition in mouse xenograft models. MEK-IN-10 can be used in the research of RAS-driven cancers, such as colorectal cancer and pancreatic cancer .
NQO1-responsive prodrug is a prodrug of Gemcitabine (dFdC) (HY-17026) with anti-cancer effect. NQO1-responsive prodrug remains stable in plasma and liver/intestinal S9 fractions, releasing dFdC in an NQO1-dependent manner. NQO1-responsive prodrug induces S-phase arrest and apoptosis. NQO1-responsive prodrug inhibits tumor growth in an A549 xenograft mouse model. NQO1-responsive prodrug can be used for breast and non-small cell lung cancer (NSCLC) research .
Damulin B is a dammarane-type saponin found in Gynostemma pentaphyllum. Damulin B can inhibit cancer cell apoptosis, decrease mitochondrial membrane potential, inhibit ROS production and cause G0/G1 phase arrest. Damulin B can prevent Cisplatin (HY-17394)-induced acute kidney injury and induce hair growth. Damulin B shows anti-inflammation anti-diabetic and anti-obesity effect. Damulin B can be used for the researches of cancer, inflammation, metabolic disease, such as lung cancer, osteoarthritis and diabetes .
SL-176 is a PPM1D (Wip1) inhibitor. SL-176 inhibits lipid droplet formation, downregulates the mRNA and protein expression of PPARγ and C/EBPα, and blocks adipocyte differentiation. SL-176 induces G2/M cell cycle arrest, apoptosis and inhibits cell proliferation in breast cancer cells overexpressing PPM1D, and activates components of the p53 pathway. SL-176 suppresses tumor growth in a zebrafish model of neuroblastoma. SL-176 is applicable to research related to obesity, breast cancer and neuroblastoma .
Zavondemstat (QC8222; TACH 101) sodium is an orally active pan-KDM4 inhibitor, with a IC50 ≤ 0.08 μM against human KDM4A-D and a Kᵢ of 0.52 μM against human KDM4C. Zavondemstat sodium induces cell apoptosis, causes S-phase cell cycle arrest, reduces the population of tumor-initiating cells and inhibits cancer cell proliferation. Zavondemstat sodium suppresses tumor growth and induces tumor regression in mouse xenograft models. Zavondemstat sodium can be used for the research of various cancers including colorectal cancer, esophageal squamous cell carcinoma and triple-negative breast cancer .
Top/HDAC-IN-3 (Compound 31) is an orally active dual inhibitor of Topoisomerase and HDAC. Top/HDAC-IN-3 increases reactive oxygen species (ROS) levels, leading to DNA damage, thereby inhibiting cancer cell colony formation and migration, inducing cancer cell Apoptosis, and causing cell cycle arrest. In the NSCLC model, Top/HDAC-IN-3 exhibited significant antitumor effects, with a tumor growth inhibition (TGI) of 77.5% at 100 mg/kg, surpassing the efficacy of the HDAC inhibitor SAHA (HY-10221) and the combination of SAHA (HY-10221) with the topoisomerase inhibitor Irinotecan (HY-16562) .
YN11 is a STAT3 inhibitor (Kd=11.9 μM). YN11 directly binds to the SH2 domain of STAT3, inhibits the phosphorylation of STAT3, and reduces the expression of downstream target proteins. YN11 induces cell cycle arrest, promotes apoptosis, and inhibits cell invasion and migration in prostate cancer cells. YN11 suppresses tumor growth in a prostate cancer xenograft mouse model. YN11 does not cause significant body weight loss or obvious histopathological changes in major organs in xenograft mice. YN11 is applicable to relevant research on prostate cancer .
NCT02 is a molecular glue degrader based on the E3 ubiquitin ligase DDB1 that targets CDK12 and its binding partner CCNK. NCT02 triggers the ubiquitination and proteasomal degradation of CCNK, thereby downregulating CDK12 protein levels and inhibiting its downstream signaling pathways. NCT02 can induce tumor cell apoptosis, arrest the cell cycle, and selectively inhibit the proliferation of colorectal cancer cells carrying TP53 defects or belonging to the consensus molecular subtype CMS4. NCT02 has the potential to inhibit tumor growth in in vitro and in vivo models .
Cinosulfuron (Standard) is the analytical standard of Cinosulfuron. This product is intended for research and analytical applications. Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
NGI‑189 is a selective OST‑A inhibitor. NGI‑189 inhibits the STT3A catalytic subunit of the OST complex and reduces N‑glycosylation of target glycoproteins. NGI‑189 blocks oncogenic and bypass signaling, reduces phosphorylation of EGFR, AKT, p70S6K and S6RP, and induces cell cycle arrest and apoptosis. NGI‑189 markedly suppresses tumor growth and induces tumor regression in non‑small cell lung cancer (NSCLC) xenograft models. NGI‑189 can be used for the research of EGFR‑mutant non‑small cell lung cancer .
VEGFR-2/AKT-IN-2 (Compound 5) is a VEGFR-2/AKT inhibitor (IC50: 0.061 μM for VEGFRin HepG2 cell). VEGFR-2/AKT-IN-2 reduces total and phosphorylated AKT as well as up-regulates BAX and Caspase-3 and down-regulates Bcl-2 in cells, thereby promoting Apoptosis. VEGFR-2/AKT-IN-2 causes cell cycle arrest in S phase. VEGFR-2/AKT-IN-2 inhibits the growth of human liver tumor cells .
NL13 is a Polo-like kinase 4 (PLK4) inhibitor with an IC50 value of 2.32 μM. NL13 can inhibit the viability of PC3 and DU145 prostate cancer cells, with IC50 values of 3.51 μM and 2.53 μM, respectively. NL13 can lead to the inactivation of the AKT signaling pathway by downregulating CCNB1/CDK1, inducing G2/M cell cycle arrest, and triggering apoptosis through the cleavage of caspase-9/caspase-3. In prostate cancer mice, NL13 can inhibit tumor growth .
Zavondemstat (QC8222 free base; TACH 101 free base) is an orally active pan-KDM4 inhibitor, with a IC50 ≤ 0.08 μM against human KDM4A-D and a Kᵢ of 0.52 μM against human KDM4C. Zavondemstat induces cell apoptosis, causes S-phase cell cycle arrest, reduces the population of tumor-initiating cells and inhibits cancer cell proliferation. Zavondemstat suppresses tumor growth and induces tumor regression in mouse xenograft models. Zavondemstat can be used for the research of various cancers including colorectal cancer, esophageal squamous cell carcinoma and triple-negative breast cancer .
PROTAC c-Met Degrader-4 (compound D15) is a potent orally active PROTAC c-MET degrader. PROTAC c-Met Degrader-4 demonstrates excellent intracellular degradation potency with a DC50 < 0.5 nM. PROTAC c-Met Degrader-4 induces cell cycle arrest and apoptosis, inhibits cell invasion and migration, thereby suppressing cell proliferation. PROTAC c-Met Degrader-4 inhibits the growth of Hs746T xenograft tumors in nude mice. PROTAC c-Met Degrader-4 can be used for cancer research, such as non-small cell lung cancer and gastric cancer .
Periplogenin (Standard) is the analytical standard of Periplogenin (HY-N2414). Periplogenin is an orally active cardiac glycoside found in Cortex periplocae. Periplogenin can induce ROS production and necroptosis and cause G0/G1 phase arrest. Periplogenin can inhibit pyroptosis by regulating the NLRP3/Caspase-1/GSDMD signaling. Periplogenin suppresses growth of prostate carcinoma cells by docking to an ATP1A1 protein pocket and forming a hydrogen bond with T804. Periplogenin can be used for the researches of cancer, inflammation and immunology, such as prostate carcinoma, rheumatoid arthritis and psoriasis .
Microtubule destabilizing agent-2 (Compound 21) is an orally active and selective antitumor compound targeting microtubule protein. Microtubule destabilizing agent-2 destabilizes microtubule proteins and inhibits microtubule polymers. Microtubule destabilizing agent-2 arrests the G0/G1 phase in human tumor cells. Microtubule destabilizing agent-2 induces Apoptosis by activating the cascade pathway of caspases. Microtubule destabilizing agent-2 has anti-inflammatory activity, as inhibiting TNF-α and IL-6 in vitro. Microtubule destabilizing agent-2 reduces tumors growth in xenograft mice .
YCH3292, a derivative of YCH189 (HY-155993) is a potent, selective and orally active PARP1/2 inhibitor with IC50 both <0.001 nM. YCH3292 can increase the stability of PARP-DNA complexes. YCH3292 exhibits robust antiproliferative activity. YCH3292 can induce double-strand breaks in DNA, increase the protein levels of γH2AX, P-RPA32, and P-Chk1 and induce tumor cells S or G2/M phase arrest and apoptosis. YCH3292 can inhibit tumor growth in MC38 xenograft model .
Anticancer agent 311 is an apoptosis inducer and p53 modulator. Anticancer agent 311 increases p53 levels, activates cleaved caspase-3, reduces p-Cdc25C levels, and disrupts p-p44/42 MAPK phosphorylation. Anticancer agent 311 induces G2/M phase arrest, inhibits cancer cell viability in a concentration- and time-dependent manner, and exhibits low toxicity to non-cancer cells. Anticancer agent 311 prevents tumor growth and angiogenesis in mouse xenograft models without detectable toxicity. Anticancer agent 311 can be used for the research of lung cancer .
KRC-00715 is an effective oral c-Met inhibitor with an IC50 of 9.0 nM, demonstrating high selectivity in gastric cancer cells. KRC-00715 specifically inhibits the growth of c-Met-highly expressed cell lines by inducing G1/S phase arrest, leading to a reduction in downstream signaling pathways, including Akt and Erk, as well as c-Met activity. KRC-00715, in the gastric cancer cell line Hs746, is characterized by an IC50 of 39 nM, and it selectively inhibits the proliferation of c-Met-highly expressed cell lines. KRC-00715 reduces tumor size in Hs746T xenograft mouse models .
QW-5-70 is a potent colchicine‑site tubulin inhibitor that blocks tubulin polymerization. QW-5-70 induces mitotic and G2/M cell cycle arrest, triggers mitochondrial apoptosis, and suppresses cancer cell colony formation and migration. QW-5-70 overcomes P‑glycoprotein‑mediated multidrug resistance and inhibits drug‑resistant tumor growth. QW-5-70 demonstrates strong in vitro and in vivo antitumor efficacy in neuroblastoma and prostate cancer models. QW-5-70 can be used for the research of high-risk neuroblastoma and castration-resistant prostate cancer .
T7 Peptide is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide induces G0/G1 cell cycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
SF-9-2 is a PD-L1/PD-1 binding inhibitor (IC50 = 24.9 nM). SF-9-2 inhibits epithelial-mesenchymal transition, migration, invasion, and proliferation of SK-N-SH cells, and also induces apoptosis and cell cycle arrest. SF-9-2 blocks PD-L1-induced SK-N-SH cell growth through the MAPK signaling pathway. SF-9-2 restores GSK-3β activity and enhances PD-L1 degradation through the ubiquitin-proteasome pathway. SF-9-2 inhibits tumor growth in the SK-N-SH NOG mouse model without significant toxicity. SF-9-2 also acts as an immune checkpoint inhibitor, blocking PD-L1 to restore T cell function. SF-9-2 can be used in neuroblastoma research .
Molibresib besylate (GSK 525762C; I-BET 762 besylate) is an orally active pan-BET inhibitor that targets and binds to BRD2, BRD3, BRD4 and BRDT. By competitively occupying acetylated lysine binding sites, Molibresib besylate disrupts the interaction between BET proteins and chromatin, thereby effectively inhibiting MYC expression and target gene transcription. Molibresib besylate exhibits broad antiproliferative activity, which not only inhibits cancer cell growth and induces growtharrest, but also downregulates mitosis-related genes and upregulates the level of p-ERK1/2. When combined with MEK inhibitors, Molibresib besylate shows a significant synergistic effect, reduces tumor burden in mouse models of leukemia, modulates the immune microenvironment and prolongs survival. Molibresib besylate is widely applicable to research related to acute myeloid leukemia, multiple myeloma, triple-negative breast cancer, small-cell lung cancer and various advanced refractory solid tumors .
YZ-836P is a Protein arginine methyltransferase 5 (PRMT5) targeting agent. YZ-836P promotes ubiquitination and proteasomal degradation of PRMT5 in a cereblon (CRBN)-dependent manner, which in turn reduces levels of its downstream target KLF5. YZ-836P induces G1 phase cell cycle arrest in triple-negative breast cancer cells. YZ-836P induces Apoptosis in triple-negative breast cancer cells. YZ-836P exerts cytotoxic effects on triple-negative breast cancer cells. YZ-836P inhibits the growth of triple-negative breast cancer patient-derived organoids. YZ-836P inhibits the growth of triple-negative breast cancer xenografts in nude mice. YZ-836P can be used for the research of triple-negative breast cancer .
HSB401 is an orally active FLT3 inhibitor (IC50: 28, 5, 72, 51 nM for FLT3-WT, FLT3-D835Y, FLT3-ITD-F691L, FLT3-ITD, respectively). HSB401 downregulates FLT3 signaling and induces cell cycle arrest and apoptosis. HSB401 spares c-KIT inhibition, thereby reducing the risk of myelosuppression. HSB401 significantly suppresses tumor growth in the MV4-11 xenograft mouse model. HSB401 can be used for the research of acute myeloid leukemia .
T7 Peptide TFA is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide TFA inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide TFA induces G0/G1 cell cycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide TFA is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
PROTAC BET Degrader-17 is a potent BET protein PROTAC degrader. By recruiting the VHLE3 ligase, PROTAC BET Degrader-17 specifically degrades BRD2, BRD3 (DC50=0.09 nM) and BRD4 (IC50=4.3 nM). PROTAC BET Degrader-17 exhibits strong anti-tumor activity in acute myeloid leukemia (AML) studies; it not only inhibits cancer cell proliferation, induces cell cycle arrest and apoptosis, but also effectively suppresses tumor growth in xenograft mouse models. PROTAC BET Degrader-17 can be used to explore targeted therapies for acute myeloid leukemia .
WK500B is a potent and orally active BCL6 inhibitor with a KD of 1.61 μM. WK500B engages intracellular BCL6 and disrupts BCL6‑corepressor interactions to reactivate BCL6 target genes. WK500B exerts cytotoxicity against diffuse large B‑cell lymphoma cells and induces apoptosis and cell cycle arrest. WK500B suppresses germinal center formation in C57BL/6 mice and DLBCL tumor growth in SCID xenograft models without observable toxicity. WK500B can be used for the study of diffuse large B‑cell lymphoma (DLBCL) .
AZ1366 is an orally active tankyrase inhibitor. AZ1366 stabilizes Axin2, reduces NuMA levels, disrupts the interaction between tankyrase and NuMA, induces G2/M phase arrest, inhibits the Wnt pathway, and downregulates the expression of β-catenin-dependent genes. AZ1366 inhibits tumor growth in colorectal cancer xenograft models. AZ1366 synergistically inhibits the proliferation of non-small cell lung cancer cells, improves tumor control and significantly prolongs survival in orthotopic non-small cell lung cancer mouse models. AZ1366 is applicable to research related to colorectal cancer and non-small cell lung cancer .
IKKβ-IN-7 is an IKKβ inhibitor with an IC50 value of 9.44 μM. IKKβ-IN-7 induces DNA damage, S-phase cell cycle arrest, ROS accumulation, mitochondrial membrane potential loss, and apoptosis. IKKβ-IN-7 inhibits phosphorylation of p65 and IκBα, suppresses p65 nuclear translocation, and regulates NF-κB-controlled genes. IKKβ-IN-7 suppresses tumor growth in xenograft models and shows activity against colorectal cancer with low normal cell cytotoxicity. IKKβ-IN-7 can be used for the research of colorectal cancer .
BRD4-IN-41 is a BRD4 inhibitor with an IC50 of 34 nM. BRD4-IN-41 also inhibits JAK2, FLT3, RET, ROS1, NTRK3, PDGFRb, and FGFR1 kinases with IC50 values ranging from 0.9 nM to 43 nM. BRD4-IN-41 inhibits acetyl-lysine binding site of BRD4, downregulates c-MYC, reduces phosphorylated STAT3 levels, induces G1 cell cycle arrest and apoptosis, thereby inhibiting cancer cells growth. BRD4-IN-41 can be used for the research of cancer, such as multiple myeloma and acute myeloid leukemia .
Zavondemstat (QC8222; TACH 101) L-lysine is an orally active pan-KDM4 inhibitor, with a IC50 ≤ 0.08 μM against human KDM4A-D and a Kᵢ of 0.52 μM against human KDM4C. Zavondemstat L-lysine induces cell apoptosis, causes S-phase cell cycle arrest, reduces the population of tumor-initiating cells and inhibits cancer cell proliferation. Zavondemstat L-lysine suppresses tumor growth and induces tumor regression in mouse xenograft models. Zavondemstat L-lysine can be used for the research of various cancers including colorectal cancer, esophageal squamous cell carcinoma and triple-negative breast cancer .
WB-308 is a novel small molecule that was identified as an inhibitor of EGFR by an in vitro EGFR kinase activity system. WB-308 was able to reduce the proliferation and clonogenicity of NSCLC cells, causing G2/M phase arrest and apoptosis. In addition, WB-308 inhibited tumor growth in two in vivo animal models (lung orthotopic transplantation model and patient-derived clonal mouse model). WB-308 impaired the phosphorylation of EGFR, AKT, and ERK1/2 proteins. Compared with Gefitinib, WB-308 had lower cytotoxicity. This study showed that WB-308 is a new EGFR-TKI that may be considered as an alternative to Gefitinib in the clinical treatment of NSCLC.
C6 Ceramide (C6-Cer) is a short-chain, cell-permeable ceramide pathway activator with anticancer activity. C6 Ceramide-mediated miR-29b expression participates in the progression of multiple myeloma through suppressing the proliferation, migration and angiogenesis of endothelial cells by targeting Akt signal pathway. C6 Ceramide exhibits multiple anti-cancer properties including cell cycle arrest, Apoptosis, inhibition of tumor growth and enhances the effects of chemotherapy in drug-resistant cancer cells. C6-ceramide can be used as an adjuvant for chemotherapeutic agents, to enhance anti-tumor effects .
Kevetrin (3-Cyanopropyl carbamimidothioate; 4-Isothioureidobutyronitrile) is an apoptosis inducer that exhibits p53-dependent and p53-independent antitumor activity. In TP53 wild-type models, Kevetrin activates and stabilizes the p53 protein by altering the processing of MDM2, thereby inducing cell cycle arrest and apoptosis. Kevetrin shows higher sensitivity in mutant models. Kevetrin is applicable for the research of various cancers including acute myeloid leukemia and breast cancer .
EGFR/CA-IX-IN-1 (Compound 14) is a dual inhibitor against epidermal growth factor receptor (EGFR) and carbonic anhydrase IX (CA-IX) with IC50 values of 5.92 nM and 63 nM, respectively. EGFR/CA-IX-IN-1 shows strong cytotoxicity against breast cancer cells (MDA-MB-231, MCF-7) with IC50 values of 5.78 μM and 8.05 μM, respectively. EGFR/CA-IX-IN-1 inhibits the catalytic activity of CA-IX, up-regulates BAX/Bcl-2, activates caspases, and arrests the cell cycle at the G1 phase. EGFR/CA-IX-IN-1 is promising for research of breast cancer .
13C20, 15N10-Cyclic di-GMP ( 13C20, 15N10-c-di-GMP) is 13C and 15N labeled Cyclic-di-GMP (disodium). Cyclic-di-GMP disodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP disodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP disodium can be used in cancer research .
Aurora A-IN-5 is a potent and highly selective Aurora A inhibitor (IC50 = 0.02 μM), showing 362-fold selectivity for over Aurora B. Aurora A-IN-5 shows its selectivity through unique C−H/π interactions, enhanced hydrophobic contacts, an open binding pocket, and tighter protein packing. Aurora A-IN-5 suppresses Aurora A autophosphorylation, thereby inhibiting cancer cell proliferation by inducing G2/M phase arrest, triggering apoptosis, and suppressing colony formation. Aurora A-IN-5 inhibits tumor growth in MDA-MB-231 xenograft mouse models. Aurora A-IN-5 can be used for breast, cervical, prostate, and lymphoma cancer research .
Aurora kinase/HDAC-IN-1 is an orally active dual Aurora kinase and HDAC inhibitor that inhibits Aurora A (IC50 = 116 nM), Aurora B (IC50 = 225 nM), HDAC1 (IC50 = 164 nM), and HDAC2 (IC50 = 346 nM).Aurora kinase/HDAC-IN-1 promotes histone H3 acetylation, inhibits Aurora A phosphorylation and downstream signaling, and induces apoptosis via G2/M cell-cycle arrest. Aurora kinase/HDAC-IN-1 exhibits potent antiproliferative activity in colorectal cancer cells, with an IC50 value of 30.2 nM in HCT-116 cells.Aurora kinase/HDAC-IN-1 significantly suppresses tumor growth in an HCT-116 colorectal cancer xenograft mouse model .
Antitumor agent-200 (Compound 2g) is a microtubule synthesis inhibitor. By binding to the colchicine site of tubulin, it causes G2/M cell cycle arrest and generates reactive oxygen species (ROS). Antitumor agent-200 exhibits significant inhibitory activity against MCF7/ADR and KBV200 cell lines with overexpression of P-glycoprotein (P-gp), with drug resistance indices (DRI) of 0.83 and 0.58 respectively. In the MCF-7 xenograft model, Antitumor agent-200 (25 mg/kg, intraperitoneal injection) can achieve a tumor growth inhibition rate of 57.2%. Antitumor agent-200 can be used in the research of the anti-cancer field .
PAWI-2 is a p53-Activator and Wnt Inhibitor. PAWI-2 inhibits β3-KRAS signaling independent of KRAS. PAWI-2 selectively inhibits phosphorylation of TBK1. PAWI-2 activates apoptosis (activation of caspase-3/7), and induces PARP cleavage. PAWI-2 promotes optineurin translocation into the nucleus and causes G2/M arrest. PAWI-2 reverses cancer stemness and overcomes drug resistance in an integrin β3 KRAS-dependent human pancreatic cancer stem cells (hPCSCs). PAWI-2 inhibits growth of tumors from hPCSCs in orthopic xenograft mice model .
R9-caPep (RRRRRRRRRCCLGIPEQEY) is a cell-penetrating peptide derived from proliferating cell nuclear antigen (PCNA). R9-caPep selectively blocks the interactions between PCNA and FEN1, as well as between PCNA and LIGI, while preserving the binding of POLD3 to PCNA. R9-caPep interferes with DNA synthesis and homologous recombination-mediated double-strand DNA break repair, inducing S-phase arrest, DNA damage accumulation, and apoptosis. R9-caPep inhibits the growth of tumor volume and weight of neuroblastoma in nude mice . R9-caPep can be used in research related to neuroblastoma and triple-negative breast cancer .
Tubulin polymerization-IN-92, an analog of KX-01 (HY-10340), is a potent orally active tubulinpolymerization inhibitor that binds tubulin with a Ka of 1.29 μM. Tubulin polymerization-IN-92 simultaneously occupies the colchicine site in β-tubulin and a cavity in α-tubulin. Tubulin polymerization-IN-92 exerts antiproliferative activity, induces G2/M cell cycle arrest and apoptosis in cancer cells. Tubulin polymerization-IN-92 inhibits tumor growth in mouse xenograft models. Tubulin polymerization-IN-92 can be used for the research of colon cancer, cervical cancer, and Paclitaxel (HY-B0015)-resistant ovarian cancer .
S9-CMC1 TFA is a covalent peptide lysine-specific demethylase 1 (LSD1) inhibitor with an IC50 value of 2.53 μM. S9-CMC1 TFA specifically recognizes Cys360 in the enzyme-active region. S9-CMC1 TFA inhibits LSD1 activity, increasing H3K4me1 and H3K4me2 levels, leading to G1 cell cycle arrest and apoptosis and inhibiting cell proliferation. S9-CMC1 TFA significantly inhibits tumor growth in A549 xenograft animal models .
XAN-5 is a mitochondrial DNA G-quadruplex (mtG4) ligand with a Kd of 3.8 μM. XAN-5 selectively binds and stabilizes mtG4 structures, disrupting mitochondrial gene transcription and DNA replication. XAN-5 triggers mitochondrial dysfunction, ROS overproduction, G0 phase arrest and caspase-dependent apoptosis. XAN-5 inhibits autophagy and induces immunogenic cell death. XAN-5 inhibits tumor growth in a mouse liver cancer model while enhancing tumor-infiltrating CD4 + and CD8 + T cells. XAN-5 targets two cancer resistance mechanisms simultaneously. XAN-5 can be used for the research of liver cancer .
PROTAC c-Met degrader-1 is a selective and orally active c-Met PROTAC degrader with a DC50 of 6.21 nM against c-Met. PROTAC c-Met degrader-1 induces CRBN-dependent ubiquitination and proteasomal degradation of c-Met. PROTAC c-Met degrader-1 induces G0/G1 phase arrest in c-Met-dependent cancer cells. PROTAC c-Met degrader-1 kills c-Met-dependent cancer cells. PROTAC c-Met degrader-1 inhibits tumor growth in animal models. PROTAC c-Met degrader-1 can be used for the research of gastric cancer .
c-Fos-IN-1 (Compound P16) is a c-Jun inhibitor, and decreases mRNA levels and protein levels of c-Fos. c-Fos-IN-1 also inhibits the phosphorylation activity of ERK and the transcriptional activity of AP-1. c-Fos-IN-1 shows anticancer activity by inhibiting ERK/c-Fos/Jun pathway. c-Fos-IN-1 inhibits the proliferation and migration of gastric cancer cells (IC50: 2.31 μM for MGC-803 cell). c-Fos-IN-1 arrests cell cycle at G2/M phase and induces cancer cell apoptosis. c-Fos-IN-1 inhibits gastric cancer tumor growth .
Sotetsuflavone is a flavonoid that can be isolated from Cycas revolute. Sotetsuflavone inhibits phosphorylation of PI3K, Akt, mTOR, JNK, and p38 MAPK; modulates expression of Cyclin D1, CDK4, Bcl-2, Bax, cleaved caspases 3/9, MMP-9, TGF-β, STAT3, and β-catenin. Sotetsuflavone induces G0/G1 cell cycle arrest, apoptosis, autophagy, and intracellular ROS elevation, inhibits cancer cell proliferation. Sotetsuflavone inhibits tumor growth in mouse tumor xenograft models. Sotetsuflavone can be used for the research of non-small cell lung cancer and Crohn’s disease .
SKLB06489 is a selective and orally active inhibitor of type I PRMT enzymes, with IC50 values of 64.55 nM (PRMT1), 4.21 nM (PRMT6), and 51.27 nM (PRMT8). SKLB06489 inhibits cell proliferation, colony formation, DNA replication, and DNA damage repair in cancer cells. SKLB06489 induces G0/G1-phase cell cycle arrest and apoptosis in cancer cells. SKLB06489 enhances intracellular cholesterol efflux via ABCA1 and ABCG1 upregulation, disrupts cholesterol metabolic homeostasis, and suppresses tumor growth in subcutaneous xenograft models. SKLB06489 can be used for the research of triple-negative breast cancer (TNBC) .
VEGFR-2-IN-67 (Compound 6b) is an inhibitor of vascular endothelial growth factor receptor 2 (VEGFR-2). Its IC50 values for MDA-231 and MCF-7 cell lines are 5.91 µM and 7.16 µM respectively, and its inhibitory effect on VEGFR-2 is comparable to that of Sorafenib (HY-10201) (IC50 is 53.63 nM). VEGFR-2-IN-67 exerts significant anti-cancer activity through mechanisms such as inducing Apoptosis (the early apoptosis rate reaches 57.20%), arresting the cell cycle at the G1 phase, upregulating pro-apoptotic markers and downregulating Bcl-2. VEGFR-2-IN-67 can be used for research in the field of cancer .
PLAGL2-IN-1 is a inhibitor of pleiomorphic adenoma-like protein 2 (PLAGL2) with a Kd of 2.23 µM. PLAGL2-IN-1 suppresses PLAGL2 transcriptional activity, induces G0/G1 cell cycle arrest, and apoptosis, thereby inhibiting hepatocellular carcinoma (HCC) cell proliferation. PLAGL2-IN-1 disrupts extracellular matrix organization and suppresses the PI3K-AKT pathway by reducing AKT phosphorylation. PLAGL2-IN-1 inhibits tumor growth in an HCCLM3 xenograft mouse model. PLAGL2-IN-1 can be used for the research of HCC .
FD274 is a highly potent PI3K/mTOR dual inhibitor with IC50s of 0.65 nM, 1.57 nM, 0.65 nM, 0.42 nM, and 2.03 nM against PI3Kα/β/γ/δ and mTOR, respectively. FD274 exhibits significant anti-proliferation of AML cell lines (HL-60 and MOLM-16). FD274 arrests HL-60 cell cycle at G1 phase and increases apoptosis. FD274 demonstrates dose-dependent inhibition of tumor growth in the HL-60 xenograft model. FD274 has the potential for acute myeloid leukemia research .
BYBC‑1 is a selective G4‑RNA‑targeting ligand with high affinity forKRAS and NRAS G4‑RNAs (Kd = 0.05-0.28 μM). BYBC‑1 stabilizes G4‑RNA structures in KRAS and NRAS mRNA, blocks thePI3K/AKT and MAPK/ERK pathways, activates the DNA damage response (DDR), suppresses energy metabolism, and induces S‑phase arrest and apoptosis. BYBC‑1 exhibits high selectivity over non‑malignant fibroblasts and significantly inhibits the growth of HCT‑116 xenograft tumors in vivo. BYBC‑1 can be used for the study of colorectal cancer .
Santamarine (Santamarin; Balchanin) is a sesquiterpene lactone found in Artemisia scoparia. Santamarine shows anti-inflammatory, antioxidant, anticancer and anti-photoaging activities. Santamarine suppresses UVA-induced phosphorylation of JNK and p38 MAPK, nuclear translocation of phosphorylated c-Fos and c-Jun, and AP-1-mediated MMP-1 transcription and secretion. Santamarine suppresses NF-κB signaling, iNOS, COX-2, TNF-α, and IL-1β production. Santamarine inhibits thioredoxin reductase activity, induces ROS production, mitochondrial apoptosis, G2/M cell cycle arrest, and DNA damage, and reduces cancer cell growth. Santamarine can be used for the photoaging, inflammatory diseases and cancer .
BBO-10203 is a potent inhibitor of PI3Kα and KRAS G12C, selectively and covalently binding to Cys242 in the RAS-Binding Domain of PI3Kα, and inhibiting both the GTP-bound and GDP-bound states of KRAS G12C with an IC50 of 0.031 nM and an EC50 of 0.02 nM. BBO-10203 disrupts the interaction between RAS isoforms and PI3Kα, leading to the inhibition of RAS-mediated PI3Kα activation, and reduces pERK expression, cell growth, and induces G1arrest and apoptosis. BBO-10203 can be used for the research of breast cancer, colorectal cancer, and non-small cell lung cancer .
BWA-6047 is an oral active PROTAC degrader targeting AR/AR-V7 and GSPT1 with DC50 values of 3.7, 3.0 and 1.2 nM in 22Rv1 cells. BWA-6047 suppresses the expression of AR downstream target genes and and transcriptional activity. BWA-6047 inhibits cancer cells proliferation, causes G1 phase cell cycle arrest and induces apoptosis. BWA-6047 increases cleaved-PARP-1 and cleaved-caspase-3 levels. BWA-6047 reduces growth of LNCaP xenograft tumors in mice models without obvious toxicity. BWA-6047 can be used for the research of prostate cancer .
CDK2-IN-42 (Compound H63) is a CDK12 inhibitor with an IC50 value of 10 nM. CDK2-IN-42 has anti-ESCC (Esophageal Squamous Cell Carcinoma) cell activity. It can block transcriptional elongation, downregulate the core genes in the G1 phase to induce cell cycle arrest, and alter the CDK12-ATM/ATR-CHEK1/CHEK2 signaling axis, resulting in DNA damage. CDK2-IN-42 can effectively inhibit tumor growth in a xenograft mouse model of human ESCC KYSE150. CDK2-IN-42 holds great promise for research in the field of cancer .
BY13 is a SRC-3PROTAC degrader with a DC50 of 0.031 μM. BY13 selectively blocks the ER signaling pathway over that of androgen receptor (AR)) through down-regulating ERα level. BY13 potently overcomes endocrine resistance in breast cancer by inducing cell cycle arrest in G1 phase and apoptosis, with superior effect over Fulvestrant (HY-13636). BY13 significantly inhibits the growth of drug-resistant breast tumors without obvious toxicity in LCC2 xenograft mice model . Pink: SRC-3 ligand (SI-2) (HY-101447); Blue: CRBN ligase ligand (HY-41547); Black: linker (HY-176226)
NSC666715 is a DNA polymerase β (Pol-β) inhibitor. NSC666715 directly and specifically interacts with Pol-β, interferes with its binding to damaged DNA, blocks its dRP lyase activity, and inhibits Pol-β-mediated SN- and LP-BER. NSC666715 induces AP site accumulation and S-phase cell cycle arrest, and triggers senescence and apoptosis (apoptosis) via the p53/p21 pathway in colorectal cancer cells. NSC666715 enhances TMZ (HY-17364)-induced DNA damage, senescence and apoptosis, and potentiates the cytotoxicity of TMZ. NSC666715 inhibits tumor growth in colon cancer xenograft models. NSC666715 can be used in research related to colorectal cancer .
Hsp90-IN-42 (Compound 13l) is a potent heat shock protein 90 (Hsp90) inhibitor (IC50=15.65 nM). Hsp90-IN-42 reduces the stability of the epidermal growth factor receptor (EGFR), suppressing the activation of the EGFR-Akt signaling pathway, inducing G0/G1 phase arrest in colorectal cancer cells (such as HT-29 cells), and slightly triggering apoptosis. Hsp90-IN-42 also inhibits cell proliferation and migration by down-regulating the expression of CDK12, CDK13, and Bcl-2 proteins, and up-regulating the expression of Bax protein. Hsp90-IN-42 is promising for research of colorectal cancer .
MMP-9-IN-14 is a MMP-9 inhibitor (IC50 = 34.46 μM). MMP-9-IN-14 induces G1-phase cell cycle arrest and caspase-dependent apoptosis in cancer cells. MMP-9-IN-14 promotes the accumulation of phosphorylated γH2AX. MMP-9-IN-14 inhibits the migration and invasion of cancer cells, and downregulates the expressions of MMP-2, MMP-9 and hTERT in cancer cells. MMP-9-IN-14 inhibits tumor growth and angiogenic spread in animal models. MMP-9-IN-14 can be used for the research of cancers such as lung adenocarcinoma, cervical cancer and colorectal cancer .
FOXM1-IN-3 is a potent FOXM1 inhibitor. FOXM1-IN-3 downregulates FOXM1 expression at protein and mRNA levels, suppressing downstream effectors CCNB1 and CDC25. FOXM1-IN-3 induces G2/M cell cycle arrest and apoptosis in colorectal cancer cells. FOXM1-IN-3 inhibits colony formation and cell migration in colorectal cancer cells. FOXM1-IN-3 targets the cancer stem cell phenotype in colorectal cancer cells, reducing cancer stem cell marker expression. FOXM1-IN-3 reduces tumor growth in a zebrafish xenograft model. FOXM1-IN-3 can be used for the research of colorectal cancer .
BI8622 is a specific inhibitor of the ubiquitin ligase HUWE1 with an IC50 of 3.1 μM. BI8622 can decrease the protein expression levels of c-myc and glycolytic markers as well as immune modulatory markers after HUWE1 inhibition in triple-negative breast cancer (TNBC) cell lines. BI8622 significantly protects against cisplatin (HY-17394)-induced acute kidney injury (AKI). BI8622 significantly reduces the growth of multiple myeloma (MM) cell lines and induces cell cycle arrest. BI8622 can prevent HUWE1-dependent TTBK2 ubiquitination. BI8622 can be studied in research for various diseases including medulloblastoma, acute kidney injury, breast cancer and MM .
Murrayafoline A is a carbazole alkaloid that can be extracted from Murraya tetramera. Murrayafoline A directly targets Specificity protein 1 (Sp1), thereby inhibiting NF-κB and MAPK signaling pathways. Murrayafoline a induces a G0/G1-phase arrest in platelet-derived growth factor (PDGF)-stimulated vascular smooth muscle cells. Murrayafoline A attenuates the Wnt/β-catenin pathway by promoting the degradation of intracellular β-catenin proteins. Murrayafoline A enhances the contraction of rat ventricular myocytes and L-type calcium current by activating protein kinase C. Murrayafoline A inhibits LPS (HY-D1056)-induced neuroinflammation in vivo. Murrayafoline A can be used for the study of inflammation, vascular complications and colon cancer .
A-800141 is an orally active, selective, sulfonamide-based MetAP2 inhibitor (IC50=12 nM) that binds reversibly to MetAP2 and interacts with its manganese ions. A-800141 induces the production of N-terminal methionine-unprocessed GAPDH variants, which in turn triggers G1-phase cell cycle arrest, elevates p21 levels, and reduces the levels of phosphorylated Rb and total cyclin A. A-800141 exhibits anti-angiogenic and tumor growth inhibitory effects, and produces synergistic effects when combined with cytotoxic inhibitors or BCL-2 inhibitors. A-800141 has been widely used in scientific research related to B-cell lymphoma, neuroblastoma, prostate cancer, colon cancer, melanoma and other fields .
COX/5-LO-IN-2 is a COX2, EGFR, COX1, 5-LOX, BRAF and FAK inhibitor with IC50 of 1.22 μM, 2.5 μM, 2.95 μM, 4.65 μM, 7.4 μM, 12.2 μM, respectively. COX/5-LO-IN-2 induces cell growtharrest at G2/M phase. COX/5-LO-IN-2 triggers apoptotic activity by up-regulating proapoptotic proteins p53, Bax, and caspase-7 and down-regulating anti-apoptotic protein Bcl-2. COX/5-LO-IN-2 can be used for the research of breast cancer .
CIRc-014 is an orally active Cyclin A/B inhibitor with a Cyclin A IC50 of 0.05 μM, Cyclin A Kd of 2.7 nM, Cyclin B IC50 of less than 0.02 μM and Cyclin B Kd of 1.0 nM. CIRc-014 activates the spindle assembly checkpoint and promotes the formation of a complex between Cyclin B and CDK2 by blocking the RxL interaction of Cyclin A/B. CIRc-014 can induce replication stress, DNA damage, mitotic arrest and apoptosis in tumor cells. CIRc-014 showed tumor growth inhibition and regression in NCI-H69 and NCI-H446 small cell lung cancer xenograft models. CIRc-014 can be used for the research of small-cell lung cancer .
DPMI-ω is a dual-specificity d-peptide antagonist of oncogenic proteins MDM2 and MDMX. DPMI-ω, upon fabrication on gold nanoparticles, efficiently traverses tumor cells and kills them by reactivating the p53 signaling pathway. DPMI-ω can disrupte the p53-MDM2/MDMX complex. DPMI-ω can inhibit B16 melanoma growth and induce cells G0/G1 phase arrest. DPMI-ω can augment the efficacy of immunotherapy by expanding CD3 +/CD8 + cytotoxic T cells and suppressing CD4 +/CD25 + regulatory T cells companied with anti-PD1 antibody. DPMI-ω can be used for research of melanoma .
LIN28-IN-2 is a Lin28 inhibitor with activity against Lin28a, Lin28b, and their zinc knuckle domain. LIN28-IN-2 blocks Lin28-RNA substrate binding, perturbs zinc knuckle domain conformation. LIN28-IN-2 inhibits cancer cell proliferation, spheroid growth, and induces G2/M phase arrest. LIN28-IN-2 suppresses cancer stem cell phenotypes, Lin28-mediated stress granule formation, let-7 target genes, cancer stem cell biomarkers, and neuroendocrine biomarkers expression in cancer cells. LIN28-IN-2 can be used for the research of cancer .
JND4135 is a Type II TRK inhibitor with IC50 values of 2.79, 3.19, and 3.01 nM against TRKA, TRKB, TRKC, respectively. JND4135 can overcome resistance from TRK xDFG and other mutant forms in the BaF3 stable model, inhibiting phosphorylation of both WT and xDFG mutant TRKs, along with their downstream signaling molecules. JND4135 can induce G0/G1 phase arrest and apoptosis in BaF3–CD74-TRKA-G667C cells. JND4135 shows tumor growth inhibition activity in the BaF3-CD74-TRKA-G667C mouse xenograft model .
SU212 is a podophyllotoxin-derived ENO1 inhibitor and AMPK activator. SU212 can selectively induce oxidative phosphorylation, reduce glycolysis activity and glucose uptake in tumor cells, and directly bind to ENO1 without affecting these pathways in normal cells. SU212 induces apoptosis and promotes ENO1 degradation via proteasomal and autophagic pathways without inhibiting the catalytic activity. SU212 leads to mitotic arrest and apoptosis in TNBC (triple-negative breast cancer) cells by activating AMPK, demonstrating potent anti-tumor activity in vitro. SU212 inhibits tumor growth and metastasis in syngeneic, xenograft, and diabetic mouse models, exhibiting an excellent safety profile. SU212 can be used in research on t TNBC, diabetes, and fatty liver disease .
Tubulin/VEGFR-2-IN-2 is an orally active tubulin and VEGFR-2 inhibitor with IC50s of 3.27 and 0.09 μM, respectively. Tubulin/VEGFR-2-IN-2 exerts the antitumor effects through multifaceted pathways, including enhancing reactive oxygen species (ROS) generation, disrupting mitochondrial membrane potential, inducing apoptosis, and arresting the cell cycle. Tubulin/VEGFR-2-IN-2 demonstrates anti-angiogenic properties by significantly impairing endothelial cell migration, invasion, and tube formation in vitro. Tubulin/VEGFR-2-IN-2 suppresses angiogenesis, tumor growth, and metastasis in vivo. Tubulin/VEGFR-2-IN-2 can be used for non-small lung cancer, breast cancer, gastric cancer and lymphoma .
WX-132-18B is a tubulin inhibitor with an IC50 of 0.45-0.99 nM. WX-132-18B selectively binds to the colchicine-binding site on tubulin, reduces microtubule content via depolymerization, and inhibits tubulin polymerization. WX-132-18B induces tumor cell cycle arrest, apoptosis and changes in nuclear membrane permeability, and decreases mitochondrial membrane potential. WX-132-18B exhibits antiproliferative activity against endothelial cells and human tumor cells, and inhibits the proliferation and growth of xenograft tumors in mice. WX-132-18B can be used in research related to sarcoma, non-small cell lung cancer, gastric cancer and breast cancer .
Multi-kinase-IN-8 is a muti-kinase inhibitor. Multi-kinase-IN-8 inhibits COX-1 (IC50 of 12.6 μM), COX-2 (IC50 of 0.05 μM) and VEGFR-2 (IC50 of 0.12 nM). Multi-kinase-IN-8 inhibits tumor-associated carbonic anhydrases (CA IX and CA XII with Ki of 31.5 nM and 386.9 nM, respectively). Multi-kinase-IN-8 triggers cell cycle arrest and apoptosis through upregulation of Caspase 9 and Bax along with downregulation of Bcl 2. Multi-kinase-IN-8 suppresses PGE2, p-VEGFR-2, MMP-9 and HIF-1α and exhibits growth-inhibitory activity against breast cancer, lung cancer, and colorectal adenocarcinoma .
Clausenidin is a selective inhibitor targeting apoptosis-related pathways, including the mitochondrial pathway and death receptor pathway, and vascular endothelial growth factor (VEGF). Clausenidin induces mitochondrial membrane depolarization by activating caspase-3, caspase-8 and caspase-9, upregulating the pro-apoptotic protein Bax and downregulating the anti-apoptotic protein Bcl-2. Clausenidin also inhibits VEGF expression and blocks angiogenesis, exerting anti-tumor activity. Clausenidin has inhibitory effects against Mycobacterium tuberculosis (MIC=200 μg/mL). Clausenidin can induce apoptosis in liver cancer cells, arrest the cell cycle in the G2/M phase, and inhibit tumor angiogenesis. Clausenidin can be used in the research of malignant tumors such as liver cancer .
PROTAC PI3Kα/δ degrader-1 is an orally active PI3Kα/δPROTAC degrader, with an IC50 of 0.34 nM for PI3Kα and 1.85 nM for PI3Kδ. PROTAC PI3Kα/δ degrader-1 inhibits the proliferation and migration of cancer cells, induces G1-phase cell cycle arrest and PI3Kα degradation. PROTAC PI3Kα/δ degrader-1 suppresses tumor growth in breast cancer xenograft mouse models. PROTAC PI3Kα/δ degrader-1 can be used for the research of breast cancer .
Sorafenib (Bay 43-9006) is a potent oral active multikinase inhibitor. Sorafenib blocks autophosphorylation and activity of receptor tyrosine kinases (VEGFR-2, VEGFR-3) and RAF family kinases, thereby suppressing the RAF/MEK/ERK and PI3K/Akt pathways, inhibiting STAT3 phosphorylation, and selectively inhibiting the MAPK pathway in cancer cells. Sorafenib induces cell cycle arrest, autophagy, apoptosis, and PARP cleavage, reduces Bcl-2, Bcl-XL, cyclin D1 levels, and activates Bak and Bax. Sorafenib inhibits tumor growth and metastasis in mouse and rat models. Sorafenib can be used for cancer research, such as colon, breast, non-small-cell lung cancer (NSCLC), ovarian, pancreatic, melanoma, colorectal and hepatocellular carcinoma .
Aspulvinone H is an orally active inhibitor of AChE, pancreatic lipase, glutamic-oxaloacetic transaminase 1, and α-glucosidase, with IC50 values of 25.95 μM, 47.06 μM, 5.91/6.91 μM, and 4.6 μM, respectively. It has a Ka of 2.14 μM against GOT1 and a Ki of 6.58 μM against α-glucosidase. Aspulvinone H inhibits cancer cell proliferation, interferes with glutamine metabolism, elevates ROS levels, and induces cell apoptosis and S-phase arrest. Aspulvinone H exhibits antibacterial activity against Staphylococcus aureus. Aspulvinone H inhibits the growth of pancreatic ductal adenocarcinoma xenografts. Aspulvinone H reduces postprandial blood glucose in mice. Aspulvinone H can be used in research related to pancreatic ductal adenocarcinoma, diabetes, and Staphylococcus aureus infection .
IKKβ-IN-5 is an orally active and selective IKKβ inhibitor with an IC50 of 7.5 nM. IKKβ-IN-5 directly inhibits IKKβ phosphorylation and attenuates NF κB mediated inflammatory and survival signals while promoting autophagy flux. IKKβ-IN-5 exhibits a 6-fold selectivity forIKKβ over the homologous kinase IKKα. IKKβ-IN-5 exerts robust antiproliferative effects through a dual mechanism involving G₂/M phase cell cycle arrest and autophagy activation, even under inflammatory stimulation in vitro. IKKβ-IN-5 demonstrates favorable pharmacokinetics and suppresses tumor growth in vivo. IKKβ-IN-5 can be used for colorectal cancer and potentially other inflammation driven malignancies research .
KRAS G13D-IN-2 (compound 8B) is a potent orally active KRAS G13D inhibitor with IC50 values of 1.95 μM (HCT-116 G13D) and 2.16 μM (HCT-15 G13D). KRAS G13D-IN-2 induces G1-phase arrest and mitochondrial membrane depolarization. KRAS G13D-IN-2 induces senescence through CDK6/TWIST1 inhibition. KRAS G13D-IN-2 inhibits tumor growth in murine models. KRAS G13D-IN-2 can be used for KRAS G13D-mutant colorectal cancer research .
MN33-63 is a Bcl-2 inhibitor, caspase-3 activator and DNA crosslinker with broad-spectrum anticancer activity. MN33-63 improves the water solubility of SN-38 (HY-13704), inhibits tumor growth and proliferation in a dose-dependent manner, and causes no obvious toxicity. MN33-63 relieves the inhibition of the mitochondrial apoptotic pathway, initiates the apoptosis program, inhibits Topo I activity, and promotes its degradation via the ubiquitin-proteasome and autophagy-lysosome pathways. MN33-63 induces DNA crosslinking, G2/M cell cycle arrest, inhibition of cancer cell migration, and cancer cell apoptosis through the mitochondrial pathway. MN33-63 can be used in the research of colorectal cancer, cervical cancer, hepatocellular carcinoma, lung adenocarcinoma and gastric cancer .
Chlorpromazine is an orally active, blood-brain barrier-transparent antipsychotic agent that effectively antagonises D2 dopamine receptors and 5-HT2A, which is widely used in schizophrenia and other psychiatric disorders. Chlorpromazine exerts anti-cancer activity through a variety of pathways, including anti-proliferation, induction of autophagy and cycle arrest (G2-M phase), inhibition of cytochrome c oxidase (CcO), inhibition of tumour growth and metastasis, and inhibition of tumour immune escape. Chlorpromazine also blocks hNav1.7 channels (IC50=25.9 μM; concentration-dependent) and HERG potassium channels (IC50=21.6 μM), which has potential for analgesic and cardiac arrhythmic studies. Chlorpromazine also can inhibit clathrin-mediated endocytosis .
DNA/TOP2A-IN-1 is an inhibitor of DNA and TOP2A. DNA/TOP2A-IN-1 selectively binds to TOP2A, not TOP2B, and interacts with DNA and TOP2A to form a stable DM1-TOP2A-DNA ternary complex. DNA/TOP2A-IN-1 induces DNA damage, increases reactive oxygen species (ROS) and triggers apoptosis in triple-negative breast cancer (TNBC) cells. DNA/TOP2A-IN-1 disrupts microtubule distribution and induces cell cycle arrest. DNA/TOP2A-IN-1 shows strong antiproliferative activity and inhibits cell migration. DNA/TOP2A-IN-1 inhibits tumor growth and can be used for TNBC research .
STAT3-D11-PROTAC-VHL (Compound D11-PROTAC) is a PROTAC degrader targeting Signal Transducer and Activator of Transcription 3 (STAT3). STAT3-D11-PROTAC-VHL exhibits anti-tumor activity with IC50 values of 1335 nM and 1973 nM against HeLa and MCF-7 cells, respectively. STAT3-D11-PROTAC-VHL binds to the DNA-binding domain of STAT3 and recruits the E3 ligase VHL to form a ternary complex, leading to the ubiquitination of STAT3 and subsequent degradation by the proteasome. STAT3-D11-PROTAC-VHL also inhibits tumor cell growth, induces cell cycle arrest and apoptosis, and suppresses tumor immune evasion .
Tubulin polymerization-IN-84 inhibits tubulin polymerization by targeting the colchicine-binding pocket, with anIC50 = 10.9 μM. Tubulin polymerization-IN-84 shows antiproliferative activity against Jurkat, B16-F10, HCT116, and MDA-MB-231 cells (IC50= 60 nM, 380 nM, 138 nM, and 1.054 μM). Tubulin polymerization-IN-84 induces G2/M-phase arrest and apoptosis in B16-F10 cells. Tubulin polymerization-IN-84 suppresses tumor growth in a B16-F10 melanoma model and potentiates anti-tumor immunity in combination with a PD-L1 mAb for the research of T-cell acute lymphoblastic leukemia, melanoma, colon cancer, and breast cancer.
ALK/PI3K/AKT-IN-1 (Compound 45) inhibits the proliferation of cancer cell A549, H1975 and PC9 with an IC50 of 0.44, 0.83 and 1.51 μM. ALK/PI3K/AKT-IN-1 increases the expression of p21 and p27, inhibits the activity of CDK2 and p-Rb, arrests the cell cycle at G1 phase. ALK/PI3K/AKT-IN-1 inhibits the ALK/PI3K/AKT signaling pathway, promotes the depolarization of mitochondrial membrane potential, and inducing apoptosis in A549 cell. ALK/PI3K/AKT-IN-1 inhibits the formation and growth of A549 cell spheroids .
2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cell cycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
Casuarinin is an orally active antiproliferative, anti-inflammatory, antifungal, virucidal and gastroprotective agent. Casuarinin upregulates the expression of p21/WAF1, Fas/APO‑1, mFasL, sFasL and HSP‑70, arrests cell cycle, induces apoptosis and inhibits cancer cell proliferation. Casuarinin inhibits TNF‑α-induced phosphorylation of MAPK and activation of NF‑κB, downregulates the expression of iNOS, NF‑κB, COX‑2 and ICAM‑1, and reduces the production of proinflammatory mediators. Casuarinin attenuates ethanol-induced activation of caspase‑3 and elevation of TNF‑α, inhibits the growth of Candida albicans, and inhibits HSV‑2. Casuarinin can be used in research related to mammary adenocarcinoma, inflammatory skin diseases, gastric ulcers, candidiasis and herpes simplex virus infections .
PROTAC EGFR degrader 14 is a potent and selective EGFR PROTACdegrader with a DC50 of about 2.9 nM and a Dmax of 93.1% for EGFR L858R/T790M/C797S. PROTAC EGFR degrader 14 selectively induces EGFR C797S degradation through a VHL and proteasome-dependent manner and downregulated EGFR-associated transcriptome and exhibits good selectivity over EGFR WT. PROTAC EGFR degrader 14 induces cell cycle arrest and apoptosis and significantly inhibits tumor growth. PROTAC EGFR degrader 14 can be used for the study of nonsmall cell lung cancer (NSCLC) (Pink: Target protein ligand: (HY-143337); Blue: E3 ligand (HY-125845); Black: Linker (HY-W004688)) .
LA-CB1 is an Abemaciclib (HY-16297A) derivative that targets CDK4/6 and promotes its degradation via the ubiquitin-proteasome pathway, thereby disrupting the CDK4/6-Cyclin D1-Rb-E2F axis and inducing G0/G1 cell cycle arrest and apoptosis. LA-CB1 exhibits antiproliferative activity against MDA-MB-231 cells, with an IC50 of 0.27 µM, and effectively inhibits epithelial-mesenchymal transition (EMT), cell migration, invasion, and angiogenesis. In highly aggressive models such as triple-negative breast cancer (TNBC), LA-CB1 significantly suppresses tumor growth in a dose-dependent manner. LA-CB1 holds potential for research in the field of breast cancer .
(2S,3R,4S)-ASX-173 is the (2S,3R,4S)-enantiomer of ASX-173 (HY-175282). ASX-173 is an orally active inhibitor of asparagine synthetase (ASNS) (IC50 = 0.113 μM, Ki = 0.4 nM). ASX-173 enhances the anticancer activity of L-asparaginase (ASNase) (HY-P1923). ASX-173 disrupts nucleotide synthesis and induces leukemia cell cycle arrest, apoptosis and autophagy in leukemia cells in combination with ASNase. ASX-173 slows the growth of OCI-AML2 xenografts in combination with ASNase. ASX-173 is indicated for the study of acute lymphoblastic leukemia, acute myeloid leukemia, colorectal cancer, and other cancers .
Chlorpromazine hydrochloride is an orally active, blood-brain barrier-transparent antipsychotic agent that effectively antagonises D2 dopamine receptors and 5-HT2A, which is widely used in schizophrenia and other psychiatric disorders. Chlorpromazine hydrochloride exerts anti-cancer activity through a variety of pathways, including anti-proliferation, induction of autophagy and cycle arrest (G2-M phase), inhibition of cytochrome c oxidase (CcO), inhibition of tumour growth and metastasis, and inhibition of tumour immune escape. Chlorpromazine hydrochloride also blocks hNav1.7 channels (IC50=25.9 μM; concentration-dependent) and HERG potassium channels (IC50=21.6 μM), which has potential for analgesic and cardiac arrhythmic studies. Chlorpromazine hydrochloride also can inhibit clathrin-mediated endocytosis .
Murrayafoline A (Standard) is the analytical standard of Murrayafoline A (HY-W100287). This product is intended for research and analytical applications. Murrayafoline A is a carbazole alkaloid that can be extracted from Murraya tetramera. Murrayafoline A directly targets Specificity protein 1 (Sp1), thereby inhibiting NF-κB and MAPK signaling pathways. Murrayafoline a induces a G0/G1-phase arrest in platelet-derived growth factor (PDGF)-stimulated vascular smooth muscle cells. Murrayafoline A attenuates the Wnt/β-catenin pathway by promoting the degradation of intracellular β-catenin proteins. Murrayafoline A enhances the contraction of rat ventricular myocytes and L-type calcium current by activating protein kinase C. Murrayafoline A inhibits LPS (HY-D1056)-induced neuroinflammation in vivo. Murrayafoline A can be used for the study of inflammation, vascular complications and colon cancer .
Sorafenib (Bay 43-9006) tosylate is a potent oral active multikinase inhibitor. Sorafenib blocks autophosphorylation and activity of receptor tyrosine kinases (VEGFR-2, VEGFR-3) and RAF family kinases, thereby suppressing the RAF/MEK/ERK and PI3K/Akt pathways, inhibiting STAT3 phosphorylation, and selectively inhibiting the MAPK pathway in cancer cells. Sorafenib tosylate induces cell cycle arrest, autophagy, apoptosis, and PARP cleavage, reduces Bcl-2, Bcl-XL, cyclin D1 levels, and activates Bak and Bax. Sorafenib tosylate inhibits tumor growth and metastasis in mouse and rat models. Sorafenib tosylate can be used for cancer research, such as colon, breast, non-small-cell lung cancer (NSCLC), ovarian, pancreatic, melanoma, colorectal and hepatocellular carcinoma .
c-MYC/BCL2 ligand 1 iodide is a dual-targeting c-MYC/Bcl-2 G4 ligand with Kd values of 0.90 μM (c-MYC G4) and 0.56 μM (Bcl-2 G4). c-MYC/BCL2 ligand 1 iodide inhibits c-MYC and Bcl-2 gene transcription by binding to G4-forming sequences and downregulates their protein expression. c-MYC/BCL2 ligand 1 iodide inhibits suppresses migration, induces caspase-dependent apoptosis, and triggers cell cycle G1 arrest in MCF-7 cells. c-MYC/BCL2 ligand 1 iodide significantly suppresses tumor growth in a 4T1 syngeneic model with no observable toxicity. c-MYC/BCL2 ligand 1 iodide can be used for the research of breast cancer.
1,4-Dihydroxy-2-naphthoic acid (Standard) is the analytical standard for 1,4-Dihydroxy-2-naphthoic acid (HY-W014701). 1,4-Dihydroxy-2-naphthoic acid is an orally active aryl hydrocarbon receptor (AhR) agonist and a bifidogenic growth stimulator. 1,4-Dihydroxy-2-naphthoic acid can improve the motor dysfunction in parkinson's disease (PD) model through AhR-dependent and -independent pathways. 1,4-Dihydroxy-2-naphthoic acid exerts anti-inflammatory effects by regulating the gut microbiota (such as promoting the proliferation of Bifidobacterium) and directly regulating the host immune system. 1,4-Dihydroxy-2-naphthoic acid induces apoptosis through G0/G1 cell cycle arrest in human keratinocyte to inhibit psoriasis .
Chlorpromazine (hydrochloride) (Standard) is the analytical standard of Chlorpromazine (hydrochloride). This product is intended for research and analytical applications. Chlorpromazine hydrochloride is an orally active, blood-brain barrier-transparent antipsychotic agent that effectively antagonises D2 dopamine receptors and 5-HT2A, which is widely used in schizophrenia and other psychiatric disorders. Chlorpromazine hydrochloride exerts anti-cancer activity through a variety of pathways, including anti-proliferation, induction of autophagy and cycle arrest (G2-M phase), inhibition of cytochrome c oxidase (CcO), inhibition of tumour growth and metastasis, and inhibition of tumour immune escape. Chlorpromazine hydrochloride also blocks hNav1.7 channels (IC50=25.9 μM; concentration-dependent) and HERG potassium channels (IC50=21.6 μM), which has potential for analgesic and cardiac arrhythmic studies. Chlorpromazine hydrochloride also can inhibit clathrin-mediated endocytosis .
PROTAC FGFR2 degrader 1 (compound N5) is a PROTAC that effectively targets FGFR2 with DC50 of 6.46 nM, the FGFR2IC50 is 0.08 nM. PROTAC FGFR2 degrader 1 has anti-proliferative activity and highly selective, induces G0/G1 arrest of KATOIII and SNU16 cell cycle and inhibits apoptosis by reducing the activation of p-ERK and p-PLCγ, the downstream proteins of FGFR2.
PROTAC FGFR2 degrader 1 inhibits gastric cancer cells remained above 50% at a concentration of 0.17 nM.
PROTAC FGFR2 degrader 1 potently inhibits the growth of SNU16 xenograft tumors in mouse model (Structure Note: Pink, FGFR2 activator: HY-18708; Blue, E3 ligase ligand: HY--10984; Black, linker: HY-163989; E3 ligase ligand + linker:HY-163986) .
Cajanol is an isoflavanone that can be isolated from the roots of Cajanus cajan (L.) Millsp. . Cajanol inhibits cancer cell proliferation and induces cancer cell apoptosis. Cajanol promotes the expression of Bax, inhibits the expression of Bcl-2, activates caspase-9 and caspase-3, induces PARP cleavage, arrests the cell cycle at the G2/M phase, generates ROS, disrupts mitochondrial membrane potential and triggers cytochrome c release. Cajanol induces bacterial DNA damage, disrupts bacterial cell membranes, and exerts antibacterial activity in vitro. Cajanol reduces the expression of PI3K, inhibits the phosphorylation of Akt and NF-κB, downregulates the expression and transport function of P-gp, restores the sensitivity of drug-resistant cancer cells to Paclitaxel, and inhibits the growth of Paclitaxel-resistant metastatic ovarian tumors. Cajanol is applicable to research related to breast cancer, ovarian cancer and bacterial infections .
C2 L-threo Ceramide (d18:1/2:0) (L-threo Cer(d18:1/2:0); L-threo Ceramide (d18:1/2:0)) is a bioactive sphingolipid and cell-permeable analog of naturally occurring ceramides. It stimulates cholesterol efflux in CHO cells expressing the human ABCA1 receptor when used at a concentration of 10 μM, however, this efflux is 50% less than that stimulated by C2 ceramide. C2 L-threo Ceramide inhibits IL-4 production by 17% in EL4 T cells stimulated with phorbol 12-myristate 13-acetate when used at a concentration of 10 μM. It also induces cell cycle arrest in the G0/G1 phase and a 7-fold increase in sphingosine accumulation as well as inhibits growth of HL-60 leukemia cells.
6-Methoxydihydrosanguinarine is an alkaloid with activity across multiple cancer cell types. 6-Methoxydihydrosanguinarine activates IRE1/JNK signaling, blocks Akt/mTOR and PI3K/AKT/mTOR pathways, reduces expression of Cdc25C, CyclinB1, Cdc2, YAP/TAZ, Survivin, GPX4, and EGFR, upregulates IRE1 and DR5, and activates JNK and caspases. 6-Methoxydihydrosanguinarine induces apoptosis, G2/M phase arrest, DNA damage, ROS generation, lipid peroxidation, ferroptosis, autophagy, and suppresses cancer cell growth. 6-Methoxydihydrosanguinarine disruptes the biofilm formation of Candida albicans (C. albicans). 6-Methoxydihydrosanguinarine can be used for the research of non-small cell lung cancer, hepatocellular carcinoma, melanoma, colon carcinoma, ovarian cancer and breast cancer .
Chlorpromazine (hydrochloride) (Standard) is the analytical standard of Chlorpromazine (hydrochloride). This product is intended for research and analytical applications. Chlorpromazine hydrochloride is an orally active, blood-brain barrier-transparent antipsychotic agent that effectively antagonises D2 dopamine receptors and 5-HT2A, which is widely used in schizophrenia and other psychiatric disorders. Chlorpromazine hydrochloride exerts anti-cancer activity through a variety of pathways, including anti-proliferation, induction of autophagy and cycle arrest (G2-M phase), inhibition of cytochrome c oxidase (CcO), inhibition of tumour growth and metastasis, and inhibition of tumour immune escape. Chlorpromazine hydrochloride also blocks hNav1.7 channels (IC50=25.9 μM; concentration-dependent) and HERG potassium channels (IC50=21.6 μM), which has potential for analgesic and cardiac arrhythmic studies. Chlorpromazine hydrochloride also can inhibit clathrin-mediated endocytosis .
6-Methoxydihydrosanguinarine hydrochloride is an alkaloid with activity across multiple cancer cell types. 6-Methoxydihydrosanguinarine hydrochloride activates IRE1/JNK signaling, blocks Akt/mTOR and PI3K/AKT/mTOR pathways, reduces expression of Cdc25C, CyclinB1, Cdc2, YAP/TAZ, Survivin, GPX4, and EGFR, upregulates IRE1 and DR5, and activates JNK and caspases. 6-Methoxydihydrosanguinarine hydrochloride induces apoptosis, G2/M phase arrest, DNA damage, ROS generation, lipid peroxidation, ferroptosis, autophagy, and suppresses cancer cell growth. 6-Methoxydihydrosanguinarine hydrochloride disruptes the biofilm formation of Candida albicans (C. albicans). 6-Methoxydihydrosanguinarine hydrochloride can be used for the research of non-small cell lung cancer, hepatocellular carcinoma, melanoma, colon carcinoma, ovarian cancer and breast cancer .
CDK9/HDAC1/HDAC3-IN-1 is dual-functional inhibitor of CDK9 and HDAC. CDK9/HDAC1/HDAC3-IN-1 inhibits the protein activity of CDK9/HDAC/HDAC3 with IC50 s of 0.17 μM, 1.73 μM and 1.11 μM for CDK9, HDAC1, and HDAC3, respectively. CDK9/HDAC1/HDAC3-IN-1 inhibits cancer cells by inducing cell apoptosis and cell cycle arrest in the G2/M phase, as well as tumor growth in a murine TNBC MDA-MB-231 xenograft model. CDK9/HDAC1/HDAC3-IN-1 has a broad-spectrum anti-cancer activity, such as breast cancer, cervical cancer, and liver cancer .
SIAIS039 is an orally active c-ros oncogene 1 (ROS1)-specific PROTAC with DC50s of 154.46 nM, 126.47 nM, 143.69 nM for HCC78 cells, Ba/F3 expressing the CD74-ROS1 fusion and Ba/F3 expressing the SDC4-ROS1 fusion, respectively. SIAIS039 suppresses cell proliferation, induces cell cycle arrest and apoptosis, and inhibits clonogenicity against ROS1-positive cells. SIAIS039 demonstrates anti-tumour effects against ROS1-driven tumor growth vivo. SIAIS039 is composed of the ALK inhibitor Brigatinib (HY-12857), a linker EM-12 (HY-138793), and a VHL ligand E3 ubiquitin ligase 1-Butyne (Red: Brigatinib; Blue: VHL ligand; Black: linker) .
2,6-Dihydroxyacetophenone (Standard) is the analytical standard of 2,6-Dihydroxyacetophenone (HY-Y0106). This product is intended for research and analytical applications. 2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cell cycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
PI3K/mTOR-IN-17 is a dual PI3K and mTOR inhibitor with IC50 values of 1.21 μM (PI3K), and 0.21 μM (mTOR). PI3K/mTOR-IN-17 induces cells caspase-mediated apoptosis by arresting their growth in the G1-phase. PI3K/mTOR-IN-17 upregulates the levels of caspases-3, 7, 8, and 9, p53 expression and Bax/Bcl-2 ratio. PI3K/mTOR-IN-17 suppresses the PI3K/mTOR signaling pathway. PI3K/mTOR-IN-17 can be used for research of cancer, such as non-small cell lung cancer (NSCLC) .
C6 Ceramide- 13C2,d2 (C6-Cer- 13C2,d2) is the deuterium labeled and 13C-labeled C6 Ceramide (HY-19542). C6 Ceramide (C6-Cer) is a short-chain, cell-permeable ceramide pathway activator with anticancer activity. C6 Ceramide-mediated miR-29b expression participates in the progression of multiple myeloma through suppressing the proliferation, migration and angiogenesis of endothelial cells by targeting Akt signal pathway. C6 Ceramide exhibits multiple anti-cancer properties including cell cycle arrest, Apoptosis, inhibition of tumor growth and enhances the effects of chemotherapy in drug-resistant cancer cells. C6-ceramide can be used as an adjuvant for chemotherapeutic agents, to enhance anti-tumor effects .
SKF-96365 is a TRPC channel antagonist and store-operated calcium entry (SOCE) inhibitor. SKF-96365 reduces calcium ion influx by inhibiting the activity and expression of TRPC6, STIM1 and Orai1. SKF-96365 inhibits voltage-gated sodium current (cardiac INa/NaV1.5) and slows myocardial conduction. SKF-96365 inhibits phosphorylation/activation of CaMKIIγ and suppresses the downstream AKT signaling pathway. SKF-96365 induces G2/M phase cell cycle arrest, apoptosis and cytoprotective autophagy in colorectal cancer cells. SKF-96365 alleviates allergic rhinitis symptoms by reducing inflammatory cytokine levels. SKF-96365 reduces intracellular calcium overload, inhibits Homer1 expression, prevents nuclear damage and suppresses apoptosis. SKF-96365 inhibits the growth of colorectal cancer xenografts in nude mice . SKF-96365 is applicable to research related to allergic rhinitis, colorectal cancer, Parkinson's disease, persistent spontaneous nociception and hyperalgesia .
LSD1/TLK1-IN-1 is an orally active LSD1, TLK1, TLK2, TTK inhibitor with an LSD1IC50 of 0.247 μM. LSD1/TLK1-IN-1 suppresses phosphorylation of Nek1 at T141 and Rad9 at S328, abrogates the TLK1>Nek1>ATR>Chk1 axis, protects H3K4me1/2 from demethylation, and does not affect LSD2, MAO-A, or MAO-B. LSD1/TLK1-IN-1 induces apoptosis, bypasses cell-cycle arrest, suppresses tumor growth, downregulates PD-L1 expression, enhances T-cell killing response, inhibits gastric cancer cell proliferation. LSD1/TLK1-IN-1 can be used for the research of prostate cancer and gastric cancer .
SKF-96365 hydrochloride is a TRPC channel antagonist and store-operated calcium entry (SOCE) inhibitor. SKF-96365 hydrochloride reduces calcium ion influx by inhibiting the activity and expression of TRPC6, STIM1 and Orai1. SKF-96365 hydrochloride inhibits voltage-gated sodium current (cardiac INa/NaV1.5) and slows myocardial conduction. SKF-96365 hydrochloride inhibits phosphorylation/activation of CaMKIIγ and suppresses the downstream AKT signaling pathway. SKF-96365 hydrochloride induces G2/M phase cell cycle arrest, apoptosis and cytoprotective autophagy in colorectal cancer cells. SKF-96365 hydrochloride alleviates allergic rhinitis symptoms by reducing inflammatory cytokine levels. SKF-96365 hydrochloride reduces intracellular calcium overload, inhibits Homer1 expression, prevents nuclear damage and suppresses apoptosis. SKF-96365 hydrochloride inhibits the growth of colorectal cancer xenografts in nude mice . SKF-96365 hydrochloride is applicable to research related to allergic rhinitis, colorectal cancer, Parkinson's disease, persistent spontaneous nociception and hyperalgesia .
Quercetin-3'-O-glucoside is an orally active flavonoid glycoside. Quercetin-3'-O-glucoside reduces liver glucose-6-phosphatase activity, alters serum insulin and glucose levels, and regulates the activities of antioxidant enzymes in the liver and kidney. Quercetin-3'-O-glucoside inhibits DNA topoisomerase II, induces S-phase cell cycle arrest and caspase-3-mediated apoptosis in hepatocellular carcinoma cells. Quercetin-3'-O-glucoside selectively inhibits EGFR-mediated signaling pathways targeting AKT, ERK1/2, FAK and MEK1/2. Quercetin-3'-O-glucoside inhibits growth factor-induced migration and invasion in pancreatic cancer cells. Quercetin-3'-O-glucoside exerts free radical scavenging effects. Quercetin-3'-O-glucoside is applicable to research related to pancreatic cancer, diabetes, hepatocellular carcinoma and malignant tumors .
Abietic acid, an orally active diterpene isolated from Colophony, displays significant anti-proliferative, anti-inflammatory, anti-obesity effect, bacteriostatic, cell cycle arresting and pro-apoptotic activities. Abietic acid inhibits lipoxygenase activity for allergy. Abietic acid enhances cell migration and tube formation in HUVECs. Abietic acid induces significant angiogenic potential, which is associated with upregulation of extracellular signal-regulated kinase (ERK) and p38 expression. Abietic acid attenuates sepsis-induced lung injury by inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway to inhibit M1 macrophage polarization. Abietic acid exhibits a positive effect against liver injury by attenuating inflammation and ferroptosis. Abietic acid shows accelerated wound closure in a mouse model of cutaneous wounds. Abietic acid significantly reduces the proliferation and growth of NSCLC cells by IKKβ inhibition.Additionally, Abietic acid ameliorates psoriasis-like inflammation and modulates gut microbiota in mice. Abietic acid is promising for research in non-small-cell lung cancer (NSCLC), liver injury-related deseases and psoriasis .
AR/AR-V7 degrader-1 is an orally active AR and AR-V7 degrader. AR/AR-V7 degrader-1 disrupts the interaction between AR/AR-V7 and HSP90, leading to their ubiquitination and degradation in castration-resistant prostate cancer cells. AR/AR-V7 degrader-1 regulates the expression of cell cycle-related proteins in prostate cancer cells (downregulates CDK4, CDK6, Cyclin D1, Cyclin E1; upregulates P21) and induces G0/G1 phase arrest. AR/AR-V7 degrader-1 inhibits the proliferation and migration of prostate cancer cells. AR/AR-V7 degrader-1 suppresses the growth of castration-resistant prostate cancer tumors in nude mice and induces the degradation of AR and AR-V7 in tumor tissues. AR/AR-V7 degrader-1 is applicable to the research of castration-resistant prostate cancer .
DHW-221 is a potent orally active dual PI3K/mTOR inhibitor, exhibiting low nanomolar potency against all four Class I PI3K isoforms and mTOR (PI3Kα, IC50 = 0.50 nM; PI3Kβ, IC50 = 1.9 nM; PI3Kγ, IC50 = 1.8 nM; PI3Kδ, IC50 = 0.74 nM; mTOR, IC50 = 3.9 nM). DHW-221 exerts antitumor effects by blocking the PI3K/Akt/mTOR pathway and inducing mitochondrialapoptosis and paraptosis (via Endoplasmic Reticulum (ER) stress and MAPK signaling) and arrests cell cycle, thereby inhibiting cell migration, invasion and angiogenesis. DHW-221 inhibits tumor growth in both the A549/Taxol (HY-B0015) and the HCC827 xenograft mouse models. DHW-221 can be used for non-small cell lung cancer (NSCLC), colon and breast cancer research .
Strictinin is an orally active phenolic compound. Strictinin reduces xanthine oxidase activity, uric acid production, and the activation of ERK1/2, JNK, NF-κB, and NLRP3 inflammasome components in hepatocytes treated with Xanthine (HY-W017389). Strictinin decreases elevated serum uric acid levels and enhanced xanthine oxidase activity in mice treated with potassium oxonate. Strictinin acts as a ROR1 inhibitor and exhibits anticancer activity against highly aggressive non-androgen-dependent prostate cancer. Strictinin induces cancer cell apoptosis (apoptosis), arrests cell cycle, and inhibits cancer cell migration, invasion, and epithelial-mesenchymal transition. Strictinin modulates gut microbiota, inhibits bacterialgrowth and biofilm formation, accelerates small intestinal transit, and blocks viral entry and replication. Strictinin can be used in research related to hyperuricemia, androgen receptor-negative non-androgen-dependent prostate cancer, triple-negative breast cancer, bacterial infections, constipation, coronavirus infections, dental caries, and infections caused by influenza A, influenza B, and human parainfluenza virus type 1 .
MN33-47 is a multi-target anti-tumor compound with broad-spectrum anti-proliferative activity. MN33-47 relieves the inhibition of the mitochondrial apoptosis pathway by downregulating the anti-apoptotic protein Bcl-2, while activating caspase-3 and inhibiting Topoisomerase I activity, thereby promoting its degradation through the ubiquitin-proteasome and autophagy-lysosome pathways. MN33-47 can also induce DNA cross-linking and G2/M cell cycle arrest, inhibit cancer cell migration and activate the mitochondrial apoptosis pathway, thus exerting potent anti-tumor effects. MN33-47 can improve the water solubility of SN-38 (HY-13704), and exhibits dose-dependent tumor growth inhibition effects in CT26 tumor-bearing mouse models without obvious toxic and side effects. MN33-47 can be used in related studies on colorectal adenocarcinoma, cervical adenocarcinoma, hepatocellular carcinoma, alveolar basal epithelial adenocarcinoma, gastric cancer and colon cancer .
FZ-AD005 is a DLL3-targeting antibody-drug conjugate (ADC) with high selectivity, composed of the anti-DLL3 antibody FZ-A038 (HY-P990896), a dipeptide linker (Val-Ala), and DXd (HY-13631D). The Kd value of FZ-AD005 for human DLL3 ranges from 13.29 to 58.3 pmol/L. After binding to DLL3 on the cell surface, FZ-AD005 mediates endocytosis, and the payload DXd is released via cleavage by lysosomal cathepsins. DXd inhibits topoisomerase TopI to induce double-strand DNA breaks, cell cycle arrest and apoptosis, and FZ-AD005 exhibits bystander killing activity against adjacent DLL3-negative cells. FZ-AD005 shows stable circulation in vivo, has good tolerance and acceptable pharmacokinetic profiles in rats and cynomolgus monkeys, and effectively inhibits the growth of DLL3-expressing tumor cells. FZ-AD005 serves as a promising candidate molecule for research on small cell lung cancer and human neuroendocrine prostate cancer .
Cytisine-Platinum(IV) Prodrug-1 is a Pt(IV) prodrug incorporating the natural compound Cytisine (HY-N0175) with antiproliferative activity against tumor cells. Cytisine-Platinum(IV) Prodrug-1 promotes calcium transfer across the IP3R1-GRP75-VDAC1 axis to drive mitochondrial calcium overload. Cytisine-Platinum(IV) Prodrug-1 initiates unfolded protein response via PERK, eIF2α, ATF4, and CHOP to modulate Bcl-2 and Bax, triggering apoptosis. Cytisine-Platinum(IV) Prodrug-1 induces mitochondrial dysfunction, ROS production, reduced ATP synthesis, DNA damage, and S-phase cell cycle arrest. Cytisine-Platinum(IV) Prodrug-1 activates the cGAS-STING pathway, reduces PD-L1 expression, drives immunogenic cell death. Cytisine-Platinum(IV) Prodrug-1 exhibits high physiological stability, efficient cellular accumulation, and enhanced platinum-DNA binding, and inhibits tumor growth in mouse models with reduced systemic toxicity. Cytisine-Platinum(IV) Prodrug-1 can be used for the research of lung cancer .
15-Deoxy-Δ12,14-Prostaglandin D2-d4 (15-Deoxy-Δ12,14-PGD2-d4) is the deuterium labeled 15-deoxy-Δ12,14-Prostaglandin D2. 15-deoxy-Δ12,14-Prostaglandin D2 (15-Deoxy-Δ12,14-PGD2) is a metabolite of prostaglandin D₂ (PGD₂) (HY-101988), which can undergo further dehydration metabolism to 15-deoxy-Δ12,14-PGJ₂. 15-deoxy-Δ12,14-Prostaglandin D2 is a highly selective agonist for DP2 receptor and PPARγ. 15-deoxy-Δ12,14-Prostaglandin D2 causes morphological changes in eosinophils and migration of type II innate lymphoid cells (ILC2). 15-deoxy-Δ12,14-Prostaglandin D2 has a growth inhibitory effect on prostate cancer cells expressing PPARγ, induces cell cycle arrest and promotes apoptosis. 15-deoxy-Δ12,14-Prostaglandin D2 can be used in related research on asthma and prostate cancer.
FZ-AD005 is a DLL3-targeting antibody-drug conjugate (ADC) with high selectivity, composed of the anti-DLL3 antibody FZ-A038 (HY-P990896), a dipeptide linker (Val-Ala), and DXd (HY-13631D). The Kd value of FZ-AD005 for human DLL3 ranges from 13.29 to 58.3 pmol/L. After binding to DLL3 on the cell surface, FZ-AD005 mediates endocytosis, and the payload DXd is released via cleavage by lysosomal cathepsins. DXd inhibits topoisomerase TopI to induce double-strand DNA breaks, cell cycle arrest and apoptosis, and FZ-AD005 exhibits bystander killing activity against adjacent DLL3-negative cells. FZ-AD005 shows stable circulation in vivo, has good tolerance and acceptable pharmacokinetic profiles in rats and cynomolgus monkeys, and effectively inhibits the growth of DLL3-expressing tumor cells. FZ-AD005 serves as a promising candidate molecule for research on small cell lung cancer and human neuroendocrine prostate cancer .
Ru (acac) 3 (Tris (acetylacetonato) ruthenium (III)) is a caspase-3 activator and Apoptosis inducer. Ru (acac) 3 exerts growth inhibitory effects on various cell lines in vitro by inhibiting DNA/RNA synthesis and inducing mild reversible S-phase cell cycle arrest. Ru (acac) 3 is commonly used in research related to ovarian cancer, osteosarcoma, cervical cancer, melanoma, and other fields .
Antifreeze Polypeptide 6 (winter flounder) is the component 6 of the winter flounder's antifreeze polypeptides. Antifreeze Polypeptide 6 (winter flounder) lowers the plasma freezing point by arresting the growth of ice nuclei .
T7 Peptide is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide induces G0/G1 cell cycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
T7 Peptide TFA is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide TFA inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide TFA induces G0/G1 cell cycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide TFA is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
DTP3 TFA is a potent and selective GADD45β/MKK7 (growtharrest and DNA-damage-inducible β/mitogen-activated protein kinase kinase 7) inhibitor. DTP3 TFA targets an essential, cancer-selective cell-survival module downstream of the NF-κB pathway .
SP1 is an α-peptide encoded by the mating pheromone MFα1 gene in Candida albicans, which can induce cell growtharrest at the mating type locus MTLa in Candida albicans. SP1 can be used in the study of the prevention and treatment of Candida albicans infection .
PKCδ substrate acts as a nuclear transporter of ERK2 and is involved in ERK2 mediated gene activation. PKCδ is involved in the regulation of cell growth, proliferation, cell cycle arrest, and apoptosis by phosphorylating hBVR and other proteins. PKCδ substrate can be used to study the development of diseases, especially cancer biology .
YVPGP is an oligopeptide exacted from Anthopleura anjunae. YVPGP has a significant antitumor activity by mediating PI3K/AKT/mTOR signaling pathway. YVPGP arrests DU-145 cells in the S phase and induces apoptosis via mitochondrial and death receptor pathways (caspase3, 7, 8, 9). YVPGP effectively inhibits tumor growth in DU-145 xenografts mice model, promising for prostate cancer research .
S9-CMC1 TFA is a covalent peptide lysine-specific demethylase 1 (LSD1) inhibitor with an IC50 value of 2.53 μM. S9-CMC1 TFA specifically recognizes Cys360 in the enzyme-active region. S9-CMC1 TFA inhibits LSD1 activity, increasing H3K4me1 and H3K4me2 levels, leading to G1 cell cycle arrest and apoptosis and inhibiting cell proliferation. S9-CMC1 TFA significantly inhibits tumor growth in A549 xenograft animal models .
R9-caPep (RRRRRRRRRCCLGIPEQEY) is a cell-penetrating peptide derived from proliferating cell nuclear antigen (PCNA). R9-caPep selectively blocks the interactions between PCNA and FEN1, as well as between PCNA and LIGI, while preserving the binding of POLD3 to PCNA. R9-caPep interferes with DNA synthesis and homologous recombination-mediated double-strand DNA break repair, inducing S-phase arrest, DNA damage accumulation, and apoptosis. R9-caPep inhibits the growth of tumor volume and weight of neuroblastoma in nude mice . R9-caPep can be used in research related to neuroblastoma and triple-negative breast cancer .
DPMI-ω is a dual-specificity d-peptide antagonist of oncogenic proteins MDM2 and MDMX. DPMI-ω, upon fabrication on gold nanoparticles, efficiently traverses tumor cells and kills them by reactivating the p53 signaling pathway. DPMI-ω can disrupte the p53-MDM2/MDMX complex. DPMI-ω can inhibit B16 melanoma growth and induce cells G0/G1 phase arrest. DPMI-ω can augment the efficacy of immunotherapy by expanding CD3 +/CD8 + cytotoxic T cells and suppressing CD4 +/CD25 + regulatory T cells companied with anti-PD1 antibody. DPMI-ω can be used for research of melanoma .
Peligifatamab is a PSMA-targeted α-radioimmunoconjugate with an EC50 of 1.2 nM against human targets. Peligifatamab induces DNA damage, DNA double-strand breaks, cell cycle arrest and apoptosis (Apoptosis) in PSMA-positive prostate cancer cells. Peligifatamab reduces cell viability in a manner dependent on cellular PSMA expression levels. Peligifatamab inhibits tumor growth and tumor-induced abnormal bone growth in prostate cancer bone metastasis models. Peligifatamab exhibits antitumor efficacy in subcutaneous prostate cancer models and xenograft models. Peligifatamab can be used for the research of metastatic castration-resistant prostate cancer .
hIMB1636 is a humanized monoclonal antibody targeting Trop2. By binding to the conformational Trop2 epitope, hIMB1636 regulates related signaling pathways, triggers lysosomal endocytosis, and further induces apoptosis, cell cycle arrest, and antibody-dependent cellular cytotoxicity. hIMB1636 effectively inhibits tumor cell proliferation, migration and in vivo tumor growth, and also exerts bystander killing effect and mediates long-term retention. hIMB1636 can be conjugated with NOTA/DOTA for radiolabeling to enable immuno-PET imaging, or prepared as hIMB1636-LDP-AE to significantly inhibit the growth of breast cancer and lung cancer xenografts .
Lactacystin is a potent, orally active, irreversible, cell-permeable, selective 20S proteasome inhibitor (IC50 = 4.8 μM). Lactacystin also inhibits the lysosomal enzyme cathepsin A. Lactacystin inhibits cell growth and induces apoptosisand cell cycle arrest, and has antiviral and antioxidative activity. Lactacystin induces neurite outgrowth and hypertension. Lactacystin has the potential for the research of cancer, Neurological Disease, hypertension and Malaria, and so on [2] [6] .
Costunolide ((+)-Costunolide) is a naturally occurring sesquiterpene lactone, with antioxidative, anti-inflammatory, antiallergic, bone remodeling, neuroprotective, hair growth promoting, anticancer, and antidiabetic properties. Costunolide can induce cell cycle arrest and apoptosis on breast cancer cells .
Abietic acid, an orally active diterpene isolated from Colophony, displays significant anti-proliferative, anti-inflammatory, anti-obesity effect, bacteriostatic, cell cycle arresting and pro-apoptotic activities. Abietic acid inhibits lipoxygenase activity for allergy. Abietic acid enhances cell migration and tube formation in HUVECs. Abietic acid induces significant angiogenic potential, which is associated with upregulation of extracellular signal-regulated kinase (ERK) and p38 expression. Abietic acid attenuates sepsis-induced lung injury by inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway to inhibit M1 macrophage polarization. Abietic acid exhibits a positive effect against liver injury by attenuating inflammation and ferroptosis. Abietic acid shows accelerated wound closure in a mouse model of cutaneous wounds. Abietic acid significantly reduces the proliferation and growth of NSCLC cells by IKKβ inhibition.Additionally, Abietic acid ameliorates psoriasis-like inflammation and modulates gut microbiota in mice. Abietic acid is promising for research in non-small-cell lung cancer (NSCLC), liver injury-related deseases and psoriasis .
Cucurbitacin B belongs to a class of highly oxidized tetracyclic triterpenoids and is oral active. Cucurbitacin B inhibits tumor cell growth, migration and invasion and cycle arrest, but induces cell apoptosis. Cucurbitacin B has potent anti-inflammatory, antioxidant, antiviral, hypoglycemic, hepatoprotective, neuroprotective activity .
Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Triacetin (Glyceryl triacetate) is a synthetic compound that is a triester of glycerol and acetic acid, orally active. Triacetin increases acetate bioavailability in glioma cells. Triacetin induces glioma cell growtharrest and Apoptosis. Triacetin freely crosses the blood brain barrier/plasma membrane. Triacetin increases histone acetylation and enhances Temozolomide (HY-17364) (TMZ) chemotherapeutic efficacy .
Cyclic-di-GMP disodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP disodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP disodium can be used in cancer research .
Tamarixetin (4'-O-Methyl Quercetin) is an orally active natural flavonoid derivative of quercetin and caseinolytic protease p (ClpP) inhibitor with anti-inflammatory, antioxidant and antitumor effects. Tamarixetin inhibits the hydrolytic activity of ClpP to the fluorescent substrate Suc-LY-AMC with an IC50 of 49.73 μM, which can be used for the study of Staphylococcus aureus infection. Tamarixetin inhibits tumor cell growth, induces apoptosis, and cell cycle arrest. Tamarixetin prevents cardiac hypertrophy by inhibiting the NFAT and AKT pathways .
Terrestrosin D is an orally active apoptosis inducer. Terrestrosin D induces cell cycle arrest at the G1 and S phases, reduces mitochondrial membrane potential, and inhibits the growth of cancer cells and endothelial cells. Terrestrosin D is studied in castration-resistant prostate cancer and pulmonary fibrosis .
8-Gingerol can be found in the rhizome of ginger (Z. officinale) and has oral bioactivity. It activates TRPV1, with an EC50 value of 5.0 µM. 8-Gingerol inhibits COX-2 and also suppresses the growth of H. pylori in vitro. Additionally, 8-Gingerol exhibits anticancer, antioxidant, and anti-inflammatory properties by inhibiting the epidermal growth factor receptor (EGFR) and modulating its downstream STAT3/ERK pathway to suppress the proliferation, migration, and invasion of colorectal cancer cells. 8-Gingerol also exerts immunosuppressive effects by inhibiting oxidative stress, inducing cell cycle arrest, promoting apoptosis, and regulating autophagy. Furthermore, 8-Gingerol has cardioprotective effects. 8-Gingerol is promising for research in the fields of cancer, infection, immunosuppression, and cardiovascular diseases.
Periplogenin is an orally active cardiac glycoside found in Cortex periplocae. Periplogenin can induce ROS production and necroptosis and cause G0/G1 phase arrest. Periplogenin can inhibit pyroptosis by regulating the NLRP3/Caspase-1/GSDMD signaling. Periplogenin suppresses growth of prostate carcinoma cells by docking to an ATP1A1 protein pocket and forming a hydrogen bond with T804. Periplogenin can be used for the researches of cancer, inflammation and immunology, such as prostate carcinoma, rheumatoid arthritis and psoriasis .
Antimycin A2 is a selective inhibitor of the cytochrome b-c1 complex in the mitochondrial electron transport chain. Antimycin A2 disrupts mitochondrial membrane potential and produces reactive oxygen species (ROS) by inhibiting electron transfer between cytochrome b and c. Antimycin A2 has bactericidal and piscicidal activity, as well as tumor cell growth inhibitory effects, and can induce S-phase cell cycle arrest and apoptosis in HeLa cells. Antimycin A2 is suitable for research of cervical cancer and fisheries management. Antimycin A2 can be naturally isolated from the fermentation products of Streptomyces sp. strains .
Cyclic-di-GMP is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP can be used in cancer research .
Triptophenolide (Hypolide) is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
Quercetin-3'-O-glucoside is an orally active flavonoid glycoside. Quercetin-3'-O-glucoside reduces liver glucose-6-phosphatase activity, alters serum insulin and glucose levels, and regulates the activities of antioxidant enzymes in the liver and kidney. Quercetin-3'-O-glucoside inhibits DNA topoisomerase II, induces S-phase cell cycle arrest and caspase-3-mediated apoptosis in hepatocellular carcinoma cells. Quercetin-3'-O-glucoside selectively inhibits EGFR-mediated signaling pathways targeting AKT, ERK1/2, FAK and MEK1/2. Quercetin-3'-O-glucoside inhibits growth factor-induced migration and invasion in pancreatic cancer cells. Quercetin-3'-O-glucoside exerts free radical scavenging effects. Quercetin-3'-O-glucoside is applicable to research related to pancreatic cancer, diabetes, hepatocellular carcinoma and malignant tumors .
Cyclic-di-GMP diammonium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP diammonium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP diammonium can be used in cancer research .
Damulin B is a dammarane-type saponin found in Gynostemma pentaphyllum. Damulin B can inhibit cancer cell apoptosis, decrease mitochondrial membrane potential, inhibit ROS production and cause G0/G1 phase arrest. Damulin B can prevent Cisplatin (HY-17394)-induced acute kidney injury and induce hair growth. Damulin B shows anti-inflammation anti-diabetic and anti-obesity effect. Damulin B can be used for the researches of cancer, inflammation, metabolic disease, such as lung cancer, osteoarthritis and diabetes .
Murrayafoline A is a carbazole alkaloid that can be extracted from Murraya tetramera. Murrayafoline A directly targets Specificity protein 1 (Sp1), thereby inhibiting NF-κB and MAPK signaling pathways. Murrayafoline a induces a G0/G1-phase arrest in platelet-derived growth factor (PDGF)-stimulated vascular smooth muscle cells. Murrayafoline A attenuates the Wnt/β-catenin pathway by promoting the degradation of intracellular β-catenin proteins. Murrayafoline A enhances the contraction of rat ventricular myocytes and L-type calcium current by activating protein kinase C. Murrayafoline A inhibits LPS (HY-D1056)-induced neuroinflammation in vivo. Murrayafoline A can be used for the study of inflammation, vascular complications and colon cancer .
Casuarinin is an orally active antiproliferative, anti-inflammatory, antifungal, virucidal and gastroprotective agent. Casuarinin upregulates the expression of p21/WAF1, Fas/APO‑1, mFasL, sFasL and HSP‑70, arrests cell cycle, induces apoptosis and inhibits cancer cell proliferation. Casuarinin inhibits TNF‑α-induced phosphorylation of MAPK and activation of NF‑κB, downregulates the expression of iNOS, NF‑κB, COX‑2 and ICAM‑1, and reduces the production of proinflammatory mediators. Casuarinin attenuates ethanol-induced activation of caspase‑3 and elevation of TNF‑α, inhibits the growth of Candida albicans, and inhibits HSV‑2. Casuarinin can be used in research related to mammary adenocarcinoma, inflammatory skin diseases, gastric ulcers, candidiasis and herpes simplex virus infections .
6-Methoxydihydrosanguinarine is an alkaloid with activity across multiple cancer cell types. 6-Methoxydihydrosanguinarine activates IRE1/JNK signaling, blocks Akt/mTOR and PI3K/AKT/mTOR pathways, reduces expression of Cdc25C, CyclinB1, Cdc2, YAP/TAZ, Survivin, GPX4, and EGFR, upregulates IRE1 and DR5, and activates JNK and caspases. 6-Methoxydihydrosanguinarine induces apoptosis, G2/M phase arrest, DNA damage, ROS generation, lipid peroxidation, ferroptosis, autophagy, and suppresses cancer cell growth. 6-Methoxydihydrosanguinarine disruptes the biofilm formation of Candida albicans (C. albicans). 6-Methoxydihydrosanguinarine can be used for the research of non-small cell lung cancer, hepatocellular carcinoma, melanoma, colon carcinoma, ovarian cancer and breast cancer .
2-Hydroxy-3-methylanthraquinone (compound 1) is a natural compound isolated from a water extract of Hedyotis diffusa WILLD. 2-Hydroxy-3-methylanthraquinone shows inhibitory activity against protein tyrosine kinases v-src and pp60src, and induces growtharrest and apoptosis in the HepG2 cancer cells .
8α-Tigloyloxyhirsutinolide 13-O-acetate (8αTGH) is a potent and orally active STAT3 inhibitor. 8α-Tigloyloxyhirsutinolide 13-O-acetate induces early oxidative stress and pyroptosis, and late DNA damage, cell cycle arrest, apoptosis in the TNBC cells. 8α-Tigloyloxyhirsutinolide 13-O-acetate suppresses tumor cell growth in vitro and tumor growth in vivo .
Globulol is a terpenoid metabolite and Antimicrobial agent. Globulol can be isolated from Alpinia oxyphylla Miq. Globulol binds to PAK4, reduces the expression level of PAK4 in cancer cells, decreases the phosphorylation of AKT, and downregulates the expressions of STAT3, phosphorylated STAT3, and PD-L1. Globulol promotes the secretion of CCL4 by cancer cells. Globulol reduces the viability and proliferation ability of cancer cells, induces G0/G1 cell cycle arrest and Apoptosis in cancer cells, and inhibits cancer cell migration and the integrity of 3D tumor spheres. Globulol enhances the relevant effects of anti-PD-1 agents in the cancer cell microenvironment. Globulol exhibits anticancer activity against liver cancer. Globulol inhibits the mycelial growth of phytopathogenic fungi and the growth of phytopathogenic bacteria. Globulol can be used in studies related to hepatocellular carcinoma .
Isocryptotanshinone is a dual STAT3 and PTP1B (IC50 = 56.1 μM) inhibitor. Isocryptotanshinone inhibits STAT3 by binding to the STAT3 SH2 domain to block phosphorylation and nuclear translocation [1][2]. Isocryptotanshinone exerts its anti-proliferative effect via the induction of cell cycle arrest, apoptosis, and pro-death autophagy, through the regulation of STAT3, AKT/mTOR and MAPK signaling pathways. Isocryptotanshinone suppresses the xenograft gastric cancer (GC) tumor growth in BALB/c nude mice. Isocryptotanshinone can be used for cancer research, such as lung cancer, breast cancer and GC .
Strictinin is an orally active phenolic compound. Strictinin reduces xanthine oxidase activity, uric acid production, and the activation of ERK1/2, JNK, NF-κB, and NLRP3 inflammasome components in hepatocytes treated with Xanthine (HY-W017389). Strictinin decreases elevated serum uric acid levels and enhanced xanthine oxidase activity in mice treated with potassium oxonate. Strictinin acts as a ROR1 inhibitor and exhibits anticancer activity against highly aggressive non-androgen-dependent prostate cancer. Strictinin induces cancer cell apoptosis (apoptosis), arrests cell cycle, and inhibits cancer cell migration, invasion, and epithelial-mesenchymal transition. Strictinin modulates gut microbiota, inhibits bacterialgrowth and biofilm formation, accelerates small intestinal transit, and blocks viral entry and replication. Strictinin can be used in research related to hyperuricemia, androgen receptor-negative non-androgen-dependent prostate cancer, triple-negative breast cancer, bacterial infections, constipation, coronavirus infections, dental caries, and infections caused by influenza A, influenza B, and human parainfluenza virus type 1 .
FK-3000 is a potent anti-tumor agent that inhibits the growth of carcinoma cells through apoptosis and induction cell cycle arrest. FK-3000 also exhibit antiviral effects against HSV-1 and HIV-1 .
Costunolide (Standard) is the analytical standard of Costunolide. This product is intended for research and analytical applications. Costunolide ((+)-Costunolide) is a naturally occurring sesquiterpene lactone, with antioxidative, anti-inflammatory, antiallergic, bone remodeling, neuroprotective, hair growth promoting, anticancer, and antidiabetic properties. Costunolide can induce cell cycle arrest and apoptosis on breast cancer cells .
Santamarine (Santamarin; Balchanin) is a sesquiterpene lactone found in Artemisia scoparia. Santamarine shows anti-inflammatory, antioxidant, anticancer and anti-photoaging activities. Santamarine suppresses UVA-induced phosphorylation of JNK and p38 MAPK, nuclear translocation of phosphorylated c-Fos and c-Jun, and AP-1-mediated MMP-1 transcription and secretion. Santamarine suppresses NF-κB signaling, iNOS, COX-2, TNF-α, and IL-1β production. Santamarine inhibits thioredoxin reductase activity, induces ROS production, mitochondrial apoptosis, G2/M cell cycle arrest, and DNA damage, and reduces cancer cell growth. Santamarine can be used for the photoaging, inflammatory diseases and cancer .
2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cell cycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
Cyclic-di-GMP sodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP sodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP sodium can be used in cancer research .
(-)-β-Peltatin is an aryltetrahydronaphthalene lignan. (-)-β-Peltatin exhibits antitumor activity and cytotoxicity against pancreatic cancer cells. (-)-β-Peltatin induces G2/M cell cycle arrest and apoptosis in pancreatic cancer cells. (-)-β-Peltatin inhibits the growth of subcutaneous xenografts of pancreatic cancer cells in nude mice. (-)-β-Peltatin can be used in pancreatic cancer-related research .
Tamarixetin (Standard) is the analytical standard of Tamarixetin. This product is intended for research and analytical applications. Tamarixetin (4'-O-Methyl Quercetin) is an orally active natural flavonoid derivative of quercetin and caseinolytic protease p (ClpP) inhibitor with anti-inflammatory, antioxidant and antitumor effects. Tamarixetin inhibits the hydrolytic activity of ClpP to the fluorescent substrate Suc-LY-AMC with an IC50 of 49.73 μM, which can be used for the study of Staphylococcus aureus infection. Tamarixetin inhibits tumor cell growth, induces apoptosis, and cell cycle arrest. Tamarixetin prevents cardiac hypertrophy by inhibiting the NFAT and AKT pathways .
Asparanin A is an apoptosis inducer with anticancer activity. Asparanin A induces cell cycle arrest in the G0/G1 phase through mitochondria and PI3K/AKT signaling pathways, inhibiting cancer cell growth. Asparanin A also demonstrated in vivo efficacy in a mouse xenograft model of Ishikawa endometrial carcinoma, significantly inhibiting tumor growth .
Cucurbitacin B (Standard) is the analytical standard of Cucurbitacin B. This product is intended for research and analytical applications. Cucurbitacin B belongs to a class of highly oxidized tetracyclic triterpenoids and is oral active. Cucurbitacin B inhibits tumor cell growth, migration and invasion and cycle arrest, but induces cell apoptosis. Cucurbitacin B has potent anti-inflammatory, antioxidant, antiviral, hypoglycemic, hepatoprotective, neuroprotective activity .
Phoyunbene B is a similar substance to Resveratrol (HY-16561). Phoyunbene B exhibits stronger growth inhibitory activity against human liver cancer cells HepG2 compared to Resveratrol. Phoyunbene B induces G2/M phase cell cycle arrest and apoptosis. Phoyunbene B increases Bax/Bcl-2 and activates Caspase-3. Phoyunbene B inhibits the invasion and migration of cancer cells. Phoyunbene B can be used for research on liver cancer .
Lepadin H is a ferroptosis inducer and apoptosis inducer with in vitro cytotoxicity and in vivo antitumor efficacy against cancer cells. Lepadin H reduces GPX4 and SLC7A11 levels, increases p53 and ACSL4 expression, drives lipid hydroperoxide production, elevates reactive oxygen species (ROS) levels, reduces cellular glutathione (GSH) levels, induces lipid peroxidation and G2/M phase cell cycle arrest, and suppresses clonogenic growth and migration of cancer cells.Lepadin H can be used for the research of melanoma .
Sotetsuflavone is a flavonoid that can be isolated from Cycas revolute. Sotetsuflavone inhibits phosphorylation of PI3K, Akt, mTOR, JNK, and p38 MAPK; modulates expression of Cyclin D1, CDK4, Bcl-2, Bax, cleaved caspases 3/9, MMP-9, TGF-β, STAT3, and β-catenin. Sotetsuflavone induces G0/G1 cell cycle arrest, apoptosis, autophagy, and intracellular ROS elevation, inhibits cancer cell proliferation. Sotetsuflavone inhibits tumor growth in mouse tumor xenograft models. Sotetsuflavone can be used for the research of non-small cell lung cancer and Crohn’s disease .
Azadirachtin D is an insect growth regulator targeting ecdysone receptor (EcR) in insects. Azadirachtin D blocks the 20-hydroxyecdysone signaling pathway, leading to developmental arrest or death in insects. Azadirachtin D is promising for research of agricultural pests .
Reticulol (K 251-1) is an inhibitor of cyclic adenosine 3', 5'-monophosphate phosphodiesterase. Reticulol shows antitumor activity independent with cell cycle arrest or apoptosis. Reticulol inhibits cell growth of murine melanoma cells and human lung tumor cells. Reticulol protects its lung metastasis via the bloodstream by inhibiting the growth of B16F10 melanoma .
Cinobufagin (Standard) is the analytical standard of Cinobufagin. This product is intended for research and analytical applications. Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
5,8-Epidioxyergosta-6,9(11),22-trien-3-ol (9,11-Dehydroergosterol peroxide), an important steroid from medicinal mushroom, exerts antitumor activity in several tumor types. 5,8-Epidioxyergosta-6,9(11),22-trien-3-ol inhibits HT29 cell growth by inducing CDKN1A expression, thus causing cell cycle arrest and apoptosis .
Cernuumolide J (TMJ-105) is an JAK2/STAT3 inhibitor. Cernuumolide J induces G2/M phase arrest and apoptosis in HEL leukemia cells by downregulating the phosphorylation of JAK2, STAT3, and Erk, and activating the phosphorylation of JNK and p38 MAPK. Cernuumolide J inhibits HEL leukemia cell growth in a time- and concentration-dependent manner, with an IC50 value of 1.79 μM. Cernuumolide J can be used for research in the field of anti-cancer therapy .
Triptophenolide (Standard) (Hypolide) is the analytical standard of Triptophenolide (HY-N0475). This product is intended for research and analytical applications. Triptophenolide is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
Cinosulfuron (Standard) is the analytical standard of Cinosulfuron. This product is intended for research and analytical applications. Gypsogenin is a selective mixed-type BChE inhibitor (Ki=19.99 μM) that also exhibits significant cytotoxicity against various human cancer cell lines. Gypsogenin inhibits tumor growth by inducing cell cycle arrest and triggering apoptosis. Gypsogenin displays antibacterial activity against bacteria such as Bacillus subtilis and Bacillus thuringiensis, and often serves as a key parent nucleus for the synthesis of anticancer compounds. Gypsogenin is widely used in research on Alzheimer's disease and various cancers including colon cancer, melanoma, and leukemia .
Periplogenin (Standard) is the analytical standard of Periplogenin (HY-N2414). Periplogenin is an orally active cardiac glycoside found in Cortex periplocae. Periplogenin can induce ROS production and necroptosis and cause G0/G1 phase arrest. Periplogenin can inhibit pyroptosis by regulating the NLRP3/Caspase-1/GSDMD signaling. Periplogenin suppresses growth of prostate carcinoma cells by docking to an ATP1A1 protein pocket and forming a hydrogen bond with T804. Periplogenin can be used for the researches of cancer, inflammation and immunology, such as prostate carcinoma, rheumatoid arthritis and psoriasis .
1,4-Dihydroxy-2-naphthoic acid (Standard) is the analytical standard for 1,4-Dihydroxy-2-naphthoic acid (HY-W014701). 1,4-Dihydroxy-2-naphthoic acid is an orally active aryl hydrocarbon receptor (AhR) agonist and a bifidogenic growth stimulator. 1,4-Dihydroxy-2-naphthoic acid can improve the motor dysfunction in parkinson's disease (PD) model through AhR-dependent and -independent pathways. 1,4-Dihydroxy-2-naphthoic acid exerts anti-inflammatory effects by regulating the gut microbiota (such as promoting the proliferation of Bifidobacterium) and directly regulating the host immune system. 1,4-Dihydroxy-2-naphthoic acid induces apoptosis through G0/G1 cell cycle arrest in human keratinocyte to inhibit psoriasis .
Chlorpromazine (hydrochloride) (Standard) is the analytical standard of Chlorpromazine (hydrochloride). This product is intended for research and analytical applications. Chlorpromazine hydrochloride is an orally active, blood-brain barrier-transparent antipsychotic agent that effectively antagonises D2 dopamine receptors and 5-HT2A, which is widely used in schizophrenia and other psychiatric disorders. Chlorpromazine hydrochloride exerts anti-cancer activity through a variety of pathways, including anti-proliferation, induction of autophagy and cycle arrest (G2-M phase), inhibition of cytochrome c oxidase (CcO), inhibition of tumour growth and metastasis, and inhibition of tumour immune escape. Chlorpromazine hydrochloride also blocks hNav1.7 channels (IC50=25.9 μM; concentration-dependent) and HERG potassium channels (IC50=21.6 μM), which has potential for analgesic and cardiac arrhythmic studies. Chlorpromazine hydrochloride also can inhibit clathrin-mediated endocytosis .
2,6-Dihydroxyacetophenone (Standard) is the analytical standard of 2,6-Dihydroxyacetophenone (HY-Y0106). This product is intended for research and analytical applications. 2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cell cycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
N-Desmethyldauricine is a NF-κB p65 inhibitor and apoptosis inducer. N-Desmethyldauricine reduces the protein expression level of p65, induces apoptosis, arrests the cell cycle at the G0/G1 phase, attenuates intercellular adhesion, and inhibits the growth of 3D spheroids of triple-negative breast cancer. N-Desmethyldauricine can be used in studies related to triple-negative breast cancer .
Clausenidin is a selective inhibitor targeting apoptosis-related pathways, including the mitochondrial pathway and death receptor pathway, and vascular endothelial growth factor (VEGF). Clausenidin induces mitochondrial membrane depolarization by activating caspase-3, caspase-8 and caspase-9, upregulating the pro-apoptotic protein Bax and downregulating the anti-apoptotic protein Bcl-2. Clausenidin also inhibits VEGF expression and blocks angiogenesis, exerting anti-tumor activity. Clausenidin has inhibitory effects against Mycobacterium tuberculosis (MIC=200 μg/mL). Clausenidin can induce apoptosis in liver cancer cells, arrest the cell cycle in the G2/M phase, and inhibit tumor angiogenesis. Clausenidin can be used in the research of malignant tumors such as liver cancer .
Aspulvinone H is an orally active inhibitor of AChE, pancreatic lipase, glutamic-oxaloacetic transaminase 1, and α-glucosidase, with IC50 values of 25.95 μM, 47.06 μM, 5.91/6.91 μM, and 4.6 μM, respectively. It has a Ka of 2.14 μM against GOT1 and a Ki of 6.58 μM against α-glucosidase. Aspulvinone H inhibits cancer cell proliferation, interferes with glutamine metabolism, elevates ROS levels, and induces cell apoptosis and S-phase arrest. Aspulvinone H exhibits antibacterial activity against Staphylococcus aureus. Aspulvinone H inhibits the growth of pancreatic ductal adenocarcinoma xenografts. Aspulvinone H reduces postprandial blood glucose in mice. Aspulvinone H can be used in research related to pancreatic ductal adenocarcinoma, diabetes, and Staphylococcus aureus infection .
Murrayafoline A (Standard) is the analytical standard of Murrayafoline A (HY-W100287). This product is intended for research and analytical applications. Murrayafoline A is a carbazole alkaloid that can be extracted from Murraya tetramera. Murrayafoline A directly targets Specificity protein 1 (Sp1), thereby inhibiting NF-κB and MAPK signaling pathways. Murrayafoline a induces a G0/G1-phase arrest in platelet-derived growth factor (PDGF)-stimulated vascular smooth muscle cells. Murrayafoline A attenuates the Wnt/β-catenin pathway by promoting the degradation of intracellular β-catenin proteins. Murrayafoline A enhances the contraction of rat ventricular myocytes and L-type calcium current by activating protein kinase C. Murrayafoline A inhibits LPS (HY-D1056)-induced neuroinflammation in vivo. Murrayafoline A can be used for the study of inflammation, vascular complications and colon cancer .
Cajanol is an isoflavanone that can be isolated from the roots of Cajanus cajan (L.) Millsp. . Cajanol inhibits cancer cell proliferation and induces cancer cell apoptosis. Cajanol promotes the expression of Bax, inhibits the expression of Bcl-2, activates caspase-9 and caspase-3, induces PARP cleavage, arrests the cell cycle at the G2/M phase, generates ROS, disrupts mitochondrial membrane potential and triggers cytochrome c release. Cajanol induces bacterial DNA damage, disrupts bacterial cell membranes, and exerts antibacterial activity in vitro. Cajanol reduces the expression of PI3K, inhibits the phosphorylation of Akt and NF-κB, downregulates the expression and transport function of P-gp, restores the sensitivity of drug-resistant cancer cells to Paclitaxel, and inhibits the growth of Paclitaxel-resistant metastatic ovarian tumors. Cajanol is applicable to research related to breast cancer, ovarian cancer and bacterial infections .
6-Methoxydihydrosanguinarine hydrochloride is an alkaloid with activity across multiple cancer cell types. 6-Methoxydihydrosanguinarine hydrochloride activates IRE1/JNK signaling, blocks Akt/mTOR and PI3K/AKT/mTOR pathways, reduces expression of Cdc25C, CyclinB1, Cdc2, YAP/TAZ, Survivin, GPX4, and EGFR, upregulates IRE1 and DR5, and activates JNK and caspases. 6-Methoxydihydrosanguinarine hydrochloride induces apoptosis, G2/M phase arrest, DNA damage, ROS generation, lipid peroxidation, ferroptosis, autophagy, and suppresses cancer cell growth. 6-Methoxydihydrosanguinarine hydrochloride disruptes the biofilm formation of Candida albicans (C. albicans). 6-Methoxydihydrosanguinarine hydrochloride can be used for the research of non-small cell lung cancer, hepatocellular carcinoma, melanoma, colon carcinoma, ovarian cancer and breast cancer .
GAS6 Protein is a secreted protein located outside the cell membrane. GAS6 Protein is a ligand for tyrosine protein kinase receptors AXL, TYRO3, and MER, and its signal transduction is related to cell growth and survival, cell adhesion, and cell migration. GAS6 is associated with tumor cell growth, metastasis, invasion, epithelial mesenchymal transition, angiogenesis, drug resistance, immune regulation, and stem cell maintenance after binding to AXL. GAS6 Protein, Human (HEK293, Fc) is a recombinant GAS6 protein with an Fc tag, expressed by HEK293.
The GAS6 protein is a ligand for AXL, TYRO3 and MER receptors and affects a variety of cellular processes.GAS6/AXL signaling promotes endothelial cell survival, aids cytokine signaling in natural killer cell development, supports liver regeneration, affects gonadotropin-releasing hormone neuron survival and migration, modulates platelet activation, and regulates thrombotic responses.GAS6 Protein, Human (HEK293, His) is the recombinant human-derived GAS6 protein, expressed by HEK293 , with C-6*His labeled tag.
The GAS6 protein is a ligand for AXL, TYRO3 and MER receptors and affects a variety of cellular processes. GAS6/AXL signaling promotes endothelial cell survival, aids cytokine signaling in natural killer cell development, supports liver regeneration, affects gonadotropin-releasing hormone neuron survival and migration, modulates platelet activation, and regulates thrombotic responses. GAS6 Protein, Mouse (HEK293, His) is the recombinant human-derived GAS6 protein, expressed by HEK293 , with C-6*His labeled tag.
GAS7 Protein potentially facilitates the maturation and morphological differentiation of cerebellar neurons. GAS7 Protein, Human (His) is the recombinant human-derived GAS7 protein, expressed by E. coli , with N-6*His labeled tag.
rHugrowtharrest and DNA damage-inducible protein GADD45 beta/GADD45B, His; growtharrest and DNA Damage-Inducible Protein GADD45 Beta; Myeloid Differentiation Primary Response Protein MyD118; Negative growth Regulatory Protein MyD118; GADD45B; MYD118
GADD45B Protein, a pivotal mediator, regulates growth and apoptosis by activating the MTK1/MEKK4 MAPKKK signaling pathway. Interacting with GADD45GIP1, it forms a complex that modulates cellular responses under stress. Its central role in growth and apoptosis underscores its significance in orchestrating cellular processes, positioning GADD45B as a key player in the intricate network of signaling pathways governing cell fate decisions. GADD45B Protein, Human (His) is the recombinant human-derived GADD45B protein, expressed by E. coli , with N-6*His labeled tag.
rHugrowtharrest and DNA damage-inducible protein GADD45 alpha/GADD45A, His; growtharrest and DNA Damage-Inducible Protein GADD45 Alpha; DNA Damage-Inducible Transcript 1 Protein; DDIT-1; GADD45A; DDIT1; GADD45
GADD45A/DDIT-1 protein regulates p38 MAPK by inhibiting p88 phosphorylation in T cells, thereby affecting cell signaling pathways. It can regulate the PCNA-CDK complex, promote DNA excision repair, and hinder S phase entry. GADD45A/DDIT-1 Protein, Human (His) is the recombinant human-derived GADD45A/DDIT-1 protein, expressed by E. coli , with N-6*His labeled tag.
Apoptosis associated protein; GADD 34; growtharrest and DNA damage inducible 34; growtharrest and DNA damage-inducible protein GADD34; Myeloid differentiation primary response protein MyD116 homolog; Ppp1r15a; PR15A_HUMAN; Protein phosphatase 1 regulatory (inhibitor) subunit 15A; Protein phosphatase 1 regulatory subunit 15A
rHugrowtharrest and DNA damage-inducible protein GADD45 gamma/GADD45G, His; growtharrest and DNA Damage-Inducible Protein GADD45 Gamma; Cytokine-Responsive Protein CR6; DNA Damage-Inducible Transcript 2 Protein; DDIT-2; GADD45G; CR6; DDIT2
GADD45G/DDIT2 protein is a key mediator that regulates growth and apoptosis through the MTK1/MEKK4 MAPKKK signaling pathway. Concentration-dependent homodimerization enhances its growth inhibitory activity and interaction with PCNA, suggesting a role in DNA repair. GADD45G/DDIT2 Protein, Human (His) is the recombinant human-derived GADD45G/DDIT2 protein, expressed by E. coli , with N-6*His labeled tag.
3-Methyl-2-quinoxalinecarboxylic acid-d4 is the deuterium labeled 3-Methyl-2-quinoxalinecarboxylic acid. 3-Methyl-2-quinoxalinecarboxylic acid (MQCA), an important N-oxide reductive metabolite of Quinocetone or Olaquindox, potently inhibits the growth of Chang liver cells through S phase arrest of the cell cycle .
Oxcarbazepine-d4 (GP 47680-D4) is the deuterium labeled Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
Oxcarbazepine-d4-1 is deuterium labeled Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Anti-cancer and anticonvulsant effects .
Sulindac Sulfone-d6 is the deuterium labeled Sulindac sulfone (HY-B1787). Sulindac sulfone is an mTORC1 pathway inhibitor and a metabolite of Sulindac. Sulindac sulfone inhibits colon cancer cell growth and induces cell cycle arrest. Sulindac sulfone is used in cancer research .
Oxcarbazepine-d8-1 is a deuterium of Oxcarbazepine. Oxcarbazepine is a sodium channel blocker . Oxcarbazepine significantly inhibits glioblastoma cell growth and induces apoptosis or G2/M arrest in glioblastoma cell lines . Oxcarbazepine-d8-1 has anti-cancer and anticonvulsant effects .
Cinobufagine-d3 is the deuterium labeled Cinobufagin (HY-N0421). Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cell cycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
13C20, 15N10-Cyclic di-GMP ( 13C20, 15N10-c-di-GMP) is 13C and 15N labeled Cyclic-di-GMP (disodium). Cyclic-di-GMP disodium is a STING agonist and a bacterial second messenger that coordinates different aspects of bacterial growth and behavior, including motility, virulence, biofilm formation, and cell cycle progression. Cyclic-di-GMP disodium has anti-cancer cell proliferation activity and also induces elevated CD4 receptor expression and cell cycle arrest. Cyclic-di-GMP disodium can be used in cancer research .
15-Deoxy-Δ12,14-Prostaglandin D2-d4 (15-Deoxy-Δ12,14-PGD2-d4) is the deuterium labeled 15-deoxy-Δ12,14-Prostaglandin D2. 15-deoxy-Δ12,14-Prostaglandin D2 (15-Deoxy-Δ12,14-PGD2) is a metabolite of prostaglandin D₂ (PGD₂) (HY-101988), which can undergo further dehydration metabolism to 15-deoxy-Δ12,14-PGJ₂. 15-deoxy-Δ12,14-Prostaglandin D2 is a highly selective agonist for DP2 receptor and PPARγ. 15-deoxy-Δ12,14-Prostaglandin D2 causes morphological changes in eosinophils and migration of type II innate lymphoid cells (ILC2). 15-deoxy-Δ12,14-Prostaglandin D2 has a growth inhibitory effect on prostate cancer cells expressing PPARγ, induces cell cycle arrest and promotes apoptosis. 15-deoxy-Δ12,14-Prostaglandin D2 can be used in related research on asthma and prostate cancer.
C6 Ceramide- 13C2,d2 (C6-Cer- 13C2,d2) is the deuterium labeled and 13C-labeled C6 Ceramide (HY-19542). C6 Ceramide (C6-Cer) is a short-chain, cell-permeable ceramide pathway activator with anticancer activity. C6 Ceramide-mediated miR-29b expression participates in the progression of multiple myeloma through suppressing the proliferation, migration and angiogenesis of endothelial cells by targeting Akt signal pathway. C6 Ceramide exhibits multiple anti-cancer properties including cell cycle arrest, Apoptosis, inhibition of tumor growth and enhances the effects of chemotherapy in drug-resistant cancer cells. C6-ceramide can be used as an adjuvant for chemotherapeutic agents, to enhance anti-tumor effects .
GAS 2 antibody; GAS-2 antibody; Gas2 antibody; GAS2_HUMAN antibody; growtharrest specific 2 antibody; growtharrest specific protein 2 antibody; growtharrest-specific protein 2 antibody; MGC32610 antibody;
WB, IHC-P
Human, Mouse, Rat
GAS2 Antibody (YA6580) is a Rabbit-derived and non-conjugated IgG monoclonal antibody, targeting to GAS2.
GAS7 (Growth Arrest Specific Protein 7) Antibody (YA8225) is a Mouse-derived and non-conjugated IgG2a monoclonal antibody, targeting to GAS7 (Growth Arrest Specific Protein 7).
GAS7 (Growth Arrest Specific Protein 7) Antibody (YA8225) is a Mouse-derived and non-conjugated IgG2a monoclonal antibody, targeting to GAS7 (Growth Arrest Specific Protein 7).
DDIT 1; DDIT-1; DDIT1; DNA damage inducible transcript 1; DNA damage-inducible transcript 1 protein; GA45A_HUMAN; GADD45; GADD45A; growtharrest and DNA damage inducible 45 alpha; growtharrest and DNA damage inducible alpha; growtharrest and DNA damage-inducible protein GADD45 alpha.
WB
Transfected
GADD45A Antibody (YA5298) is a Mouse-derived and non-conjugated monoclonal antibody, targeting to GADD45A.
GADD153; CHOP; growtharrest and DNA damage-inducible 153; C/EBP homologous protein; C/EBP Homology Protein; CEBPZ; CHOP10; DDIT 3; DNA Damage Inducible Transcript 3; GADD 153; growtharrest and DNA Damage Inducible Protein 153; growtharrest and DNA damage inducible protein GADD153; MGC4154; DDIT3_HUMAN.
WB, IHC-P, ICC/IF
Human, Mouse, Rat
DDIT3 Antibody is a Mouse-derived and non-conjugated IgG1 monoclonal antibody, targeting to DDIT3.
GADD153; CHOP; growtharrest and DNA damage-inducible 153; C/EBP homologous protein; C/EBP Homology Protein; CEBPZ; CHOP10; DDIT 3; DNA Damage Inducible Transcript 3; GADD 153; growtharrest and DNA Damage Inducible Protein 153; growtharrest and DNA damage inducible protein GADD153; MGC4154; DDIT3_HUMAN.
WB, IHC-P, ICC/IF
Human, Mouse, Rat
DDIT3 Antibody is a Mouse-derived and non-conjugated IgG1 monoclonal antibody, targeting to DDIT3.
GADD153; CHOP; growtharrest and DNA damage-inducible 153; C/EBP homologous protein; C/EBP Homology Protein; CEBPZ; CHOP10; DDIT 3; DNA Damage Inducible Transcript 3; GADD 153; growtharrest and DNA Damage Inducible Protein 153; growtharrest and DNA damage inducible protein GADD153; MGC4154; DDIT3_HUMAN.
WB, IHC-P, IHC-F, IF-Tissue
Human, Mouse, Rat
DDIT3 Antibody is a Rabbit-derived and non-conjugated IgG recombinant monoclonal antibody, targeting to DDIT3.
GADD153; CHOP; growtharrest and DNA damage-inducible 153; C/EBP homologous protein; C/EBP Homology Protein; CEBPZ; CHOP10; DDIT 3; DNA Damage Inducible Transcript 3; GADD 153; growtharrest and DNA Damage Inducible Protein 153; growtharrest and DNA damage inducible protein GADD153; MGC4154; DDIT3_HUMAN.
WB, IHC-P, IHC-F, IF-Tissue
Human, Mouse, Rat
DDIT3 Antibody is a Rabbit-derived and non-conjugated IgG recombinant monoclonal antibody, targeting to DDIT3.
SIAIS039 is an orally active c-ros oncogene 1 (ROS1)-specific PROTAC with DC50s of 154.46 nM, 126.47 nM, 143.69 nM for HCC78 cells, Ba/F3 expressing the CD74-ROS1 fusion and Ba/F3 expressing the SDC4-ROS1 fusion, respectively. SIAIS039 suppresses cell proliferation, induces cell cycle arrest and apoptosis, and inhibits clonogenicity against ROS1-positive cells. SIAIS039 demonstrates anti-tumour effects against ROS1-driven tumor growth vivo. SIAIS039 is composed of the ALK inhibitor Brigatinib (HY-12857), a linker EM-12 (HY-138793), and a VHL ligand E3 ubiquitin ligase 1-Butyne (Red: Brigatinib; Blue: VHL ligand; Black: linker) .
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1 cell cycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
Prexigebersen (BP1001) is an antisense oligonucleotide targeting Bcl-2 and Grb2. Prexigebersen exhibits antileukemic activity in cell models. Prexigebersen induces apoptosis (apoptosis), cell cycle arrest and ROS production in leukemia cells. Prexigebersen inhibits Grb2 expression, thereby suppressing tumor growth and survival. Prexigebersen can be used in studies related to acute myeloid leukemia .
GEM231 is an 18mer antisense oligonucleotide targeting the mRNA of the PKA-I (RIα regulatory subunit of cAMP dependent protein kinase type I ). GEM231 induces cell growtharrest, apoptosis, and differentiation in a variety of cancer cell lines in vitro and in tumors in vivo.
GEM231 sodium is an 18mer antisense oligonucleotide targeting the mRNA of the PKA-I (RIα regulatory subunit of cAMP dependent protein kinase type I ). GEM231 sodium induces cell growtharrest, apoptosis, and differentiation in a variety of cancer cell lines in vitro and in tumors in vivo.
Human TGFBR2 mRNA encodes the human transforming growth factor beta receptor 2 (TGFBR2) protein, a transmembrane protein that has a protein kinase domain, forms a heterodimeric complex with TGF-beta receptor type-1, and binds TGF-beta. TGFBR2/ligand complex phosphorylates proteins, which then enter the nucleus and regulate the transcription of genes related to cell proliferation, cell cycle arrest, wound healing, immunosuppression, and tumorigenesis.
GYY4137 (GMP) is the GMP-grade version of GYY4137 (HY-107632). Small molecules of GMP grade can be used as adjuvant reagents in cell therapy. GYY4137 (GMP) is a sustained-release H2S donor possessing vasodilatory, antihypertensive, and anti-inflammatory activities. GYY4137 (GMP) can inhibit cell growth, induce apoptosis, and cause cell cycle arrest by blocking the STAT3 pathway, demonstrating potent anticancer activity .
Inquiry Online
Your information is safe with us. * Required Fields.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
MedchemExpress Validation 03
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
MedchemExpress Validation 04
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedChemExpress values your privacy and your trust is important to us. We use cookies to enhance your website experience. Some cookies are necessary to run the website.
Privacy and Cookie Policy