From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
Indisulam (E 7070) is a carbonic anhydrase inhibitor with anticancer activity. Indisulam (E 7070) is a sulfonamide agent that targets the G1 phase of the cellcycle. Indisulam (E 7070) causes a blockade in the G1/S transition through inhibition of the activation of both CDK2 and cyclin E. Indisulam (E 7070) targets splicing by inducing RBM39 degradation via recruitment to DCAF15 .
Milademetan (DS-3032) is a specific and orally active MDM2 inhibitor for the research of acute myeloid leukemia (AML) or solid tumors. Milademetan (DS-3032) induces G1cellcycle arrest, senescence and apoptosis .
Danazol inhibits the proliferation of cancer cell MDA-MB-231 and MCF-7 with IC50 of 65 µg/mL and 31 µg/mL. Danazol arrests the cellcycle at G1 phase, induces apoptosis in MDA-MB-231 through PKCα signaling pathway .
Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Avadomide is an orally active cereblon modulator. Avadomide modulates cereblon E3 ligase activity, inhibits NF-κB pathway, arrests the cellcycle at G1 phase, and thus induces apoptosis in cancer cell PDAC. Avadomide exhibits potent antitumor and immunomodulatory activities .
T2AA is a monoubiquitinated proliferating cell nuclear antigen (PCNA) inhibitor that prevents DNA repair, increases double-strand break (DSB) formation and promotes necroptosis and cellcycle arrest in G1 phase .
CDK2 degrader 3 is a selective CDK2 molecular glue-like degrader. CDK2 degrader 3 induces G1cellcycle arrest in CCNE1-amplified cancer cells. CDK2 degrader 3 is applicable to breast cancer-related research .
Protosappanin B is a phenolic compound extracted from Caesalpinia sappan. Anti-cancer activity . Protosappanin B induces apoptosis and causes G1cellcycle arrest in human bladder cancer cells .
Meisoindigo (Dian III), a derivative of Indirubin (HY-N0117), halts the cellcycle at the G0/G1 phase and induces apoptosis in primary acute myeloid leukemia (AML) cells. Meisoindigo exhibits high antitumor activity .
Topotecan (SKF 104864A; NSC 609669) is an orally active and potent Topoisomerase I inhibitor. Topotecan induces cellcycle arrest in G0/G1 and S phases and promotes apoptosis. Topotecan shows anticancer activity .
Phenoxodiol (Idronoxil), a synthetic analog of Genestein, activates the mitochondrial caspase system, inhibits XIAP (an apoptosis inhibitor), and sensitizes the cancer cells to Fas-mediated apoptosis. Phenoxodiol also inhibits DNA topoisomerase II by stabilizing the cleavable complex. Phenoxodiol induces cellcycle arrest in the G1/S phase of the cellcycle and upregulates p21 WAF1 via a p53 independent manner .
Terrestrosin D is an orally active apoptosis inducer. Terrestrosin D induces cellcycle arrest at the G1 and S phases, reduces mitochondrial membrane potential, and inhibits the growth of cancer cells and endothelial cells. Terrestrosin D is studied in castration-resistant prostate cancer and pulmonary fibrosis .
Cyproheptadine hydrochloride sesquihydrate acts as a p38 MAP kinase activator, CHK2 activator, histamine H1 receptor inhibitor and serotonin receptor inhibitor. Cyproheptadine hydrochloride sesquihydrate mediates cellcycle arrest via G1 phase arrest, G1/S transition arrest, G0/G1 phase arrest, reduced expression of cyclins D1/D2/D3, upregulated expression of HBP1, p16, p21, p27, and decreased phosphorylation of retinoblastoma protein. Cyproheptadine hydrochloride sesquihydrate induces Apoptosis by increasing PARP and cleaved PARP, as well as activating the mitochondrial caspase pathway. Cyproheptadine hydrochloride sesquihydrate inhibits tumor growth with extremely low toxicity to normal cells. Cyproheptadine hydrochloride sesquihydrate can be used in research related to hepatocellular carcinoma, multiple myeloma and acute myeloid leukemia .
XEN445 is a potent, selective and orally active endothelial lipase (EL) inhibitor with an IC50 value of 0.237 μM. XEN445 selectively inhibits phospholipase enzymatic activity of LIPG. XEN445 raises plasma HDL and cholesterol levles. XEN445 induces G1cellcycle arrest, reduces cell viability, suppresses cancer stem cell self-renewal, and inhibits tumor formation in LIPG-expressing triple-negative breast cancer cells, while showing no inhibitory effect on invasiveness or cancer stem cell stemness in these cells. XEN445 can be used for the research of cancer and metabolic disease, such as triple-negative breast cancer .
Illudin S, a cytotoxic Illudin, is a natural sesquiterpene with strong anti-tumour and antiviral activities. Illudin S has genotoxic activities. Illudin S blocks the G1-S phase interface of the cellcycle in human leukemia cells .
N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cellapoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma - .
PROTAC CDK4/6 degrader 1 is a CDK4/CDK6 PROTAC degrader, with DC50 values of 10.5 nM and 2.5 nM, respectively. PROTAC CDK4/6 degrader 1 inhibits the proliferation of Jurkat cells (IC50 = 0.18 μM), arrests the cellcycle at the G1 phase, and induces apoptosis. PROTAC CDK4/6 degrader 1 can be used for cancer research.
CVT-11127 is a potent SCD inhibitor. CVT-11127 induces apoposis and arrests the cellcycle at the G1/S phase. CVT-11127 has the potential for the research of lung cancer .
Ganoderic acid DM, a natural triterpenoid isolated from Ganoderma lucidum, induces DNA damage, G1cellcycle arrest and apoptosis in human breast cancer cells. Ganoderic acid DM as a specific inhibitor of osteoclastogenesis .
Dehydrozingerone is a ginger-derived component and cyclin D1 inhibitor that downregulates cyclin D1 expression and induces cellcycleG1 phase arrest. Dehydrozingerone reduces the proliferative capacity of castration-resistant prostate cancer cells under in vitro conditions. Dehydrozingerone reduces subcutaneous tumor growth by inhibiting cell proliferation and angiogenesis. Dehydrozingerone exerts antibacterial and antifungal activities via its α,β-unsaturated carbonyl conjugated system. Dehydrozingerone can be used in studies related to castration-resistant prostate cancer, bacterial infections, and food spoilage fungal infections .
DC-CPin711 is a potent and selective inhibitor of CREB-binding protein (CBP) bromodomain with an IC50 of 0.0626 μM. DC-CPin711 arrests cellcycle at G1 phase and induces apoptosis .
HNHA is a potent HDAC inhibitor with an IC50 of 100 nM. HNHA arrests the cellcycle at the G1/S phase via p21 induction. HNHA inhibits tumor growth and tumor neovascularization. HNHA may be a potent anti-cancer agent against breast cancer .
MIR002 is a potent and orally active DNA polymerase α (POLA1) and HDAC 11 dual inhibitor. MIR002 induces acetylation of p53, activation of p21, G1/Scellcycle arrest, and apoptosis. MIR002 shows significant antitumor activity in vivo .
Suberosin, isolated from Plumbago zeylanica, exhibits anti-inflammatory and anticoagulant activity. Suberosin suppresses PHA-induced PBMC proliferation and arrested cellcycle progression from the G1 transition to the S phase through the modulation of the transcription factors NF-AT and NF-κB .
A-800141 is an orally active, selective, sulfonamide-based MetAP2 inhibitor (IC50=12 nM) that binds reversibly to MetAP2 and interacts with its manganese ions. A-800141 induces the production of N-terminal methionine-unprocessed GAPDH variants, which in turn triggers G1-phasecellcycle arrest, elevates p21 levels, and reduces the levels of phosphorylated Rb and total cyclin A. A-800141 exhibits anti-angiogenic and tumor growth inhibitory effects, and produces synergistic effects when combined with cytotoxic inhibitors or BCL-2 inhibitors. A-800141 has been widely used in scientific research related to B-cell lymphoma, neuroblastoma, prostate cancer, colon cancer, melanoma and other fields .
ZC0109 is a dual inhibitor of IDO1 and thioredoxin reductase 1 (TrxR1) with IC50s of 50 nM and 3.0 μM, respectively. ZC0109 induces ROS accumulation and cellcycle arrest at G1/S phase, thus leads to cancer cellsapoptosis .
SIAIS001 is a CRBN-dependent ALK PROTAC degrader with a DC50 of 3.9 nM. SIAIS001 induces ALK protein degradation via the ubiquitin-proteasome system. SIAIS001 induces G1/S phase cellcycle arrest and inhibits proliferation of cancer cells. SIAIS001 can be used for the research of anaplastic large-cell lymphomas .
(-)-Hinesol (Hinesol) is a potent anticancer agent. (-)-Hinesol induces apoptosis and cellcycle arrest at G0/G1 phase. (-)-Hinesol downregulates MEK/ERK pathway and NF-κB pathway and mediates theexpression of cyclin D1, Bax and Bcl-2. (-)-Hinesol has the potential for the research of non–small cell lung cancer .
Milademetan (DS-3032) tosylate hydrate is a specific and orally active MDM2 inhibitor for the research of acute myeloid leukemia (AML) or solid tumors. Milademetan (DS-3032) tosylate hydrate induces G1cellcycle arrest, senescence and apoptosis .
(+)-Perillyl alcohol is the enantiomer of S-(-)-Perillyl alcohol (HY-116514). (+)-Perillyl alcohol can inhibit the growth of polypeptides and block the cellcycle in the G0/G1 phase. (+)-Perillyl alcohol induces cell signaling that is associated with changes in cytoskeletal actin organization and reduced protein expression of growth regulatory proteins such as Ras and CDC2 kinase .
Ganoderic acid Mf is an antitumor triterpenoid. Ganoderic acid Mf causes cellcycle arrest in the G1 phase. Ganoderic acid Mf shows high selectivity between normal and cancer cells and induces cellapoptosis via mitochondria mediated pathway .
Kazusamycin B is an antibiotic that could be isolated from the fermentation broth of Streptomyces sp. No. 81-484. Kazusamycin B inhibits cell growth and arrests cellcycle at G1 phase. Kazusamycin B can be used in research of cancer .
Autophagy inducer 7 (Compound SSA) is an Autophagy and Apoptosis inducer. Autophagy inducer 7 activates autophagy by inhibiting Akt/mTOR signaling and the expression of downstream proteins. Autophagy inducer 7 suppresses DNA synthesis and causes a G0-G1cell-cycle arrest. Autophagy inducer 7 inhibits tumor cell growth .
cis,trans-Germacrone is a isomer of Germacrone (HY-N0440). Germacrone exhibits a wide range of antitumor, antioxidant and anti-inflammatory effects. Germacrone inhibits lung cancer cell proliferation and alters the Akt/MDM2/p53. Germacrone also arrests cellcycle at G1/S phase .
YS-363 is a potent, selective, and orally active EGFR inhibitor, with IC50s of 0.96 nM and 0.67 nM for wild-type and L858R mutant forms of EGFR, respectively. YS-363 can induce G0/G1cellcycle arrest and apoptosis .
CDK3 is a major player driving retinoblastoma (Rb) phosphorylation during the G0/G1 transition and in the early G1 phase of the cellcycle. CDK3 interacts with various transcription factors involved in cell proliferation, differentiation, and transformation driven by the EGFR/Ras signaling pathway. CDK3/CycE1 Recombinant Human Active Protein Kinase is an ortholog of CDK3 .
p-Tolylmaleimide (compound 9) is a naphthalimide derivative that has cytotoxic effects on cancer cells. p-Tolylmaleimide can arrest the cellcycle of human acute myeloid leukemia cells K562 in the sub-G0/G1 phase and induce apoptosis .
Apiole is an anti-tumor agent that induces apoptosis and inhibits human colon cancer cells by inducing G0/G1cellcycle arrest. Apiole also significantly inhibited colon tumor development in an in vivo mouse xenograft model .
Acrofolione A is an acetophenone dimer isolated from Acronychia pendunculata with anticancer effects. Acrofolione A induces G0/G1 phase cellcycle arrest and apoptosis in human NALM-6 pre-B cell leukaemia cells. Acrofolione A can be used for leukaemia research .
AP23464 is an ATP-based inhibitor for Kit, that inhibits the phosphorylation of Kit wildtype and mutants, with IC50 of 5-85 nM. AP23464 inhibits the proliferation of Kit mutated cells (IC50 is 3-20 nM), arrests the cellcycle at G0/G1 phase, and induces apoptosis in Kit mutated cells .
Indisulam (Standard) is the analytical standard of Indisulam. This product is intended for research and analytical applications. Indisulam (E 7070) is a carbonic anhydrase inhibitor with anticancer activity. Indisulam (E 7070) is a sulfonamide agent that targets the G1 phase of the cellcycle. Indisulam (E 7070) causes a blockade in the G1/S transition through inhibition of the activation of both CDK2 and cyclin E. Indisulam (E 7070) targets splicing by inducing RBM39 degradation via recruitment to DCAF15 .
Dalpiciclib (isethionate) is a CDK4/6 inhibitor. Dalpiciclib (isethionate) inhibits the kinase activity of CDK4 and CDK6, thereby blocking the G1-to-S phase transition of the cellcycle and suppressing abnormal cell proliferation. Dalpiciclib (isethionate) can be used for the study of breast cancer .
Maleimide-Val-Ala-PAB-SNS032 is a conjugate of ADC toxin and linker. SNS032 is an inhibitor for CDK, inhibiting the cellcycle at G1/S phase and cell viability of cancer cells. Maleimide-Val-Ala-PAB is a cleavable ADC linker. Maleimide-Val-Ala-PAB-SNS032 can be utilized for the synthesis of ADC molecules .
Multi-kinase-IN-6 (compound 10e) is a multikinase inhibitor that shows good enzyme inhibitory activity against TrkA, ALK2, c-KIT, EGFR, PIM1, CK2α, CHK1, and CDK2. Multi-kinase-IN-6 reveals antiproliferative activity against MCF7, HCT116 and EKVX with IC50 values of 3.36 μM, 1.40 μM and 3.49 μM, respectively. Multi-kinase-IN-6 shows cellcycle arrest at the G1/S phase and G1 phase in MCF7 and HCT116 cells with good apoptotic effect .
Aurora kinase-IN-1 (Compound 9) is a potent inhibitor of aurora kinase. Aurora kinase-IN-1 upregulates the expression of G1cellcycle inhibitory proteins including p21 and p27, and G1 progressive cyclin D1, and downregulates G1-to-S progressive cyclins, resulting in cellcycle arrest at the G1/S boundary. Aurora kinase-IN-1 also induces apoptosis. Aurora kinase-IN-1 is a lead compound for chemotherapeutic agents .
S9-CMC1 TFA is a covalent peptide lysine-specific demethylase 1 (LSD1) inhibitor with an IC50 value of 2.53 μM. S9-CMC1 TFA specifically recognizes Cys360 in the enzyme-active region. S9-CMC1 TFA inhibits LSD1 activity, increasing H3K4me1 and H3K4me2 levels, leading to G1cellcycle arrest and apoptosis and inhibiting cell proliferation. S9-CMC1 TFA significantly inhibits tumor growth in A549 xenograft animal models .
Typhatifolin B (Compd 2), an anti-cancer agent, could remarkably induce cellapoptosis and G0/G1cycle arrest, as well as block cell migration and invasion .
Antiproliferative agent-43 (Compound e4 ) has notable cytotoxic effects against cancer cell lines and causes apoptosis by stopping the cellcycle at G1 phase .
CDK3 is a major player driving retinoblastoma (Rb) phosphorylation during the G0/G1 transition and in the early G1 phase of the cellcycle. CDK3 interacts with various transcription factors involved in cell proliferation, differentiation, and transformation driven by the EGFR/Ras signaling pathway. CDK3/CycC Recombinant Human Active Protein Kinase is an ortholog of CDK3 .
CDK3 is a major player driving retinoblastoma (Rb) phosphorylation during the G0/G1 transition and in the early G1 phase of the cellcycle. CDK3 interacts with various transcription factors involved in cell proliferation, differentiation, and transformation driven by the EGFR/Ras signaling pathway. CDK3/CycO Recombinant Human Active Protein Kinase is an ortholog of CDK3 .
Topotecan hydrochloride hydrate is an orally active and potent Topoisomerase I inhibitor. Topotecan hydrochloride hydrate induces cellcycle arrest in G0/G1 and S phases and promotes apoptosis. Topotecan hydrochloride hydrate shows anticancer activity .
YM281 is a potent EZH2 inhibitor. YM281 induces cellapoptosis and cellcycle arrest at the G0/G1 phase. YM281 shows antitumor effects in vivo. YM281 has the potential for the research of lymphoma .
CDK6 is a cell-cycle kinases that regulate exit from the G1 phase of the cellcycle. CDK6 is directly involved in transcription in tumor cells and in hematopoietic stem cells. CDK6/CycD2 Recombinant Human Active Protein Kinase is an ortholog of CDK6 .
CDK6 is a cell-cycle kinases that regulate exit from the G1 phase of the cellcycle. CDK6 is directly involved in transcription in tumor cells and in hematopoietic stem cells. CDK6/CycD1 Recombinant Human Active Protein Kinase is an ortholog of CDK6 .
CDK6 is a cell-cycle kinases that regulate exit from the G1 phase of the cellcycle. CDK6 is directly involved in transcription in tumor cells and in hematopoietic stem cells. CDK6/CycD3 Recombinant Human Active Protein Kinase is an ortholog of CDK6 .
Antitumor agent-92, an Icaritin (HY-N0678) derivative, causes arrest at the G0/G1 phase in the cellcycle and induces cellapoptosis. Antitumor agent-92 has the potential for hepatocellular carcinoma (HCC) research .
CDK7-IN-27 (Compound 37) is a selective inhibitor for cyclin-dependent kinase 7 (CDK7), with Ki of 3 nM. CDK7-IN-27 arrests the cellcycle at G0/G1 phase .
Apoptosis inducer 21 (Compound 5h) inhibits the proliferation of lung cancer cell H69AR with an IC50 of 1.58 μM. Apoptosis inducer 21 arrests the cellcycle at G0/G1 phase, induces apoptosis in H69AR .
Antitumor agent-111 (compound 46) is an c-Met kinase inhibitor (IC50=46 nM) with antitumor and antiproliferative activity. Antitumor agent-111 arrests cellcycle at G0/G1 phase, and induces apoptosis .
Topoisomerase IIα-IN-3 (Compound 12c) is a DNA intercalative topoisomerase-IIα inhibitor. Topoisomerase IIα-IN-3 arrests cellcycle at the G0/G1 phase and induces apoptosis .
Antitumor agent-103 (compound 24l) is an apoptosis inducer with antiproliferative and anti-clony formation activities. Antitumor agent-103 arrests cellcycle at G0/G1 phase, enhances NO production, and exhibits anti-tumor activity .
Anticancer agent 204 (Compound 6), a cinnamide fluorinated derivative, possesses anticancer activity. Anticancer agent 204 can arrest the cellcycle of HepG2 cells in the G1 phase and induce apoptosis by reducing the level of mitochondrial membrane polarization (MMP) .
C2 L-Erythro ceramide (d18:1/2:0) is a cell-permeable sphingolipid. C2 L-Erythro ceramide (d18:1/2:0) induces cellcycle arrest in the G0/G1 phase and inhibits cell growth .
4-TM.P binds to the minor groove of DNA, inhibits proliferation of cancer cell K562 with an IC50 of 25 µM, arrests the cellcycle at G0/G1 phase, and induces apoptosis in cell K562. 4-TM.P can be used in anti-leukemia research .
SK-7041 is a HDAC inhibitor with the IC50 of 172 nM. SK-7041 induces the hyperacetylation of histones H3 and H4 .SK-7041 inhibits tumor cell growth in vivo and in vitro, induces cellapoptosis, and arrests cellcycle at the G1 phase .
EGFR/DHFR-IN-1 (Compound 10e) is a dual inhibitor of EGFR and DHFR with IC50s of 0.151 and 0.541 µM, respectively. EGFR/DHFR-IN-1 arrests the cellcycle at both G0-G1 and S phases .
HR488B is an efficient HDAC1 inhibitor. HR488B specifically suppressed the growth of CRC cells by inducing cellcycleG0/G1 arrest and apoptosis. HR488B causes mitochondrial dysfunction, reactive oxygen species (ROS) generation, and DNA damage accumulation .
FW-1 is a type I inhibitor for FLT3 with IC50 of ca. 1 μM. FW-1 exhibits cytotoxicity in FLT3 mutated AML cell. FW-1 arrests the cellcycle at G0/G1 phase, and induces apoptosis in cell MV4-11 and MOLM-13 .
Apiole (Standard) is the analytical standard of Apiole. This product is intended for research and analytical applications. Apiole is an anti-tumor agent that induces apoptosis and inhibits human colon cancer cells by inducing G0/G1cellcycle arrest. Apiole also significantly inhibited colon tumor development in an in vivo mouse xenograft model .
Meisoindigo (Standard) is the analytical standard of Meisoindigo. This product is intended for research and analytical applications. Meisoindigo (Dian III), a derivative of Indirubin (HY-N0117), halts the cellcycle at the G0/G1 phase and induces apoptosis in primary acute myeloid leukemia (AML) cells. Meisoindigo exhibits high antitumor activity .
Danazol standard inhibits the proliferation of cancer cell MDA-MB-231 and MCF-7 with IC50 of 65 µg/mL and 31 µg/mL. Danazol arrests the cellcycle at G1 phase, induces apoptosis in MDA-MB-231 through PKCα signaling pathway .
FAK-IN-25 (4c) is a FAK inhibitor with an IC50 of 50.98 nM. FAK-IN-25 (4c) induces apoptosis and causes cellcycle arrest at G1 phase. FAK-IN-25 (4c) can be used in cancer research .
Cr-ACP1 is an anti-cancerous peptide. Cr-ACP1 binds to DNA, inducing cellcycle arrest in the G0-G1 phase, leading to the initiation of Apoptosis mechanisms. Cr-ACP1 exhibits anticancer effects against colon cancer and epidermoid carcinoma .
Angustilongine M is a microtubule-targeting antitumor alkaloid (IC50=0.2 μM against HT-29 cells). Angustilongine M induces G0/G1cellcycle arrest and mitochondrial apoptosis via tubulin polymerization promotion. Angustilongine M is promising for research of colorectal cancer and other solid tumors .
Protosappanin B (Standard) is the analytical standard of Protosappanin B. This product is intended for research and analytical applications. Protosappanin B is a phenolic compound extracted from Caesalpinia sappan. Anti-cancer activity . Protosappanin B induces apoptosis and causes G1cellcycle arrest in human bladder cancer cells .
DNA topoisomerase II inhibitor 1 (compound 8ed) is a potent DNA topoisomerase II inhibitor. DNA topoisomerase II inhibitor 1 shows anti-proliferative activity. DNA topoisomerase II inhibitor 1 induces apoptosis and cellcycle arrest at sub G1 phase .
VMY-1-103 is an inhibitor for cyclin/Cdk complex, that arrests the cellcycle at G1 phase. VMY-1-103 reduces mitochondrial membrane potential, induces p53 phosphorylation and and PARP cleavage, activates caspase-3, and thus induces apoptosis in prostate cancer cell LNCaP .
Anticancer agent 70 (Compound 21), an anticancer agent, exhibits remarkable cytotoxic activity against numerous human cancer cell lines. Anticancer agent 70 results in the G0/G1-cellcycle arrest with a concomitant increase in p53 and p21 protein levels. Anticancer agent 70 leads to ATP depletion and disruption of the mitochondrial membrane potential .
Phenoxodiol (Standard) is the analytical standard of Phenoxodiol. This product is intended for research and analytical applications. Phenoxodiol (Idronoxil), a synthetic analog of Genestein, activates the mitochondrial caspase system, inhibits XIAP (an apoptosis inhibitor), and sensitizes the cancer cells to Fas-mediated apoptosis. Phenoxodiol also inhibits DNA topoisomerase II by stabilizing the cleavable complex. Phenoxodiol induces cellcycle arrest in the G1/S phase of the cellcycle and upregulates p21WAF1 via a p53 independent manner .
[1,1′:4′,1′′-Terphenyl]-3,4′′,5-triol (compound 13g) is a terphenyl derivative that blocks the cellcycle in the G0-G1 phase and induces differentiation in leukemia cells. [1,1′:4′,1′′-Terphenyl]-3,4′′,5-triol shows a potent apoptotic effect .
Apiol analog-1 (Compound 2b) is an analog of Apiol (HY-135217). Apiole is an anti-tumor agent that induces apoptosis and inhibits human colon cancer cells by inducing G0/G1cellcycle arrest. Apiole also significantly inhibited colon tumor development in an in vivo mouse xenograft model .
XSJ110 is a potent irreversible topoisomerase I (Topo I) inhibitor with an IC50 value of 0.133 μM. XSJ110 blocks DNA topoisomerization, induces DNA double-strand breaks, and triggers cellcycle arrest at G0/G1 phase, inducing tumor cellapoptosis. XSJ110 is promising for research of ampullary carcinoma (AC) .
Anticancer agent 322 is a spirocyclohexane-chroman-4-one derivative. Anticancer agent 322 induces apoptosis and G1cellcycle arrest in breast cancer cells. Anticancer agent 322 can be used for the research of breast cancer .
eALM1137 is a mTOR inhibitor with an IC50 of 4.8 nM. eALM1137 mediates dual inhibition of the mTORC1 and mTORC2 signaling pathways, and inhibits DNA-PK (IC50=77 nM). eALM1137 exhibits antiproliferative and cytostatic activities, and induces G1cellcycle arrest. eALM1137 is applicable to the research of glioblastoma multiforme .
Leptolstatin is an inhibitor of the progression of G1 and G2 phases of the mammalian cellcycle. Leptolstatin can be extracted from the fermentation broth of Streptomyces sp. SAM1595 .
CDK3 is a major player driving retinoblastoma (Rb) phosphorylation during the G0/G1 transition and in the early G1 phase of the cellcycle. CDK3 interacts with various transcription factors involved in cell proliferation, differentiation, and transformation driven by the EGFR/Ras signaling pathway. CDK3/CycE2 Recombinant Human Active Protein Kinase is an ortholog of CDK3 .
HDAC11-IN-5 is a selective, potent and orally active HDAC11 inhibitor with an IC50 of 0.021 μM. HDAC11-IN-5 increases fatty acylation levels of substrate SHMT2 in AML cells. HDAC11-IN-5 induces apoptosis, G1 phase cellcycle arrest, ferroptosis, ROS production and terminal myeloid differentiation in AML cells. HDAC11-IN-5 demonstrates anti-tumor potency in an MLL-AF9-induced mouse AML model. HDAC11-IN-5 can be used for the research of cancer, such as acute myeloid leukemia .
FLT3-IN-27 (compound 49) is a FLT3-ITD inhibitor with the IC50 of 174 nM. FLT3-IN-27 inhibits cell growth and increases the number of cells in the G1 phase of the cellcycle and can be used for study of acute myeloid leukemia .
EGFR-IN-210 is a EGFR kinase inhibitor with an IC50 of 0.198 μM. EGFR-IN-210 induces antiproliferative, pro-apoptotic, G0/G1cellcycle arrest, DNA synthesis inhibition and anti-migratory effects in cancer cells. EGFR-IN-210 can be used for the research of various cancers including colorectal cancer .
Esterbut-3 is a potent anticancer agent. Esterbut-3 inhibits cell proliferation. Esterbut-3 decreases the antigen expression of 115D8, 140C1 and increases the antigen expression of 123C3. Esterbut-3 induces cellcycle arrest at G0/G1 phase .
SHP2-IN-30 (compound 14i) is an allosteric SHP2 inhibitor with an IC50 of 104 nM. SHP2-IN-30 shows low inhibitory effect on SHP2-PTP. SHP2-IN-30 induces cellapoptosis and arrests the cellcycle at the G0/G1 phase .
GEM144 is a potent and orally active DNA polymerase α (POLA1) and HDAC 11 dual inhibitor. GEM144 induces acetylation of p53, activation of p21, G1/Scellcycle arrest, and apoptosis. GEM144 has significant antitumor activity in human orthotopic malignant pleural mesothelioma xenografts .
Cucurbitacin C is a triterpenoid calabinoid that can be isolated from Cucurbitaceae plants. Cucurbitacin C has anti-cancer activity in vivo and in vitro. Cucurbitacin C can induce cellcycle arrest in G1 or G2/M phase and apoptosis by inhibiting Akt signaling .
BK50164 is a potent CD73 inhibitor with an IC50 value of 13.089 μM. BK50164 binds to CD99 with a KD value of 1.5 μM. BK50164 shows antiproliferative activity. BK50164 induced Apoptosis and cellcycle arrest at Sub-G1 phase .
ALC67 is a cytotoxic thiazolidine compound with an IC50 of approximately 5 μM against liver, breast cancer, colon cancer, and endometrial cancer cell lines. ALC67 induces apoptosis in cancer cells by activating caspase-9 and causing cellcycle arrest at the SubG1/G1 phase, via a pathway that is independent of death receptors. ALC67 can be used in cancer research .
rel-Levormeloxifene (rel-L-Centchroman) is the relative configuration of Levormeloxifene (HY-122359). rel-Levormeloxifene is a selective estrogen receptor modulator (SERM). rel-Levormeloxifene inhibits proliferation of leukemia cells with IC50 about 7 μM, arrests cellcycle at G0/G1 phase, and induces apoptosis. rel-Levormeloxifene induces differentation of myelogenesis leukemia, and enhances ROS production in K562 cells .
BHA536 is an orally active selective inhibitor for PKCα/β and NF-kB signaling pathway. BHA536 inhibits the proliferation of CD79-mutated ABC DLBCL cell, arrests cellcycle at G1 phase, and induces apoptosis in TMD8 cell. BHA536 exhibits antitumor efficacy in mice .
SYHA1815 is an orally active RET inhibitor (IC50=0.9 nmol/L) with antitumor activity. SYHA1815 is more selective for RET than KDR (IC50=15.9 nmol/L). SYHA1815 arrests the G1cellcycle and inhibits RET-driven cell proliferation by downregulating c-Myc .
TMLZ-G46 is an orally active ZNF207 inhibitor with blood-brain barrier penetration ability, with a Kd value of 68 nM. TMLZ-G46 inhibits cancer cell proliferation, stemness, migration and invasion, induces G0/G1cellcycle arrest and apoptosis, and suppresses colony formation. TMLZ-G46 can be used in glioma research .
HSP90-IN-23 (Comp 12-1) is an inhibitor of heat shock protein 90(HSP90) with an IC50 of 9 nM. HSP90-IN-23 induces apoptosis of tumor cells and arrests the tumor cellcycle in G0/G1 phase. HSP90-IN-23 can be used for cancer research .
Asparanin A is an apoptosis inducer with anticancer activity. Asparanin A induces cellcycle arrest in the G0/G1 phase through mitochondria and PI3K/AKT signaling pathways, inhibiting cancer cell growth. Asparanin A also demonstrated in vivo efficacy in a mouse xenograft model of Ishikawa endometrial carcinoma, significantly inhibiting tumor growth .
VEGFR-2-IN-83 is a VEGFR-2 kinase inhibitor with an IC50 of 0.037 μM. VEGFR-2-IN-83 induces G0/G1cellcycle arrest and apoptosis in breast cancer cells, and increases the production of reactive oxygen species (ROS). VEGFR-2-IN-83 is applicable to relevant research on breast cancer .
EP26 is a potent and orally active EGFR and PD-L1 inhibitor with IC50 values of 48.6 nM, 1.77 µM, respectively. EP26 decreased the protein expression of p-EGFR. EP26 induces cellcycle arrest at G0/G1 phase. EP26 has the potential for the research of glioblastoma .
EGFR-IN-131 (compound 3a) is a potent and cross the blood-brain barrier EGFR inhibitor with an IC50 value of 272.9 nM. EGFR-IN-131 shows antiproliferative activity. EGFR-IN-131 induces apoptosis and cellcycle arrest at G0/G1 phase. EGFR-IN-131 decreases the protein expression of p-EGFR .
J208 is a dual inhibitor for histone deacetylase (HDAC) and DNA methyltransferase (DNMT). J208 inhibits proliferation of cancer cells, as well as the migration/invasion of triple-negative breast cancer (TNBC) cells. J208 induces apoptosis, arrests the cellcycle at G0/G1 phase. J2008 activates the innate immune signalling pathway in TNBC, by inducing the expression of endogenous retroviruses (ERVs) .
c-Met/HDAC-IN-4 is a dual inhibitor of c-Met/HDAC. The IC50 value of c-Met/HDAC-IN-4 for c-Met is 28.92 nM. c-Met/HDAC-IN-4 can induce G0/G1 phase cellcycle arrest and apoptosis in MDA-MB-231 breast cancer cells, and it inhibits the proliferation and invasion of breast cancer cell lines .
PAK4-IN-3(compound 27e) is aPAK4inhibitor, with theIC50of 10 nM.PAK4-IN-3shows antiproliferative activity against A549 cells with anIC50value of 0.61μM, and inducesapoptosisof A549 cells in a concentration-dependent manner and blocked the cellcycle at phase G0/G1 .
TM471-1 is an orally active and covalent Bruton's tyrosine kinase (BTK) inhibitor with IC50 values of 1.3 nM (BTK WT), >40,000 nM (BTK C481S), 7.9 nM (TEC) and 12.4 nM (TXK). TM471-1 inhibits cell growth in vivo and in vitro, arrests cellcycle at G0/G1 phase and induces cellapoptosis .
Lobaplatin (D-19466) is a diastereometric mixture of platinum(II) complexe. Lobaplatin arrests cellcycle at G1 and G2/M phase. Lobaplatin induces apoptosis by increasing expressions of caspase and Bax, decreasing expression of Bcl-2. Lobaplatin can be used for research of cancer .
VEGFR-2-IN-23 (compound 11b) is a potent and selective VEGFR-2 inhibitor with an IC50 value of 0.34 nM. VEGFR-2-IN-23 shows antitumor activity. VEGFR-2-IN-23 induces apoptosis and cellcycle arrest at G1 phase .
INX-315 is an orally active and selective CDK2 inhibitor that induces cellcycle arrest in the G1 phase. INX-315 reduces CDK2 substrate phosphorylation and inhibits tumor growth in a dose-dependent manner in xenograft mouse models. INX-315 may be used in cancer research .
Amentoflavone (Didemethyl-ginkgetin) is a potent and orally active GABA(A) negative modulator. Amentoflavone also shows anti-inflammatory, antioxidative, anti-viral, anti-tumor, anti-radiation, anti-fungal, antibacterial activity. Amentoflavone induces apoptosis and cellcycle arrest at sub-G1 phase .
BET-IN-20 (compound 10) is an inhibitor of BRD4 BD1 (IC50=1.9 nM) with anticancer activity. BET-IN-20 can promote acute myeloid leukemia (AML) cell apoptosis and arrest the cellcycle in the G0/G1 phase. BET-IN-20 also inhibits c-Myc and CDK6 and enhances PARP cleavage .
Antiparasitic agent-27 (Compound 2) is a potent antiparasitic agent targeting Leishmania infantum (IC50=3.1 μM). Antiparasitic agent-27 induces G0/G1cellcycle arrest and reactive oxygen species (ROS) generation to trigger programmed cell death. Antiparasitic agent-27 is promising for research of visceral leishmaniasis (VL) .
Cantharidic acid is a selective inhibitor for protein phosphatase 2 (PP2A) and protein phosphatase 1 (PP1). Cantharidic acid inhibits cell viability and arrest cellcycle at sub G1 phase, induces apoptosis in cells NPC-39 and HONE-1 through the upregulation of ERK1/2, p38, and JNK1/2 pathway .
Secalonic acid D is a toxic compound against tumor cells. Secalonic acid D can be isolated from the metabolites of Aspergillus aculeatus. Secalonic acid D activates GSK3-β, and degrades β-catenin. Thus, Secalonic acid D down-regulates c-Myc expression, arrests cellcycle at G1 phase, induces cellapoptosis .
Milademetan tosylate is the tosylate salt form of Milademetan (HY-101266). Milademetan tosylate is an orally active inhibitor for MDM2. Milademetan tosylate arrests the cellcycle at G1 pahse, induces the apoptosis Milademetan tosylate restores the p53 activity by targeting the p53-MDM2 interaction, and exhibits anticancer activity against Merkel cell carcinoma (MCC) .
Antiproliferative agent-63 (compound 4d), a ring-annulated analogue of Cannabidiol, is an anticancer agent. Antiproliferative agent-63 demonstrates a promising activity against breast and colorectal cancer. Antiproliferative agent-63 arrests the G1 phase of the cellcycle and induces apoptosis via the mitochondrial pathway in breast cancer cell lines .
Moreollic acid is a PTP1B inhibitor, with an IC50 of 4.44 μM and a Ki of 1.75 μM against human targets. Moreollic acid blocks cellcycle progression from the G1 to S phase and inhibits tumor cell proliferation. Moreollic acid can be used in research related to type 2 diabetes, obesity, colon cancer and other cancers .
Avadomide hydrochloride (CC 122 hydrochloride) is the hydrochloride form of Avadomide (HY-100507). Avadomide hydrochloride is an orally active cereblon modulator. Avadomide hydrochloride modulates cereblon E3 ligase activity, inhibits NF-κB pathway, arrests the cellcycle at G1 phase, and thus induces apoptosis in cancer cell PDAC. Avadomide hydrochloride exhibits potent antitumor and immunomodulatory activities .
DH-18 is a matrix metalloproteinase-2 (MMP-2) inhibitor with the IC50 values of 139.45 nM, 518.11 nM and 833.34 nM for MMP-2, MMP-9 and MMP-8, respectively. DH-18 induces cellapoptosis and arrests cellcycle in the G0/G1 phase. DH-18 inhibits cell growth and can be used for study of chronic myeloid leukemia .
Diosbulbin C is a diterpene lactone component, which can be extracted from traditional Chinese medicine Dioscorea bulbifera L.. Diosbulbin C possesses high anticancer activity in non-small cell lung cancer (NSCLC). Diosbulbin C could induce cellcycle arrest at G0/G1 phase in NSCLC. Diosbulbin C also inhibits the proliferation of NSCLC cells, possibly by downregulating the expression/activation of AKT, DHFR, and TYMS .
FB49 is a highly selective inhibitor of Bcl-2-associated athanogene 3 (BAG3), with the Ki of 45 μM. FB49 inhibits the cell growth in human tumoral cells, but has no toxicity in human peripheral mononuclear cells. FB49 block cellcycle in G1 phase and to induce apoptosis as well as autophagy in medulloblastoma HD-MB03 treated cells .
Apoptosis inducer 36 (Compound 42) exhibits anti-leukemic activity through reduction of leukemia stem cells (LSCs) and induction of differentiation. Apoptosis inducer 36 inhibits the proliferation of AML cells, arrests cellcycle at G1 phase, and induces PANoptosis including apoptosis, pyroptosis and necrosis. Prodrug of apoptosis inducer 36 exhibits orally active antitumor efficacy in mouse model .
Antiproliferative agent-26 (compound 4g) is an antiproliferative agent with much broad range of activity targeting Leukemia, CNS, Melanoma, Renal and Breast (at the concentration of 10 μM). Antiproliferative agent-26 inhibits colony forming and arrests cellcycle at G1 phase/S phase at 5 μM and 25 μM, respectively .
FGFR1 inhibitor-6 is a potent FGFR1 inhibitor with an IC50 value of 16.31 nM. FGFR1 inhibitor-6 shows cytotoxic activities. FGFR1 inhibitor-6 induces apoptosis and cellcycle arrest at pre-G1 and G2/M phase .
1,4-Dimethylnaphthalen is a non-competitive potato tuber sprouting inhibitor. 1,4-Dimethylnaphthalen blocks cellcycle progression (G1/S arrest) by inducing PP2A phosphatase and oxygen metabolism-related genes. 1,4-Dimethylnaphthalen is promising for research of fungistatic activity during potato storage and sprout control under abiotic stress .
EGFR-IN-11 is a fourth-generation EGFR-tyrosine kinase inhibitor (EGFR-TKI) with an IC50 of 18 nM for triple mutant EGFR L858R/T790M/C797S. EGFR-IN-11 significantly suppresses the EGFR phosphorylation, induce the apoptosis, and arrest cellcycle at G0/G1 .
CHMFL-FLT3-122 is a potent, selective and orally active FLT3 kinase with an IC50 of 40 nM. CHMFL-FLT3-122 shows selectivity for FLT3 over BTK (IC50 of 421 nM) and c-KIT (IC50 of 559 nM) kinases. CHMFL-FLT3-122 induces apoptosis by arresting the cellcycle into the G0/G1 phase .
AKT-IN-12 (compound 3e) is a potent Akt kinase inhibitor with an IC50 value of 0.55 μM. AKT-IN-12 induces G0/G1cellcycle arrest and apoptosis. AKT-IN-12 also inhibits p-AKT, p-ERK, and activates p-JNK, JNK. AKT-IN-12 can be used for researching leukemia .
SHP2-IN-42 is a src homology 2 domain-containing phosphatase 2 (SHP2) inhibitor with an IC50 of 15 nM. SHP2-IN-42 inhibits cell proliferation and induces apoptosis and G1 phase cellcycle arrest. SHP2-IN-42 can be used for the research of cancer, such as acute myeloid leukemia (AML) .
RET/TRKA-IN-1 (Compound 13) is a dual inhibitor for RET (IC50=0.375 µM) and TRKA. RET/TRKA-IN-1 inhibits cell viability of LC-2 and KM12, with GI50 of 0.72 and 0.25 μM. RET/TRKA-IN-1 arrests the cellcycle at G1 phase .
Pelargonidin chloride is an anthocyanidin and also is a scavenger of nitric oxide radical and has antioxidant activities. Pelargonidin inhibits cell viability and induces cellcycle arrest at sub-G1 phase. Pelargonidin chloride increases the mRNA and protein expression of HO-1, NQO1, Nrf2. Pelargonidin chloride improves Aβ-induced memory and learning impairment .
Avadomide (Standard) is the analytical standard of Avadomide. This product is intended for research and analytical applications. Avadomide is an orally active cereblon modulator. Avadomide modulates cereblon E3 ligase activity, inhibits NF-κB pathway, arrests the cellcycle at G1 phase, and thus induces apoptosis in cancer cell PDAC. Avadomide exhibits potent antitumor and immunomodulatory activities .
SIAIS164018 is a PROTAC-based ALK and EGFR degrader, with IC50 value of 2.5 nM and 6.6 nM for ALK and ALK G1202R, respectively. SIAIS164018 strongly inhibits cancer cells migration and invasion, causes G1cellcycle arrest and induces apoptosis. SIAIS164018 exhibits better property than Brigatinib (HY-12857) .
SIAIS164018 hydrochloride is a PROTAC-based ALK and EGFR degrader, with IC50 value of 2.5 nM and 6.6 nM for ALK and ALK G1202R, respectively. SIAIS164018 hydrochloride strongly inhibits cancer cells migration and invasion, causes G1cellcycle arrest and induces apoptosis. SIAIS164018 hydrochloride exhibits better property than Brigatinib (HY-12857) .
Anticancer agent 25 is an anticancer agent. Anticancer agent 25 can arrest the cellcycle at the G1 phase, and significantly inhibit tumor cell colony forming and migration even at low concentrations. Anticancer agent 25 can significantly induce cytoplasmic vacuolation. Anticancer agent 25 can be used for the study of prostate cancer, breast cancer, and colon cancer .
ALK/EGFR-IN-2 is a potent dual inhibitor of ALK and EGFR. ALK/EGFR-IN-2 induces apoptosis and G0/G1cellcycle arrest in cancer cells. ALK/EGFR-IN-2 significantly inhibits the cell proliferation of H1975, PC9, and Baf3-EML4-ALK cancer cell lines with IC50s of 0.0034, 0.0065, and 0.0018 μM, respectively .
PRMT5-IN-48 (compound D3) is an orally effective PRMT5 inhibitor (IC50=20.7 nM) with anticancer activity. PRMT5-IN-48 can inhibit the growth of various cancer cells, induce apoptosis (apoptosis), and arrest the cellcycle at the G0/G1 phase. PRMT5-IN-48 can be used for non-Hodgkin lymphoma (NHL) research .
ER covalent antagonist-1 (Compound 39D) is the antagonist for estrogen receptor α (ERα). ER covalent antagonist-1 inhibits the proliferation of ERα-positive cell MCF-7 with an IC50 of 0.98 μM, arrests the cellcycle at G0/G1 phase, and induces apoptosis. ER covalent antagonist-1 exhibits antitumor efficacy in mouse model .
HDACs/mTOR Inhibitor 1 is a dual HDACs and mTOR inhibitor, with IC50s of 0.19 nM, 1.8 nM, 1.2 nM for HDAC1, HDAC6, mTOR, respectively. HDACs/mTOR Inhibitor 1 stimulates cellcycle arrest in G0/G1 phase and induces tumor cellapoptosis with low toxicity in vivo. HDACs/mTOR Inhibitor 1 can be used in the research of hematologic malignancies .
LDHA-IN-10 (Compound HP19) is a Lactate Dehydrogenase-A (LDHA) inhibitor with an IC50 value of 5.2 μM. LDHA-IN-10 reduces lactate production and ATP levels, inhibiting the proliferation of pancreatic cancer cell line PANC-1. LDHA-IN-10 induces G1/Scellcycle arrest and promotes apoptosis. LDHA-IN-10 is promising for research of pancreatic ductal adenocarcinoma (PDAC) .
N-Desmethyldauricine is a NF-κB p65 inhibitor and apoptosis inducer. N-Desmethyldauricine reduces the protein expression level of p65, induces apoptosis, arrests the cellcycle at the G0/G1 phase, attenuates intercellular adhesion, and inhibits the growth of 3D spheroids of triple-negative breast cancer. N-Desmethyldauricine can be used in studies related to triple-negative breast cancer .
Methylstat is a potent histone demethylases inhibitor. Methylstat shows anti-proliferative activity with low cytotoxicity. Methylstat induces apoptosis and cellcycle arrest at G0/G1 phase. Methylstat increases the expression of p53 and p21 protein levels. Methylstat inhibits angiogenesis induced by various cytokines. Methylstat can be used as a chemical probe for addressing its role in angiogenesis .
Aromatase-IN-5 (Compound 10) is a potent inhibitor of aromatase with an IC50 value of 0.06 μM. Aromatase-IN-5 effectively blocks estrogen production. Aromatase-IN-5 inhibits the proliferation of breast cancer cell lines like MCF-7, arrests the cellcycle at the G1 phase, and induces apoptosis. Aromatase-IN-5 is promising for research of breast cancer .
Sirtuin-IN-1 (Compound 18) is a SIRT1/2 inhibitor (IC50s: 6.2 μM and 4.2μM for SIRT1 and SIRT2, respectively). Sirtuin-IN-1 induces G1cellcycle arrest. Sirtuin-IN-1 exhibits potent anti-cancer activity against glioma .
Milademetan (Standard) is the analytical standard of Milademetan (HY-101266). This product is intended for research and analytical applications. Milademetan (DS-3032) is a specific and orally active MDM2 inhibitor for the research of acute myeloid leukemia (AML) or solid tumors. Milademetan (DS-3032) induces G1cellcycle arrest, senescence and apoptosis .
Aristoforin, a hypericin derivative, inhibits the activities of SIRT1 and SIRT2. Aristoforin induces G1 phase cellcycle arrest, scavenges hydroxyl free radicals, and exhibits protective activity against Fe 2+-induced DNA breakage. Aristoforin can be used in studies related to breast cancer and colon adenocarcinoma.
LQFM030 is a novel small molecule MDM2 inhibitor. LQFM030 exhibits concentration dependent cytotoxicity in K562 cells (IC50 = 0.28 mM). LQFM030 induces cellapoptosis through G0/G1 phase cellcycle arrest and increased Caspase activity. LQFM030 downregulates the mRNA expression of MDM2, MDMX, p73, MYC, and NF-κB. LQFM030 is commonly used in research on cancers such as leukemia .
14-3-3η Protein inhibitor 1 (Compound C11) is a 14-3-3η protein inhibitor with a KD of 35 µM. 14-3-3η Protein inhibitor 1 shows inhibitory activities against several typical human liver cancer cell lines. 14-3-3η Protein inhibitor 1 induces cellapoptosis and G1-Scellcycle arrest with good metabolic stability .
c-Myc inhibitor 16 iodide (Compound W11) is a selective c-Myc G-quadruplex (c-Myc G4) inhibitor. c-Myc inhibitor 16 iodide inhibits the transcription and translation of the c-Myc gene, disrupts the tumor cellcycle, arrests cell growth in the G0/G1 phase and activates the mitochondrial apoptosis pathway to induce early apoptosis of cancer cells. c-Myc inhibitor 16 iodide is promising for research of breast cancer .
FLT3-ITD-IN-3 (13v), an orally active FLT3-ITD (FLT3 internal tandem duplication) inhibitor, disrupts FLT3 signal transduction and induced G0/G1cellcycle arrest and apoptosis. FLT3-ITD-IN-3 (13v) is used in the research of acute myeloid leukemia (AML) .
Menin-MLL inhibitor-25 (compound A6) is a potent Menin-MLL interaction inhibitor with an IC50 value of 0.38 µM. Menin-MLL inhibitor-25 shows anti-proliferative activity. Menin-MLL inhibitor-25 induces apoptosis and cellcycle arrest at G0/G1 phase. Menin-MLL inhibitor-25 reverses the differentiation arrest .
Topoisomerase I inhibitor 5 is an effective topoisomerase inhibitor with IC50 value of. Topoisomerase I inhibitor 5 can interfere with DNA and significantly inhibit the activity of Topoisomerase I. Topoisomerase I inhibitor 5 can arrest cellcycle at the G1 phase and induce MCF-7 cellsapoptosis. Topoisomerase I inhibitor 5 has potency in reversing P-gp-mediated resistance to Adriamycin .
PROTAC BRD9 Degrader-8 is a selective, orally active BRD9 PROTAC degrader with a DC50 of 16 pM.\nPROTAC BRD9 Degrader-8 induces cellcycle arrest at the G1 phase and promotes apoptosis. PROTAC BRD9 Degrader-8 can be used for research on acute myeloid leukemia and diffuse large B-cell lymphoma .
Cinobufagin (Standard) is the analytical standard of Cinobufagin. This product is intended for research and analytical applications. Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Cinobufagine-d3 is the deuterium labeled Cinobufagin (HY-N0421). Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
TS-IN-6 (Compound 10) is a thymidylate synthase (TS) inhibitor with an IC50 value of 0.54 μM, demonstrating significant antiproliferative activity. TS-IN-6 induces G1 phase cellcycle arrest and apoptosis (marked by substantial increases in both early and late apoptotic rates) and shows potential for research on cancers such as colon, breast, and liver cancers .
Milademetan (tosylate hydrate) (Standard) is the analytical standard of Milademetan (tosylate hydrate) (HY-101266B). This product is intended for research and analytical applications. Milademetan (DS-3032) tosylate hydrate is a specific and orally active MDM2 inhibitor for the research of acute myeloid leukemia (AML) or solid tumors. Milademetan (DS-3032) tosylate hydrate induces G1cellcycle arrest, senescence and apoptosis .
Dendrogenin A (DDA) is a ligand for liver X receptor (LXR), that induces the expression of sodium/iodine symporter, and increases iodine uptake. Dendrogenin A induces cell differentiation of MCF-7, and reactivates the function of lactating cells. Dendrogenin A induces the expressions of the TSH receptor, thyroid peroxidase, and thyroglobulin, and affects thyroid hormone generation. Dendrogenin A exhibits cytotoxicity in cancer cell B-CPAP and 8505c with IC50 of 4.1 and 6.2 µM. Dendrogenin A arrests the cellcycle at G0/G1 phase .
UR-1505 is a nuclear factor of activated T cells (NF-AT) inhibitor. UR-1505 can suppress CD3/CD28 induced T cell proliferation, increase p27 KIP levels, and induce G1/Scellcycle arrest. UR-1505 can also inhibit the production of IL-5 and IFN-γ in activated T cells. UR-1505 has immunomodulatory properties and can be used in the study of atopic dermatitis .
ICSN3250 hydrochloride is a halitulin analogue and a mTORC1 inhibitor. ICSN3250 hydrochloride directly binds to mTOR's FRB domain and displaces phosphatidic acid (PA), reversing mTORC1 activation. ICSN3250 hydrochloride shows high cytotoxicity in cancer cells (nanomolar concentration) through a caspase-independent cell death mechanism. ICSN3250 hydrochloride specifically inhibits the mTORC1 pathway, inducing autophagy and G0-G1cell-cycle arrest in cancer cells. ICSN3250 hydrochloride can be used for the study of cancer .
ICSN3250 is a halitulin analogue and specific mTORC1 inhibitor. ICSN3250 directly binds to mTOR's FRB domain and displaces phosphatidic acid (PA), reversing mTORC1 activation. ICSN3250 shows high cytotoxicity in cancer cells (nanomolar concentration) through a caspase-independent cell death mechanism. ICSN3250 specifically inhibits the mTORC1 pathway, inducing autophagy and G0-G1cell-cycle arrest in cancer cells. ICSN3250 can be used for the study of cancer .
Apoptosis inducer 30 (Compound 15a) is an anticancer agent. Apoptosis inducer 30 induces MCF-7 cellsapoptosis through mitochondrial pathway. Apoptosis inducer 30 induces intracellular reactive oxygen species levels and decreases mitochondrial membrane potential, and blocks the cellcycle in the G0/G1 phase. Apoptosis inducer 30 inhibits cell growth, with an IC50 value of 0.32 μM against MCF-7 cells, and inhibits tumor growth in a mouse model of breast cancer .
NA-17 is a naphthalimide compound with anti-tumor activity and lower toxicity to normal cells like HL-7702 and WI-38. NA-17 exhibits a p53-dependent selective inhibition in various NSCLC cells, inducing the accumulation of active p53 in the mitochondria and nuclei of NSCLC cells. NA-17 can cause cellcycle arrest in the G1 phase, leading to apoptosis and cell death .
GP130-IN-1 (compound 49) is a potent GP130 inhibitor with significant in vitro antitumor activity and higher selectivity than Bazedoxifene (HY-A0031). GP130-IN-1 induces ultrastructural changes in cells, causing cellcycle arrest in the G0/G1 phase in a time-dependent manner and triggering apoptosis and autophagy. GP130-IN-1 can be used in the study of triple-negative breast cancer .
PI3K/mTOR Inhibitor-8 (Compound 18b) is a PI3K and mTOR dual inhibitor with IC50 values of 0.46 nM and 12 nM against PI3Kα and mTOR, respectively. PI3K/mTOR Inhibitor-8 induces HCT-116 cellsapoptosis and arrests cellcycle at the G1/S phase .
EGFR-IN-144 (Compound 4B) exhibits inhibitory activities against EGFR (IC50=0.639 µg/mL) and tubulin polymerization (IC50=7.339 µg/mL). EGFR-IN-144 exhibits cytotoxicity in multiple cancer cell with GI50 of nanomolare levels. EGFR-IN-144 downregulates the expressions of mTOR, TNF-α, and IL-6, arrests the cellcycle at G1/S phase, and induces apoptosis .
HDAC6-IN-48 (compound 5i) is a potent and selective HDAC6 inhibitor with IC50 values of 5.16, 396.72, 638.08 nM for HDAC6, HDAC3, HDAC1, respectively. HDAC6-IN-48 induces apoptosis and cellcycle arrest at G0/G1 phase. HDAC6-IN-48 increases the protein expression of acetylated α-tubulin .
Antitumor agent-182 (Compound 12a) decreases mitochondrial membrane potential (MMP) and enhances ROS levels. Antitumor agent-182 arrests the cellcycle at G0/G1 phase, induces apoptosis in HeLa. Antitumor agent-182 inhibits the proliferation of HeLa, PC-3 and HCT-15 with IC50s of 8.83, 10.07 and 7.84 μM, respectively .
Antileishmanial agent-40 is an orally active and selective antileishmanial agent. Antileishmanial agent-40 elevates intracellular reactive oxygen species (ROS) levels in Leishmania donovani promastigotes. Antileishmanial agent-40 induces cellcycle arrest at the sub-G0/G1 phase in Leishmania donovani promastigotes, indicative of programmed-like parasite death. Antileishmanial agent-40 can be used for the research of leishmaniasis .
HSP90-IN-33 (compound 24e) is a potent Hsp90 inhibitor with Kd values of ≥200, 7.3 µM for Hsp90α, Hsp90β, respectively. HSP90-IN-33 induces apoptosis and cellcycle arrest at G0/G1 phase. HSP90-IN-33 decreases the protein expression of ERα, CDK4 and Akt .
HS-438 is a potent and selective BCR-ABL inhibitor. HS-438 shows antiproliferative activity. HS-438 decreases the expression of phosphorylation of BCR-ABL (Tyr177). HS-438 induces apoptosis and cellcycle arrest at G0/G1 phase. HS-438 shows antitumor activity. HS-438 has the potential for the research of chronic myeloid leukemia (CML) .
Zardaverine is an orally active and selective PDE3/4 inhibitor (IC50)=0.58 uM/0.17 uM) with potent bronchodilator activity. Zardaverine also selectively inhibits the proliferation of HCC cells and induces apoptosis and cycle arrest (G0/G1 phase). Zardaverine has good antitumor potential and is effective in both bronchial relaxation and reduction of inflammation in asthma .
hCAIX-IN-16 (Compound 12d) is hCA IX inhibitor, with Ki values of 190.0 and 187.9 nM for hCA IX and hCA XII, respectively. hCAIX-IN-16 can arrest the cellcycle of breast cancer MDA-MB-468 in G0-G1 and S phase and induce apoptosis. hCAIX-IN-16 shows good broad-spectrum anticancer activity and can be used for cancer research .
VEGFR-2/BRAF-IN-1 (Compound 4b) is a dual VEGFR-2 and BRAF kinases inhibitor with IC50 values of 0.049, 0.063 and 0.005 µM against VEGFR-2, BRAF V600E and BRAF WT, respectively. VEGFR-2/BRAF-IN-1 induces apoptosis and arrests the cellcycle mainly in the G1/S phase .
Broussoflavonol F is a HER2-RAS-MEK-ERK signaling pathway modulator and mushroom tyrosinase inhibitor, with an IC50 of 82.3 μM against mushroom tyrosinase. Broussoflavonol F reduces the protein expression levels of RAS, HER2, phosphorylated BRAF, phosphorylated MEK and phosphorylated Erk. It induces cell apoptosis, triggers G0/G1 phase cellcycle arrest, and exhibits cytotoxicity against colon cancer cells. Broussoflavonol F is applicable to related research on colon cancer .
Apoptosis inducer 43 is an apoptosis inducer. Apoptosis inducer 43 can induce apoptosis, SubG0-G1cellcycle arrest, secondary necrosis, and upregulate caspase-3, p53, and Bax/Bcl-2 expression in HCT116 cells. Apoptosis inducer 43 can inhibit tumor growth in a solid Ehrlich carcinoma (SEC) mouse model. Apoptosis inducer 43 can be used to study cancers such as colon cancer, leukemia, and non-small cell lung cancer .
K783-0308 is a potent and selective dual inhibitor of FLT3 and MNK2 with IC50 values of 680 and 406 nM, respectively. K783-0308 inhibits the growth of MOLM-13 (IC50=10.5 µM) and MV-4-11 (IC50=10.4 µM) cells. K783-0308 promotes acute myeloid leukemia (AML) cellapoptosis and cellcycle arrests in the G0/G1 phase .
PRMT7-IN-2 (A33) is a selective PRMT7 inhibitor with an IC50 of 0.50 μM. PRMT7-IN-2 arrests cellcycle at G0/G1 phase, induces cellapoptosis, and inhibits cell growth in vivo and in vitro. PRMT7-IN-2 decreases the monomethylarginine level of PRMT7, increases expression of epithelial marker (E-cadherin, and reduces expression of mesenchymal markers such as N-cadherin, Vimentin, and ZEB2 .
AZD8421 is a selective CDK2 inhibitor (IC50 = 9 nM) as well as achieving CDK family selectivity in cells versus key off-targets (CDK1, CDK4/6, CDK9), AZD8421 had no significant kinase inhibition outside the CDK family. AZD8421 inhibits cancer cell proliferation by inhibiting pRB phosphorylation, inducing cellcycle arrest in G1/S phase and senescence. AZD8421 can be studied in research for breast cancer and ovarian cancer .
Angelylalkannin is a naphthoquinone compound isolated from the root of Alkanna tinctoria. Angelylalkannin showed significant inhibitory effects in the antiproliferative effect test on human colon cancer cells HCT-116 and SW-480. The median inhibitory concentration (IC50) of Angelylalkannin was 4.76 mM for HCT-116 cells and 7.03 mM for SW-480 cells. Like alkannin, Angelylalkannin can arrest the cellcycle in the G1 phase and induce apoptosis at a concentration of 1-10 mM.
Atractylenolide II (Asterolide) is a sesquiterpenoid compound. Atractylenolide II can induce G1 phase cellcycle arrest and apoptosis in B16 melanoma cells. Atractylenolide II is an orally effective anticancer agent that can exert anti-melanoma effects by inhibiting the STAT3 signaling pathway. In addition, Atractylenolide II has been shown to ameliorate myocardial fibrosis, oxidative stress, and neuroprotective activity .
Antitumor agent-159 (Compound 13b) targets the mitochondria and downregulates cardiolipin levels. Antitumor agent-159 inhibits the proliferation of cancer cell MDA-MB-231, arrests the cellcycle at sub-G1 phase, and induces apoptosis in MDA-MB-231. Antitumor agent-159 exhibits antitumor efficacy in MDA-MB-231 xenograft mouse models .
Tunicamycin is a mixture of homologous nucleoside antibiotic that inhibits N-linked glycosylation and blocks GlcNAc phosphotransferase (GPT). Tunicamycin causes accumulation of unfolded proteins in cell endoplasmic reticulum (ER) and induces ER stress, and causes blocking of DNA synthesis and cellcycle arrest in G1 phase. Tunicamycin inhibits gram-positive bacteria, yeasts, fungi, and viruses and has anti-cancer activity .Tunicamycin increases exosome release in cervical cancer cells .
YCH3124 (compound Z33) is a USP7 inhibitor (IC50=41.9 nM) with antitumor activity. YCH3124 has good in vitro antiproliferative activity against various tumor cells including LNCaP (IC50=3.6 nM) and CHP-212 (IC50=9.9 nM). In addition, YCH3124 disrupts cellcycle progression by restricting G1 phase and induces apoptosis in CHP-212 cells .
EGFR-IN-152 (compound D4) is a potent EGFR tyrosine kinase inhibitor, exhibiting potent EGFR L858R/T790M/C797S inhibition activity (IC50 = 40 nM). EGFR-IN-152 induces G0/G1 phase cellcycle arrest and apoptosis, thereby inhibiting colony formation and cell proliferation of non-small cell lung cancer (NSCLC) cells. EGFR-IN-152 can be used for NSCLC research .
BG11 induces the accumulation of Fe 2+ and intracellular lipid peroxides, induces ferroptosis. BG11 regulates the expression of Bax and Bcl-2 proteins, and induces apoptosis in MDA-MB-231 cell. BG11 arrests the cellcycle at G0/G1 and S phase, inhibits the proliferation of TNBC cancer cell (IC50 for MDA-MB-231 and BT549 is 0.49 μM and 0.52 μM), and inhibits the cell migration and invasion. BG11 exhibits antitumor efficacy in mouse models .
MM927 is a potent NVL inhibitor, with an IC50of 0.053 μM. MM927 blocks 60S ribosomal subunit biogenesis in the nucleolus. MM927 induces half-mer polysomes, cellcycle arrest at G1/S and G2/M and apoptosis in cells. MM927 demonstrates antitumor efficacy in MOLM-13 AML and HCT116 CRC xenograft models. MM927 can be used for the study of cancers such as acute myeloid leukemia (AML) and colorectal cancer (CRC) .
EGFR-IN-137 (Compound 4c) is an inhibitor for aromatase and EGFR with IC50s of 1.67 μg/mL and 0.08 μg/mL. EGFR-IN-137 inhibits the proliferation of cancer cell MCF-7 and MDA-MB-231 with IC50s of 1.62 µM and 4.14 µM. EGFR-IN-137 arrests the cellcycle at G0/G1 phase in MDA-MB-231, and induces apoptosis through caspase-dependent pathway .
Apoptosis inducer 52 is a derivative of flutamide that induces apoptosis in prostate cancer cells. Apoptosis inducer 52 promotes apoptosis by activating both intrinsic and extrinsic apoptotic pathways, without generating reactive oxygen species (ROS). Apoptosis inducer 52 triggers caspase 3/7 activity and the externalization of phosphatidylserine, leading to cellcycle arrest at the G0/G1 phase. Apoptosis inducer 52 can be used for the research of androgen receptor (AR)-dependent and -independent prostate cancers and leukemia .
BRAF V600E/CRAF-IN-1 (Compound 8b) is a potent inhibitor of BRAF V600E/CRAF. BRAF V600E/CRAF-IN-1 triggers apoptosis and cellcycle arrest at G0/G1 phase in HCT-116 colon cancer cell. BRAF V600E/CRAF-IN-1 has the potential for the research of cancer diseases .
MXC-017 is a blood-brain barrier (BBB)-penetrant apoptosis inducer that directly targets Vimentin (VIM). MXC-017 prevents radiation-induced glioma stem cell (GSC) formation, while promoting G0/G1cellcycle arrest and apoptosis. MXC-017 exhibits minimal off-target effects and shows no significant cytotoxicity. MXC-017 significantly prolongs median survival when used in combination with radiation therapy in glioblastoma (GBM) mouse models.
CDK2-IN-45 (Compound 8f) is a CDK2 inhibitor (IC50: 0.64 μM). CDK2-IN-45 inhibits the proliferation of DU-145 and PC-3 cell lines with IC50 of 2.20 μM and 4.17 μM, respectively. CDK2-IN-45 induces G0/G1cellcycle arrest and apoptosis. CDK2-IN-45 can be used in prostate cancer research .
HDAC1/6-IN-1 (compound D7) is a potent multitarget inhibitor of GLP, HDAC6 and HDAC1, with IC50 values of 1.3, 13, and 89 nM, respectively. HDAC1/6-IN-1 can inhibit the methylation and deacetylation of H3K9 on protein level. HDAC1/6-IN-1 induces cancer cellapoptosis, G0/G1cellcycle arrest, and blocks migration and invasion .
Amentoflavone (Standard) is the analytical standard of Amentoflavone. This product is intended for research and analytical applications. Amentoflavone (Didemethyl-ginkgetin) is a potent and orally active GABA(A) negative modulator. Amentoflavone also shows anti-inflammatory, antioxidative, anti-viral, anti-tumor, anti-radiation, anti-fungal, antibacterial activity. Amentoflavone induces apoptosis and cellcycle arrest at sub-G1 phase .
EGFR/HER2/DHFR-IN-1 is a potent anticancer agent with high selectivity against MCF-7 breast cancer cells. EGFR/HER2/DHFR-IN-1 is a multiple inhibitor of EGFR/HER2 kinase and DHFR, with IC50s of 0.153 μM, 0.108 μM, 0.291 μM, respectively. EGFR/HER2/DHFR-IN-1 arrests cellcycle at G1/S and induces cellsapoptosis .
EGFR/VEGFR2-IN-6 (Compound 3k) is a dual-functional inhibitor of EGFR and VEGFR2 with IC50s of 10.53 and 3.37 μM for EGFR and VEGFR2, respectively. EGFR/VEGFR2-IN-6 has significant anti-proliferation activity against breast cancer cells, and induces G0/G1cellcycle arrest and cellapoptosis, especially early apoptosis. EGFR/VEGFR2-IN-6 can be used for cancers research .
SILA-123 is a FLT3 inhibitor (FLT3-WT: IC50=2.1 nM; FLT3-ITD: IC50=1.0 nM). SILA-123 inhibits FLT3 phosphorylation and downstream signaling pathways, leading to apoptosis by arresting cellcycle progression at the G0/G1 phase. SILA-123 can be used in the study of acute myeloid leukemia .
BRD4 Inhibitor-18 is a highly potent BRD4 inhibitor with an IC50 value of 110 nM. BRD4 Inhibitor-18 has a hydrophobic acetylcyclopentanyl side chain. BRD4 Inhibitor-18 can significantly suppress the proliferation of MV-4-11 cells with high BRD4 level. BRD4 Inhibitor-18 has apoptosis-promoting and G0/G1cycle-arresting activity .
Geiparvarin is an anticancer agent and an inhibitor of MAO-B (pIC50 = 6.84 μM). Geiparvarin exerts anti-tumor effects by downregulating COX2 expression and inhibiting angiogenesis. Geiparvarin blocks the cellcycle at the G1 phase and induces apoptosis of cancer cells. Geiparvarin has anti-microtubule activity and destroys the cytoskeleton to exert anti-proliferative effects. Geiparvarin has research significance for lung cancer, leukemia, and breast cancer .
Methoxyfenoterol is a β2-adrenergic receptor agonist. Methoxyfenoterol stimulates intracellular cAMP accumulation, inhibits tumor cell proliferation, induces G1cellcycle arrest, upregulates cyclin-dependent kinase inhibitor p27, downregulates cyclin D1 and cyclin A, and inhibits Akt phosphorylation. Methoxyfenoterol crosses the blood-brain barrier and inhibits growth of astrocytoma xenografts. Methoxyfenoterol can be used for the research of astrocytoma, glioblastoma .
Wnt/β-catenin activator 1 (Compound 5m) is the orally active activator for Wnt/β-catenin signaling pathway, that arrests cellcycle at G1 phase, inhibits early proliferation of adipocytes, and inhibits adipogenesis in cell 3T3-L1 with an IC50 of 330 nM. Wnt/β-catenin activator 1 exhibits anti-adipogenic and anti-dyslipidemic activities in high-fat diet fed Syrian golden hamster model .
CTL-06 is an inhibitor of Fatty Acid Synthase (FASN) (IC50: 3 μM) and can induce apoptosis. CTL-12 blocks the cellcycle in the Sub-G1/S phase, upregulates the expression of caspase-9 and the apoptosis marker Bax, and downregulates the anti-apoptotic marker Bcl-xL. CTL-12 inhibits de novo lipogenesis, blocks the metabolic demands of tumor cells, and is commonly used in breast and colorectal cancer research .
CDK4/6-IN-15 hydrochloride is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 hydrochloride potently inhibits cancer cells growth. CDK4/6-IN-15 hydrochloride arrests cellcycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
CDK4/6-IN-15 is an orally active and selective CDK4/6 inhibitor. CDK4/6-IN-15 potently inhibits cancer cells growth. CDK4/6-IN-15 arrests cellcycle at G1 phase and suppresses retinoblastoma tumour suppressor protein (Rb) phosphorylation at S780 and E2 factor (E2F)-regulated gene expression .
Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities .
Apoptosis Inducer 28 (Compound X1) is an apoptosis-inducing agent with anticancer activity in vitro. Apoptosis Inducer 28 can arrest the cellcycle at the G1 phase, promote cell death, and induce apoptosis by disrupting mitochondrial membrane potential. Apoptosis inducer 28 can also decrease the production of reactive oxygen species, downregulate the gene expression of BAX, Bcl-xL, and Bcl-2, while upregulating the gene expression of PAR-4 .
ZDLD20, a β-carboline, is orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 6.51 μM. ZDLD20 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cellcycle. ZDLD20 exhibits potent anticancer activity .
VEGFR-2/BRAF-IN-2 (Compound 4a) is a dual VEGFR-2 and BRAF kinases inhibitor with IC50 values of 0.111, 0.089 and 0.071 µM against VEGFR-2, BRAF V600E and BRAF WT, respectively. VEGFR-2/BRAF-IN-2 induces apoptosis and arrests the cellcycle mainly in the G1 phase .
FLT3-IN-18 is a potent and selective FLT3 inhibitor with an IC50 value of 0.003 μM. FLT3-IN-18 induces apoptosis and cellcycle arrest at G1 phase. FLT3-IN-18 inhibits FLT3 and STAT5 phosphorylation. FLT3-IN-18 has the potential for the research of acute myeloid leukemia (AML) .
Autophagy agonist-1 (compound 22) is an Autophagy agonist. Autophagy agonist-1 exhibits significant anticancer activity against HepG2 cells and normal cells with IC50s of 8.8 μM and > 50 μM. Autophagy agonist-1 induces G1/S phase cellcycle arrest and inhibits CDK4 and CyclinD1 expression while upregulating P21. Autophagy agonist-1 promotes the accumulation of autophagosomes and the proteins LC3 and PINK1, enhancing autophagy and mitophagy in HepG2 cells .
PD-166285 is a PKMYT1 inhibitor, with an IC50 value of 17 nM. PD166285 shows strong antiproliferative effects against CCNE1-amplified OVCAR3 cells (IC50= 0.14 μM) and HCC1569 cells (IC50 = 0.21 μM). PD166285 induces apoptosis and arrests CCNE1-amplified HCC1569 cells at the G1/S phase of the cellcycle. PD166285 can be used for the research of PKMYT1-targeted therapies for CCNE1-amplified cancers .
VEGFR-2-IN-70 is a potent VEGFR-2 inhibitor with an IC50 of 18.04 nM. VEGFR-2-IN-70 exhibits cytotoxicity against A549 and MCF-7 cancer cells with IC50 values of 0.43 μM and 3.8 μM, respectively. VEGFR-2-IN-70 induces G1cellcycle arrest and apoptosis in lung cancer cells. VEGFR-2-IN-70 is useful in cancer research .
AKT-IN-25 (Compound 14a) is an inhibitor for Akt, that inhibits the phosphorylation of Akt, and thereby inhibits the PI3K/Akt/mTOR signaling pathway. AKT-IN-25 arrests the cellcycle at G1 phase, inhibits the cell migration of PANC-1, and inhibits the proliferation of cancer cells PANC-1, PATU-T, and SUIT-2 with IC50s of 3.05, 1.32, and 3.85 μM, respectively .
ZINC13000658 is a METTL inhibitor. ZINC13000658 exhibits significant anti proliferative activity in various cells and can induce G1 phase cellcycle arrest and apoptosis such as HepG2 (IC50 = 5.632 µM) and SNU-449 (IC50 = 6.184 µM) cells. ZINC13000658 may be related to the inhibition of the activity of multiple methyltransferases such as METTL1, 3, 6, 16, 18, etc. ZINC13000658 can be used for research on various types of cancer .
N6-Benzyladenosine (Standard) (Benzyladenosine (Standard)) is the analytical standard of N6-Benzyladenosine (HY-N7844). This product is intended for research and analytical applications. N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cell apoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma - .
MFDCH016 is a potent HDAC1/6 (IC50 = 38/59 nM) and CDK4/6 (IC50 = 680/720 nM) inhibitor. MFDCH016 induces apoptosis and cellcycle arrest in G2/M and G0/G1 phases in MCF-7 cells. MFDCH016 can modulate the HDAC-p21-CDK signaling pathway, increasing the levels of acetylated H3 and p21. MFDCH016 can be used for the study of breast cancer .
Dac590 is an orally active and selective obesity-associated protein (FTO) inhibitor with an IC50 of 6.06 nM. Dac590 shows highly selective over ALKBH5 and ALKBH3. Dac590 suppresses oncogenic FTO signaling, induces myeloid differentiation, G1-phasecellcycle arrest, and apoptosis in acute myeloid leukemia (AML) cells. Dac590 inhibits xenograft tumor growth and prolongs survival in acute myeloid leukemia mouse models with no observed toxicity. Dac590 can be used for the research of AML .
Dihydroaeruginoic acid ((Rac)-CGP 52547), an antifungal antibiotic, is a thiazoline iron chelator. Dihydroaeruginoic acid is the condensation product of salicylate and one cysteine residue. Dihydroaeruginoic acid chelates Fe(III), inhibits DNA replication via ribonucleotide reductase, induces G1/Scellcycle block, reduces leukemia cell clonogenic viability. Dihydroaeruginoic acid inhibits phytopathogenic fungi and bacteria, suppresses Candida albicans development, and inhibits Agrobacterium tumefaciens biofilm formation via extracellular iron sequestration. Dihydroaeruginoic acid can be used for the research of phytopathogenic fungal and bacterial infections, and leukemia .
JAK2-IN-7 is a selective JAK2 inhibitor with IC50s of 3, 11.7, and 41 nM for JAK2, SET-2, and Ba/F3 V617Fcells, respectively. JAK2-IN-7 possesses >14-fold selectivity over JAK1, JAK3, FLT3. JAK2-IN-7 stimulates cellcycle arrest in the G0/G1 phase and induces tumor cellapoptosis. Antitumor activities .
3,4,5-Tricaffeoylquinic acid (3,4,5-triCQA) inhibits tumor necrosis factor-α-stimulated production of inflammatory mediators in keratinocytes via suppression of Akt- and NF-κB-pathways. 3,4,5-Tricaffeoylquinic acid induces cellcycle arrest at G0/G1, actin cytoskeleton organization, chromatin remodeling, neuronal differentiation, and bone morphogenetic protein signaling in human neural stem cells. 3,4,5-Tricaffeoylquinic acid has the potential for the research of aging-associated diseases .
N6-Benzyladenosine-d5 (Benzyladenosine-d5) is deuterium labeled N6-Benzyladenosine. N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cellapoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma .
Triphenyltin(IV) diisopropyl dithiocarbamate (Compound OC2) is an anti-leukemia agent. Triphenyltin(IV) diisopropyl dithiocarbamate exhibits extremely strong cytotoxicity towards Jurkat cells, with an IC₅₀ value of 0.1 μM. Triphenyltin(IV) diisopropyl dithiocarbamate causes DNA damage, which subsequently leads to mitochondrial dysfunction, a large amount of ROS production, and ultimately results in the activation of the mitochondrial apoptosis pathway (involving the activation of Caspase-9/-3) and the G0/G1 phase cellcycle arrest, all of which jointly lead to the death of leukemia cells .
TQFL13 is derivative of Thymoquinone (TQ) (HY-D0803) with potent anti-breast cancer activity. TQFL13 exhibits higher cytotoxicity against breast cancer cells (BT549, MDA-MB-231, 4T1). TQFL13 increases apoptosis and blocks the cellcycle at S and G2/G1 phases in breast cancer cells. TQFL13 shows dose-dependent anti-tumor efficacy in mouse breast cancer allograft model. TQFL13 can be used for the study of breast cancer .
SJL2-1 is a PRMT5 inhibitor, with an IC50 of 1.56 μM. SJL2-1 suppresses proliferation, migration, and invasion in prostate cancer cells. SJL2-1 promotes apoptosis and blocks the cellcycle at the G0/G1 phase. SJL2-1 can target the binding of PRMT5 in cells and inhibit the methylation and expression of the androgen receptor. SJL2-1 can be used for the study of early androgen-sensitive prostate cancer and advanced castration-resistant prostate cancer (CRPC) .
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1cellcycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
Carvacrol is an orally active and blood-brain barrier-permeable monoterpenic phenol that can be extract from an abundant number of aromatic plants, including thyme and oregano, possessing antioxidant, antibacterial, antifungal, anticancer, anti-inflammatory, hepatoprotective, spasmolytic, and vasorelaxant properties. Carvacrol also causes cellcycle arrest in G0/G1, downregulates Notch-1, and Jagged-1, and induces apoptosis. Carvacrol is used in low concentrations as a food flavoring ingredient and preservative, as well as a fragrance ingredient in cosmetic formulations .
EGFR/DHFR-IN-2 (9b) is a dual h-DHFR/EGFR TK inhibitor, with IC50 values of 0.192 μM and 0.109 μM for h-DHFR and EGFR, respectively. EGFR/DHFR-IN-2 (9b) halts the cellcycle at the G1/S phase and induces apoptosis. EGFR/DHFR-IN-2 (9b) is a potential inhibitor of CYP2C9 and CYP3A4. EGFR/DHFR-IN-2 (9b) can be used in the cancer research .
Anticancer agent 205 (compound 9) is a potent anticancer agent. Anticancer agent 205 binds to G4-mtDNA target and inhibits the replication, transcription, and translation of mtDNA (mitochondrial genome). Anticancer agent 205 causes mitochondrial dysfunction, increases ROS production, induces DNA damage and cellular senescence. Anticancer agent 205 induces apoptosis and cellcycle arrests at G0/G1 phase. Anticancer agent 205 has the potential for the research of colorectal cancer .
Annexuzlimab is a humanised IgG1 monoclonal antibody which specifically binds to ANXA1 disrupting its interaction with formyl peptide receptors 1 and 2 (FPR1/2). Annexuzlimab arrests cellcycle progression with cancer cells accumulating in the G1 phase. Annexuzlimab targets secreted ANXA1, preventing FPR1/2 activation and reducing cancer progression. Annexuzlimab can be used for the research of triple negative breast cancer, pancreatic cancer and osteosarcoma .
HTR2A antagonist 1 (Compound 15f) is a HTR2A antagonist, with an IC50 of 42.79 nM. HTR2A antagonist 1 induces sub-G1cellcycle arrest and apoptosis in colorectal cancer cells via the activation of p53/p21/caspase 3 signaling. HTR2A antagonist 1 has good liver microsomal stability. HTR2A antagonist 1 can be used for the research of colorectal cancer .
HIT211504993 is a selective histone deacetylase 6 (HDAC6) inhibitor with an IC50 of 0.070 μM. HIT211504993 suppresses cancer cell proliferation, cause G1 phase cellcycle arrest and induces apoptosis. HIT211504993 inhibits Myc-driven tumorigenesis via nucleocytoplasmic acetylation, p53 modulation, and Wnt/β-catenin signaling modulation. HIT211504993 inhibits tumor growth in a colon cancer xenograft mouse model. HIT211504993 can be used for the research of colon cancer .
EGFR-IN-207 (Compound 5h) is an epidermal growth factor receptor (EGFR) kinase inhibitor with an IC50 of 0.21 μM. EGFR-IN-207 induces cellcycle arrest at the Sub-G1 phase and promotes Apoptosis. EGFR-IN-207 exhibits anticancer activity against lung cancer. EGFR-IN-207 shows extremely low toxicity in non-cancerous cell lines. EGFR-IN-207 can be used in lung cancer-related research .
CTL-12 is an inhibitor of fatty acid synthase (FASN) (IC50: 2.5 μM) and can induce apoptosis. CTL-12 blocks the cellcycle in the Sub-G1/S phase, upregulates the expression of caspase-9 and the apoptosis marker Bax, and downregulates the anti-apoptotic marker Bcl-xL. CTL-12 inhibits de novo lipogenesis, blocks the metabolic demands of tumor cells, and is commonly used in breast and colorectal cancer research .
Topoisomerase I-IN-21 is a promising topoisomerase I inhibitor with an IC50 of 18.79 μM. Topoisomerase I-IN-21 shows higher selectivity toward cancer cells over normal CD8 +cells. Topoisomerase I-IN-21 induces G0/G1cellcycle arrest and apoptosis. Topoisomerase I-IN-21 activates the cGAS-STING pathway, leading to enhanced immune gene expression. Topoisomerase I-IN-21 can be used for research on leukemia, non-small-cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate, and breast cancers .
JUN-1111 is an irreversible and selective Cdc25 phosphatase inhibitor with IC50 values of 0.38, 1.8, 0.66, 28, 37 µM for Cdc25A, Cdc25B, Cdc25C, VHR, PTP1B, respectively. JUN-1111 induces cellcycle arrest at G1 and G2/M phases. JUN-1111 decreases the expression of phosphoCdk1 .
ZDLD13, a β-carboline, is an orally active and selective CDK4/CycD3 inhibitor with an IC50 value of 0.38 μM. ZDLD13 exhibits potent anti-HCT116 activity including inhibition of colony formation, inhibition of invasion and migration, inducing of apoptosis, and arresting of G1 phase in cellcycle. ZDLD13 shows significant tumor growth inhibition in HCT116 tumor xenograft model .
MR24, a G-5555 (HY-19635) derivative, is a mammalian STE20-like (MST) kinase inhibitor. MR24 shows selectively to MST3/4 over MST1/2, with EC50 values of 57 and 583 nM, respectively. MR24 induces G1 phase cellcycle arrest. MR24 can be used for cancer research, such as breast, liver and lung cancers .
Topoisomerase II inhibitor 23 is a potent topoisomerase II inhibitor (IC50= 0.94 μM). Topoisomerase II inhibitor 23 shows high selectivity and exceptional cytotoxic activity in MCF-7, HepG2, and HCT116 cells. Topoisomerase II inhibitor 23 induces cellcycle arrest at the G1 phase, leading to inhibition of cell proliferation. Topoisomerase II inhibitor 2 induces apoptosis by up-regulating the pro-apoptotic Bax level and down-regulating the anti-apoptotic Bcl-2 level.
A-357300 is a reversible and selective MetAP2 inhibitor with IC50s of 0.12 and 57 μM against MetAP2 and MetAP1. A-357300 induces cytostasis by cellcycle arrest at the G1 phase selectively in endothelial cells and in a subset of tumor cells. A-357300 inhibits angiogenesis both in vitro and in vivo and shows potent antitumor efficacy in carcinoma, sarcoma, and neuroblastoma murine models. A-357300 can be used for the studies of neuroblastoma, fibrosarcoma and breast cancer .
KRAS G12C-IN-74 is an orally active, selective KRAS G12C inhibitor with a target IC50 of 43.18 nM. KRAS G12C-IN-74 induces G0/G1cellcycle arrest and apoptosis in KRAS G12C-mutant cancer cells. KRAS G12C-IN-74 is applicable for the research of KRAS G12C-mutant pancreatic cancer, colorectal cancer and lung cancer .
WWZ-11-098 is a selective CDK6 PROTAC degrader with DC50 of 2.6 nM. WWZ-11-098 induces degradation of CDK6 in a CRBN-dependent manner, while sparing CDK1, CDK2, CDK4, and CDK9. WWZ-11-098 induces apoptosis, G1-Scellcycle arrest and shows anti-proliferative activity in cancer cells. WWZ-11-098 exhibits antitumor efficacy in a xenograft model without signs of toxicity. WWZ-11-098 can be used for the research of leukemia .
BRAF V600E/CRAF-IN-2 (Compound 9c) is a potent inhibitor of BRAF V600E/CRAF with IC50s of 0.888 and 0.229 μM, respectively. BRAF V600E/CRAF-IN-2 triggers apoptosis and cellcycle arrest at G0/G1 phase in HCT-116 colon cancer cell. BRAF V600E/CRAF-IN-2 has the potential for the research of cancer diseases .
MTHFD2-IN-8 is a selective MTHFD2 inhibitor with an IC50 of 0.066 μM. MTHFD2-IN-8 directly binds to intracellular mitochondrially localized protein MTHFD2 and accumulates selectively in tumor mitochondria. MTHFD2-IN-8 increases intracellular ROS levels, induces mitochondrial membrane potential depolarization, arrests cellcycle at G0/G1 phase, promotes apoptosis in cancer cells. MTHFD2-IN-8 inhibits tumor growth in a mouse colon cancer graft model .
TOPOI/PARP-1-IN-1 (Compound B6) is an orally active, low cytotoxic TOPOI/PARP dual inhibitor with an IC50 value of 0.09 μM for PARP1. TOPOI/PARP-1-IN-1 can effectively inhibit the proliferation and migration of cancer cells. TOPOI/PARP-1-IN-1 also causes cellcycle arrest in the G0/G1 phase and induces apoptosis. The tumor growth inhibition rate (TGI) of TOPOI/PARP-1-IN-1 in mice is 75.4% .
BML-278 is a SIRT1 activator (EC150: 1 μM). BML-278 increases H3K9 methylation and inhibits H3K9 acetylation in both the paternal and maternal pronucleus. BML-278 improves early embryonic development. BML-278 arrests the cellcycle at the G1/S phase, and reduces senescence in primary human mesenchymal cells. BML-278 reduces tubulin acetylation in U937 cells. BML-278 also increases mitochondrial density in murine C2C12 myoblasts .
INNO-220 is an orally active, CRBN-dependent molecular glue degrader targeting CK1α. INNO-220 induces cellcycle arrest at G0/G1 phase and triggers apoptosis by degrading CK1α. INNO-220 disrupts the assembly and function of the CARD11/BCL10/MALT1 complex, thereby inhibiting NF-κB signaling in stimulated T cells and lymphoma cells that harbor an activating mutation in CARD11. INNO-220 provides a new direction for lymphoma research.
CDK4-IN-5 is a potent, orally active and selective CDK4 inhibitor. CDK4-IN-5 suppresses CDK4 expression and downregulates the CDK4/CyclinD1 complex. CDK4-IN-5 induces G0/G1 phase cellcycle arrest in bladder cancer cells via CyclinD1 expression suppression. CDK4-IN-5 selectively exerts activity against bladder cancer cells. CDK4-IN-5 can be used for the research of bladder cancer .
NF-κB-IN-5 (compound 4d) is an orally active and potent NF-κB inhibitor by interacting directly with NF-κB. NF-κB-IN-5 shows antitumor activity against human cancer cell lines (HCT116, U87-MG, HepG2, BGC823, PC9), with IC50 values of 5.35, 2.81, 2.83, 2.02 and 3.90 μM, respectively. NF-κB-IN-5 induces apoptosis in U87-MG tumor cell and cellcycle arrest in G0/G1 phase .
4'-Bromo-resveratrol (4′‐BR) is a dual SIRT1/SIRT3 inhibitor with an IC50 of 0.2 mM for both targets. 4'-Bromo-resveratrol induces caspase-dependent apoptosis, induces G0/G1cellcycle arrest, and inhiibits proliferation. 4'-Bromo-resveratrol reduces lactate production, glucose uptake, and NAD +/NADH ratio, and downregulates lactate dehydrogenase A and glucose transporter 1 (GLUT1). 4'-Bromo-resveratrol can be used for the research of melanoma .
DHODH-IN-32 (Compound A1) is a DHODH inhibitor. DHODH-IN-32 shows significant cytotoxicity against NCI-60 cell lines, especially being sensitive to breast cancer, prostate cancer and leukemia cell lines. DHODH-IN-32 can induce cellapoptosis by activating the Caspase pathway. DHODH-IN-32 causes G0/G1 phase cellcycle arrest and inhibits cellular metabolism by ROS. DHODH-IN-32 exhibits significant anti-tumor properties in mouse breast cancer models. DHODH-IN-32 can be used for the study of breast cancer .
RK-682 is the inhibitor for protein tyrosine phosphatase (PTPase), heparanase, phospholipase A2 and HIV-1 protease. RK-682 inhibits the dephosphorylation of CD45 (IC50 is 54 μM) and VHR (IC50 is 2.0 μM), and thereby inhibits the ERK signaling pathway. RK-682 inhibits the cell viability of cancer cell MGH-U3, T24 and UROtsa with IC50s of 78.2, 43.2 and 145 nM, respectively, arrests the cellcycle at G1/S phase, inhibits the cell migration and autophagy in MGH-U3 and T24 .
Decursin (Standard) is the analytical standard of Decursin. This product is intended for research and analytical applications. Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities [4].
CDK2-IN-47 is a potent CDK2 inhibitor with an IC50 of 0.21 μM. CDK2-IN-47 exhibits outstanding anticancer activity against MCF-7, HCT-116, and MGC-803 cell lines. CDK2-IN-47 effectively induces G1cellcycle arrest, retinoblastoma protein (Rb) dephosphorylation, and significant apoptosis. CDK2-IN-47 can be used for the studies of breast cancer, colorectal cancer and gastric cancer .
Atractylenolide II (Standard) is the analytical standard of Atractylenolide II. This product is intended for research and analytical applications. Atractylenolide II (Asterolide) is a sesquiterpenoid compound. Atractylenolide II can induce G1 phase cellcycle arrest and apoptosis in B16 melanoma cells. Atractylenolide II is an orally effective anticancer agent that can exert anti-melanoma effects by inhibiting the STAT3 signaling pathway. In addition, Atractylenolide II has been shown to ameliorate myocardial fibrosis, oxidative stress, and neuroprotective activity .
Panaxatriol is an orally active insulin sensitizer. Panaxatriol enhances the phosphorylation levels of Akt, insulin receptor and p70S6K in skeletal muscle. Panaxatriol reduces the mRNA expression level of Atrogin1 in skeletal muscle. Panaxatriol induces apoptosis, pre-G1cellcycle arrest and increased intracellular ROS levels in prostate cancer cells, decreases mitochondrial membrane potential, inhibits cell migration and reduces colony formation. Panaxatriol can be used in research related to insulin resistance, myocardial ischemia/reperfusion injury and prostate cancer .
RO5068760 is a potent, orally active and selective non-ATP-competitive MEK1/2 inhibitor with an IC50 of 0.025 μM for MEK1. RO5068760 significantly inhibits MAPK pathway activity, thereby inducing G1cellcycle arrest and apoptosis to inhibit cancer cell growth. RO5068760 exhibits significant efficacy in a broad spectrum of tumors with aberrant MAPK pathway activation. RO5068760 can be used for melanoma, colorectal cancer, non-small cell lung cancer (NSCLC), and pancreatic cancer research .
CLPP-2068 is the orally active activator for human caseinolytic protease P (HsClpP) with an EC50 of 50.4 nM. CLPP-2068 exhibits anti-proliferative efficacy in OCI-LY10 cancer cell with an IC50 of 5.2 nM. CLPP-2068 decreases mitochondrial membrane potential, increases mitochondrial ROS levels, and induces mitochondrial dysfunction. CLPP-2068 arrests the cellcycle at G1 phase, and induces apoptosis in cell OCI-LY10. CLPP-2068 exhibits antitumor activity in mouse xenograft models .
TRK-IN-30 (Compound C11) is the inhibitor for tropomyosin receptor kinase (TRK) that inhibits TRKA, TRKB and TRKC and drug resistant mutant TRKA G595R with an IC50 of 1.8, 0.98, 3.8, and 54 nM, respectively. TRK-IN-30 inhibits the activation of the downstream PI3K/AKT and MEK/ERK signaling pathways. TRK-IN-30 inhibits the colony formation and cell migration of Km-12, arrests the cellcycle at G0/G1 phase, and induces apoptosis in Km-12 .
Chikusetsusaponin IVa methyl ester (CME) is a natural triterpenoid saponin compound. Chikusetsusaponin IVa methyl ester induces G0/G1cellcycle arrest and apoptosis in colon cancer cells by inhibiting the Wnt/β-catenin signaling pathway. By inhibiting the NF-κB and AP-1 signaling pathways, Chikusetsusaponin IVa methyl ester significantly reduces the production of NO, PGE₂ and pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), and downregulates the levels of iNOS and COX-2. Chikusetsusaponin IVa methyl ester can be used in researches on colorectal cancer and inflammation .
PD-L1/HDAC3-IN-1 (PH3) is a dual PD-L1/HDAC3 Inhibitor with IC50 values of 89.4 nM and 107 nM for PD-1/PD-L1 and HDAC3, respectively. PD-L1/HDAC3-IN-1 induces cellapoptosis and arrests cellcycle at G0/G1 phase. PD-L1/HDAC3-IN-1 shows anticancer activity both in vivo and in vitro .
Contragestazol (DL111-IT) is a non-hormonal antifertility agent. Contragestazol reduces the expression of Cyclin D1 and CDK4, increases the expression of total retinoblastoma protein (pRb), and decreases the level of hyperphosphorylated pRb. Contragestazol induces G0/G1 phase cellcycle arrest. Contragestazol inhibits embryonic development by inducing luteal cellapoptosis and reducing intrauterine polyamine levels. Contragestazol exhibits antitumor activity against prostate cancer, S180 tumor and H22 tumor. Contragestazol shows extremely potent activity in terminating early pregnancy in animals .
FLT3-IN-14 is a potent FLT3 inhibitor with IC50s of 5.6 nM and 1.4 nM for FLT3-WT and FLT3-ITD. FLT3-IN-14 reduces the phosphorylation of FLT3 (Y591), induces cellcycle arrest at G1 phase and apoptosis. FLT3-IN-14 significantly reduces the tumor growth in an MV4-11 xenograft mouse model .
Cyclin K degrader 2 is a molecular glucose degrading agent that targets the cyclin K protein. Cyclin K degrader 2 has inhibitory activity against CDK1 and CDK9. Cyclin K degrader 2 causes a decrease in RNA polymerase II Ser2 phosphorylation levels, downregulation of DNA damage response gene expression, accumulation of DNA damage, G1 phase arrest of the cellcycle, and apoptosis. Cyclin K degrader 2 can be used for cancer research .
MR24 TFA, a G-5555 (HY-19635) derivative, is a mammalian STE20-like (MST) kinase inhibitor. MR24 TFA shows selectively to MST3/4 over MST1/2, with EC50 values of 57 and 583 nM, respectively. MR24 TFA induces G1 phase cellcycle arrest. MR24 TFA can be used for cancer research, such as breast, liver and lung cancers .
CDK4/6-IN-28 is a potent, orally active, and selective CDK4/6 inhibitor IC50 values of 14.02 and 10.03 nM, respectively. CDK4/6-IN-28 inhibits breast cancer cell colony formation, migration, and proliferation. CDK4/6-IN-28 induces G1-phasecellcycle arrest and apoptosis in breast cancer cells. CDK4/6-IN-28 exhibits tumor inhibitory activity in breast cancer xenograft mouse models. CDK4/6-IN-28 can be used for the research of breast cancer .
EGFR/VEGFR2-IN-12 (compound 11a) is a dual EGFR/VEGFR2 inhibitor, with an IC50 value of 64 nM against human EGFR and an IC50 value of 74 nM against human VEGFR2. EGFR/VEGFR2-IN-12 inhibits the phosphorylation of EGFR and VEGFR2, induces cellcycle arrest at the G1/S phase, activates apoptotic pathways, promotes PARP-1 cleavage, exhibits low micromolar antiproliferative activity, and shows much higher selectivity for cancer cells than normal cells. EGFR/VEGFR2-IN-12 is applicable for cancer-related research .
Lotusine is an orally active signaling pathway modulator and enzyme inhibitor, with an IC50 of 30.60 μg/mL against α-amylase and an IC50 of 36.15 μg/mL against α-glucosidase. Lotusine inhibits the EGFR-Akt-ERK signaling pathway by reducing the levels of phosphorylated EGFR, Akt and ERK. Lotusine induces apoptosis, triggers G0/G1cellcycle arrest and inhibits cancer cell proliferation. Lotusine reduces lipid peroxidation and increases the activities of SOD, CAT and GPx. Lotusine is applicable to researches related to non-small cell lung cancer, type 2 diabetes and autism spectrum disorder.
FGFR3-IN-11(compound B11) is a Fibroblast growth factor receptor 3 (FGFR3) inhibitor with a Ka value of 4.8 μM. FGFR3-IN-11 induces apoptosis, suppresses colony formation, and causes dose-dependent G0/G1cellcycle arrest in cancer cells. FGFR3-IN-11 exerts anticancer activity against cancer cells with minimal toxicity toward normal hepatocytes and demonstrates tumor growth suppression in xenograft mouse models. FGFR3-IN-11 can be used for the research of hepatocellular carcinoma .
T7 Peptide is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide induces G0/G1cellcycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
Oximidine III is an anti-tumor antibiotic. Oximidine III can selectively inhibit the growth of 3Y1 in rat fibroblasts with degeneration of various tumor genes. Oximidine III inhibits v-H-ras-3Y1, v-src-3Y1 cells and the normal 3Y1 cells with IC50s (nM) of 14, 4.5 and 140, respectively. Oximidine III stops RAS-or SRC-denatured cells at G1 phase of the cellcycle and increases p21WAF1 expression .
DNMT1-IN-3 (compound 7t-S) is an effective DNA methyltransferase 1 (DNMT1) inhibitor with an IC50 value of 0.777 μM and a KD value of 0.183 μM. DNMT1-IN-3 can bind to the methyl donor S-adenosyl-l-methionine (SAM) site in DNMT1. DNMT1-IN-3 can inhibit cell proliferation in K562 cells by inducing cells apoptosis and arresting cellcycle at G0 / G1 phase, which has the potential to be used for the research of hematologic tumor .
HDAC1-IN-13 is an orally active HDAC1 inhibitor with IC50 values of 91, 185, 170, and 280 nM against HDAC1, HDAC2, HDAC3, and HDAC10, respectively, and shows no activity against HDAC4, HDAC5, HDAC6, HDAC7, and HDAC9. HDAC1-IN-13 induces extrinsic apoptosis by activating the caspase-8 pathway and triggers G0/G1cellcycle arrest. HDAC1-IN-13 can be used for the research of leukemia .
VEGFR-2-IN-52 (compound 14d) is a potent VEGFR-2 inhibitor with an IC50 value of 191.1 nM. VEGFR-2-IN-52 decreases the protein expression of p-VEGFR-2, MMP9, p-ERK1/2 and p-MEK1. VEGFR-2-IN-52 shows cytotoxicity. VEGFR-2-IN-52 induces apoptosis and cellcycle arrest at G0/G1 phase. VEGFR-2-IN-52 increases the levels of ROS .
EGFR-IN-151 (Compound 10) inhibits EGFR and its downstream signaling pathways ERK/STAT3. EGFR-IN-151 inhibits the proliferation of a variety lung cancer cells (IC50s for NCI-H1781, HCC827, NCI-H3255 and NCI-H1975 is 11.7, 5.19, 7.32 and 1.53 μM, respectively), inhibits the colony formation and migration of H1975, arrests the cellcycle at G1 phase, and induces apoptosis in H1975 .
FLT3-IN-21 (compound LC-3) is a potent FLT3 inhibitor (IC50: 8.4 nM) and induces apoptosis. FLT3-IN-21 can arrest the cellcycle in the G1 phase and inhibit the proliferation of FLT3-ITD-positive AML cells MV-4-11 (IC50: 5.3 nM). In mice, FLT3-IN-21 (10 mg/kg/d) inhibited tumor growth in the MV-4-11 xenograft model (TGI=92.16%) .
EGFR-IN-57 (Compound 25a) is a potent, orally active EGFR-TK inhibitor with an IC50 of 0.054 µM. EGFR-IN-57 also inhibits VEGFR-2, CK2α, topoisomerase IIβ and tubulin polymerization with IC50 values of 0.087, 0.171, 0.13 and 3.61 µM, respectively. EGFR-IN-57 induces cellcycle arrest at G2/M and pre-G1 phases. EGFR-IN-57 induces cancer cellapoptosis .
Gomisin G is a lignin from S. chinesis with anti-HIV (EC50 = 0.006 μg/mL), anti-liver cancer and anti-inflammatory activities. Gomisin G has an AKT-cyclin D1 dependent mechanism against triple-negative breast cancer (TNBC) cells through suppressing phosphorylation rather than inducing apoptosis. Gomisin G can inhibit AKT phosphorylation. Gomisin G can cause cellcycle arrest in the G1 phase. Gomisin G can be studied in research for diseases such as HIV, breast and liver cancers .
Decursin-d6 ((+)-Decursin-d6) is the deuterium labeled Decursin (HY-18981). Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities .
SL43 is an orally active and potent SOS1 inhibitor with a Kd of 0.16 μM. SL43 disrupts SOS1-KRAS interaction, inhibits SOS1-mediated nucleotide exchange on KRAS mutants, and suppresses RAS-MAPK signaling. SL43 exerts antiproliferative activity against KRAS-mutant cancer cells, induces early apoptosis and G1 phase cellcycle arrest, and reduces phosphorylated MEK and ERK levels. SL43 suppresses tumor growth in a colorectal cancer xenograft model .
PAK4-IN-5 (Compound 12i) is a PAK4 inhibitor (IC50: 7.68 nM for PAK4, 1872.01 nM for PAK1). PAK4-IN-5 binds to PAK4 stably via multiple interactions. PAK4-IN-5 inhibits the proliferation and the migratory potential of MDA-MB-231 cells by inhibiting the phosphorylation of PAK4 and LIMK1. PAK4-IN-5 arrests cellcycle in the G0/G1 phase, induces apoptosis and ROS production. LD50: >500 mg/kg for mice (p.o.) .
Hsp90-Cdc37-IN-2 (Compound 41) is an inhibitor for the interaction between heat shock protein 90 (Hsp90) and cyclin 37 (Cdc37). Hsp90-Cdc37-IN-2 inhibits the proliferation of cancer cell A549, MCF-7, HOS and HepG2 with IC50 of 0.41-0.94 μM. Hsp90-Cdc37-IN-2 decreases the mitochondrial membrane potential, induces apoptosis, and arrest cellcycle at G0/G1 phase in A549 .
T7 Peptide TFA is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide TFA inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide TFA induces G0/G1cellcycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide TFA is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
CDK2-IN-51 is a pyrazolopyridine derivative, a CDK2 inhibitor with an IC50 of 23.47 nM. CDK2-IN-51 does not have a pro-apoptotic effect and had no significant effect on CDK2 protein expression. CDK2-IN-51 reduces expression of DNA replication factors (Polα, MCM7, ORC2, and ORC4) and pre-G1cellcycle arrest. CDK2-IN-51 can be used for the research of colorectal cancer .
SKLB06489 is a selective and orally active inhibitor of type I PRMT enzymes, with IC50 values of 64.55 nM (PRMT1), 4.21 nM (PRMT6), and 51.27 nM (PRMT8). SKLB06489 inhibits cell proliferation, colony formation, DNA replication, and DNA damage repair in cancer cells. SKLB06489 induces G0/G1-phasecellcycle arrest and apoptosis in cancer cells. SKLB06489 enhances intracellular cholesterol efflux via ABCA1 and ABCG1 upregulation, disrupts cholesterol metabolic homeostasis, and suppresses tumor growth in subcutaneous xenograft models. SKLB06489 can be used for the research of triple-negative breast cancer (TNBC) .
PPIA-IN-1 is a PPIA inhibitor with a Kd value of 0.52 μM. PPIA-IN-1 inhibits the PPIA/MAPK signaling pathway to exert antiproliferative activity. PPIA-IN-1 induces G0/G1cellcycle arrest in cancer cells. PPIA-IN-1 upregulates the expression of Bax and caspase-3, downregulates Bcl-2 expression, and induces apoptosis in cancer cells. PPIA-IN-1 induces increased ROS levels, DNA damage, endoplasmic reticulum stress, and mitochondrial dysfunction in cancer cells. PPIA-IN-1 exhibits antitumor activity in a mouse colon cancer xenograft model. PPIA-IN-1 can be used for the research of colorectal cancer .
USP30-IN-20 is an orally active USP30 inhibitor (Kd = 1.61 μM, IC50 = 12.8 μM). USP30-IN-20 induces ferroptosis by promoting ubiquitination-mediated degradation of GPX4. USP30-IN-20 inhibits the proliferation, migration, invasion, and stemness of prostate cancer cells. USP30-IN-20 induces G0/G1cellcycle arrest and ROS levels in prostate cancer cells. USP30-IN-20 exhibits significant anti-tumor efficacy in PC3 cell subcutaneous xenografts in mice. USP30-IN-20 can be used for the study of advanced prostate cancer .
EGFR/HER2-IN-16 (compound 12K) is an effective dual-target inhibitor of EGFR (IC50=6.15 nM) and HER-2 (IC50=9.78 nM) with anti-tumor activity. EGFR/HER2-IN-16 can inhibit the migration of SK-BR-3 cells, arrest the cellcycle in the G0/G1 phase, and induce apoptosis. EGFR/HER2-IN-16 exhibits good anti-proliferative activity against tumor cell models and has little damage to healthy cells. EGFR/HER2-IN-16 can be used in breast cancer research .
MMP-9-IN-14 is a MMP-9 inhibitor (IC50 = 34.46 μM). MMP-9-IN-14 induces G1-phasecellcycle arrest and caspase-dependent apoptosis in cancer cells. MMP-9-IN-14 promotes the accumulation of phosphorylated γH2AX. MMP-9-IN-14 inhibits the migration and invasion of cancer cells, and downregulates the expressions of MMP-2, MMP-9 and hTERT in cancer cells. MMP-9-IN-14 inhibits tumor growth and angiogenic spread in animal models. MMP-9-IN-14 can be used for the research of cancers such as lung adenocarcinoma, cervical cancer and colorectal cancer .
Carvacrol (Standard) is the analytical standard of Carvacrol. This product is intended for research and analytical applications. Carvacrol is an orally active and blood-brain barrier-permeable monoterpenic phenol that can be extract from an abundant number of aromatic plants, including thyme and oregano, possessing antioxidant, antibacterial, antifungal, anticancer, anti-inflammatory, hepatoprotective, spasmolytic, and vasorelaxant properties. Carvacrol also causes cellcycle arrest in G0/G1, downregulates Notch-1, and Jagged-1, and induces apoptosis. Carvacrol is used in low concentrations as a food flavoring ingredient and preservative, as well as a fragrance ingredient in cosmetic formulations .
FLT3-IN-29 (Compound MY-10) is a FLT3 inhibitor (IC50s: 6.5 and 10.3 nM for FLT3-ITD and FLT3-D835Y mutants). FLT3-IN-29 arrests cellcycle at the G0/G1 phase and efficiently induces Apoptosis. FLT3-IN-29 also reduces reactive oxygen species (ROS) production and mitochondrial membrane potential (MMP). FLT3-IN-29 displays antileukemic activity .
HDAC-IN-50 is a potent and orally active FGFR and HDAC dual inhibitor with IC50 values of 0.18, 1.2, 0.46, 1.4, 1.3, 1.6, 2.6, 13 nM for FGFR1, FGFR2, FGFR3, FGFR4, HDAC1, HDAC2, HDAC6, HDAC8, respectively. HDAC-IN-50 induces Apoptosis and cellcycle arrest at G0/G1 phase. HDAC-IN-50 decreases the expression of pFGFR1, pERK, pSTAT3. HDAC-IN-50 shows anti-tumor activity .
DFX117 is a selective, orally active inhibitor for PI3Kα and c-Met tyrosine kinase. DFX117 inhibits PI3K/Akt/mTOR pathway, inhibits the proliferation of NCI-H1975, NCI-H1993, and HCC827 with IC50s 0.02-0.08 µM. DFX117 arrests cellcycle at G0/G1 phase, induces apoptosis in A549 and NCI-H1975. DFX117 exhibits antitumor efficacy in mice .
EGFR-IN-165 is a potent EGFR inhibitor. EGFR-IN-165 demonstrates superior potency with IC50s of 17.18 and 64.74 nM against EGFR L858R/T790M and EGFR WT; 2.17 and 6.2 μM against NCI-H1975 cells and A431 cells. EGFR-IN-165 significantly inhibits the migration and induces G1 phase cellcycle arrest and cellapoptosis. EGFR-IN-165 can be used for the study of cancers such as non-small cell lung cancer, colorectal cancer, and head and neck squamous cell carcinoma .
FD223 is a potent and selective phosphoinositide 3-kinase delta (PI3Kδ) inhibitor. FD223 displays high potency (IC50=1 nM) and good selectivity over other isoforms (IC50s of 51 nM, 29 nM and 37 nM, respectively for α, β and γ). FD223 exhibits efficient inhibition of the proliferation of acute myeloid leukemia (AML) cell lines by suppressing p-AKT Ser473 thus causing G1 phase arrest during the cellcycle. FD223 has potential for the research of leukemia such as AML .
EGFR-IN-169 is an epidermal growth factor (EGFR) (IC50 = 5.19 μM) inhibitor form panaxadiol. EGFR-IN-169 interferes with the migration and growth of colorectal cancer cells by inhibiting EGFR-mediated RalA/EMT pathway. EGFR-IN-169 shows an IC50 value of 4.46 μM and SI of 16.92 for HCT-116 cells. EGFR-IN-169 inhibits CDKs activity, induces G0/G1cycle arrest and inhibits migration and invasion. EGFR-IN-169 reduces mitochondrial membrane potential and induces apoptosis and ROS production. EGFR-IN-169 can be used for the research of cancer, such as colorectal cancer .
Lotusine hydroxide is an orally active signaling pathway modulator and enzyme inhibitor, with an IC50 of 30.60 μg/mL against α-amylase and an IC50 of 36.15 μg/mL against α-glucosidase. Lotusine hydroxide inhibits the EGFR-Akt-ERK signaling pathway by reducing the levels of phosphorylated EGFR, Akt and ERK. Lotusine hydroxide induces apoptosis, triggers G0/G1cellcycle arrest and inhibits cancer cell proliferation. Lotusine hydroxide reduces lipid peroxidation and increases the activities of SOD, CAT and GPx. Lotusine hydroxide is applicable to researches related to non-small cell lung cancer, type 2 diabetes and autism spectrum disorder .
Dibenzo (a,i) pyrene is a polycyclic aromatic hydrocarbon and also a carcinogenic ligand of the TCDD (Ah) receptor. Dibenzo (a,i) pyrene binds to the TCDD (Ah) receptor in rat liver. Dibenzo (a,i) pyrene induces DNA adduct formation and upregulates the protein levels of p53 and p21 WAF1 in diploid lung fibroblasts. Dibenzo (a,i) pyrene alters the cellcycle distribution of diploid lung fibroblasts, increasing the proportion of cells in the S phase, decreasing the proportions of cells in the G0/G1 and G2/M phases, and causing S phase delay/arrest. Dibenzo (a,i) pyrene is applicable for cancer research .
Sotetsuflavone is a flavonoid that can be isolated from Cycas revolute. Sotetsuflavone inhibits phosphorylation of PI3K, Akt, mTOR, JNK, and p38 MAPK; modulates expression of Cyclin D1, CDK4, Bcl-2, Bax, cleaved caspases 3/9, MMP-9, TGF-β, STAT3, and β-catenin. Sotetsuflavone induces G0/G1cellcycle arrest, apoptosis, autophagy, and intracellular ROS elevation, inhibits cancer cell proliferation. Sotetsuflavone inhibits tumor growth in mouse tumor xenograft models. Sotetsuflavone can be used for the research of non-small cell lung cancer and Crohn’s disease .
VEGFR/PD-L1-IN-1 is a VEGFR2 and PD-L1 inhibitor, with IC50 values of 0.383 μM and 134.407 pg/mL against VEGFR2 and PD-L1, respectively. VEGFR/PD-L1-IN-1 enhances the secretion of INF-γ, induces G0/G1cellcycle arrest in cancer cells, and triggers cancer cellapoptosis. VEGFR/PD-L1-IN-1 upregulates the expression of BAX and Caspase-3, and downregulates the expression of Bcl-2. VEGFR/PD-L1-IN-1 can be used in research related to hepatocellular carcinoma, prostate cancer, and colorectal cancer .
ZW-49 is an orally active pan-EGFR inhibitor with IC50 values at 0.03-1.5 nM. ZW-49 inhibits all subgroups of EGFR mutations with selectivity over wild-type EGFR and other target families. ZW-49 blocks the ATP-binding pocket, occupies a conserved hydrophobic subpocket, avoids steric conflicts with PACC mutation P loops. ZW-49 inhibits cancer cells proliferation, induces G0/G1 phase cell-cycle arrest and apoptosis, and demonstrates anti-proliferative activity in xenograft mice models. ZW-49 can be used for the research of cancer, such as non-small cell lung cancer .
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cellcycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
(+)-Columbianetin ((S)-Columbianetin) acts as an inhibitor of JNK/ERK. (+)-Columbianetin inhibits UVA-induced phosphorylation of JNK and ERK, reduces the production of MMP-1, reverses UVA-induced Collagen (HY-NP003) degradation, and alleviates UVA-mediated inhibition of Smad2/3 phosphorylation and translocation. (+)-Columbianetin regulates the AP-1 and ASK1-MAPK signaling pathways, inhibits the production of ROS and blocks sub-G1cellcycle arrest. (+)-Columbianetin is applicable to research related to skin aging .
WJ35435 is a dual-targeted anticancer hybrid that induces anti-HDAC (in particular HDAC1 and HDAC6) and anti-topoisomerase I activities that causes DNA damage associated with a low DNA repair capability and induces cellcycle arrest at G1- and G2-phase to apoptosis. WJ35435 induces histone H3 acetylation and phosphorylation, α-tubulin acetylation and γ-H2AX formation to achieve anti-HDAC effect. WJ35435 is promising for research of cancer .
EGFR-IN-134 (compound 3f), a triazolo[3,4-a]isoquinoline derivative, is a potent EGFR inhibitor with an IC50 of 0.023 µM. EGFR-IN-134 induces apoptosis and necrosis. EGFR-IN-134 initiates cellcycle arrest at the G2/M and pre-G1 phases, downregulates anti-apoptotic protein Bcl2 and upregulates pro-apoptotic proteins: p53, Bax, and caspases 3, 8, and 9. EGFR-IN-134 shows antiproliferative and anticancer activity .
PROTAC IKKβ degrader-1 is a IKKβ PROTAC degrader (DC50 = 7.15 μM). PROTAC IKKβ degrader-1 induces apoptosis (Apoptosis) in triple-negative breast cancer cells. PROTAC IKKβ degrader-1 induces G1 phase cellcycle arrest in triple-negative breast cancer cells. PROTAC IKKβ degrader-1 exhibits antiproliferative activity against a variety of cells. PROTAC IKKβ degrader-1 is applicable for research related to cancers such as triple-negative breast cancer, colon cancer, liver cancer, pancreatic cancer .
FLT3/TrKA-IN-1 is a potent FLT3/TrKA dual kinase inhibitor with the IC50s of 43.8 nM, 97.2 nM, 92.5 nM and 23.6 nM for FLT3, FLT3-ITD, FLT3-TKD and TrKA, respectively. FLT3/TrKA-IN-1 induces cellcycle arrest at the G0/G1 phase as well as apoptosis and shows antiproliferative activity in vitro. FLT3/TrKA-IN-1 has the potential for the research of Acute myeloid leukemia (AML) .
FOXM1-IN-4 is a selective 5-HT7 receptor inhibitor with a Ki of 92 nM. FOXM1-IN-4 blocks 5-HT7 receptor signaling to reduce FOXM1, p-FOXM1, cyclin B1, and cdc25B levels. FOXM1-IN-4 acts as an antiproliferative, clonogenic inhibitor, and cellcycle inhibitor that induces G2/M arrest, reduces G0/G1 population. FOXM1-IN-4 can be used for the research of triple-negative breast cancer .
FOXM1-IN-4 hydrochloride is a selective 5-HT7 receptor inhibitor with a Ki of 92 nM. FOXM1-IN-4 hydrochloride blocks 5-HT7 receptor signaling to reduce FOXM1, p-FOXM1, cyclin B1, and cdc25B levels. FOXM1-IN-4 hydrochloride acts as an antiproliferative, clonogenic inhibitor, and cellcycle inhibitor that induces G2/M arrest, reduces G0/G1 population. FOXM1-IN-4 hydrochloride can be used for the research of triple-negative breast cancer .
PLAGL2-IN-1 is a inhibitor of pleiomorphic adenoma-like protein 2 (PLAGL2) with a Kd of 2.23 µM. PLAGL2-IN-1 suppresses PLAGL2 transcriptional activity, induces G0/G1cellcycle arrest, and apoptosis, thereby inhibiting hepatocellular carcinoma (HCC) cell proliferation. PLAGL2-IN-1 disrupts extracellular matrix organization and suppresses the PI3K-AKT pathway by reducing AKT phosphorylation. PLAGL2-IN-1 inhibits tumor growth in an HCCLM3 xenograft mouse model. PLAGL2-IN-1 can be used for the research of HCC .
FLT3-IN-39 (Compound W4) is a selective FLT3 inhibitor, with an IC50 value of 16.0 nM against FLT3-ITD and an IC50 value of 20.4 nM against FLT3-D835Y. FLT3-IN-39 inhibits FLT3-ITD and FLT3-D835Y mutant kinases. FLT3-IN-39 induces G0/G1cellcycle arrest and Apoptosis in cancer cells, and reduces intracellular ROS levels. FLT3-IN-39 exhibits anti-tumor activity against acute myeloid leukemia .
EGFR-IN-201 is a potent EGFR inhibitor, with an IC50 of 0.091 μM against wild-type EGFR; for mutant EGFR variants, the IC50 values of EGFR T790M, EGFR L858R and EGFR C797S are 0.147 μM, 0.221 μM and 0.703 μM, respectively. EGFR-IN-201 inhibits EGFR downstream signaling proteins AKT1 (IC50 = 0.225 μg/mL) and ERK1 (IC50 = 0.705 μg/mL). EGFR-IN-201 induces G0/G1cellcycle arrest, apoptosis and low-level necrosis in cancer cells. EGFR-IN-201 is applicable to research on cancers such as colon cancer .
Germacrone is a sesquiterpene compound with multiple biological activities. Germacrone inhibits the H1N1 and H3N2influenza A virus and the influenza B virus. Germacrone blocks the progressionof arthritis by regulating Th1/Th2 balance and inhibiting NF-κB signaling. Germacrone can arrest the cellcycle at G0/G1 and G2/M phases and induce apoptosis in breast cancer cells. Germacrone inhibits 5α-reductase and has anti-androgenic effect. Germacrone has neuroprotective functions and can be used for the study of traumatic brain injury (TBI). Germacrone also has antioxidant activity .
SHP2-IN-33 (Compound D13) is an allosteric inhibitor of SHP2 with an IC50 of 1.2 μM. In cellular studies, SHP2-IN-33 demonstrates antiproliferative activity with an IC50 of 38 μM against Huh7 cells by arresting the G0/G1cellcycle, promoting apoptosis (Apoptosis), and suppressing the MAPK signaling pathway. In an in vivo Huh7 xenograft mouse model, SHP2-IN-33 exhibits significant antitumor activity and favorable pharmacokinetics, including 54% oral bioavailability and a half-life of 10.57 hours. SHP2-IN-33 is a promising compound for studying tumor diseases associated with SHP2 .
Cyclo (phg-isoDGR-k)-PEG4-non-cleavable-SAR405838 is a dual MDM2 and α5β1 integrin modulator. Cyclo (phg-isoDGR-k)-PEG4-non-cleavable-SAR405838 acts as an antiproliferative agent, apoptosis inducer and cellcycle regulator, induces reactivation of p53 and upregulation of p21, redistributes glioblastoma cells from the G0/G1 phase to the G2/M phase, and enhances apoptosis. Cyclo (phg-isoDGR-k)-PEG4-non-cleavable-SAR405838 is applicable to the research of glioblastoma .
Veratramine (NSC17821; NSC23880) is an orally active inhibitor of the PI3K/Akt/mTOR signaling pathway and a SIGMAR1 modulator. Veratramine induces autophagic apoptosis of tumor cells, arrests the cellcycle at the G0/G1 phase, and inhibits epithelial-mesenchymal transition (EMT)-related proteins to reduce tumor migration. Veratramine reduces spinal cord and sciatic nerve pathological damage in a neuropathy model by inhibiting SIGMAR1 binding to NMDAR and phosphorylation of NMDAR Ser896. Veratramine has anti-tumor proliferation, apoptosis induction, anti-inflammatory and neuroprotective activities, and can be used in the study of cancers such as liver cancer and osteosarcoma, as well as diabetic peripheral neuropathy .
MPT0L145 is a PIK3C3/FGFR inhibitor, with a Kd value of 0.53 nM for PIK3C3. MPT0L145 decreases the phosphorylation of FGFR1, FGFR3 and their downstream proteins (FRS2, ERK and Akt). MPT0L145 induces G0/G1cellcycle arrest and decreased protein levels of cyclin E. MPT0L145 promotes mitochondrial dysfunction, ROS production, and DNA damage. MPT0L145 is an autophagy inhibitor. MPT0L145 significantly sensitizes cancer cells to targeted or chemotherapeutic agents. MPT0L145 can be used for cancer research, such as bladder cancer and NSCLC .
Atopaxar hydrochloride (E5555 hydrochloride) is the hydrochloride salt form of Atopaxar (HY-18200). Atopaxar hydrochloride is an orally active, selective and reversible antagonist for thrombin receptor protease-activated receptor-1 (PAR-1). Atopaxar hydrochloride is the inhibitor for Janus kinase 1 (JAK1) and JAK2, which inhibits the JAK-STAT with EC50 of 5.90 μM in A549. Atopaxar hydrochloride inhibits the cell viability of A549 (IC50=7.02 μM), arrests the cellcycle at G1 phase and induces apoptosis. Atopaxar hydrochloride exhibits antiplatelet and antitumor activities. Atopaxar hydrochloride can be used for the research of atherothrombotic disease .
Sulindac sulfone is an orally active metabolite of Sulindac (HY-B0008). Sulindac sulfone activates PPARγ and drives transcriptional induction of SSAT by binding to the PPRE-2 element. Sulindac sulfone induces Apoptosis. Sulindac sulfone negatively regulates the function of VDAC1/2 to inhibit the mTORC1 pathway, reduces Cyclin D1 levels, and induces G1cellcycle arrest in colon cancer cells. Sulindac sulfone exerts colon cancer preventive effects through a COX-independent mechanism. Sulindac sulfone can be used in research related to colon cancer .
PROTAC XPO1 degrader-1 (Compound 2c) is an XPO1 degrader. PROTAC XPO1 degrader-1 exhibits anti-proliferative effects, can induce cellapoptosis, inhibit NF-κB activity, and cause cellcycle arrest in the G1 phase. PROTAC XPO1 degrader-1 can be used in research on hematological malignancies (Pink: Target Protein Ligand (HY-170672); Black: Linker (HY-W010525); Blue: E3 Ligase Ligand (HY-170671); E3 Ligase Ligand-Linker Conjugate (HY-170673)) .
RK-682 hemicalcium is the hemicalcium salt form of RK-682 (HY-135564A). RK-682 hemicalcium is the inhibitor for protein tyrosine phosphatase (PTPase), heparanase, phospholipase A2 and HIV-1 protease. RK-682 hemicalcium inhibits the dephosphorylation of CD45 (IC50 is 54 μM) and VHR (IC50 is 2.0 μM), and thereby inhibits the ERK signaling pathway. RK-682 hemicalcium inhibits the cell viability of cancer cell MGH-U3, T24 and UROtsa with IC50s of 78.2, 43.2 and 145 nM, respectively, arrests the cellcycle at G1/S phase, inhibits the cell migration and autophagy in MGH-U3 and T24 [2] .
Gemcitabine triphosphate (dFdCTP) is the active metabolite of Gemcitabine (HY-17026). The mechanism of Gemcitabine triphosphate cell-killing is its competition with cytidine triphosphate during DNA replication, which results in the inhibition of chain elongation. Gemcitabine triphosphate shows a Ki of 11.2 μM against DNA polymerase α and 14.4 μM against DNA polymerase ε. Gemcitabine triphosphate partially inhibits dCMP deaminase and acts as a substrate for DNA synthesis to incorporate into cellular DNA and RNA. Gemcitabine triphosphate disrupts DNA and RNA synthesis, arrests cellcycle in G0/G1 and S phases, triggers apoptosis, reduces tumor cell proliferation. Gemcitabine triphosphate can be used for the research of pancreatic cancer and non-small cell lung cancer .
Gemcitabine triphosphate (dFdCTP) trisodium is the active metabolite of Gemcitabine (HY-17026). The mechanism of Gemcitabine triphosphate trisodium cell-killing is its competition with cytidine triphosphate during DNA replication, which results in the inhibition of chain elongation. Gemcitabine triphosphate trisodium shows a Ki of 11.2 μM against DNA polymerase α and 14.4 μM against DNA polymerase ε. Gemcitabine triphosphate trisodium partially inhibits dCMP deaminase and acts as a substrate for DNA synthesis to incorporate into cellular DNA and RNA. Gemcitabine triphosphate trisodium disrupts DNA and RNA synthesis, arrests cellcycle in G0/G1 and S phases, triggers apoptosis, reduces tumor cell proliferation. Gemcitabine triphosphate trisodium can be used for the research of pancreatic cancer and non-small cell lung cancer .
HDAC1-IN-14 is an indole-based benzamide selective HDAC1 inhibitor with an IC50 of 77 nM. HDAC1-IN-14 acts as an antiproliferative agent, with GI50 values ranging from nanomolar to low micromolar levels in various cancer cells. HDAC1-IN-14 induces G0-G1cellcycle arrest in colon cancer cells. HDAC1-IN-14 upregulates the expression of Caspase-3, Cyto-C and Bax, and downregulates the expression of AKT-1. HDAC1-IN-14 can be used in research related to leukemia, non-small cell lung cancer, colon cancer, central nervous system cancer, melanoma, ovarian cancer, renal cancer, prostate cancer and breast cancer .
HDAC-IN-56 ((S)-17b) is an orally active class I histone deacetylase (HDAC) inhibitor with IC50 values of 56.0 ± 6.0, 90.0 ± 5.9, 422.2 ± 105.1, >10000 nM for HDAC1, HDAC2, HDAC3, and HDAC4-11, respectively. HDAC-IN-56 has potent inhibitory activity while strongly increasing intracellular levels of acetylhistone H3 and P21 and effectively inducing G1cellcycle arrest and apoptosis.HDAC-IN-56 has antitumor activity .
HDAC1-IN-8 (compound 5c) is a potent and selective HDAC1 inhibitor with IC50 values of 11.94, 22.95, >500 µM for HDAC1, HDAC6, HDAC8, respectively. HDAC1-IN-8 shows antiproliferative activity. HDAC1-IN-8 induces cellcycle arrest at G1 and G2/M. HDAC1-IN-8 induces autophagy. HDAC1-IN-8 shows anticancer activity and has the potential for the research of lung cancer .
GDC-4198 (RGT-419B) is an orally active CDK4/2 inhibitor with desired degrees of selectivity against kinases such as CDK6, CDK9 and GSK3β. GDC-4198 inhibits the activity of cyclin-CDK complexes, blocks phosphorylation of the retinoblastoma protein (pRb), arresting the cellcycle transition from G1 to S phase. GDC-4198 is promising for research of cancers, such as breast cancer (especially hormone receptor-positive, HER2-negative type), lung cancer, and colorectal cancer .
MK-2206 is an orally active pan-AKT inhibitor, with IC50 values of 8 nM, 12 nM and 65 nM against AKT1, AKT2 and AKT3, respectively. MK-2206 inhibits the Akt/mTOR signaling pathway and reduces the levels of downstream GSK3β and Mcl-1 via proteasomal degradation. MK-2206 induces G1-phasecellcycle arrest, apoptosis, epithelial-mesenchymal transition, fibroblast activation and extracellular matrix deposition. MK-2206 causes transient hyperglycemia and hyperinsulinemia in animals. MK-2206 can be used in research related to solid tumors, renal fibrosis and hypercholesterolemia .
PROTAC c-Met degrader-5 (Compound D19) is an orally active c-MetPROTAC degrader with DC50s of 0.42 and 0.32 nM in EBC-1 and Hs746T cells, respectively. PROTAC c-Met degrader-5 significantly induces cellapoptosis, G1cellcycle arrest, and inhibits cell migration and invasion. PROTAC c-Met degrader-5 has potent antiproliferative and degradation efficacy against c-Met-addicted cancer cells and Tepotinib (HY-14721)-resistant cancer cells . Pink: c-Met ligand (HY-W425461); Blue: CRBN ligase ligand (HY-14658); Black: linker
Globulol is a terpenoid metabolite and Antimicrobial agent. Globulol can be isolated from Alpinia oxyphylla Miq. Globulol binds to PAK4, reduces the expression level of PAK4 in cancer cells, decreases the phosphorylation of AKT, and downregulates the expressions of STAT3, phosphorylated STAT3, and PD-L1. Globulol promotes the secretion of CCL4 by cancer cells. Globulol reduces the viability and proliferation ability of cancer cells, induces G0/G1cellcycle arrest and Apoptosis in cancer cells, and inhibits cancer cell migration and the integrity of 3D tumor spheres. Globulol enhances the relevant effects of anti-PD-1 agents in the cancer cell microenvironment. Globulol exhibits anticancer activity against liver cancer. Globulol inhibits the mycelial growth of phytopathogenic fungi and the growth of phytopathogenic bacteria. Globulol can be used in studies related to hepatocellular carcinoma .
PROTAC c-Met degrader-6 is a potent and orally active c-Met PROTACdegrader. PROTAC c-Met degrader-6 significantly induces the degradation of the c-Met protein with DC50s of 0.52 nM and 0.45 nM in EBC-1 and Hs746T. PROTAC c-Met degrader-6 almost abrogates the migratory and invasion abilities of tumor cells and significantly induces the apoptosis and blocks the cellcycle in the G0/G1 phase. PROTAC c-Met degrader-6 can be used for the study of various cancers such as non-small cell lung cancer and stomach cancer (Pink: c-Met ligand (HY-W425461); Blue: E3 ligand (HY-14658); Black: Linker (HY-20797)) .
S116836, a potent, orally active BCR-ABL tyrosine kinase inhibitor, blocks both wild-type as well as T315I Bcr-Abl. S116836 arrests the cells in the G0/G1 phase of cellcycle, induces apoptosis, increases ROS production, and decreases GSH production in BaF3/WT and BaF3/T315I cells. S116836 also inhibits SRC, LYN, HCK, LCK and BLK, and receptor tyrosine kinases such as FLT3, TIE2, KIT, PDGFR-β. Antitumor activies . S116836 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
L 741742 is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 induces G0/G1cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cellsin vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 can be used for the research of schizophrenia and glioblastoma .
L 741742 hydrochloride is a highly selective and brain-penetrant D4 dopamine receptor antagonist, with Ki values of 3.5 nM, 770 nM and >1700 nM for human D4, D3 and D2 receptors, respectively. L 741742 hydrochloride suppresses PDGFRβ, ERK1/2, and mTOR signaling pathways, and impairs autophagic flux while disrupting lysosomal function.L 741742 hydrochloride induces G0/G1cell-cycle arrest and apoptosis, promotes neuronal differentiation of normal human neural stem cells, selectively inhibits growth and clonogenic potential of glioblastoma neural stem cells and primary glioblastoma tumor cells, exerts synergistic effects with Temozolomide (TMZ) (HY-17364) against glioblastoma neural stem cellsin vitro, and inhibits glioblastoma neural stem cell xenograft growth in immunocompromised mice. L 741742 hydrochloride can be used for the research of schizophrenia and glioblastoma .
Gomisin G (Standard) is the analytical standard of Gomisin G. This product is intended for research and analytical applications. Gomisin G is a lignin from S. chinesis with anti-HIV (EC50 = 0.006 μg/mL), anti-liver cancer and anti-inflammatory activities. Gomisin G has an AKT-cyclin D1 dependent mechanism against triple-negative breast cancer (TNBC) cells through suppressing phosphorylation rather than inducing apoptosis. Gomisin G can inhibit AKT phosphorylation. Gomisin G can cause cellcycle arrest in the G1 phase. Gomisin G can be studied in research for diseases such as HIV, breast and liver cancers .
Methylcarbamyl PAF C-8 is resistant to the degradation function of platelet-activating factor acetylhydrolase (PAF-AH). It has a half-life of more than 100 minutes in platelet-poor plasma and possesses the activity of inducing platelet aggregation. In NRK-49 cells overexpressing the PAF receptor, Methylcarbamyl PAF C-8 can induce the expression of c-myc and c-fos, and activate mitogen-activated protein kinase (MAPK). Additionally, Methylcarbamyl PAF C-8 can induce cellcycle arrest in the G1 phase. Methylcarbamyl PAF C-8 holds promise for research in the fields of cardiovascular diseases and anti-cancer therapy .
PIM-1/HDAC-IN-1 (compound 4d) is a PIM-1 inhibitor, with an IC50 of 343.87 nM. PIM-1/HDAC-IN-1 has strong inhibitory activity and selectivity against HDAC 1 and HDAC 6, with IC50 values of 63.65 and 62.39 nM, respectively. PIM-1/HDAC-IN-1 exhibits apoptosis inducing potential in MCF-7 cell lines. PIM-1/HDAC-IN-1 shows pre-G1 apoptosis and cellcycle arrest at G2/M phase .
DP-15 is the degrader for GSPT1 and BRD4 with DC50s of 5.25 nM and 0.48 nM. DP-15 exhibits anti-proliferative activity of AML cells and NHL cells with an IC50 of nanomolar levels, arrests the cellcycle at G1 phase, and induces apoptosis in MOLM13. DP-15 exhibits anti-leukemia activity in MOLM-13 xenograft mouse models . (Pink: ligand for target protein JQ-1 carboxylic acid (HY-78695); Black: linker (HY-W262798); Blue: ligand for E3 ligase Cereblon Thalidomide-5-OH (HY-23095))
Rafutrombopag (tautomerism) (Hetrombopag) is an orally active nonpeptide thrombopoietin receptor (TPOR/MPL) agonist. Rafutrombopag can chelate iron and alleviate iron overload while promoting haematopoiesis. Rafutrombopag specifically stimulates proliferation and differentiation of human TPOR‐expressing cells, including 32D‐ MPL and human hematopoietic stem cells through stimulation of STAT, PI3K and ERK signalling pathways. Rafutrombopag effectively up-regulates G1-phase-related proteins, including p-RB, Cyclin D1 and CDK4/6, normalizes progression of the cellcycle, and prevents apoptosis by modulating BCL-XL/BAK expression in 32D-MPL cells. Rafutrombopag protects cardiomyocyte survival from oxidative stress damage as an enhancer of stem cells. Rafutrombopag can be used for the study of immune thrombocytopenia and oxidative stress-related cardiovascular disease .
Rafutrombopag (Hetrombopag) is an orally active nonpeptide thrombopoietin receptor (TPOR/MPL) agonist. Rafutrombopag can chelate iron and alleviate iron overload while promoting haematopoiesis. Rafutrombopag specifically stimulates proliferation and differentiation of human TPOR-expressing cells, including 32D-MPL and human hematopoietic stem cells through stimulation of STAT, PI3K and ERK signalling pathways. Rafutrombopag effectively up-regulates G1-phase-related proteins, including p-RB, Cyclin D1 and CDK4/6, normalizes progression of the cellcycle, and prevents apoptosis by modulating BCL-XL/BAK expression in 32D-MPL cells. Rafutrombopag protects cardiomyocyte survival from oxidative stress damage as an enhancer of stem cells. Rafutrombopag can be used for the study of immune thrombocytopenia and oxidative stress-related cardiovascular disease .
2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cellcycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
PROTAC CDK9 degrader-11 (Compound C3) is an orally active PROTAC degrader for CDK9 with DC50 of 1.09 nM. PROTAC CDK9 degrader-11 exhibits cytotoxicity in multi small cell lung cancer cell with IC50 of nanomolar levels. PROTAC CDK9 degrader-11 arrests cellcycle at G0/G1 phase, inhibits the cell invasion in DMS114 and DMS53 cell. PROTAC CDK9 degrader-11 exhibits antitumor efficacy in NCI-H446 xenograft mouse models .(Pink: ligand for target protein CDK9 ligand 3 (HY-170979); Black: linker; Blue: ligand for E3 ligase Cereblon E3 ligase Ligand 56 (HY-W247437))
PIM-1/HDAC-IN-2 is a robust PIM/HDAC inhibitor (IC50 = 0.11 μM in MV4-11cells), which exerts a synergistic antiproliferative effect through a dual mechanism of inhibiting PIM1 kinase and selectively inhibiting HDAC6. PIM-1/HDAC-IN-2 induces cellapoptosis. PIM-1/HDAC-IN-2 remarkably induces the cleavage of PARP, thereby initiating the arrest of the cellcycle in G1 phase and a reduction in S phase. PIM-1/HDAC-IN-2 demonstrates significant anticancer efficacyin the MV4-11 xenograft model without notable toxicity[1].
MK-2206 dihydrochloride (MK-2206 2HCl) is an orally active pan-AKT inhibitor, with IC50 values of 8 nM, 12 nM and 65 nM against AKT1, AKT2 and AKT3, respectively. MK-2206 dihydrochloride inhibits the Akt/mTOR signaling pathway and reduces the levels of downstream GSK3β and Mcl-1 via proteasomal degradation. MK-2206 dihydrochloride induces G1-phasecellcycle arrest, apoptosis, epithelial-mesenchymal transition, fibroblast activation and extracellular matrix deposition. MK-2206 dihydrochloride causes transient hyperglycemia and hyperinsulinemia in animals. MK-2206 dihydrochloride can be used in research related to solid tumors, renal fibrosis and hypercholesterolemia .
Hsp90-Cdc37-IN-4, a Celastrol (HY-13067) derivative, inhibits the Hsp90-Cdc37 protein-protein interaction (PPI). Hsp90-Cdc37-IN-4 selectively inhibits casein kinase 2 (CK2), reducing phosphorylation of its substrate Cdc37 at Serine 13. Hsp90-Cdc37-IN-4 induces G0/G1cellcycle arrest and triggers apoptosis via the mitochondrial pathway. Hsp90-Cdc37-IN-4 demonstrates potent anti-breast cancer activity .
PROTAC CDK6 Degrader 1 (compound 48a) is a potent and selective PROTAC CDK6 degrader with a DC50 of 0.037 μM. PROTAC CDK6 Degrader 1 exhibits selectivity over CDK4 (DC50 > 10 μM). PROTAC CDK6 Degrader 1 induces G0/G1cell-cycle arrest and apoptosis through inhibition of CDK6 downstream signaling. PROTAC CDK6 Degrader 1 reduces tumor burden and CDK6 levels in a MOLM-14 xenograft mouse model. PROTAC CDK6 Degrader 1 can be used for CDK6-driven cancers research, such as acute myeloid leukemia (AML) .
PARP1/BRD4-IN-3 (compound HF4) is a potent BRD4 and PARP1 inhibitor with IC50 values of 1210, 2019 nM for BRD4, PARP1, respectively. PARP1/BRD4-IN-3 shows antiproliferative activities. PARP1/BRD4-IN-3 induces apoptosis and cellcycle arrest at G0/G1 phase. PARP1/BRD4-IN-3 causes DNA damage and reduces the protein expression of Rad51. PARP1/BRD4-IN-3 shows antitumor efficacy .
PROTAC SMARCA2/4 degrader-38 is a degrader SMARCA2/4 PROTAC (DC50: 3.0 nM and 4.0 nM respectively). PROTAC SMARCA2/4 degrader-38 promotes the ubiquitination and degradation of SMARCA2/4. PROTAC SMARCA2/4 degrader-38 blocks the G0/G1cellcycle and induces apoptosis. PROTAC SMARCA2/4 degrader-38 can be used in acute myeloid leukemia (AML) research. (Pink: SMARCA2/4 ligand; Blue: VHL ligand (HY-112078); Black: linker; Target Protein Ligand-Linker Conjugates (HY-173343)) .
FLT3-IN-31 (compound 10q) is a potent FLT3 inhibitor with IC50 values of 0.16, 2.4 nM for FLT3, FLT3-D835Y, resprctively. FLT3-IN-31 shows antiproliferation activity. FLT3-IN-31 decreases the protein expression of p-FLT3, P-STAT5, P-ERK. FLT3-IN-31 induces apoptosis and cellcycle arrest at G1 phase. FLT3-IN-31 shows antitumor activity .
[Ala92]-p16 (84-103) is a peptide derived from the p16CDKN2/INK4a (p16) tumor suppressor protein. [Ala92]-p16 (84-103) binds to both cdk4 and cdk6 and inhibits cdk4-cyclin D1 kinase activity in vitro (IC50: 1.5 μM). [Ala92]-p16 (84-103) blocks cellcycle progression through the G1 phase .
PI3K/AKT-IN-4 (compound 3) is a diterpenoid that can be isolated from the roots and rhizomes of Salvia castanea Dielsf. PI3K/AKT-IN-4 has antitumor activity, inhibiting cell viability and proliferation (IC50=4.72 μM) and promoting apoptosis by blocking the G0/G1 phase of the Hep3B cellcycle, inducing mitochondrial dysfunction and oxidative stress. In addition, PI3K/AKT-IN-4 inhibits hepatocellular carcinoma by inhibiting the PI3K-Akt signaling pathway and binding to PARP1 and CDK2 targets .
HEMTAC CDK4/6 degrader 1 is a PROTAC connected by ligands for HSP90 and CDK4/6 with a Kd value of 35.7 μM. HEMTAC CDK4/6 degrader 1 induces CDK4/6 degradation in B16F10 melanoma cells. HEMTAC CDK4/6 degrader 1 arrests cellcycle at G0/G1 phase and induces apoptosis. HEMTAC CDK4/6 degrader 1 can be used in research of cancer . HEMTAC CDK4/6 degrader 1 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
PROTAC EZH2 Degrader-3 (compound ZJ-20) is a EZH2 PROTAC degrader. PROTAC EZH2 Degrader-3 (5 μM, 24 h) not only potently inhibits the expression of EZH2 protein, but also had a strong inhibits effect on the expression levels of other subunits of PRC2 as well as H3k27me3 protein. PROTAC EZH2 Degrader-3 shows anti-proliferative activity and blocks the cellcycle in the G0-G1 phase and induces cellapoptosis((Blue: cereblon ligand Pomalidomide (HY-10984), Black: linker HY-W361751;Pink: EZH2 inhibitor Tazemetostat (HY-13803)) .
Pipoxolan is an orally active smooth muscle relaxant, anti-inflammatory agent and anticancer agent. Pipoxolan modulates PI3K/AKT signaling pathways, and reduces the levels of Ras/MEK/p-ERK, MMP-2 and MMP-9. Pipoxolan inhibits pro-inflammatory transcription factor pathways, activates Nrf2/HO-1, and suppresses the production of pro-inflammatory mediators. Pipoxolan induces ROS generation, endogenous mitochondrial Apoptosis, and G0/G1cellcycle arrest. Pipoxolan reduces cerebral infarction size and inhibits intimal hyperplasia. Pipoxolan can be used in research related to cerebral ischemia, intimal hyperplasia, oral squamous cell carcinoma, leukemia and lung cancer .
G9a-IN-4 is a G9a inhibitor with high selectivity (IC50 = 32 nM). G9a-IN-4 shows high selectivity against the other tested lysine/arginine methyltransferases. G9a-IN-4 exhibits high enzymatic activity against G9a and more potent antiproliferative effects against all tested cancer cells. G9a-IN-4 significantly suppresses the H3K9me2 level. G9a-IN-4 triggers autophagy by inducing the production of ROS, thus leading to cellapoptosis and cellcycle arrest at G0/G1 in CT26 colon cells. G9a-IN-4 can be used for the study of colon cancer .
EGFR/VEGFR2-IN-9 (Compound 9b) is an inhibitor of VEGFR-2 (IC50 = 1.325 μM) and EGFR (IC50 = 1.891 μM). EGFR/VEGFR2-IN-9 significantly inhibits the proliferation of multiple cancer cell lines, particularly leukemia cells. EGFR/VEGFR2-IN-9 upregulates the expression levels of Bax, caspase-3, and p53, while downregulating the expression of Bcl-2. EGFR/VEGFR2-IN-9 induces apoptosis and arrests the cellcycle in the G1 phase. EGFR/VEGFR2-IN-9 can be used to investigate anti-tumor angiogenesis and multi-drug resistant cancers .
EGFR-IN-156 (Compound 7f) is an EGFR inhibitor that also has inhibitory activity against mutations EGFRL858R and EGFRT790M (IC50 values are 0.186, 0.131, and 0.107 μM, respectively). EGFR-IN-156 has significant anticancer activity against human cancer cell lines HepG-29 (liver cancer), MCF-7 (breast cancer), and HCT-116 (colon cancer) (IC50 values are 1.67, 5.32, and 6.56 μM, respectively). EGFR-IN-156 inhibits cancer cell proliferation by inducing cellcycle arrest at the G/G1 phase and triggering apoptosis. EGFR-IN-156 shows promise in EGFR-related cancers .
BWA-6047 is an oral active PROTAC degrader targeting AR/AR-V7 and GSPT1 with DC50 values of 3.7, 3.0 and 1.2 nM in 22Rv1 cells. BWA-6047 suppresses the expression of AR downstream target genes and and transcriptional activity. BWA-6047 inhibits cancer cells proliferation, causes G1 phase cellcycle arrest and induces apoptosis. BWA-6047 increases cleaved-PARP-1 and cleaved-caspase-3 levels. BWA-6047 reduces growth of LNCaP xenograft tumors in mice models without obvious toxicity. BWA-6047 can be used for the research of prostate cancer .
PI3Kα-IN-14 (compound F8) is a selective PI3Kα inhibitor with an IC50 of 0.14 nM. PI3Kα-IN-14 induces a great decrease in mitochondrial membrane which caused cellcycle arrest at G1 phase and apoptosis in U87-MG cells. PI3Kα-IN-14 shows significant anti-proliferative activities against three tumor-derived cell lines (PC-3: IC50 of 0.28 μM; HCT-116: IC50 of 0.57 μM; and U87-MG: IC50 of 1.37 μM) .
CDK2-IN-42 (Compound H63) is a CDK12 inhibitor with an IC50 value of 10 nM. CDK2-IN-42 has anti-ESCC (Esophageal Squamous Cell Carcinoma) cell activity. It can block transcriptional elongation, downregulate the core genes in the G1 phase to induce cellcycle arrest, and alter the CDK12-ATM/ATR-CHEK1/CHEK2 signaling axis, resulting in DNA damage. CDK2-IN-42 can effectively inhibit tumor growth in a xenograft mouse model of human ESCC KYSE150. CDK2-IN-42 holds great promise for research in the field of cancer .
Theaflavin-3-gallate, a black tea theaflavin monomer, is regarded as the biologically important active component of black tea and provides health benefits. Theaflavin-3-gallate acts as prooxidants and induces oxidative stress in the carcinoma cells. Theaflavin-3-gallate reacts directly with reduced glutathione (GSH) in a time- and concentration-dependent manner. Theaflavin-3-gallate induces apoptosis and G1cellcycle arrest in ovarian cancer A2780/CP70 cells through p53-dependent pathways. Theaflavin-3-gallate induces DNA damage through ATM/Chk/p53 pathway .
BRD4-IN-41 is a BRD4 inhibitor with an IC50 of 34 nM. BRD4-IN-41 also inhibits JAK2, FLT3, RET, ROS1, NTRK3, PDGFRb, and FGFR1 kinases with IC50 values ranging from 0.9 nM to 43 nM. BRD4-IN-41 inhibits acetyl-lysine binding site of BRD4, downregulates c-MYC, reduces phosphorylated STAT3 levels, induces G1cellcycle arrest and apoptosis, thereby inhibiting cancer cells growth. BRD4-IN-41 can be used for the research of cancer, such as multiple myeloma and acute myeloid leukemia .
ALK degrader 1 is a potent, hydrophobic tag (HyT)-based degrader that induces ubiquitin-proteasome system (UPS)-dependent EML4-ALK degradation (DC50 = 0.13 μM). ALK degrader 1 demonstrates potent ALK degradation and antiproliferative effects in ALK-dependent cell lines, while showing minimal cytotoxicity in ALK fusion-negative cells. ALK degrader 1 triggers cellcycle arrest at the G0/G1 phase and stimulates apoptosis. ALK degrader 1 not only facilitates efficient degradation of the ALK protein but also disrupts key downstream effectors, including the STAT3 signaling axis. ALK degrader 1 mediates robust EML4-ALK degradation in vivo. ALK degrader 1 can be used for ALK-related diseases research .
CA IX-IN-2 (Compound 9o) is an inhibitor for carbonic anhydrase (CA), that inhibits CA IX, CA XII and CA II with an IC50 of 5.6, 7.4 and 430 nM, respectively. CA IX-IN-2 inhibits the proliferation of cancer cell HCT-116, SW480, MDA-MB 231 and MCF-7, with IC50s of 14.63-29.33 μM. CA IX-IN-2 intercalates DNA, arrests cellcycle at G1/S phase, and induces apoptosis in MDA-MB-231. CA IX-IN-2 affects the mitochondrial membrane potential (MMP), increases the intracellular ROS levels, causes mitochondrial damage, and inhibits the cell migration of MDA-MB-231. CA IX-IN-2 exhibits antitumor efficacy in mouse models .
LA-CB1 is an Abemaciclib (HY-16297A) derivative that targets CDK4/6 and promotes its degradation via the ubiquitin-proteasome pathway, thereby disrupting the CDK4/6-Cyclin D1-Rb-E2F axis and inducing G0/G1cellcycle arrest and apoptosis. LA-CB1 exhibits antiproliferative activity against MDA-MB-231 cells, with an IC50 of 0.27 µM, and effectively inhibits epithelial-mesenchymal transition (EMT), cell migration, invasion, and angiogenesis. In highly aggressive models such as triple-negative breast cancer (TNBC), LA-CB1 significantly suppresses tumor growth in a dose-dependent manner. LA-CB1 holds potential for research in the field of breast cancer .
Bromuconazole is a triazole fungicide with oral efficacy and blood-brain barrier permeability . Bromuconazole protects crops from various fungal contaminations. Bromuconazole exhibits cytotoxicity against a variety of cancer cells, induces G0/G1cellcycle arrest and inhibits DNA synthesis in cancer cells, and triggers cytoskeletal structural disorder, genotoxic damage, apoptotic (apoptosis) cell death, and mitochondrial membrane depolarization. Bromuconazole activates caspase-3, induces excessive production of ROS, p53 and Bax, lipid peroxidation, increased activities of SOD and CAT, and downregulates Bcl-2. By upregulating p-ERK1/2 and p-JNK, Bromuconazole disrupts the MAPK signaling pathway, impairs the cellular stress response of human trophoblast cells and endometrial cells, and damages the implantation process . Bromuconazole is applicable to research related to glioma, colon cancer, reproductive injury (implantation dysfunction), and cardiac dysfunction .
NSC756093 (SU093) is a GBP1:PIM1 interaction inhibitor. NSC756093 binds to GBP1-PIM1 with a Kd of 38 nM. NSC756093 suppresses proliferation, reduces migration, induces G1 phase cell-cycle arrest, and increases apoptoticcell death in ovarian cancer cells. NSC756093 reduces cellular proteasomal activity, induces accumulation of ubiquitinated proteins, and restrains tumor progression and lung metastasis in murine ovarian cancer xenograft models. NSC756093 increases sensitivity of prostate cancer cells to Docetaxel (HY-B0011) and sensitizes GBP1-overexpressing ovarian cancer cells to Paclitaxel (HY-B0015). NSC756093 can be used for the research of prostate cancer and ovarian cancer .
C2 L-threo Ceramide (d18:1/2:0) (L-threo Cer(d18:1/2:0); L-threo Ceramide (d18:1/2:0)) is a bioactive sphingolipid and cell-permeable analog of naturally occurring ceramides. It stimulates cholesterol efflux in CHO cells expressing the human ABCA1 receptor when used at a concentration of 10 μM, however, this efflux is 50% less than that stimulated by C2 ceramide. C2 L-threo Ceramide inhibits IL-4 production by 17% in EL4 T cells stimulated with phorbol 12-myristate 13-acetate when used at a concentration of 10 μM. It also induces cellcycle arrest in the G0/G1 phase and a 7-fold increase in sphingosine accumulation as well as inhibits growth of HL-60 leukemia cells.
EGFR/CA-IX-IN-1 (Compound 14) is a dual inhibitor against epidermal growth factor receptor (EGFR) and carbonic anhydrase IX (CA-IX) with IC50 values of 5.92 nM and 63 nM, respectively. EGFR/CA-IX-IN-1 shows strong cytotoxicity against breast cancer cells (MDA-MB-231, MCF-7) with IC50 values of 5.78 μM and 8.05 μM, respectively. EGFR/CA-IX-IN-1 inhibits the catalytic activity of CA-IX, up-regulates BAX/Bcl-2, activates caspases, and arrests the cellcycle at the G1 phase. EGFR/CA-IX-IN-1 is promising for research of breast cancer .
Alisol B is a triterpene with diverse biological activities. Alisol B binds human soluble epoxide hydrolase (sEH) with a Ki of 5.97 μM and reduces sEH activity. Alisol B inhibits RANKL-induced JNK phosphorylation, NFATc1 and c-Fos expression, osteoclast formation, mature osteoclast pit-forming and actin ring activity, and SERCA pump activity. Alisol B induces calcium mobilization, CaMKK-AMPK-mTOR pathway activation, autophagic flux, autophagosome formation, G1 phase cellcycle arrest, endoplasmic reticulum stress, unfolded protein responses, and cancer cellapoptosis. Alisol B can be used for the research of hypercalcemia, osteoporosis, rheumatoid arthritis, periodontitis, acute kidney injury, and breast cancer .
SOS1/EGFR-IN-1 (compound SE-9) is a dual-target inhibitor for the prostate cancer. SOS1/EGFR-IN-1 inhibits effectively SOS1(IC50=42.13±1.55 nM) and EGFR(IC50=1.01±0.04 nM) by inhibiting their downstream effector molecules. SOS1/EGFR-IN-1 induces apoptosis and G1 phase cellcycle arrest, reducing angiogenesis and migration. SOS1/EGFR-IN-1 shows significant antitumor effects in prostate cancer cells PC-3 (IC50=0.45±0.03 μM) .
VEGFR-2-IN-67 (Compound 6b) is an inhibitor of vascular endothelial growth factor receptor 2 (VEGFR-2). Its IC50 values for MDA-231 and MCF-7 cell lines are 5.91 µM and 7.16 µM respectively, and its inhibitory effect on VEGFR-2 is comparable to that of Sorafenib (HY-10201) (IC50 is 53.63 nM). VEGFR-2-IN-67 exerts significant anti-cancer activity through mechanisms such as inducing Apoptosis (the early apoptosis rate reaches 57.20%), arresting the cellcycle at the G1 phase, upregulating pro-apoptotic markers and downregulating Bcl-2. VEGFR-2-IN-67 can be used for research in the field of cancer .
FD274 is a highly potent PI3K/mTOR dual inhibitor with IC50s of 0.65 nM, 1.57 nM, 0.65 nM, 0.42 nM, and 2.03 nM against PI3Kα/β/γ/δ and mTOR, respectively. FD274 exhibits significant anti-proliferation of AML cell lines (HL-60 and MOLM-16). FD274 arrests HL-60 cellcycle at G1 phase and increases apoptosis. FD274 demonstrates dose-dependent inhibition of tumor growth in the HL-60 xenograft model. FD274 has the potential for acute myeloid leukemia research .
MK-2206 free base is an orally active pan-AKT inhibitor, with IC50 values of 8 nM, 12 nM and 65 nM against AKT1, AKT2 and AKT3, respectively. MK-2206 free base inhibits the Akt/mTOR signaling pathway and reduces the levels of downstream GSK3β and Mcl-1 via proteasomal degradation. MK-2206 free base induces G1-phasecellcycle arrest, apoptosis, epithelial-mesenchymal transition, fibroblast activation and extracellular matrix deposition. MK-2206 free base causes transient hyperglycemia and hyperinsulinemia in animals. MK-2206 free base can be used in research related to solid tumors, renal fibrosis and hypercholesterolemia .
KB-15 is a STAT3 inhibitor. KB-15 exhibits potent anti-proliferative activity against AGS gastric cancer cells (IC50 = 0.29 μM) and BGC-823 gastric cancer cells (IC50 = 0.65 μM). KB-15 exerts anti-tumor effects by inhibiting STAT3 phosphorylation, downregulating HO-1 expression, and promoting intracellular ROS accumulation. KB-15 induces G0/G1 phase cellcycle arrest and apoptosis, as well as suppresses colony formation and migration of gastric cancer cells. KB-15 demonstrates excellent anti-tumor efficacy in BGC-823 subcutaneous xenograft model. KB-15 can be used for the study of gastric cancer .
STAT-3/c-Src-IN-1 (Compound 12d) is a dual STAT-3 (IC50=0.844 μM) and c-Src (IC50=0.268 μM) inhibitor. STAT-3/c-Src-IN-1 blocks tumor cell proliferation, migration, and angiogenesis signaling pathways. STAT-3/c-Src-IN-1 exhibits potent anti-proliferative activity against melanoma (SK-MEL-2) and CNS cancer (SNB-75) cell lines (GI50=-5.75 μM and -5.63 μM), inducing tumor cell apoptosis via G0/G1 and G2/M phase cellcycle arrest. STAT-3/c-Src-IN-1 is promising for research of melanoma and glioblastoma .
PROTAC PI3Kα/δ degrader-1 is an orally active PI3Kα/δPROTAC degrader, with an IC50 of 0.34 nM for PI3Kα and 1.85 nM for PI3Kδ. PROTAC PI3Kα/δ degrader-1 inhibits the proliferation and migration of cancer cells, induces G1-phasecellcycle arrest and PI3Kα degradation. PROTAC PI3Kα/δ degrader-1 suppresses tumor growth in breast cancer xenograft mouse models. PROTAC PI3Kα/δ degrader-1 can be used for the research of breast cancer .
PDGFRA/CAIX/XII-IN-1 is an inhibitor of PDGFRA, CA IX and CA XII, with an IC50 of 20 nM against PDGFRA, a Ki of 93.3 nM against CA IX, and a Ki of 80.0 nM against CA XII. PDGFRA/CAIX/XII-IN-1 binds to the ATP-binding pocket of PDGFRA and blocks the downstream STAT3, AKT and ERK1/2 signaling pathways. PDGFRA/CAIX/XII-IN-1 induces G0/G1cellcycle arrest and endogenous apoptosis (Apoptosis), including cleavage of PARP-1, caspase-9 and caspase-3, activation of caspase 3/7, and down-regulation of Mcl-1. PDGFRA/CAIX/XII-IN-1 exhibits antiproliferative activity in eosinophilic leukemia cells. PDGFRA/CAIX/XII-IN-1 can be used for the research of leukemia .
CDK4/6-IN-26 is a carbamate derivative that targets CDK4/CDK6. CDK4/6-IN-26 reduces CDK4/CDK6 levels, resulting in cellcycle arrest in the G0/G1 phase and in the S phase. CDK4/6-IN-26 exhibits high potency against SW480 cells (IC50 = 6.3 μM). CDK4/6-IN-26 affects ROS levels by increasing the expression of SOD2/MnSOD. CDK4/6-IN-26 establishes several interactions with the amino acids of the CDK6 active site. CDK4/6-IN-26 can be used for the research of colorectal cancer .
EGFR/VEGFR2-IN-10 is a selective EGFR, VEGFR2 and COX2 inhibitor with IC50s of 8.5, 68 and 158 nM, respectively. EGFR/VEGFR2-IN-10 induces G1-phasecellcycle arrest in MCF-7cells. EGFR/VEGFR2-IN-10 increases the Bax/Bcl-2 ratio, upregulates caspase-8, and elevates caspase-9 protein levels, confirming activation of the intrinsic apoptotic pathway. EGFR/VEGFR2-IN-10 demonstrates exceptional therapeutic potential by simultaneously inhibiting tumor proliferation, angiogenesis, and inflammation pathways while maintaining a favorable selectivity profile. EGFR/VEGFR2-IN-10 can be used as a research tool for cervical, liver, colon, and breast cancer studies .
2,6-Dihydroxyacetophenone (Standard) is the analytical standard of 2,6-Dihydroxyacetophenone (HY-Y0106). This product is intended for research and analytical applications. 2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cellcycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
EGFR/CDK2-IN-5 is a potent dual EGFR and CDK2 inhibitor with IC50s of 17.30 and 212.10 nM, respectively. EGFR/CDK2-IN-5 also inhibits EGFR T790M with an IC50 of 123.8 nM. EGFR/CDK2-IN-5 exhibits potent anticancer activity. EGFR/CDK2-IN-5 induces G1 and S cellcycle arrest and apoptosis, accompanied by increased levels of caspase-3/9 and Bax, as well as decreased Bcl-2 levels. EGFR/CDK2-IN-5 can be used for the research of cancers, such as lung cancer, breast cancer, and leukemia .
EGFR/BRAFV600E-IN-1 (Compound 23) is a potent EGFR and BRAF V600E dual inhibitor with IC50s of 0.08 and 0.15 µM, respectively. EGFR/BRAFV600E-IN-1 induces apoptosis and cellcycle arrest in both pre-G1 and G2/M phases. EGFR/BRAFV600E-IN-1 exhibits antiproliferative activity againist A-549, MCF-7, Panc-1, HT-29 with IC50s of 1.2, 0.79, 1.3, and 1.23 µM, respectively .
BY13 is a SRC-3PROTAC degrader with a DC50 of 0.031 μM. BY13 selectively blocks the ER signaling pathway over that of androgen receptor (AR)) through down-regulating ERα level. BY13 potently overcomes endocrine resistance in breast cancer by inducing cellcycle arrest in G1 phase and apoptosis, with superior effect over Fulvestrant (HY-13636). BY13 significantly inhibits the growth of drug-resistant breast tumors without obvious toxicity in LCC2 xenograft mice model . Pink: SRC-3 ligand (SI-2) (HY-101447); Blue: CRBN ligase ligand (HY-41547); Black: linker (HY-176226)
BRD4 Inhibitor-40 (Compound 23) is the inhibitor for BRD that inhibits BRD4-BD1, BRD4-BD2, BRD2-BD1 and BRD2-BD2 with IC50s of 16.1, 142.18, 29.35 and 302.35 nM, respectively. BRD4 Inhibitor-40 modulates the expression of c-Myc and p21, arrests cellcycle at G1 phase, inhibits Pkd1-null (PN) renal cystic epithelial cells, and blocks the renal cysts formation in Madin-Darby canine kidney and embryonic kidney vesicle models. BRD4 Inhibitor-40 exhibits renal cysts inhibitory activity in mouse models .
BRAFV600E/ABL2-IN-2 is a dual BRAFV 600E and ABL2 kinase inhibitor, with an IC50 of 0.088 μM against human BRAFV 600E and an IC50 of 0.3 μM against human ABL2. BRAFV600E/ABL2-IN-2 reduces the phosphorylation levels of downstream ERK1/2 and CrkL in melanoma cells. BRAFV600E/ABL2-IN-2 decreases the expression of P-glycoprotein (P-glycoprotein) in melanoma cells. BRAFV600E/ABL2-IN-2 induces G1cellcycle arrest and apoptosis (Apoptosis) in melanoma cells. BRAFV600E/ABL2-IN-2 is applicable to relevant research on melanoma .
ALK/PI3K/AKT-IN-1 (Compound 45) inhibits the proliferation of cancer cell A549, H1975 and PC9 with an IC50 of 0.44, 0.83 and 1.51 μM. ALK/PI3K/AKT-IN-1 increases the expression of p21 and p27, inhibits the activity of CDK2 and p-Rb, arrests the cellcycle at G1 phase. ALK/PI3K/AKT-IN-1 inhibits the ALK/PI3K/AKT signaling pathway, promotes the depolarization of mitochondrial membrane potential, and inducing apoptosis in A549 cell. ALK/PI3K/AKT-IN-1 inhibits the formation and growth of A549 cell spheroids .
MK-2206 dihydrochloride (MK-2206 2HCl) (Standard) is the analytical standard of MK-2206 dihydrochloride (HY-10358). This product is intended for research and analytical applications. MK-2206 dihydrochloride is an orally active pan-AKT inhibitor, with IC50 values of 8 nM, 12 nM and 65 nM against AKT1, AKT2 and AKT3, respectively. MK-2206 dihydrochloride inhibits the Akt/mTOR signaling pathway and reduces the levels of downstream GSK3β and Mcl-1 via proteasomal degradation. MK-2206 dihydrochloride induces G1-phasecellcycle arrest, apoptosis, epithelial-mesenchymal transition, fibroblast activation and extracellular matrix deposition. MK-2206 dihydrochloride causes transient hyperglycemia and hyperinsulinemia in animals. MK-2206 dihydrochloride can be used in research related to solid tumors, renal fibrosis and hypercholesterolemia .
HDAC-IN-88 (Compound HJ-9) is the inhibitor for HDAC that inhibits HDAC6, HDAC1, HDAC2, HDAC8 and HDAC3 with IC50s of 0.226, 1.103, 2.308, 3.255 and 3.864 μM, respectively. HDAC-IN-88 inhibits the proliferation of cancer cell HepG2, HCT116 and MV4-11 with IC50 of 5.47, 9.78 and 0.38 μM, inhibits the migration of HCT116, arrests the cellcycle at G0/G1 phase, and induces apoptosis and autophagy in MV4-11. HDAC-IN-88 reduces ROS level and mitochondrial membrane potential. HDAC-IN-88 exhibits antimalarial activity that inhibits P. falciparum 3D7 with EC50 of 165 nM. HDAC-IN-88 also exhibits anti-angiogenic activity .
PROTAC FGFR2 degrader 1 (compound N5) is a PROTAC that effectively targets FGFR2 with DC50 of 6.46 nM, the FGFR2IC50 is 0.08 nM. PROTAC FGFR2 degrader 1 has anti-proliferative activity and highly selective, induces G0/G1 arrest of KATOIII and SNU16 cellcycle and inhibits apoptosis by reducing the activation of p-ERK and p-PLCγ, the downstream proteins of FGFR2.
PROTAC FGFR2 degrader 1 inhibits gastric cancer cells remained above 50% at a concentration of 0.17 nM.
PROTAC FGFR2 degrader 1 potently inhibits the growth of SNU16 xenograft tumors in mouse model (Structure Note: Pink, FGFR2 activator: HY-18708; Blue, E3 ligase ligand: HY--10984; Black, linker: HY-163989; E3 ligase ligand + linker:HY-163986) .
Veratramine (NSC17821; NSC23880) (Standard) is the analytical standard of Veratramine (HY-N0837). This product is intended for research and analytical applications. Veratramine (NSC17821; NSC23880) is an orally active inhibitor of the PI3K/Akt/mTOR signaling pathway and a SIGMAR1 modulator. Veratramine induces autophagic apoptosis of tumor cells, arrests the cellcycle at the G0/G1 phase, and inhibits epithelial-mesenchymal transition (EMT)-related proteins to reduce tumor migration. Veratramine reduces spinal cord and sciatic nerve pathological damage in a neuropathy model by inhibiting SIGMAR1 binding to NMDAR and phosphorylation of NMDAR Ser896. Veratramine has anti-tumor proliferation, apoptosis induction, anti-inflammatory and neuroprotective activities, and can be used in the study of cancers such as liver cancer and osteosarcoma, as well as diabetic peripheral neuropathy .
Germacrone (Standard) is an analytical standard of Gemmacrone (HY-N0440). This product is intended for research and analytical applications. Germacrone (Standard) is a sesquiterpene compound with multiple biological activities. Germacrone (Standard) inhibits the H1N1 and H3N2 influenza A viruses and the influenza B virus. Germacrone (Standard) blocks the progressionof arthritis by regulating Th1/Th2 balance and inhibiting NF-κB signaling. Germacrone (Standard) can arrest the cellcycle at G0/G1 and G2/M phases and induce apoptosis in breast cancer cells. Germacrone (Standard) inhibits 5α-reductase and has anti-androgenic effect. Germacrone (Standard) has neuroprotective functions and can be used for the study of traumatic brain injury (TBI). Germacrone (Standard) also has antioxidant activity .
PI3Kδ/HDAC6-IN-1 (Compound 22E) is an orally active and dual inhibitor of PI3Kδ and HDAC6 with IC50 values of 2.4 nM and 6.2 nM, respectively. PI3Kδ/HDAC6-IN-1 exhibits potent antiproliferative effects on non-Hodgkin lymphoma (NHL) cells and possesses in vivo antitumor activity without significant toxicity. PI3Kδ/HDAC6-IN-1 arrests the cellcycle at the G0/G1 phase and induces apoptosis. PI3Kδ/HDAC6-IN-1 blocks the PI3K/AKT/mTOR signaling pathway and increases the acetylation levels of α-tubulin and histone H3 .
[Cu2Cl2(4'-(4-Methoxy-1-naphthyl)-terpy)2](PF6)2 (Compound 3) is a copper complex, which inhibits cell viability of HCT116, HCT116DoxR, A2780 and fibroblasts, with IC50s of 0.13, 0.15, 0.66 and 6.24 μM, respectively. [Cu2Cl2(4'-(4-Methoxy-1-naphthyl)-terpy)2](PF6)2 induces apoptosis and autophagy, and arrests cellcycle at G0/G1 phase in HCT116DoxR. [Cu2Cl2(4'-(4-Methoxy-1-naphthyl)-terpy)2](PF6)2 exhibits antimetastatic efficacy .
BRD4/AKT-IN-1 is a BRD4/AKT inhibitor with BRD4IC50 66.12 nM and AKT1IC50 143.81 nM. BRD4/AKT-IN-1 blocks BRD4-mediated c-Myc transcriptional regulation, modulates AKT1 signaling, decouples AKT phosphorylation from pro-survival effectors. BRD4/AKT-IN-1 induces G0/G1cellcycle arrest via downregulated phosphorylated RB, cyclin E1, CDK2. BRD4/AKT-IN-1 elevates LC3B levels to promote autophagy. BRD4/AKT-IN-1 promotes apoptosis in cancer cells. BRD4/AKT-IN-1 can be used for the research of metastatic castration-resistant prostate cancer .
Theaflavin-3-gallate (Standard) is the analytical standard of Theaflavin-3-gallate. This product is intended for research and analytical applications. Theaflavin-3-gallate, a black tea theaflavin monomer, is regarded as the biologically important active component of black tea and provides health benefits. Theaflavin-3-gallate acts as prooxidants and induces oxidative stress in the carcinoma cells. Theaflavin-3-gallate reacts directly with reduced glutathione (GSH) in a time- and concentration-dependent manner. Theaflavin-3-gallate induces apoptosis and G1cellcycle arrest in ovarian cancer A2780/CP70 cells through p53-dependent pathways. Theaflavin-3-gallate induces DNA damage through ATM/Chk/p53 pathway .
JAK1/CDK7-IN-1 (compound 11) is a JAK1/CDK7 inhibitor and cytotoxic agent.JAK1/CDK7-IN-1 forms a stable, tightly bound complex with JAK1.JAK1/CDK7-IN-1 disrupts cellcycle, induces G2/M phase arrest, and increases Pre-G1 phase cell proportion.JAK1/CDK7-IN-1 induces apoptosis, elevates caspase 1, 3, and 9 levels, and triggers apoptotic and necrotic cell death.JAK1/CDK7-IN-1 can be used for the research of breast cancer, prostate cancer .
EGFR T790M/L858R-IN-2 is a potent and selective EGFRT790M/L858R inhibitor with IC50 values of 3.5, 1290 nM for EGFRT790M/L858R, EGFR WT, respectively. EGFR T790M/L858R-IN-2 decreases the expression of p-EGFR, P-AKT, P-ERK1/2. EGFR T790M/L858R-IN-2 induces Apoptosis and cellcycle arrest in the G1 phase. EGFR T790M/L858R-IN-2 shows anti-cancer activity .
Alisol B (Standard) is the analytical standard of Alisol B (HY-N0805A). This product is intended for research and analytical applications. Alisol B is a triterpene with diverse biological activities. Alisol B binds human soluble epoxide hydrolase (sEH) with a Ki of 5.97 μM and reduces sEH activity. Alisol B inhibits RANKL-induced JNK phosphorylation, NFATc1 and c-Fos expression, osteoclast formation, mature osteoclast pit-forming and actin ring activity, and SERCA pump activity. Alisol B induces calcium mobilization, CaMKK-AMPK-mTOR pathway activation, autophagic flux, autophagosome formation, G1 phase cellcycle arrest, endoplasmic reticulum stress, unfolded protein responses, and cancer cell apoptosis. Alisol B can be used for the research of hypercalcemia, osteoporosis, rheumatoid arthritis, periodontitis, acute kidney injury, and breast cancer.
PROTAC MNK1 degrader-1 is a selective MNK1PROTAC degrader with a DC50 of 11.92 nM, and a Dmax > 96% in MV4-11 cells. PROTAC MNK1 degrader-1 significantly reduces p-eIF4E (IC50: 22.07 nM), induces apoptosis, and arrests the cellcycle at the G1 phase. PROTAC MNK1 degrader-1 has potent antitumor activity. PROTAC MNK1 degrader-1 has robust antileukemic efficacy in MV4-11 xenograft mice model with acceptable drug safety . Pink: MNK1 ligand (HY-176429); Blue: CRBN ligase ligand (HY-A0003); Black: linker (HY-Y1139); CRBN + linker: HY-176430
Ly101-4B is an apoptosis inducer and multi-target inhibitor with antiproliferative, antitumor and cycytotoxic effects. Ly101-4B reduces HSF1 expression, inhibits microRNA-214 synthesis, downregulates HSP27, HSP70 and HSP90 expression, while suppressing E2F-dependent transcriptional activity and downregulating its target genes. Ly101-4B induces caspase 3/7-mediated apoptosis by reducing DNA synthesis, inhibiting the cellcycle and G1/S phase transition, without affecting RNA synthesis or inducing necrosis. Ly101-4B is selective for pancreatic ductal adenocarcinoma cells with different genotypes and varying degrees of E2F dependence. Ly101-4B can be used in research related to epithelial ovarian cancer and pancreatic ductal adenocarcinoma .
PI3K/mTOR-IN-19 is an orally active, potent, selective PI3K (IC50 = 4.23 nM) and mTOR (IC50 = 2.3 nM) inhibitor. PI3K/mTOR-IN-19 significantly inhibits Eca109 cell viability and induces apoptosis. PI3K/mTOR-IN-19 causes G0/G1cellcycle arrest, decreased mitochondrial membrane potential, and demonstrates marked telomerase inhibitory activity. PI3K/mTOR-IN-19 modulates the expression of key apoptotic regulators (Bcl-2, Bax, and p53) and downregulates the PI3K/Akt/mTOR signaling pathway. PI3K/mTOR-IN-19 can be used for the study of esophageal cancer .
Cephalotaxine-ester-(R)-1-ethoxy-3-mercaptopropan-2-ol-Ph (3,4OMe) is an anti-leukemic agent with potent ribosome-targeting protein synthesis inhibition. Cephalotaxine-ester-(R)-1-ethoxy-3-mercaptopropan-2-ol-Ph (3,4OMe) downregulates short-lived oncoproteins, including c-Myc and Mcl-1, by inhibiting protein synthesis. Cephalotaxine-ester-(R)-1-ethoxy-3-mercaptopropan-2-ol-Ph (3,4OMe) induces cellcycle arrest at the G0/G1 phase and triggers mitochondrial pathway-mediated apoptosis in acute myeloid leukemia (AML) cells. Cephalotaxine-ester-(R)-1-ethoxy-3-mercaptopropan-2-ol-Ph (3,4OMe) is applicable for research on leukemia .
Kukoamine A, a spermine alkaloid, is an orally active and brain-penetrant component found in the root barks of Lycium chinense (L. chinense) Miller. Kukoamine A inhibits purified Crithidia fasciculata trypanothione reductase and soybean lipoxygenase, activates μ-opioid receptor. Kukoamine A can inhibt cancer cell proliferation, migration and invasion, cause G0/G1 phase cellcycle arrest and induce apoptosis. Kukoamine A exerts neuroprotective effect and can induce autophagy . Kukoamine A inhibits LPS (HY-D1056)-induced NO, ROS, PGE2, TNF-α, IL-1β, IL-6 production and COX-2 activity. Kukoamine A reverses palmitic acid-induced insulin resistance, lipid accumulation, and oxidative stress via downregulation of Srebp-1c. Kukoamine A can be used for the research of cancer, infection, inflammation, metabolic and neurological disease, such as glioblastoma and Parkinson's disease .
1,4-Dihydroxy-2-naphthoic acid (Standard) is the analytical standard for 1,4-Dihydroxy-2-naphthoic acid (HY-W014701). 1,4-Dihydroxy-2-naphthoic acid is an orally active aryl hydrocarbon receptor (AhR) agonist and a bifidogenic growth stimulator. 1,4-Dihydroxy-2-naphthoic acid can improve the motor dysfunction in parkinson's disease (PD) model through AhR-dependent and -independent pathways. 1,4-Dihydroxy-2-naphthoic acid exerts anti-inflammatory effects by regulating the gut microbiota (such as promoting the proliferation of Bifidobacterium) and directly regulating the host immune system. 1,4-Dihydroxy-2-naphthoic acid induces apoptosis through G0/G1cellcycle arrest in human keratinocyte to inhibit psoriasis .
AR/AR-V7 degrader-1 is an orally active AR and AR-V7 degrader. AR/AR-V7 degrader-1 disrupts the interaction between AR/AR-V7 and HSP90, leading to their ubiquitination and degradation in castration-resistant prostate cancer cells. AR/AR-V7 degrader-1 regulates the expression of cellcycle-related proteins in prostate cancer cells (downregulates CDK4, CDK6, Cyclin D1, Cyclin E1; upregulates P21) and induces G0/G1 phase arrest. AR/AR-V7 degrader-1 inhibits the proliferation and migration of prostate cancer cells. AR/AR-V7 degrader-1 suppresses the growth of castration-resistant prostate cancer tumors in nude mice and induces the degradation of AR and AR-V7 in tumor tissues. AR/AR-V7 degrader-1 is applicable to the research of castration-resistant prostate cancer .
IU1 is a selective, reversible USP14 inhibitor with an IC50 of 4-5 μM. IU1 binds USP14’s catalytic cleft to block deubiquitinase activity. IU1 induces calpain-dependent Tau cleavage, causes ATP deficits, reduces E1~Ub thioester levels and 26S proteasome assembly. IU1 enhances 26S proteasome chymotrypsin-like activity, modulates LC3B-dependent autophagy flux, reduces cancer cell proliferation and migration, and blocks G0/G1 to S phase cellcycle transition in follicular thyroid cancer cells. IU1 activates autophagy-lysosomal and ubiquitin-proteasome pathways, triggers apoptosis, and reduces cervical cancer cell growth. IU1 enhances degradation of proteasome substrates linked to neurodegenerative disease, accelerates oxidized protein degradation, and increases oxidative stress resistance. IU1 can be used for the research of Alzheimer’s disease, follicular thyroid cancer, ischemic stroke, cervical cancer, and neurodegenerative disease .
YZ-836P is a Protein arginine methyltransferase 5 (PRMT5) targeting agent. YZ-836P promotes ubiquitination and proteasomal degradation of PRMT5 in a cereblon (CRBN)-dependent manner, which in turn reduces levels of its downstream target KLF5. YZ-836P induces G1 phase cellcycle arrest in triple-negative breast cancer cells. YZ-836P induces Apoptosis in triple-negative breast cancer cells. YZ-836P exerts cytotoxic effects on triple-negative breast cancer cells. YZ-836P inhibits the growth of triple-negative breast cancer patient-derived organoids. YZ-836P inhibits the growth of triple-negative breast cancer xenografts in nude mice. YZ-836P can be used for the research of triple-negative breast cancer .
c-MYC/BCL2 ligand 1 iodide is a dual-targeting c-MYC/Bcl-2 G4 ligand with Kd values of 0.90 μM (c-MYC G4) and 0.56 μM (Bcl-2 G4). c-MYC/BCL2 ligand 1 iodide inhibits c-MYC and Bcl-2 gene transcription by binding to G4-forming sequences and downregulates their protein expression. c-MYC/BCL2 ligand 1 iodide inhibits suppresses migration, induces caspase-dependent apoptosis, and triggers cellcycleG1 arrest in MCF-7 cells. c-MYC/BCL2 ligand 1 iodide significantly suppresses tumor growth in a 4T1 syngeneic model with no observable toxicity. c-MYC/BCL2 ligand 1 iodide can be used for the research of breast cancer.
PROTAC PI3Kδ degrader-1 is a Lysine-targeted covalent PI3KδPROTAC degrader with a DC50 of 3.98 nM. PROTAC PI3Kδ degrader-1 has a potent antiproliferative activity and selective PI3Kδ inhibition (IC50: 8 nM). PROTAC PI3Kδ degrader-1 also significantly degrades p-AKT, induces cellcycle arrest in G1 phase and prompts cellapoptosis and autophagy. PROTAC PI3Kδ degrader-1 effectively inhibits the tumor growth in SU-DHL-6 xenograft mice model . Pink: PI3Kδ ligand (HY-169983); Blue: VHL ligase ligand (HY-112078); Black: linker (HY-W013381)
9-cis-UAB30 is a rexinoid agonist. 9-cis-UAB30 significantly decreases the proliferation, viability, and motility of both patient-derived xenografts (PDXs). 9-cis-UAB30 induced cell-cycle arrest as demonstrated by the significant increase in the percentage of cells in G1 and a decrease in the percentage of cells in S phase by downregulating SKP2 and/or 20S proteasome activity, which leads to increased p27kip1 protein stability. 9-cis-UAB30 downregulates the abundance of stem cell marker mRNAs (Oct4, Nanog, Sox2, nestin) and upregulates the abundance of differentiation marker mRNAs (β3-tubulin, NSE, HOXC9, GAP43). 9-cis-UAB30 has no adverse effects on the central nervous system and cardiovascular system at the tested dose. 9-cis-UAB30 can be used for the study of neuroblastoma, cutaneous T-cell lymphomas, and breast cancer .
SKLB-D18 is an orally active ERK1/2/ERK5 inhibitor, with an IC50 of 38.69 nM and a Kd of 126.9 nM against human ERK1, an IC50 of 40.12 nM and a Kd of 209.8 nM against ERK2, and an IC50 of 59.72 nM and a Kd of 468.2 nM against ERK5. SKLB-D18 inhibits cancer cell proliferation, induces G0/G1cellcycle arrest and apoptosis. SKLB-D18 reduces the levels of p-ERK5, p-RSKp90, p-c-Myc and c-Myc, and upregulates the level of p-ERK1/2, thereby inhibiting the ERK1/2/5 pathway in cells. SKLB-D18 increases LC3B-II accumulation, and decreases the levels of p62, p-mTOR and p-p70S6K. SKLB-D18 elevates the levels of ROS, lipid peroxidation and free ferrous ions, reduces the levels of NCOA4 and GPX4, and induces ferritin autophagy-dependent ferroptosis in cancer cells. SKLB-D18 exhibits antitumor activity in a triple-negative breast cancer xenograft mouse model. SKLB-D18 can be used in research related to triple-negative breast cancer .
CDK6/9-IN-1 (compound 66) is an orally active active and dual CDK 6 and CDK 9 inhibitor, with IC50 values of 40.5 nM and 39.5 nM for CDK6 anmd CDK9, respectively .
23-Hydroxybetulinic acid (Anemosapogenin) is an orally active triterpenoid with broad-spectrum anticancer activity. 23-Hydroxybetulinic acid reduces the levels of Bcl-2 and survivin, elevates the level of Bax, promotes the cleavage/activation of caspase-3 and caspase-9, and induces apoptosis via the endogenous mitochondrial pathway involving cytochrome C release and mitochondrial membrane potential disruption. 23-Hydroxybetulinic acid arrests the cellcycle at S and G1 phases, inhibits cancer cell proliferation, blocks the MAPK signaling pathway, regulates MMP2, and induces autophagic apoptosis by upregulating beclin-1. 23-Hydroxybetulinic acid inhibits the activity and efflux function of P-gp, increases the intracellular accumulation of chemotherapeutic drugs, and synergistically enhances cytotoxicity with Doxorubicin (HY-15142). 23-Hydroxybetulinic acid inhibits the phosphorylation and nuclear translocation of STAT6, blocks M2 macrophage polarization, and reduces M2 macrophage-mediated apoptosis resistance of colon cancer cells. 23-Hydroxybetulinic acid can be used in related studies on chronic myeloid leukemia, hepatocellular carcinoma, sarcoma 180, multidrug-resistant breast cancer, leukemia, Doxorubicin-induced cardiotoxicity, and colorectal cancer .
23-Hydroxybetulinic acid (Standard) is the analytical standard of 23-Hydroxybetulinic acid (HY-N0566). This product is intended for research and analytical applications. 23-Hydroxybetulinic acid (Anemosapogenin) is an orally active triterpenoid with broad-spectrum anticancer activity. 23-Hydroxybetulinic acid reduces the levels of Bcl-2 and survivin, elevates the level of Bax, promotes the cleavage/activation of caspase-3 and caspase-9, and induces apoptosis via the endogenous mitochondrial pathway involving cytochrome C release and mitochondrial membrane potential disruption. 23-Hydroxybetulinic acid arrests the cellcycle at S and G1 phases, inhibits cancer cell proliferation, blocks the MAPK signaling pathway, regulates MMP2, and induces autophagic apoptosis by upregulating beclin-1. 23-Hydroxybetulinic acid inhibits the activity and efflux function of P-gp, increases the intracellular accumulation of chemotherapeutic drugs, and synergistically enhances cytotoxicity with Doxorubicin (HY-15142). 23-Hydroxybetulinic acid inhibits the phosphorylation and nuclear translocation of STAT6, blocks M2 macrophage polarization, and reduces M2 macrophage-mediated apoptosis resistance of colon cancer cells. 23-Hydroxybetulinic acid can be used in related studies on chronic myeloid leukemia, hepatocellular carcinoma, sarcoma 180, multidrug-resistant breast cancer, leukemia, Doxorubicin-induced cardiotoxicity, and colorectal cancer.
GLUT4-IN-2 is a potent and selective GLUT4 inhibitor with IC50s of 11.4 µM and 6.8 µM for GLUT1 and GLUT4, respectively. GLUT4-IN-2 induces cellapoptosis and cellcycle arrest at G0/G1phase. GLUT4-IN-2 shows potent antitumor activity .
Ethylenediaminediacetic acid (N,N-Ethylenediglycine) is an important ligand that enhances the antiproliferative activity of metal complexes. The complexes formed by ethylenediacetic acid and metal ions exhibited significant antiproliferative properties in MCF-7 cancer cell line. The metal complexes of ethylenediacetic acid were able to interact with DNA and were studied by CD and EPR spectroscopy techniques. Ethylenediaminediacetic acid and its metal complexes were able to induce cellcycle arrest at the G(0)/G(1) phase. The crystal structure analysis of ethylenediacetic acid provided important structural information for understanding its biological activity .
OKI-005 is an orally active inhibitor of Class I HDACs, with primary targeting of HDAC1, HDAC2 and HDAC3. OKI-005 is a prodrug of OKI-006 (HY-144893). OKI-005 increases histone acetylation levels, induces apoptosis and inhibits cancer cell proliferation. OKI-005 can be used in research related to triple-negative breast cancer and colorectal cancer .
Safrole oxide is a p53 modulator that upregulates the expression of the p53 tumor suppressor protein, linking cellcycle arrest to the apoptotic process. Safrole oxide induces apoptosis in lung cancer cells without triggering necrosis. Safrole oxide can be used in lung cancer-related research .
MI-773 TFA is an orally active, selective MDM2-p53 interactioninhibitor with a Ki of 0.88 nM for MDM2.MI-773 TFA blocks the MDM2-TP53 interaction. MI-773 TFA potently activates p53. MI-773 TFA induces Apoptosis. MI-773 TFA causes tumor regression in xenograft models of adenoid cystic carcinoma. MI-773 TFA exhibits anticancer effects in neuroblastoma. MI-773 TFA can be used for the research of adenoid cystic carcinoma .
CVT-11127 is a potent SCD inhibitor. CVT-11127 induces apoposis and arrests the cellcycle at the G1/S phase. CVT-11127 has the potential for the research of lung cancer .
T7 Peptide is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide induces G0/G1cellcycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
T7 Peptide TFA is a protein synthesis inhibitor and anti-angiogenic agent, with a Kd of 10 nM for human transferrin receptor. T7 Peptide TFA inhibits the phosphorylation of focal adhesion kinase, the activation of phosphatidylinositol 3-kinase and Akt, the kinase activity of mTOR, as well as the phosphorylation of 4E-BP1 in endothelial cells. T7 Peptide TFA induces G0/G1cellcycle arrest, apoptosis and protective autophagy in hepatocellular carcinoma cells, and suppresses tumor growth in mouse models. T7 Peptide TFA is applicable to research related to cancer, glioblastoma, hepatocellular carcinoma and glioma .
[Ala92]-p16 (84-103) is a peptide derived from the p16CDKN2/INK4a (p16) tumor suppressor protein. [Ala92]-p16 (84-103) binds to both cdk4 and cdk6 and inhibits cdk4-cyclin D1 kinase activity in vitro (IC50: 1.5 μM). [Ala92]-p16 (84-103) blocks cellcycle progression through the G1 phase .
S9-CMC1 TFA is a covalent peptide lysine-specific demethylase 1 (LSD1) inhibitor with an IC50 value of 2.53 μM. S9-CMC1 TFA specifically recognizes Cys360 in the enzyme-active region. S9-CMC1 TFA inhibits LSD1 activity, increasing H3K4me1 and H3K4me2 levels, leading to G1cellcycle arrest and apoptosis and inhibiting cell proliferation. S9-CMC1 TFA significantly inhibits tumor growth in A549 xenograft animal models .
Cr-ACP1 is an anti-cancerous peptide. Cr-ACP1 binds to DNA, inducing cellcycle arrest in the G0-G1 phase, leading to the initiation of Apoptosis mechanisms. Cr-ACP1 exhibits anticancer effects against colon cancer and epidermoid carcinoma .
TAT-p16 (p16INK4a peptide) is a peptide mimic of p16INK4a that can induce an early G phase cellcycle arrest in the absence of active cyclin E:Cdk2 complex .
Annexuzlimab is a humanised IgG1 monoclonal antibody which specifically binds to ANXA1 disrupting its interaction with formyl peptide receptors 1 and 2 (FPR1/2). Annexuzlimab arrests cellcycle progression with cancer cells accumulating in the G1 phase. Annexuzlimab targets secreted ANXA1, preventing FPR1/2 activation and reducing cancer progression. Annexuzlimab can be used for the research of triple negative breast cancer, pancreatic cancer and osteosarcoma .
Tunicamycin is a mixture of homologous nucleoside antibiotic that inhibits N-linked glycosylation and blocks GlcNAc phosphotransferase (GPT). Tunicamycin causes accumulation of unfolded proteins in cell endoplasmic reticulum (ER) and induces ER stress, and causes blocking of DNA synthesis and cellcycle arrest in G1 phase. Tunicamycin inhibits gram-positive bacteria, yeasts, fungi, and viruses and has anti-cancer activity .Tunicamycin increases exosome release in cervical cancer cells .
Amentoflavone (Didemethyl-ginkgetin) is a potent and orally active GABA(A) negative modulator. Amentoflavone also shows anti-inflammatory, antioxidative, anti-viral, anti-tumor, anti-radiation, anti-fungal, antibacterial activity. Amentoflavone induces apoptosis and cellcycle arrest at sub-G1 phase .
Carvacrol is an orally active and blood-brain barrier-permeable monoterpenic phenol that can be extract from an abundant number of aromatic plants, including thyme and oregano, possessing antioxidant, antibacterial, antifungal, anticancer, anti-inflammatory, hepatoprotective, spasmolytic, and vasorelaxant properties. Carvacrol also causes cellcycle arrest in G0/G1, downregulates Notch-1, and Jagged-1, and induces apoptosis. Carvacrol is used in low concentrations as a food flavoring ingredient and preservative, as well as a fragrance ingredient in cosmetic formulations .
Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Pelargonidin chloride is an anthocyanidin and also is a scavenger of nitric oxide radical and has antioxidant activities. Pelargonidin inhibits cell viability and induces cellcycle arrest at sub-G1 phase. Pelargonidin chloride increases the mRNA and protein expression of HO-1, NQO1, Nrf2. Pelargonidin chloride improves Aβ-induced memory and learning impairment .
Atractylenolide II (Asterolide) is a sesquiterpenoid compound. Atractylenolide II can induce G1 phase cellcycle arrest and apoptosis in B16 melanoma cells. Atractylenolide II is an orally effective anticancer agent that can exert anti-melanoma effects by inhibiting the STAT3 signaling pathway. In addition, Atractylenolide II has been shown to ameliorate myocardial fibrosis, oxidative stress, and neuroprotective activity .
Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities .
Veratramine (NSC17821; NSC23880) is an orally active inhibitor of the PI3K/Akt/mTOR signaling pathway and a SIGMAR1 modulator. Veratramine induces autophagic apoptosis of tumor cells, arrests the cellcycle at the G0/G1 phase, and inhibits epithelial-mesenchymal transition (EMT)-related proteins to reduce tumor migration. Veratramine reduces spinal cord and sciatic nerve pathological damage in a neuropathy model by inhibiting SIGMAR1 binding to NMDAR and phosphorylation of NMDAR Ser896. Veratramine has anti-tumor proliferation, apoptosis induction, anti-inflammatory and neuroprotective activities, and can be used in the study of cancers such as liver cancer and osteosarcoma, as well as diabetic peripheral neuropathy .
Protosappanin B is a phenolic compound extracted from Caesalpinia sappan. Anti-cancer activity . Protosappanin B induces apoptosis and causes G1cellcycle arrest in human bladder cancer cells .
Meisoindigo (Dian III), a derivative of Indirubin (HY-N0117), halts the cellcycle at the G0/G1 phase and induces apoptosis in primary acute myeloid leukemia (AML) cells. Meisoindigo exhibits high antitumor activity .
Terrestrosin D is an orally active apoptosis inducer. Terrestrosin D induces cellcycle arrest at the G1 and S phases, reduces mitochondrial membrane potential, and inhibits the growth of cancer cells and endothelial cells. Terrestrosin D is studied in castration-resistant prostate cancer and pulmonary fibrosis .
Gemcitabine triphosphate (dFdCTP) trisodium is the active metabolite of Gemcitabine (HY-17026). The mechanism of Gemcitabine triphosphate trisodium cell-killing is its competition with cytidine triphosphate during DNA replication, which results in the inhibition of chain elongation. Gemcitabine triphosphate trisodium shows a Ki of 11.2 μM against DNA polymerase α and 14.4 μM against DNA polymerase ε. Gemcitabine triphosphate trisodium partially inhibits dCMP deaminase and acts as a substrate for DNA synthesis to incorporate into cellular DNA and RNA. Gemcitabine triphosphate trisodium disrupts DNA and RNA synthesis, arrests cellcycle in G0/G1 and S phases, triggers apoptosis, reduces tumor cell proliferation. Gemcitabine triphosphate trisodium can be used for the research of pancreatic cancer and non-small cell lung cancer .
Theaflavin-3-gallate, a black tea theaflavin monomer, is regarded as the biologically important active component of black tea and provides health benefits. Theaflavin-3-gallate acts as prooxidants and induces oxidative stress in the carcinoma cells. Theaflavin-3-gallate reacts directly with reduced glutathione (GSH) in a time- and concentration-dependent manner. Theaflavin-3-gallate induces apoptosis and G1cellcycle arrest in ovarian cancer A2780/CP70 cells through p53-dependent pathways. Theaflavin-3-gallate induces DNA damage through ATM/Chk/p53 pathway .
Kukoamine A, a spermine alkaloid, is an orally active and brain-penetrant component found in the root barks of Lycium chinense (L. chinense) Miller. Kukoamine A inhibits purified Crithidia fasciculata trypanothione reductase and soybean lipoxygenase, activates μ-opioid receptor. Kukoamine A can inhibt cancer cell proliferation, migration and invasion, cause G0/G1 phase cellcycle arrest and induce apoptosis. Kukoamine A exerts neuroprotective effect and can induce autophagy . Kukoamine A inhibits LPS (HY-D1056)-induced NO, ROS, PGE2, TNF-α, IL-1β, IL-6 production and COX-2 activity. Kukoamine A reverses palmitic acid-induced insulin resistance, lipid accumulation, and oxidative stress via downregulation of Srebp-1c. Kukoamine A can be used for the research of cancer, infection, inflammation, metabolic and neurological disease, such as glioblastoma and Parkinson's disease .
Illudin S, a cytotoxic Illudin, is a natural sesquiterpene with strong anti-tumour and antiviral activities. Illudin S has genotoxic activities. Illudin S blocks the G1-S phase interface of the cellcycle in human leukemia cells .
N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cellapoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma - .
Broussoflavonol F is a HER2-RAS-MEK-ERK signaling pathway modulator and mushroom tyrosinase inhibitor, with an IC50 of 82.3 μM against mushroom tyrosinase. Broussoflavonol F reduces the protein expression levels of RAS, HER2, phosphorylated BRAF, phosphorylated MEK and phosphorylated Erk. It induces cell apoptosis, triggers G0/G1 phase cellcycle arrest, and exhibits cytotoxicity against colon cancer cells. Broussoflavonol F is applicable to related research on colon cancer .
3,4,5-Tricaffeoylquinic acid (3,4,5-triCQA) inhibits tumor necrosis factor-α-stimulated production of inflammatory mediators in keratinocytes via suppression of Akt- and NF-κB-pathways. 3,4,5-Tricaffeoylquinic acid induces cellcycle arrest at G0/G1, actin cytoskeleton organization, chromatin remodeling, neuronal differentiation, and bone morphogenetic protein signaling in human neural stem cells. 3,4,5-Tricaffeoylquinic acid has the potential for the research of aging-associated diseases .
Gomisin G is a lignin from S. chinesis with anti-HIV (EC50 = 0.006 μg/mL), anti-liver cancer and anti-inflammatory activities. Gomisin G has an AKT-cyclin D1 dependent mechanism against triple-negative breast cancer (TNBC) cells through suppressing phosphorylation rather than inducing apoptosis. Gomisin G can inhibit AKT phosphorylation. Gomisin G can cause cellcycle arrest in the G1 phase. Gomisin G can be studied in research for diseases such as HIV, breast and liver cancers .
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cellcycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
Germacrone is a sesquiterpene compound with multiple biological activities. Germacrone inhibits the H1N1 and H3N2influenza A virus and the influenza B virus. Germacrone blocks the progressionof arthritis by regulating Th1/Th2 balance and inhibiting NF-κB signaling. Germacrone can arrest the cellcycle at G0/G1 and G2/M phases and induce apoptosis in breast cancer cells. Germacrone inhibits 5α-reductase and has anti-androgenic effect. Germacrone has neuroprotective functions and can be used for the study of traumatic brain injury (TBI). Germacrone also has antioxidant activity .
Gemcitabine triphosphate (dFdCTP) is the active metabolite of Gemcitabine (HY-17026). The mechanism of Gemcitabine triphosphate cell-killing is its competition with cytidine triphosphate during DNA replication, which results in the inhibition of chain elongation. Gemcitabine triphosphate shows a Ki of 11.2 μM against DNA polymerase α and 14.4 μM against DNA polymerase ε. Gemcitabine triphosphate partially inhibits dCMP deaminase and acts as a substrate for DNA synthesis to incorporate into cellular DNA and RNA. Gemcitabine triphosphate disrupts DNA and RNA synthesis, arrests cellcycle in G0/G1 and S phases, triggers apoptosis, reduces tumor cell proliferation. Gemcitabine triphosphate can be used for the research of pancreatic cancer and non-small cell lung cancer .
Ganoderic acid DM, a natural triterpenoid isolated from Ganoderma lucidum, induces DNA damage, G1cellcycle arrest and apoptosis in human breast cancer cells. Ganoderic acid DM as a specific inhibitor of osteoclastogenesis .
Secalonic acid D is a toxic compound against tumor cells. Secalonic acid D can be isolated from the metabolites of Aspergillus aculeatus. Secalonic acid D activates GSK3-β, and degrades β-catenin. Thus, Secalonic acid D down-regulates c-Myc expression, arrests cellcycle at G1 phase, induces cellapoptosis .
(+)-Columbianetin ((S)-Columbianetin) acts as an inhibitor of JNK/ERK. (+)-Columbianetin inhibits UVA-induced phosphorylation of JNK and ERK, reduces the production of MMP-1, reverses UVA-induced Collagen (HY-NP003) degradation, and alleviates UVA-mediated inhibition of Smad2/3 phosphorylation and translocation. (+)-Columbianetin regulates the AP-1 and ASK1-MAPK signaling pathways, inhibits the production of ROS and blocks sub-G1cellcycle arrest. (+)-Columbianetin is applicable to research related to skin aging .
Alisol B is a triterpene with diverse biological activities. Alisol B binds human soluble epoxide hydrolase (sEH) with a Ki of 5.97 μM and reduces sEH activity. Alisol B inhibits RANKL-induced JNK phosphorylation, NFATc1 and c-Fos expression, osteoclast formation, mature osteoclast pit-forming and actin ring activity, and SERCA pump activity. Alisol B induces calcium mobilization, CaMKK-AMPK-mTOR pathway activation, autophagic flux, autophagosome formation, G1 phase cellcycle arrest, endoplasmic reticulum stress, unfolded protein responses, and cancer cellapoptosis. Alisol B can be used for the research of hypercalcemia, osteoporosis, rheumatoid arthritis, periodontitis, acute kidney injury, and breast cancer .
23-Hydroxybetulinic acid (Anemosapogenin) is an orally active triterpenoid with broad-spectrum anticancer activity. 23-Hydroxybetulinic acid reduces the levels of Bcl-2 and survivin, elevates the level of Bax, promotes the cleavage/activation of caspase-3 and caspase-9, and induces apoptosis via the endogenous mitochondrial pathway involving cytochrome C release and mitochondrial membrane potential disruption. 23-Hydroxybetulinic acid arrests the cellcycle at S and G1 phases, inhibits cancer cell proliferation, blocks the MAPK signaling pathway, regulates MMP2, and induces autophagic apoptosis by upregulating beclin-1. 23-Hydroxybetulinic acid inhibits the activity and efflux function of P-gp, increases the intracellular accumulation of chemotherapeutic drugs, and synergistically enhances cytotoxicity with Doxorubicin (HY-15142). 23-Hydroxybetulinic acid inhibits the phosphorylation and nuclear translocation of STAT6, blocks M2 macrophage polarization, and reduces M2 macrophage-mediated apoptosis resistance of colon cancer cells. 23-Hydroxybetulinic acid can be used in related studies on chronic myeloid leukemia, hepatocellular carcinoma, sarcoma 180, multidrug-resistant breast cancer, leukemia, Doxorubicin-induced cardiotoxicity, and colorectal cancer .
Suberosin, isolated from Plumbago zeylanica, exhibits anti-inflammatory and anticoagulant activity. Suberosin suppresses PHA-induced PBMC proliferation and arrested cellcycle progression from the G1 transition to the S phase through the modulation of the transcription factors NF-AT and NF-κB .
Panaxatriol is an orally active insulin sensitizer. Panaxatriol enhances the phosphorylation levels of Akt, insulin receptor and p70S6K in skeletal muscle. Panaxatriol reduces the mRNA expression level of Atrogin1 in skeletal muscle. Panaxatriol induces apoptosis, pre-G1cellcycle arrest and increased intracellular ROS levels in prostate cancer cells, decreases mitochondrial membrane potential, inhibits cell migration and reduces colony formation. Panaxatriol can be used in research related to insulin resistance, myocardial ischemia/reperfusion injury and prostate cancer .
(-)-Hinesol (Hinesol) is a potent anticancer agent. (-)-Hinesol induces apoptosis and cellcycle arrest at G0/G1 phase. (-)-Hinesol downregulates MEK/ERK pathway and NF-κB pathway and mediates theexpression of cyclin D1, Bax and Bcl-2. (-)-Hinesol has the potential for the research of non–small cell lung cancer .
Dendrogenin A (DDA) is a ligand for liver X receptor (LXR), that induces the expression of sodium/iodine symporter, and increases iodine uptake. Dendrogenin A induces cell differentiation of MCF-7, and reactivates the function of lactating cells. Dendrogenin A induces the expressions of the TSH receptor, thyroid peroxidase, and thyroglobulin, and affects thyroid hormone generation. Dendrogenin A exhibits cytotoxicity in cancer cell B-CPAP and 8505c with IC50 of 4.1 and 6.2 µM. Dendrogenin A arrests the cellcycle at G0/G1 phase .
Lotusine is an orally active signaling pathway modulator and enzyme inhibitor, with an IC50 of 30.60 μg/mL against α-amylase and an IC50 of 36.15 μg/mL against α-glucosidase. Lotusine inhibits the EGFR-Akt-ERK signaling pathway by reducing the levels of phosphorylated EGFR, Akt and ERK. Lotusine induces apoptosis, triggers G0/G1cellcycle arrest and inhibits cancer cell proliferation. Lotusine reduces lipid peroxidation and increases the activities of SOD, CAT and GPx. Lotusine is applicable to researches related to non-small cell lung cancer, type 2 diabetes and autism spectrum disorder.
Globulol is a terpenoid metabolite and Antimicrobial agent. Globulol can be isolated from Alpinia oxyphylla Miq. Globulol binds to PAK4, reduces the expression level of PAK4 in cancer cells, decreases the phosphorylation of AKT, and downregulates the expressions of STAT3, phosphorylated STAT3, and PD-L1. Globulol promotes the secretion of CCL4 by cancer cells. Globulol reduces the viability and proliferation ability of cancer cells, induces G0/G1cellcycle arrest and Apoptosis in cancer cells, and inhibits cancer cell migration and the integrity of 3D tumor spheres. Globulol enhances the relevant effects of anti-PD-1 agents in the cancer cell microenvironment. Globulol exhibits anticancer activity against liver cancer. Globulol inhibits the mycelial growth of phytopathogenic fungi and the growth of phytopathogenic bacteria. Globulol can be used in studies related to hepatocellular carcinoma .
Germacrone (Standard) is an analytical standard of Gemmacrone (HY-N0440). This product is intended for research and analytical applications. Germacrone (Standard) is a sesquiterpene compound with multiple biological activities. Germacrone (Standard) inhibits the H1N1 and H3N2 influenza A viruses and the influenza B virus. Germacrone (Standard) blocks the progressionof arthritis by regulating Th1/Th2 balance and inhibiting NF-κB signaling. Germacrone (Standard) can arrest the cellcycle at G0/G1 and G2/M phases and induce apoptosis in breast cancer cells. Germacrone (Standard) inhibits 5α-reductase and has anti-androgenic effect. Germacrone (Standard) has neuroprotective functions and can be used for the study of traumatic brain injury (TBI). Germacrone (Standard) also has antioxidant activity .
2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cellcycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
Veratramine (NSC17821; NSC23880) (Standard) is the analytical standard of Veratramine (HY-N0837). This product is intended for research and analytical applications. Veratramine (NSC17821; NSC23880) is an orally active inhibitor of the PI3K/Akt/mTOR signaling pathway and a SIGMAR1 modulator. Veratramine induces autophagic apoptosis of tumor cells, arrests the cellcycle at the G0/G1 phase, and inhibits epithelial-mesenchymal transition (EMT)-related proteins to reduce tumor migration. Veratramine reduces spinal cord and sciatic nerve pathological damage in a neuropathy model by inhibiting SIGMAR1 binding to NMDAR and phosphorylation of NMDAR Ser896. Veratramine has anti-tumor proliferation, apoptosis induction, anti-inflammatory and neuroprotective activities, and can be used in the study of cancers such as liver cancer and osteosarcoma, as well as diabetic peripheral neuropathy .
Ganoderic acid Mf is an antitumor triterpenoid. Ganoderic acid Mf causes cellcycle arrest in the G1 phase. Ganoderic acid Mf shows high selectivity between normal and cancer cells and induces cellapoptosis via mitochondria mediated pathway .
1,4-Dimethylnaphthalen is a non-competitive potato tuber sprouting inhibitor. 1,4-Dimethylnaphthalen blocks cellcycle progression (G1/S arrest) by inducing PP2A phosphatase and oxygen metabolism-related genes. 1,4-Dimethylnaphthalen is promising for research of fungistatic activity during potato storage and sprout control under abiotic stress .
Apiole is an anti-tumor agent that induces apoptosis and inhibits human colon cancer cells by inducing G0/G1cellcycle arrest. Apiole also significantly inhibited colon tumor development in an in vivo mouse xenograft model .
Acrofolione A is an acetophenone dimer isolated from Acronychia pendunculata with anticancer effects. Acrofolione A induces G0/G1 phase cellcycle arrest and apoptosis in human NALM-6 pre-B cell leukaemia cells. Acrofolione A can be used for leukaemia research .
Cucurbitacin C is a triterpenoid calabinoid that can be isolated from Cucurbitaceae plants. Cucurbitacin C has anti-cancer activity in vivo and in vitro. Cucurbitacin C can induce cellcycle arrest in G1 or G2/M phase and apoptosis by inhibiting Akt signaling .
Asparanin A is an apoptosis inducer with anticancer activity. Asparanin A induces cellcycle arrest in the G0/G1 phase through mitochondria and PI3K/AKT signaling pathways, inhibiting cancer cell growth. Asparanin A also demonstrated in vivo efficacy in a mouse xenograft model of Ishikawa endometrial carcinoma, significantly inhibiting tumor growth .
Angelylalkannin is a naphthoquinone compound isolated from the root of Alkanna tinctoria. Angelylalkannin showed significant inhibitory effects in the antiproliferative effect test on human colon cancer cells HCT-116 and SW-480. The median inhibitory concentration (IC50) of Angelylalkannin was 4.76 mM for HCT-116 cells and 7.03 mM for SW-480 cells. Like alkannin, Angelylalkannin can arrest the cellcycle in the G1 phase and induce apoptosis at a concentration of 1-10 mM.
Amentoflavone (Standard) is the analytical standard of Amentoflavone. This product is intended for research and analytical applications. Amentoflavone (Didemethyl-ginkgetin) is a potent and orally active GABA(A) negative modulator. Amentoflavone also shows anti-inflammatory, antioxidative, anti-viral, anti-tumor, anti-radiation, anti-fungal, antibacterial activity. Amentoflavone induces apoptosis and cellcycle arrest at sub-G1 phase .
Geiparvarin is an anticancer agent and an inhibitor of MAO-B (pIC50 = 6.84 μM). Geiparvarin exerts anti-tumor effects by downregulating COX2 expression and inhibiting angiogenesis. Geiparvarin blocks the cellcycle at the G1 phase and induces apoptosis of cancer cells. Geiparvarin has anti-microtubule activity and destroys the cytoskeleton to exert anti-proliferative effects. Geiparvarin has research significance for lung cancer, leukemia, and breast cancer .
Carvacrol (Standard) is the analytical standard of Carvacrol. This product is intended for research and analytical applications. Carvacrol is an orally active and blood-brain barrier-permeable monoterpenic phenol that can be extract from an abundant number of aromatic plants, including thyme and oregano, possessing antioxidant, antibacterial, antifungal, anticancer, anti-inflammatory, hepatoprotective, spasmolytic, and vasorelaxant properties. Carvacrol also causes cellcycle arrest in G0/G1, downregulates Notch-1, and Jagged-1, and induces apoptosis. Carvacrol is used in low concentrations as a food flavoring ingredient and preservative, as well as a fragrance ingredient in cosmetic formulations .
Sotetsuflavone is a flavonoid that can be isolated from Cycas revolute. Sotetsuflavone inhibits phosphorylation of PI3K, Akt, mTOR, JNK, and p38 MAPK; modulates expression of Cyclin D1, CDK4, Bcl-2, Bax, cleaved caspases 3/9, MMP-9, TGF-β, STAT3, and β-catenin. Sotetsuflavone induces G0/G1cellcycle arrest, apoptosis, autophagy, and intracellular ROS elevation, inhibits cancer cell proliferation. Sotetsuflavone inhibits tumor growth in mouse tumor xenograft models. Sotetsuflavone can be used for the research of non-small cell lung cancer and Crohn’s disease .
Typhatifolin B (Compd 2), an anti-cancer agent, could remarkably induce cellapoptosis and G0/G1cycle arrest, as well as block cell migration and invasion .
Apiole (Standard) is the analytical standard of Apiole. This product is intended for research and analytical applications. Apiole is an anti-tumor agent that induces apoptosis and inhibits human colon cancer cells by inducing G0/G1cellcycle arrest. Apiole also significantly inhibited colon tumor development in an in vivo mouse xenograft model .
Meisoindigo (Standard) is the analytical standard of Meisoindigo. This product is intended for research and analytical applications. Meisoindigo (Dian III), a derivative of Indirubin (HY-N0117), halts the cellcycle at the G0/G1 phase and induces apoptosis in primary acute myeloid leukemia (AML) cells. Meisoindigo exhibits high antitumor activity .
Protosappanin B (Standard) is the analytical standard of Protosappanin B. This product is intended for research and analytical applications. Protosappanin B is a phenolic compound extracted from Caesalpinia sappan. Anti-cancer activity . Protosappanin B induces apoptosis and causes G1cellcycle arrest in human bladder cancer cells .
Diosbulbin C is a diterpene lactone component, which can be extracted from traditional Chinese medicine Dioscorea bulbifera L.. Diosbulbin C possesses high anticancer activity in non-small cell lung cancer (NSCLC). Diosbulbin C could induce cellcycle arrest at G0/G1 phase in NSCLC. Diosbulbin C also inhibits the proliferation of NSCLC cells, possibly by downregulating the expression/activation of AKT, DHFR, and TYMS .
Cinobufagin (Standard) is the analytical standard of Cinobufagin. This product is intended for research and analytical applications. Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
Decursin (Standard) is the analytical standard of Decursin. This product is intended for research and analytical applications. Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities [4].
Atractylenolide II (Standard) is the analytical standard of Atractylenolide II. This product is intended for research and analytical applications. Atractylenolide II (Asterolide) is a sesquiterpenoid compound. Atractylenolide II can induce G1 phase cellcycle arrest and apoptosis in B16 melanoma cells. Atractylenolide II is an orally effective anticancer agent that can exert anti-melanoma effects by inhibiting the STAT3 signaling pathway. In addition, Atractylenolide II has been shown to ameliorate myocardial fibrosis, oxidative stress, and neuroprotective activity .
Oximidine III is an anti-tumor antibiotic. Oximidine III can selectively inhibit the growth of 3Y1 in rat fibroblasts with degeneration of various tumor genes. Oximidine III inhibits v-H-ras-3Y1, v-src-3Y1 cells and the normal 3Y1 cells with IC50s (nM) of 14, 4.5 and 140, respectively. Oximidine III stops RAS-or SRC-denatured cells at G1 phase of the cellcycle and increases p21WAF1 expression .
Lotusine hydroxide is an orally active signaling pathway modulator and enzyme inhibitor, with an IC50 of 30.60 μg/mL against α-amylase and an IC50 of 36.15 μg/mL against α-glucosidase. Lotusine hydroxide inhibits the EGFR-Akt-ERK signaling pathway by reducing the levels of phosphorylated EGFR, Akt and ERK. Lotusine hydroxide induces apoptosis, triggers G0/G1cellcycle arrest and inhibits cancer cell proliferation. Lotusine hydroxide reduces lipid peroxidation and increases the activities of SOD, CAT and GPx. Lotusine hydroxide is applicable to researches related to non-small cell lung cancer, type 2 diabetes and autism spectrum disorder .
Gomisin G (Standard) is the analytical standard of Gomisin G. This product is intended for research and analytical applications. Gomisin G is a lignin from S. chinesis with anti-HIV (EC50 = 0.006 μg/mL), anti-liver cancer and anti-inflammatory activities. Gomisin G has an AKT-cyclin D1 dependent mechanism against triple-negative breast cancer (TNBC) cells through suppressing phosphorylation rather than inducing apoptosis. Gomisin G can inhibit AKT phosphorylation. Gomisin G can cause cellcycle arrest in the G1 phase. Gomisin G can be studied in research for diseases such as HIV, breast and liver cancers .
2,6-Dihydroxyacetophenone (Standard) is the analytical standard of 2,6-Dihydroxyacetophenone (HY-Y0106). This product is intended for research and analytical applications. 2,6-Dihydroxyacetophenone, a polyphenolic derivative of Acetophenone (HY-Y0989), is an orally active mTOR inhibitor. 2,6-Dihydroxyacetophenone shows antioxidant activity. 2,6-Dihydroxyacetophenone inhibits cell growth and proliferation in CRC cells. 2,6-Dihydroxyacetophenone arrests at G0/G1 phase of cellcycle, induces apoptosis and suppresses cell migration in CRC cells. 2,6-Dihydroxyacetophenone inhibits xanthine oxidase (XOD) with an IC50 of 1.24 mM. 2,6-dihydroxyacetophenone improves uric acid metabolism in hyperuricemia mice, reduces plasma cholesterol in hypercholesterolemic rats, and inhibits lipid accumulation in HFD-induced obese mice. 2,6-Dihydroxyacetophenone can be used for the study of colorectal cancer (CRC), hyperuricemia and hypercholesterolemia .
Theaflavin-3-gallate (Standard) is the analytical standard of Theaflavin-3-gallate. This product is intended for research and analytical applications. Theaflavin-3-gallate, a black tea theaflavin monomer, is regarded as the biologically important active component of black tea and provides health benefits. Theaflavin-3-gallate acts as prooxidants and induces oxidative stress in the carcinoma cells. Theaflavin-3-gallate reacts directly with reduced glutathione (GSH) in a time- and concentration-dependent manner. Theaflavin-3-gallate induces apoptosis and G1cellcycle arrest in ovarian cancer A2780/CP70 cells through p53-dependent pathways. Theaflavin-3-gallate induces DNA damage through ATM/Chk/p53 pathway .
Alisol B (Standard) is the analytical standard of Alisol B (HY-N0805A). This product is intended for research and analytical applications. Alisol B is a triterpene with diverse biological activities. Alisol B binds human soluble epoxide hydrolase (sEH) with a Ki of 5.97 μM and reduces sEH activity. Alisol B inhibits RANKL-induced JNK phosphorylation, NFATc1 and c-Fos expression, osteoclast formation, mature osteoclast pit-forming and actin ring activity, and SERCA pump activity. Alisol B induces calcium mobilization, CaMKK-AMPK-mTOR pathway activation, autophagic flux, autophagosome formation, G1 phase cellcycle arrest, endoplasmic reticulum stress, unfolded protein responses, and cancer cell apoptosis. Alisol B can be used for the research of hypercalcemia, osteoporosis, rheumatoid arthritis, periodontitis, acute kidney injury, and breast cancer.
1,4-Dihydroxy-2-naphthoic acid (Standard) is the analytical standard for 1,4-Dihydroxy-2-naphthoic acid (HY-W014701). 1,4-Dihydroxy-2-naphthoic acid is an orally active aryl hydrocarbon receptor (AhR) agonist and a bifidogenic growth stimulator. 1,4-Dihydroxy-2-naphthoic acid can improve the motor dysfunction in parkinson's disease (PD) model through AhR-dependent and -independent pathways. 1,4-Dihydroxy-2-naphthoic acid exerts anti-inflammatory effects by regulating the gut microbiota (such as promoting the proliferation of Bifidobacterium) and directly regulating the host immune system. 1,4-Dihydroxy-2-naphthoic acid induces apoptosis through G0/G1cellcycle arrest in human keratinocyte to inhibit psoriasis .
23-Hydroxybetulinic acid (Standard) is the analytical standard of 23-Hydroxybetulinic acid (HY-N0566). This product is intended for research and analytical applications. 23-Hydroxybetulinic acid (Anemosapogenin) is an orally active triterpenoid with broad-spectrum anticancer activity. 23-Hydroxybetulinic acid reduces the levels of Bcl-2 and survivin, elevates the level of Bax, promotes the cleavage/activation of caspase-3 and caspase-9, and induces apoptosis via the endogenous mitochondrial pathway involving cytochrome C release and mitochondrial membrane potential disruption. 23-Hydroxybetulinic acid arrests the cellcycle at S and G1 phases, inhibits cancer cell proliferation, blocks the MAPK signaling pathway, regulates MMP2, and induces autophagic apoptosis by upregulating beclin-1. 23-Hydroxybetulinic acid inhibits the activity and efflux function of P-gp, increases the intracellular accumulation of chemotherapeutic drugs, and synergistically enhances cytotoxicity with Doxorubicin (HY-15142). 23-Hydroxybetulinic acid inhibits the phosphorylation and nuclear translocation of STAT6, blocks M2 macrophage polarization, and reduces M2 macrophage-mediated apoptosis resistance of colon cancer cells. 23-Hydroxybetulinic acid can be used in related studies on chronic myeloid leukemia, hepatocellular carcinoma, sarcoma 180, multidrug-resistant breast cancer, leukemia, Doxorubicin-induced cardiotoxicity, and colorectal cancer.
Leptolstatin is an inhibitor of the progression of G1 and G2 phases of the mammalian cellcycle. Leptolstatin can be extracted from the fermentation broth of Streptomyces sp. SAM1595 .
N-Desmethyldauricine is a NF-κB p65 inhibitor and apoptosis inducer. N-Desmethyldauricine reduces the protein expression level of p65, induces apoptosis, arrests the cellcycle at the G0/G1 phase, attenuates intercellular adhesion, and inhibits the growth of 3D spheroids of triple-negative breast cancer. N-Desmethyldauricine can be used in studies related to triple-negative breast cancer .
N6-Benzyladenosine (Standard) (Benzyladenosine (Standard)) is the analytical standard of N6-Benzyladenosine (HY-N7844). This product is intended for research and analytical applications. N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cell apoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma - .
Chikusetsusaponin IVa methyl ester (CME) is a natural triterpenoid saponin compound. Chikusetsusaponin IVa methyl ester induces G0/G1cellcycle arrest and apoptosis in colon cancer cells by inhibiting the Wnt/β-catenin signaling pathway. By inhibiting the NF-κB and AP-1 signaling pathways, Chikusetsusaponin IVa methyl ester significantly reduces the production of NO, PGE₂ and pro-inflammatory cytokines (TNF-α, IL-6, IL-1β), and downregulates the levels of iNOS and COX-2. Chikusetsusaponin IVa methyl ester can be used in researches on colorectal cancer and inflammation .
CDK1; cyclin-dependent kinase 1; CDC2, cell division cycle 2, G1 to S and G2 to M; CDC28A; p34 protein kinase; cellcycle controller CDC2; cell division protein kinase 1; cell division control protein 2 homolog; cell division cycle 2, G1 to S and G2 to M; CDC2; P34CDC2; MGC111195; DKFZp686L20222
The CDK1/CDC2-cyclin-B complex is a key regulator in the cell cycle. It plays an important role in cell division and development and regulates a variety of cellular activities, including chromosome segregation, nuclear membrane rupture, cytokinesis, etc. The activity of CDK1 is regulated by a variety of phosphorylation and dephosphorylation, thereby controlling the progression of the cell cycle and the DNA repair process. CDK1-CCNB1 Protein, Human (sf9, GST, Flag, His) is the recombinant human-derived CDK1-CCNB1, expressed by Sf9 insect cells, with N-GST, N-Flag, N-His labeled tag.
CDK1; cyclin-dependent kinase 1; CDC2, cell division cycle 2, G1 to S and G2 to M; CDC28A; p34 protein kinase; cellcycle controller CDC2; cell division protein kinase 1; cell division control protein 2 homolog; cell division cycle 2, G1 to S and G2 to M; CDC2; P34CDC2; MGC111195; DKFZp686L20222
The CDK1/CDC2-cyclin-B complex is a key regulator in the cell cycle. It plays an important role in cell division and development and regulates a variety of cellular activities, including chromosome segregation, nuclear membrane rupture, cytokinesis, etc. The activity of CDK1 is regulated by a variety of phosphorylation and dephosphorylation, thereby controlling the progression of the cell cycle and the DNA repair process. CDK1-CCNA2 Protein, Human (sf9, GST, His) is the recombinant human-derived CDK1-CCNA2, expressed by Sf9 insect cells, with N-His, N-GST labeled tag.
CDK1; cyclin-dependent kinase 1; CDC2, cell division cycle 2, G1 to S and G2 to M; CDC28A; p34 protein kinase; cellcycle controller CDC2; cell division protein kinase 1; cell division control protein 2 homolog; cell division cycle 2, G1 to S and G2 to M; CDC2; P34CDC2; MGC111195; DKFZp686L20222
The CDK1/CDC2-cyclin-B complex is a key regulator in the cell cycle. It plays an important role in cell division and development and regulates a variety of cellular activities, including chromosome segregation, nuclear membrane rupture, cytokinesis, etc. The activity of CDK1 is regulated by a variety of phosphorylation and dephosphorylation, thereby controlling the progression of the cell cycle and the DNA repair process. CDK1-CCNE2 Protein, Human (sf9, GST, His, Flag) is the recombinant human-derived CDK1-CCNE2, expressed by Sf9 insect cells, with N-His, N-GST, N-Flag labeled tag.
CDK1; cyclin-dependent kinase 1; CDC2, cell division cycle 2, G1 to S and G2 to M; CDC28A; p34 protein kinase; cellcycle controller CDC2; cell division protein kinase 1; cell division control protein 2 homolog; cell division cycle 2, G1 to S and G2 to M; CDC2; P34CDC2; MGC111195; DKFZp686L20222
The CDK1/CDC2-cyclin-B complex is a key regulator in the cell cycle. It plays an important role in cell division and development and regulates a variety of cellular activities, including chromosome segregation, nuclear membrane rupture, cytokinesis, etc. The activity of CDK1 is regulated by a variety of phosphorylation and dephosphorylation, thereby controlling the progression of the cell cycle and the DNA repair process. CDK1-CCNE1 Heterodimer Protein, Human (sf9, GST, His, Flag) is the recombinant human-derived CDK1-CCNE1, expressed by Sf9 insect cells, with N-GST, N-Flag, N-His labeled tag.
Cinobufagine-d3 is the deuterium labeled Cinobufagin (HY-N0421). Cinobufagin is an anticancer agent that can be secreted by the Asiatic toad Bufo gargarizans. Cinobufagin induces the cellcycle arrests in the G1 phase or G2/M phase, leading to apoptosis in cancer cells. Cinobufagin inhibits tumor growth in melanoma and glioblastoma multiforme xenograft mouse models .
N6-Benzyladenosine-d5 (Benzyladenosine-d5) is deuterium labeled N6-Benzyladenosine. N6-Benzyladenosine is an adenosine receptor agonist, has a cytoactive activity. N6-Benzyladenosine arrests cellcycle at G0/G1 phase and induces cellapoptosis. N6-Benzyladenosine also exerts inhibitory effect on T. gondii adenosine kinase and glioma .
TAT-p16 (p16INK4a peptide) is a peptide mimic of p16INK4a that can induce an early G phase cellcycle arrest in the absence of active cyclin E:Cdk2 complex .
Decursin-d6 ((+)-Decursin-d6) is the deuterium labeled Decursin (HY-18981). Decursin ((+)-Decursin) is a potent anti-tumor agent. Decursin also is a cytotoxic agent and a potent protein kinase C activator. Decursin induces apoptosis and cellcycle arrest at G1 phase. Decursin decreases the expression of CDK2, CDK4, CDK6, cyclin D1 protein at 48 h. Decursin inhibits cell proliferation and migration. Decursin shows anti-tumor, anti-inflammatory and analgesic activities .
Danazol inhibits the proliferation of cancer cell MDA-MB-231 and MCF-7 with IC50 of 65 µg/mL and 31 µg/mL. Danazol arrests the cellcycle at G1 phase, induces apoptosis in MDA-MB-231 through PKCα signaling pathway .
HEMTAC CDK4/6 degrader 1 is a PROTAC connected by ligands for HSP90 and CDK4/6 with a Kd value of 35.7 μM. HEMTAC CDK4/6 degrader 1 induces CDK4/6 degradation in B16F10 melanoma cells. HEMTAC CDK4/6 degrader 1 arrests cellcycle at G0/G1 phase and induces apoptosis. HEMTAC CDK4/6 degrader 1 can be used in research of cancer . HEMTAC CDK4/6 degrader 1 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
EGFR/HER2-IN-16 (compound 12K) is an effective dual-target inhibitor of EGFR (IC50=6.15 nM) and HER-2 (IC50=9.78 nM) with anti-tumor activity. EGFR/HER2-IN-16 can inhibit the migration of SK-BR-3 cells, arrest the cellcycle in the G0/G1 phase, and induce apoptosis. EGFR/HER2-IN-16 exhibits good anti-proliferative activity against tumor cell models and has little damage to healthy cells. EGFR/HER2-IN-16 can be used in breast cancer research .
RNK08954 is an orally active KRASG12D inhibitor with a Kd of 0.0395 nM. RNK08954 selectively binds the inactive GDP-bound KRASG12D form, suppresses downstream KRAS-mediated signaling pathways p-ERK1/2 experssion. RNK08954 inhibits KRASG12D-mutant cell proliferation, induces G0-G1cellcycle arrest, and inhibits tumor growth in mouse xenograft models. RNK08954 can be used for the research of non-small cell lung cancer, pancreatic ductal adenocarcinoma .
Inquiry Online
Your information is safe with us. * Required Fields.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
MedchemExpress Validation 03
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
MedchemExpress Validation 04
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedChemExpress values your privacy and your trust is important to us. We use cookies to enhance your website experience. Some cookies are necessary to run the website.
Privacy and Cookie Policy









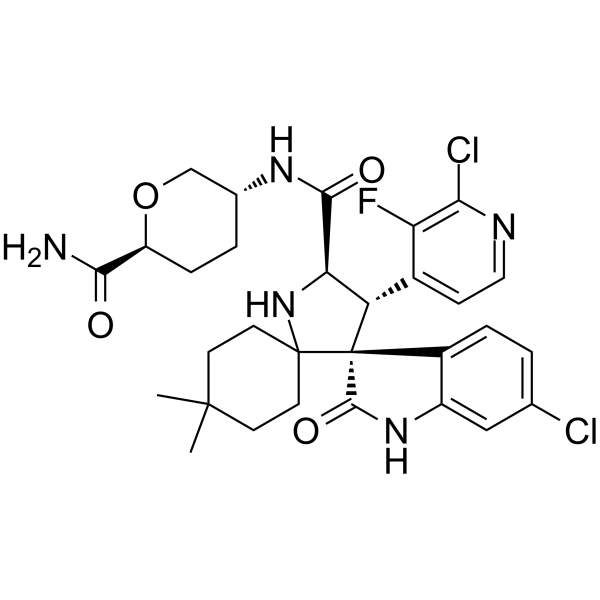
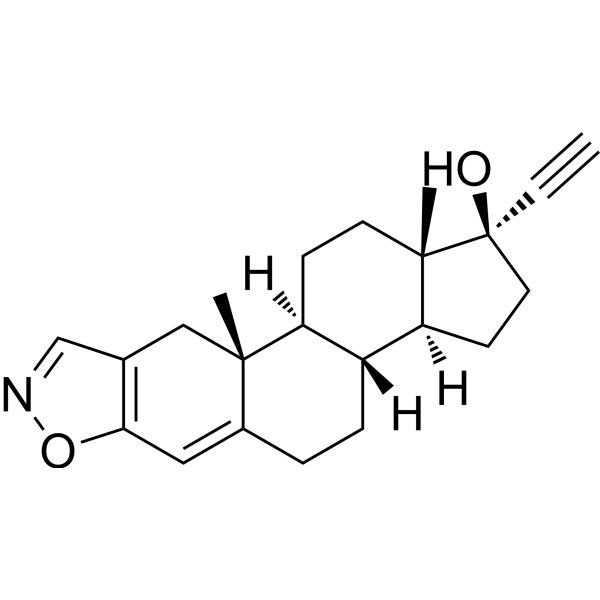
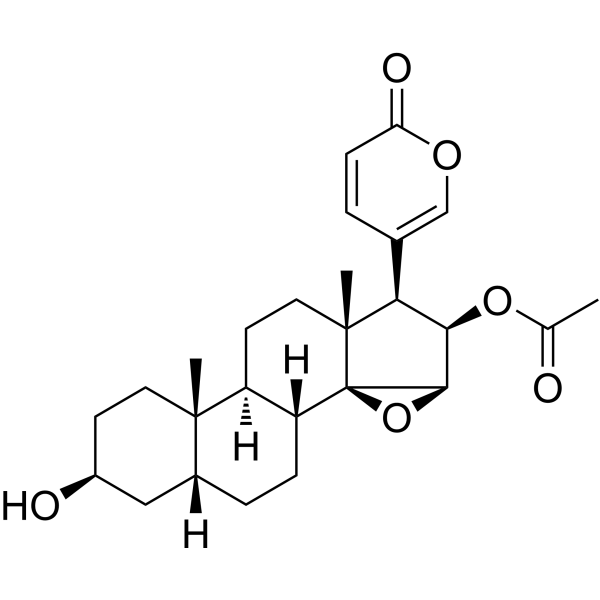
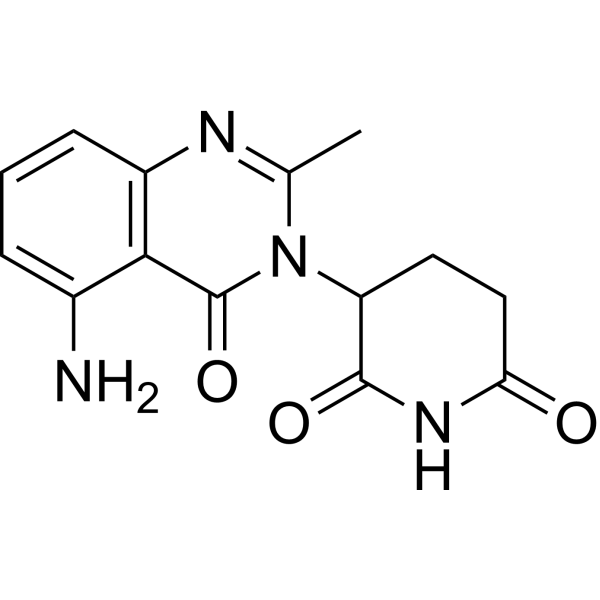

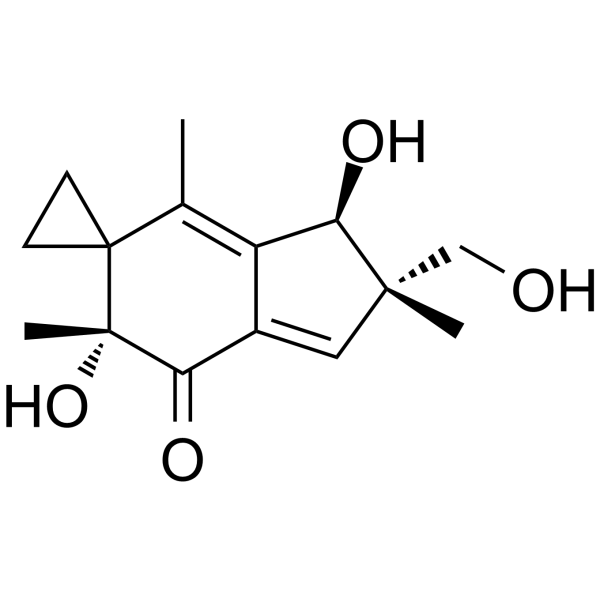
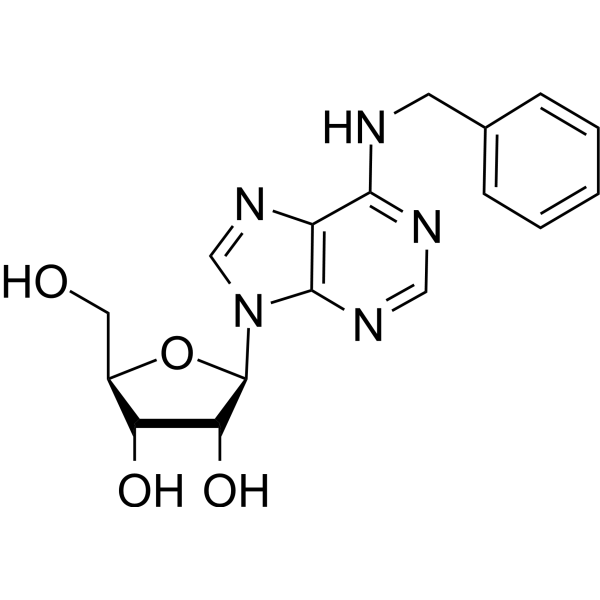









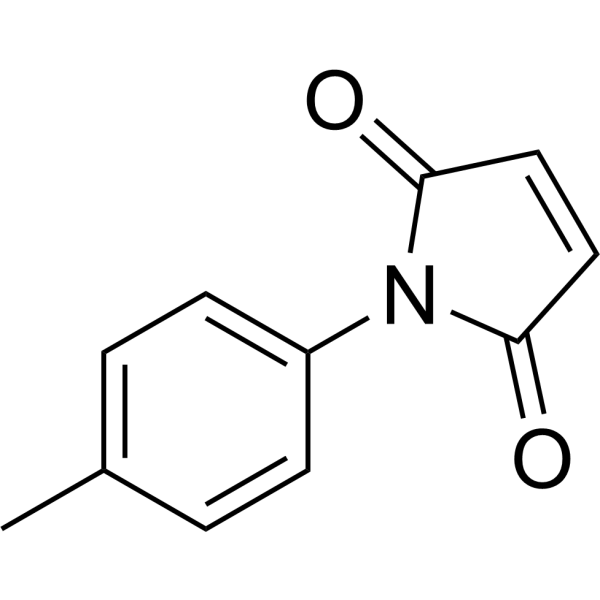



























![[1,1′:4′,1′′-Terphenyl]-3,4′′,5-triol](http://file.medchemexpress.com/product_pic/hy-120654.gif)























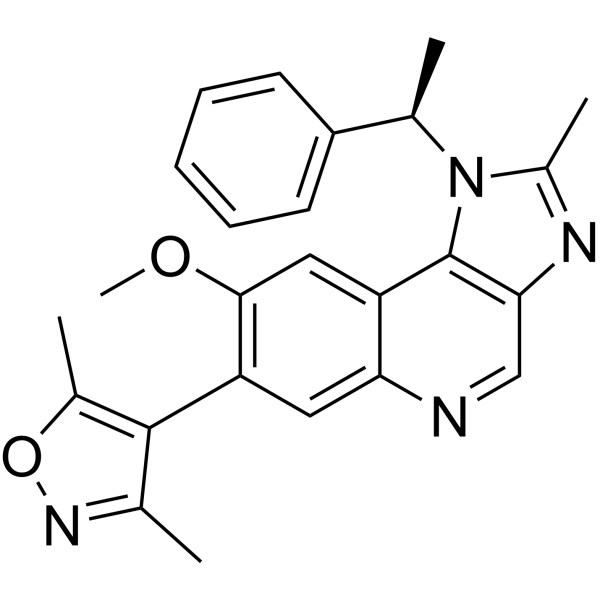


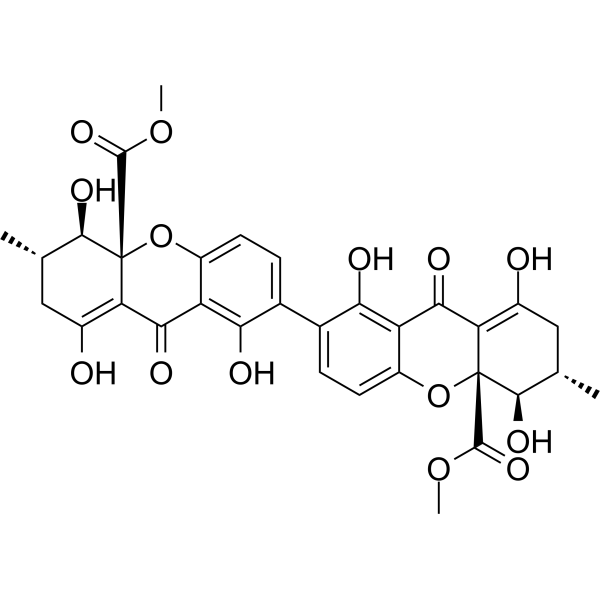






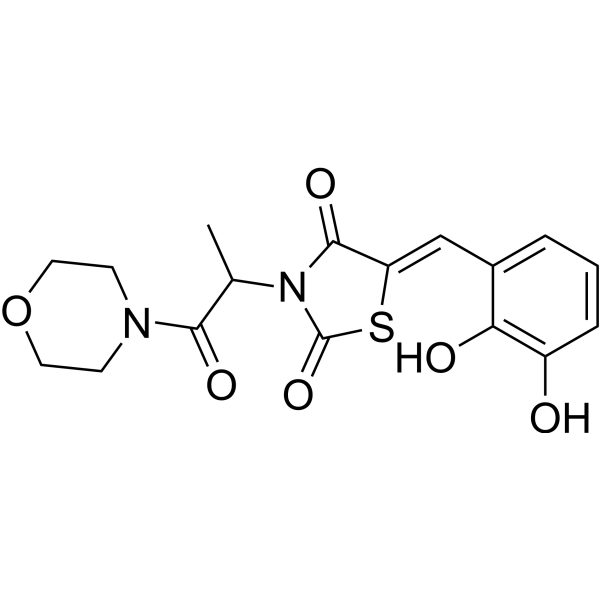

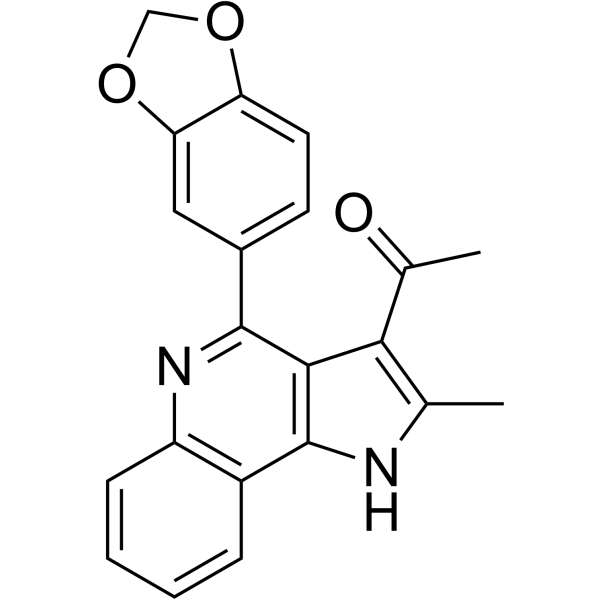
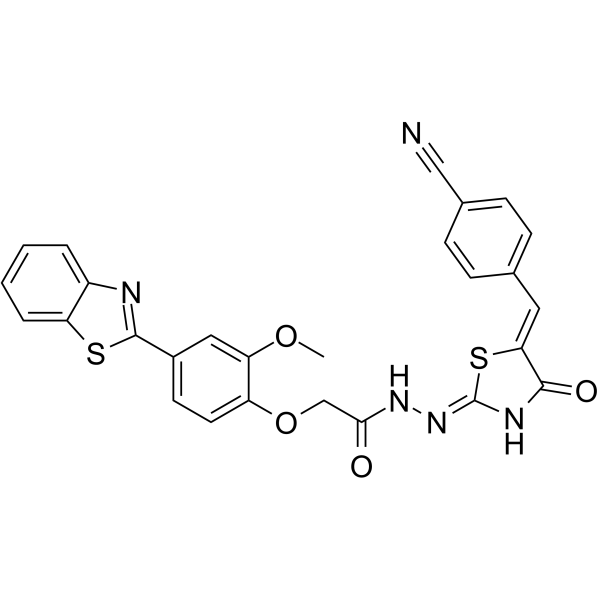

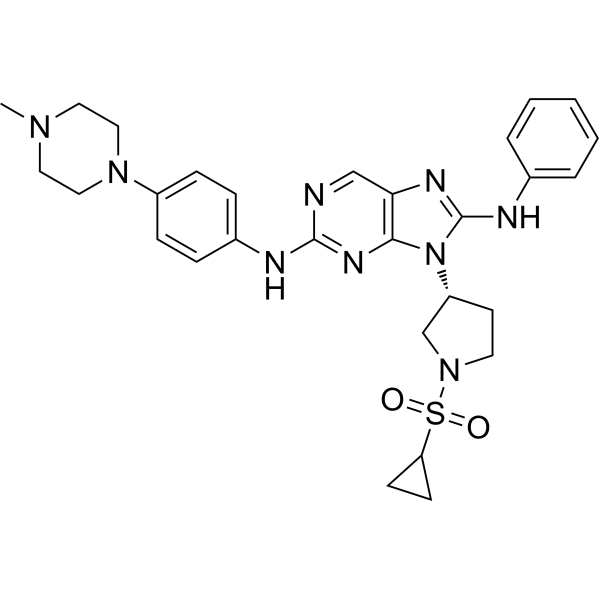

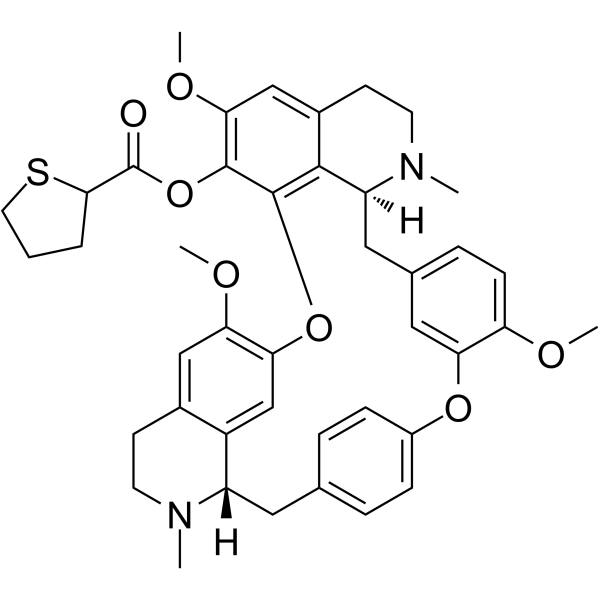


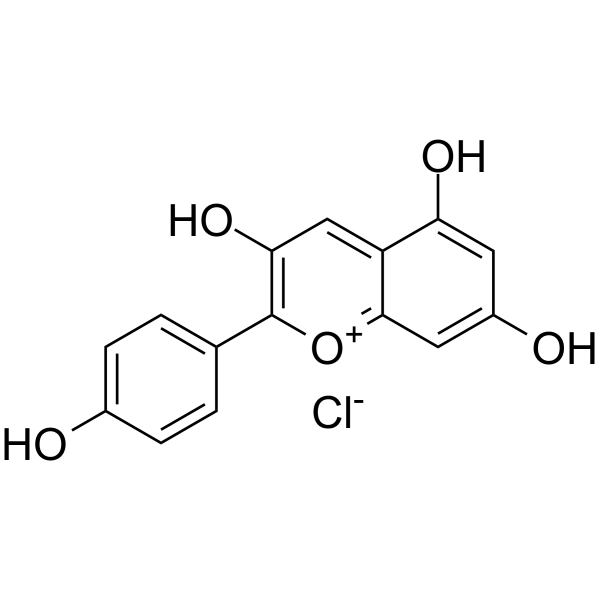

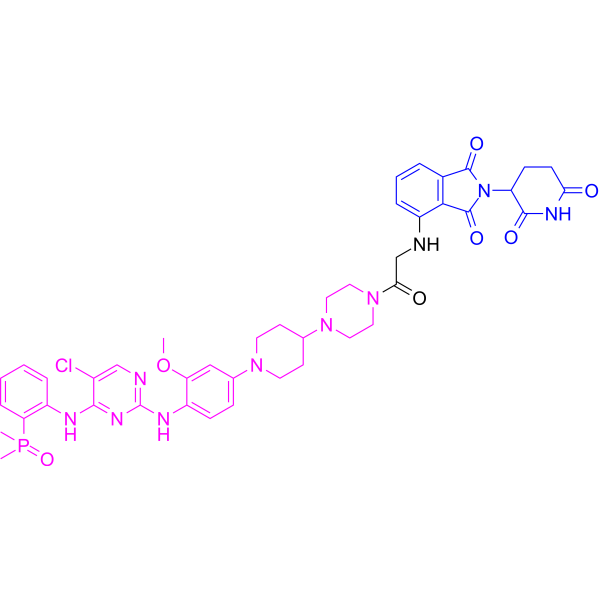
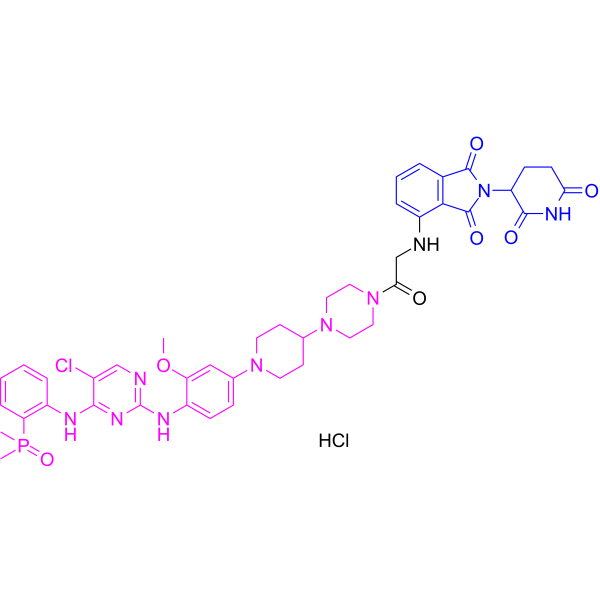
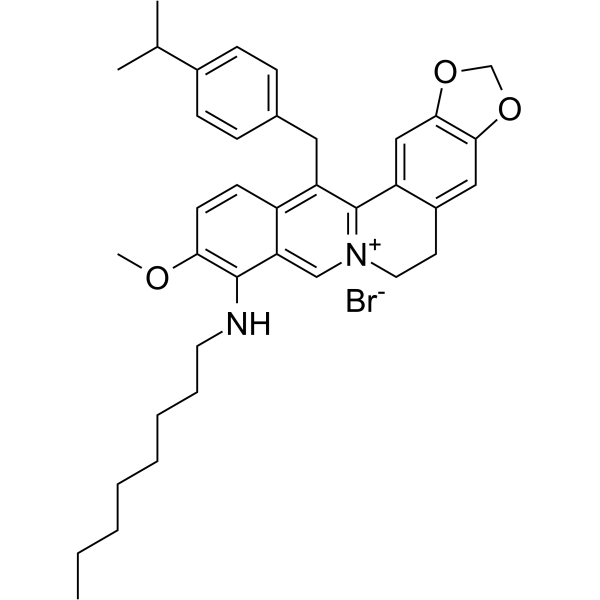
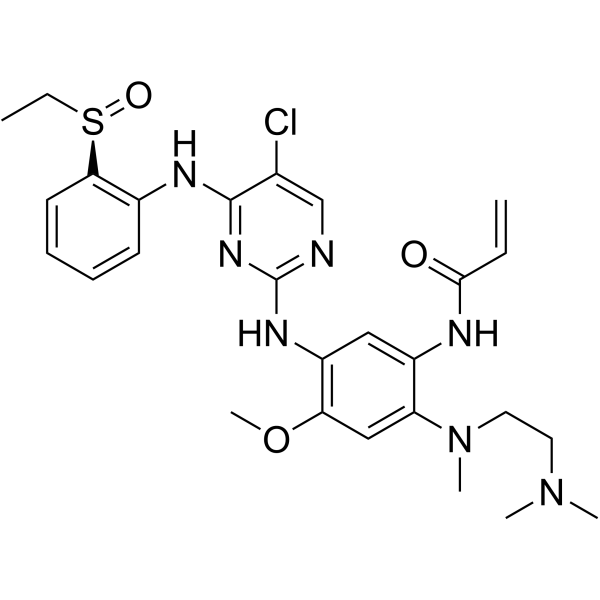
































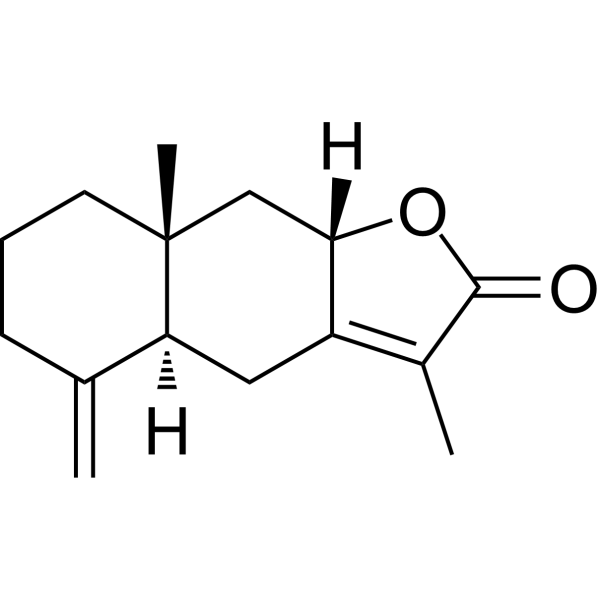
















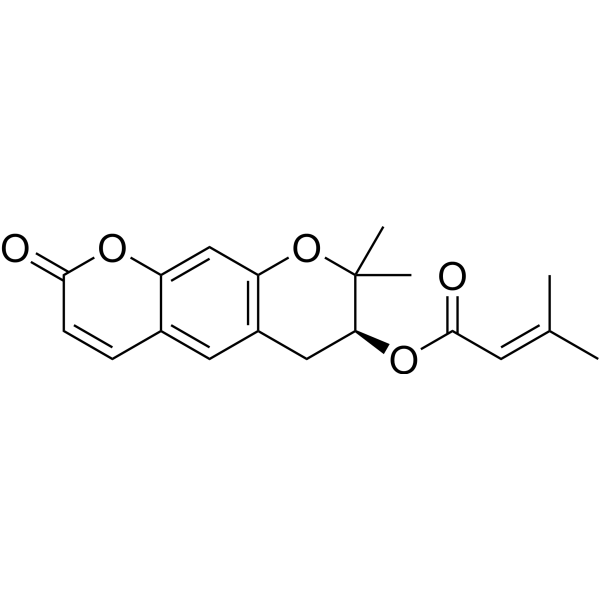












































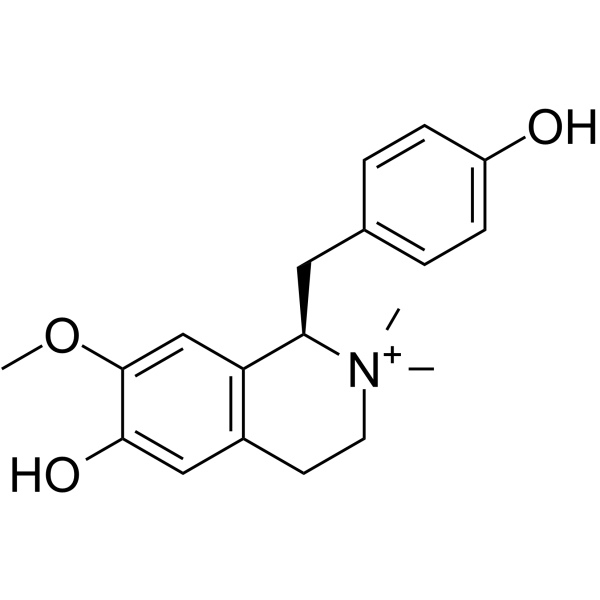







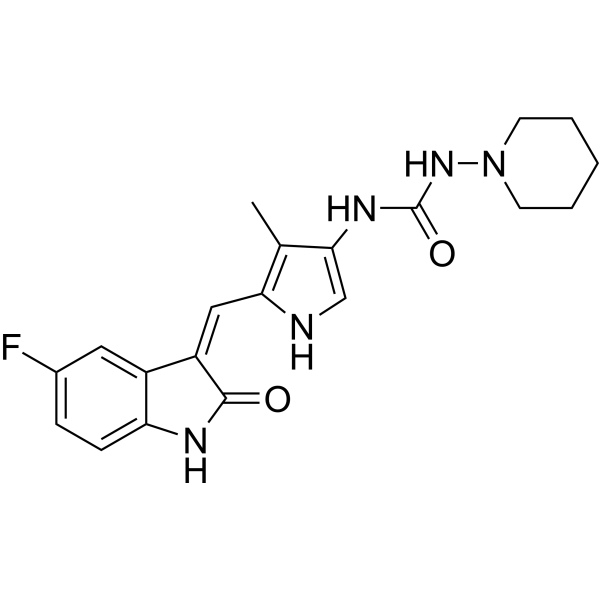
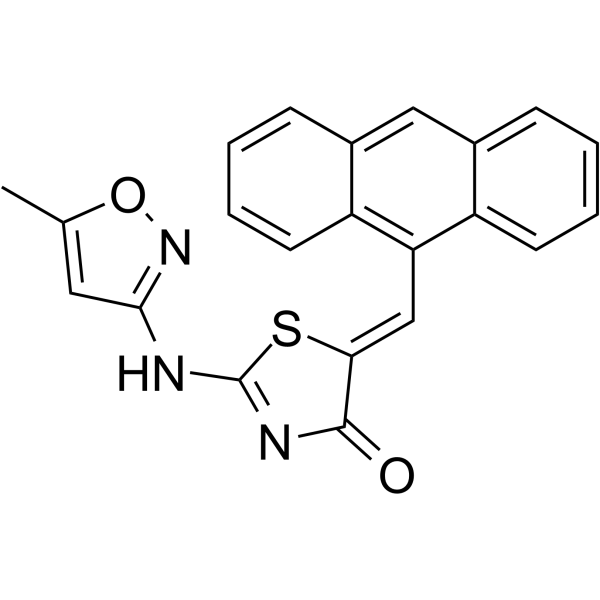
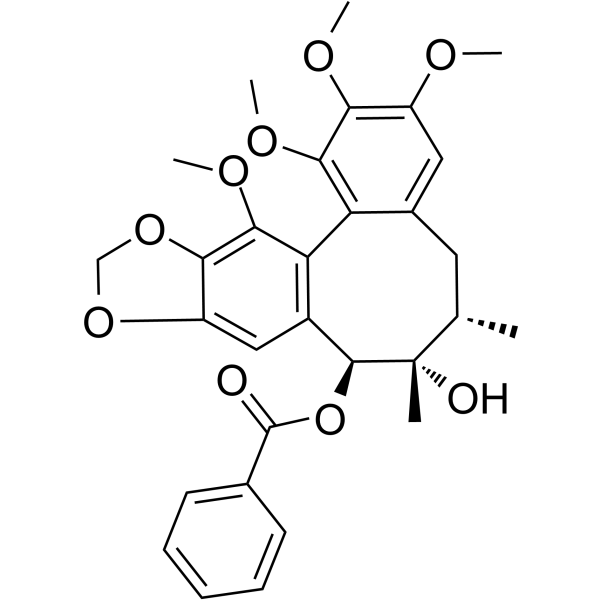










































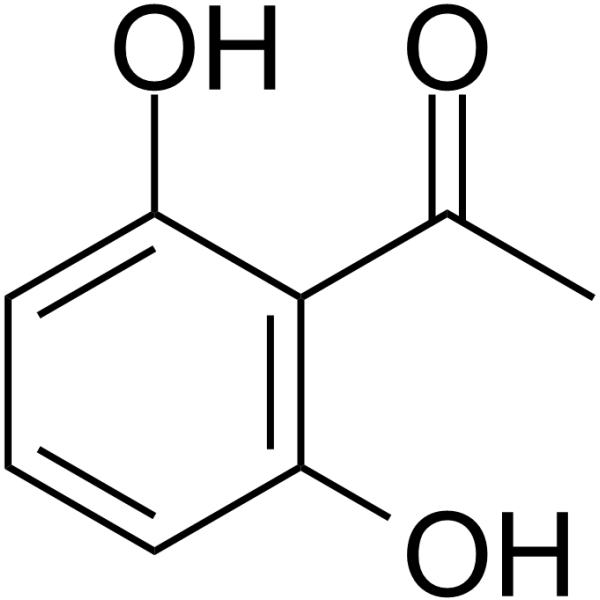


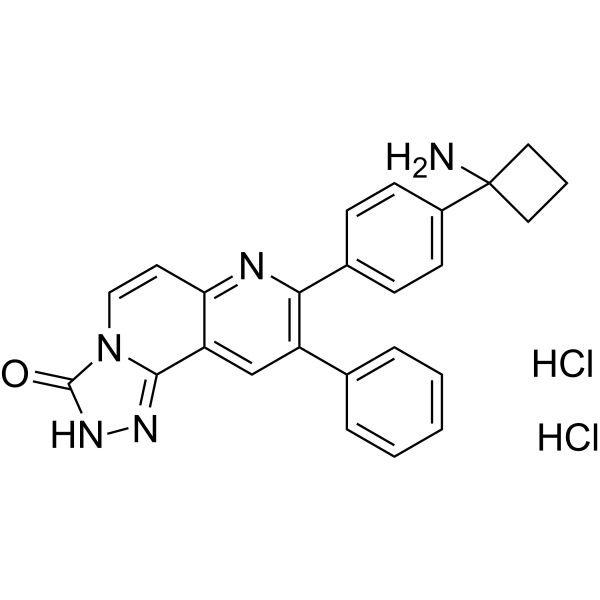





![[Ala92]-p16 (84-103)](http://file.medchemexpress.com/product_pic/hy-p1109.gif)

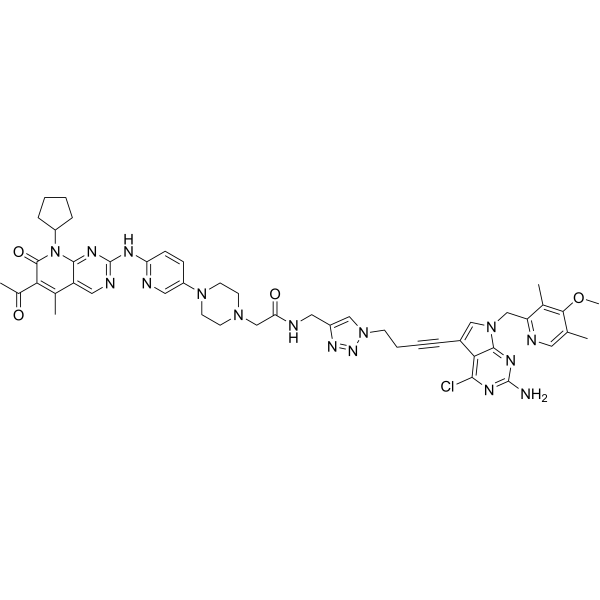






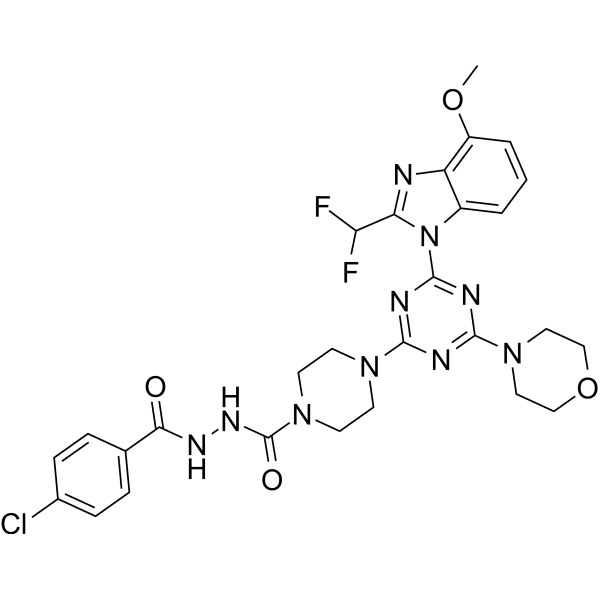




























2](http://file.medchemexpress.com/product_pic/hy-158117.gif)