Search Result
Results for "
selective competitive inhibitor
" in MedChemExpress (MCE) Product Catalog:
7
Biochemical Assay Reagents
26
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
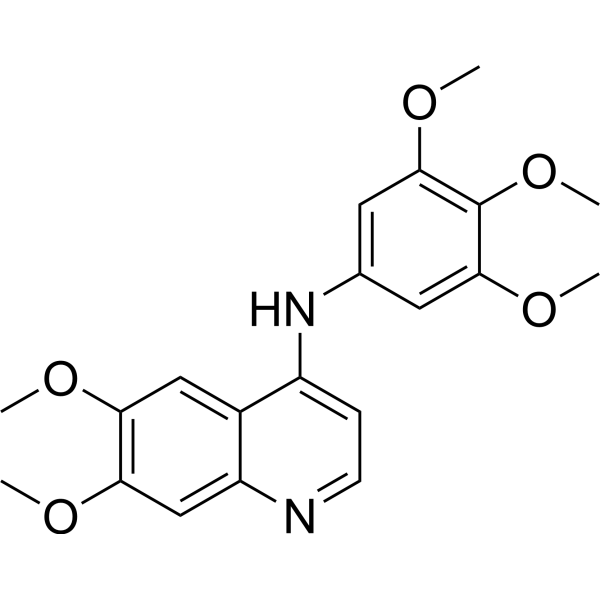
- HY-13823
-
-
-
- HY-12031A
-
|
|
MEK
Autophagy
Mitophagy
Influenza Virus
|
Cancer
|
|
U0126 is a potent, non-ATP competitive and selective MEK1 and MEK2 inhibitor, with IC50s of 72 nM and 58 nM, respectively. U0126 is an autophagy and mitophagy inhibitor .
|
-
-
- HY-10422
-
|
|
mTOR
Autophagy
Apoptosis
|
Cancer
|
|
AZD-8055 is a potent, selective, and orally bioavailable ATP-competitive mTOR kinase inhibitor with an IC50 of 0.8 nM. AZD-8055 inhibits both mTORC1 and mTORC2 .
|
-
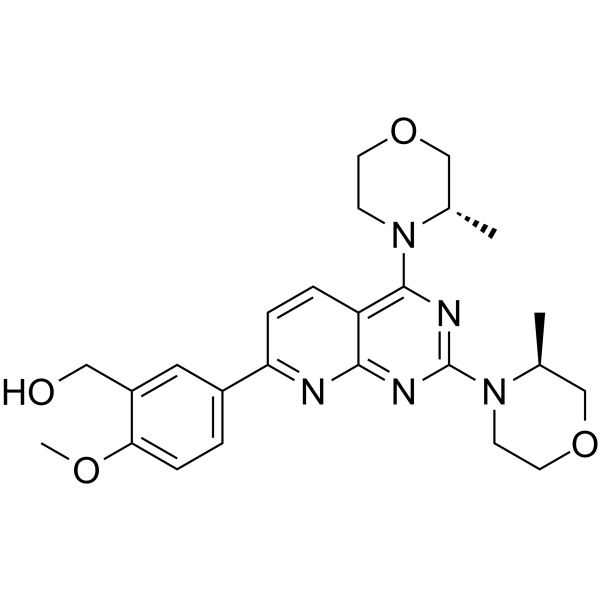
-
- HY-16985
-
|
ODM-201; BAY-1841788
|
Androgen Receptor
|
Cancer
|
|
Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist. Darolutamide has a Ki of 11 nM for rat wild-type AR (wtAR) and IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-
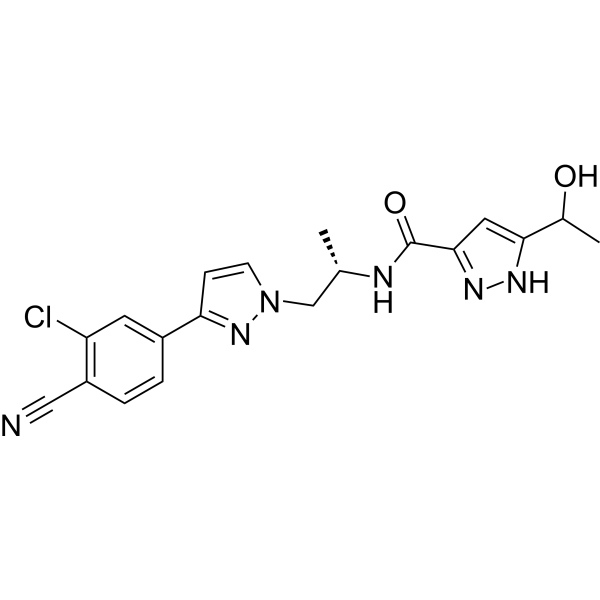
-
- HY-B0418A
-
|
R-18553 hydrochloride
|
Opioid Receptor
Autophagy
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Loperamide (hydrochloride) (R-18553 (hydrochloride)) is an opioid receptor agonist . Loperamide hydrochloride is a selective and competitive human intestinal carboxylesterases (hiCE) inhibitor. Loperamide hydrochloride has anti-diarrheal effect .
|
-
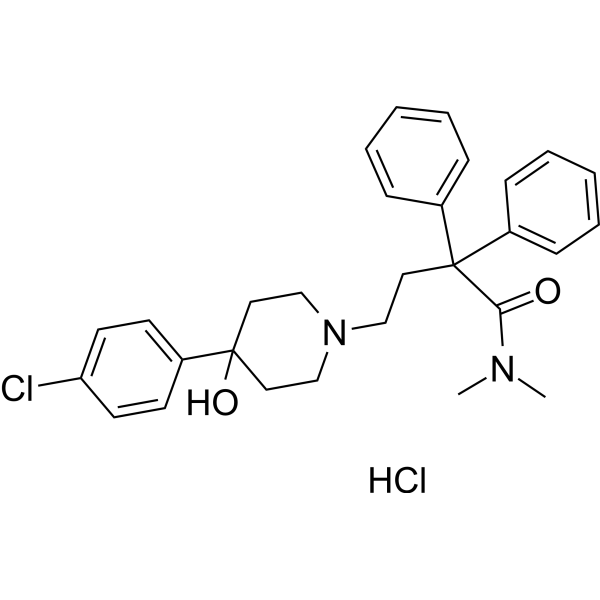
-
- HY-13820
-
|
|
PERK
Autophagy
Apoptosis
|
Cancer
|
|
GSK2656157 is a selective and ATP-competitive inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) with an IC50 of 0.9 nM.
|
-
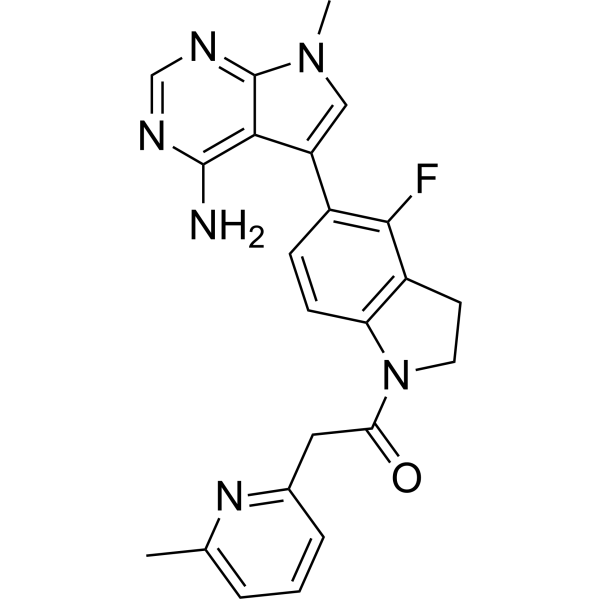
-
- HY-17353
-
|
|
Telomerase
Apoptosis
|
Cancer
|
|
BIBR 1532 is a potent, selective and non-competitive telomerase inhibitor with IC50 of 100 nM in a cell-free assay.
|
-
-
- HY-15646
-
-
-
- HY-19715
-
-
-
- HY-13462
-
|
HTS466284
|
TGF-β Receptor
|
Cancer
|
|
LY-364947 (HTS466284) is a potent ATP-competitive inhibitor of TGFβR-I with IC50 of 59 nM, and exhibits 7-fold selectivity over TGFβR-II .
|
-
-
- HY-101053
-
|
Src Kinase inhibitor 1; Src-l1
|
Src
|
Cancer
|
|
Src Inhibitor 1 is a potent, ATP-competitive and selective dual site Src tyrosine kinase inhibitor with IC50 values of 44 nM for Src and 88nM for Lck.
|
-
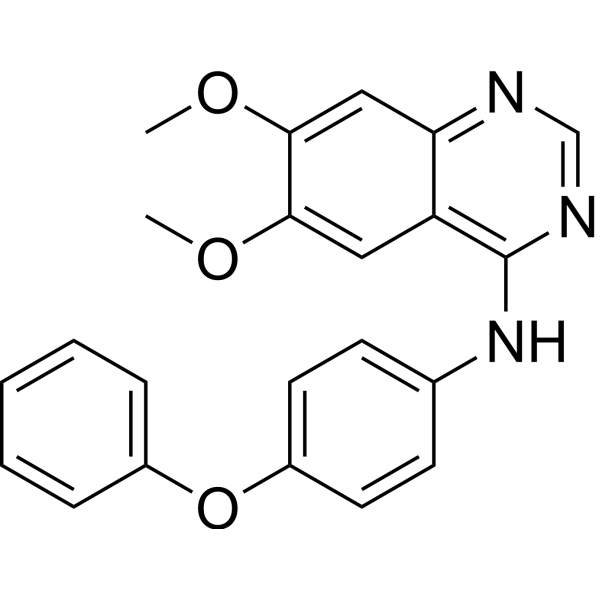
-
- HY-50877
-
|
GSK461364A
|
Polo-like Kinase (PLK)
|
Cancer
|
|
GSK461364 is a selective, reversible and ATP-competitive Polo-like kinase 1 (PLK1) inhibitor with a Ki value of 2.2 nM.
|
-
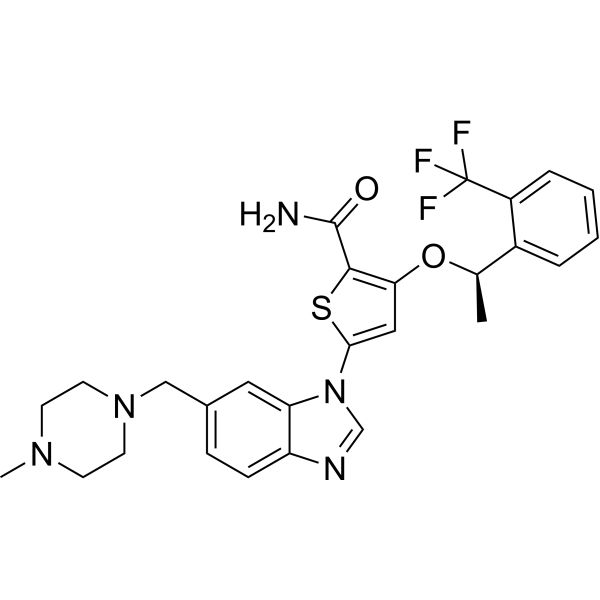
-
- HY-15663
-
|
|
PAK
|
Cancer
|
|
IPA-3 is a selective non-ATP competitive PAK1 inhibitor with IC50 of 2.5 μM, and shows no inhibition to group II PAKs (PAKs 4-6).
|
-
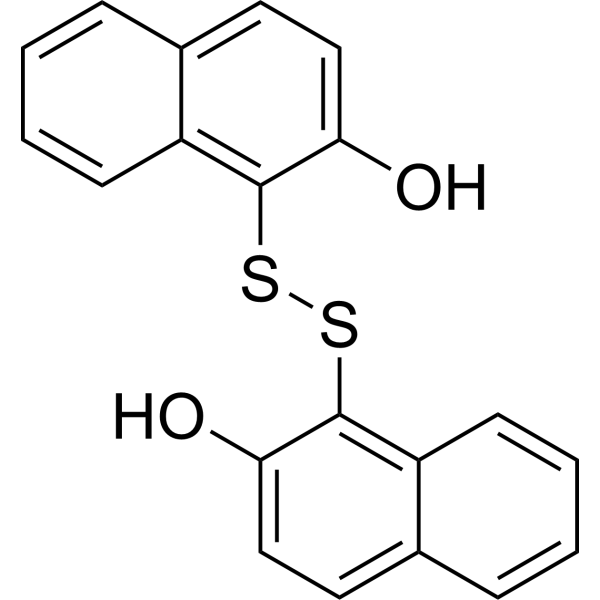
-
- HY-10474
-
|
PP 242
|
mTOR
Autophagy
Mitophagy
Apoptosis
|
Cancer
|
|
Torkinib (PP 242) is a selective and ATP-competitive mTOR inhibitor with an IC50 of 8 nM . PP242 inhibits both mTORC1 and mTORC2 with IC50s of 30 nM and 58 nM, respectively .
|
-
-
- HY-11092
-
-
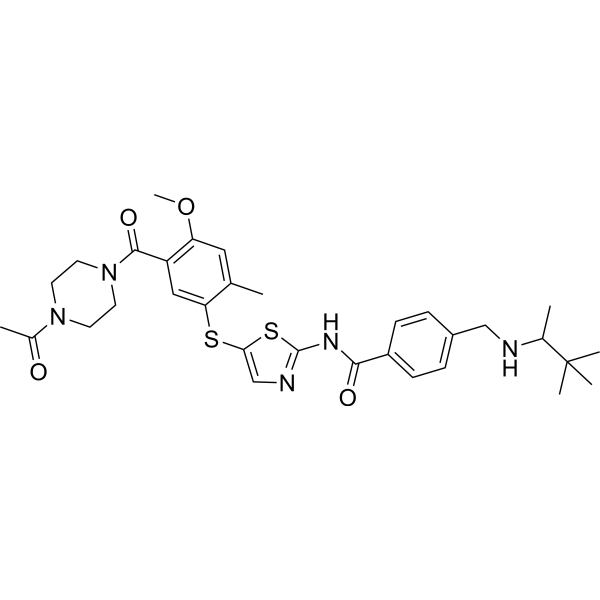
-
- HY-70044
-
|
GSK-1070916A
|
Aurora Kinase
Apoptosis
|
Cancer
|
|
GSK-1070916 is a potent and selective ATP-competitive inhibitor of aurora B and aurora C with Kis of 0.38 and 1.5 nM, respectively, and is >250- fold selective over Aurora A.
|
-
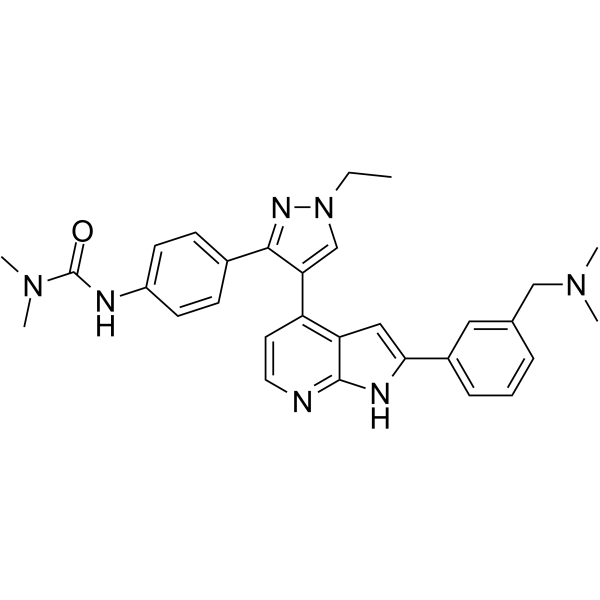
-
- HY-10512
-
|
AR 0133418; GSK 3β inhibitor VIII; AR 014418
|
GSK-3
|
Metabolic Disease
Cancer
|
|
AR-A014418 is a potent, selective and ATP-competitive GSK3β inhibitor (IC50=104 nM; Ki=38 nM) .
|
-
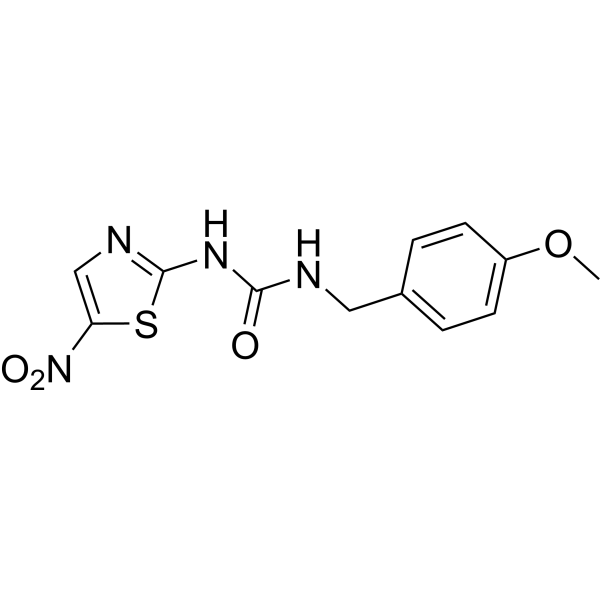
-
- HY-15339
-
|
Cdk2 inhibitor III
|
CDK
|
Cancer
|
|
CVT-313 (Cdk2 Inhibitor III) is a potent, selective, reversible, and ATP-competitive inhibitor of CDK2 with IC50 of 0.5 μM. CVT-313 inhibits CDC5L phosphorylation .
|
-
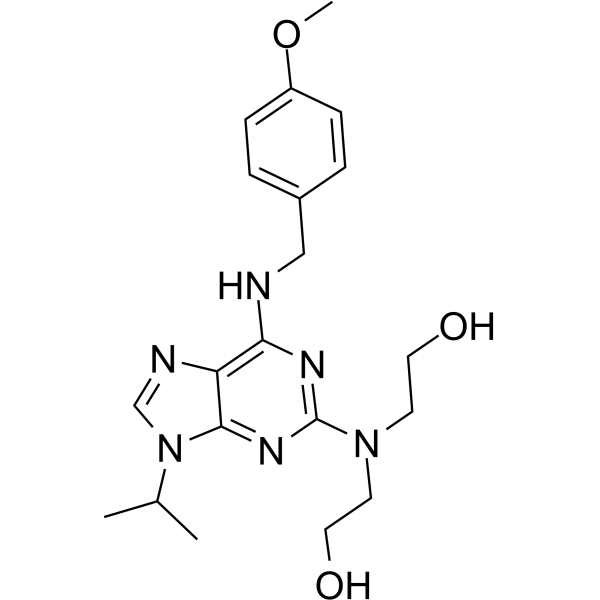
-
- HY-P1178
-
|
|
Trk Receptor
p38 MAPK
|
Neurological Disease
|
|
Cyclotraxin B is a BBB-penetrable and selective TrkB inhibitor. Cyclotraxin B inhibits BDNF-induced TrkB activity in a non-competitive manner, with an IC50 of 0.30 nM. Cyclotraxin B has analgesic and anxiolytic effects .
|
-
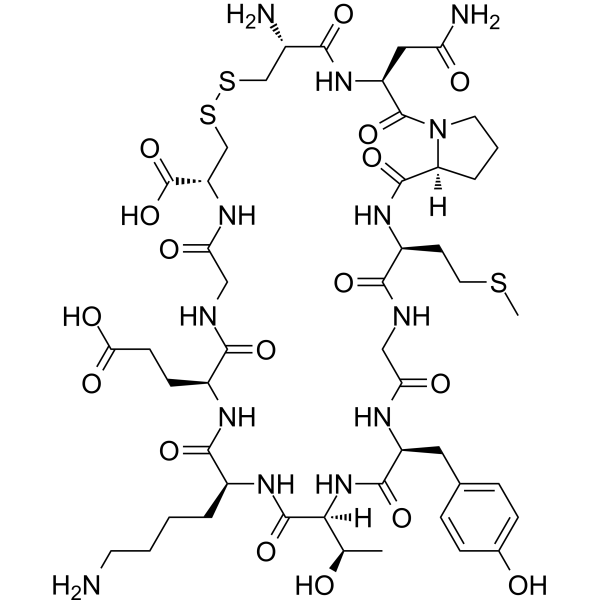
-
- HY-16015
-
|
ABC294640
|
SphK
|
Cancer
|
|
Opaganib (ABC294640) is a selective, competitive sphingosine kinase 2 (SK2) inhibitor with Ki of 9.8 μM.
|
-
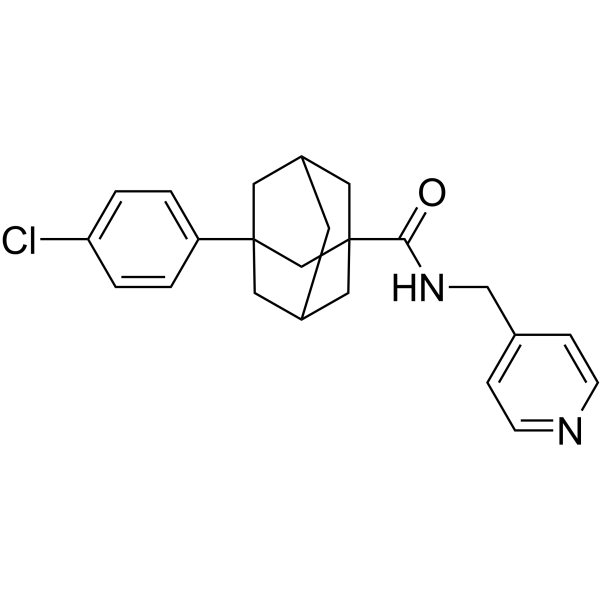
-
- HY-B0140
-
-
-
- HY-12019
-
|
|
c-Met/HGFR
|
Cancer
|
|
SGX523 is a exquisitely selective and ATP-competitive MET inhibitor. SGX523 potently inhibits MET with an IC50 of 4 nM and is >1,000-fold selective versus other protein kinases. Antitumor activity .
|
-
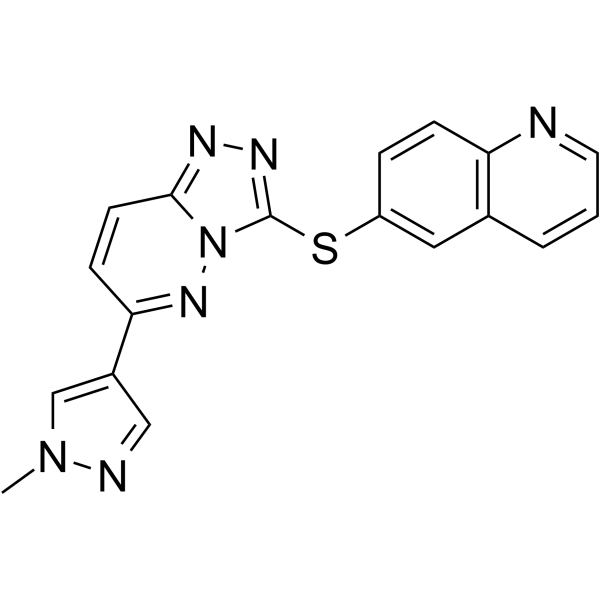
-
- HY-12042
-
|
AS703026; MSC1936369B
|
MEK
|
Cancer
|
|
Pimasertib (AS703026) is a highly selective, ATP non-competitive allosteric orally available MEK1/2 inhibitor .
|
-
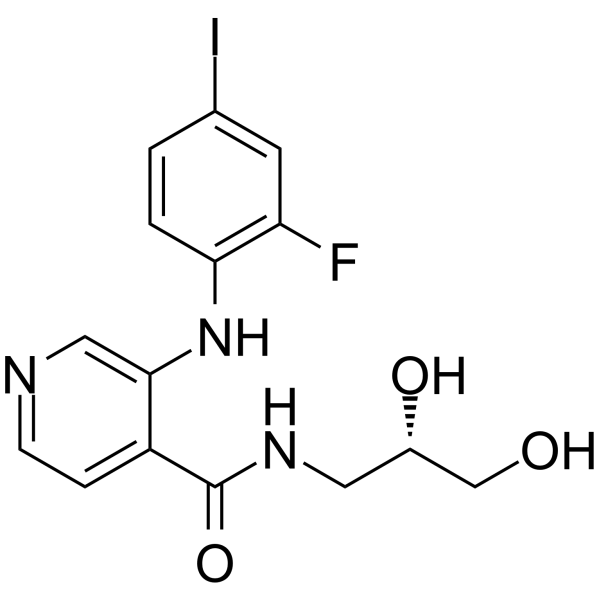
-
- HY-15513
-
|
|
DAPK
|
Cancer
|
|
TC-DAPK 6 is a potent, ATP-competitive, and highly selective DAPK inhibitor (IC50=69 and 225 nM against DAPK1 and DAPK3, respectively, with 10 μM ATP).
|
-
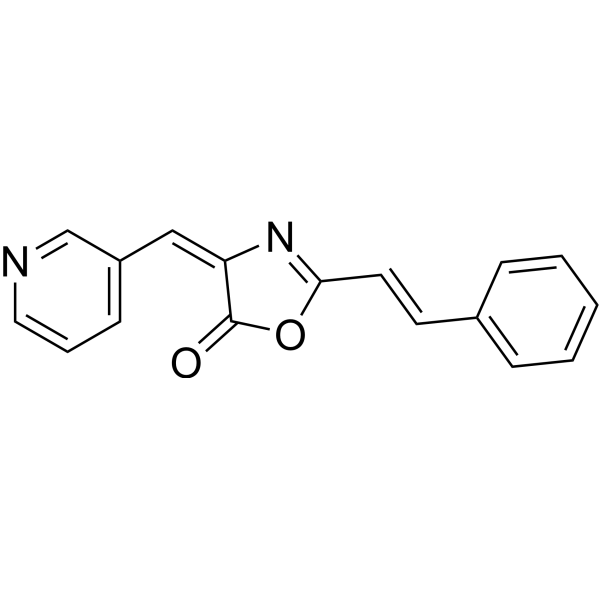
-
- HY-12382
-
|
|
Mps1
|
Cancer
|
|
NMS-P715 is a selective, ATP-competitive inhibitor of MPS1, with an IC50 of 182 nM.
|
-
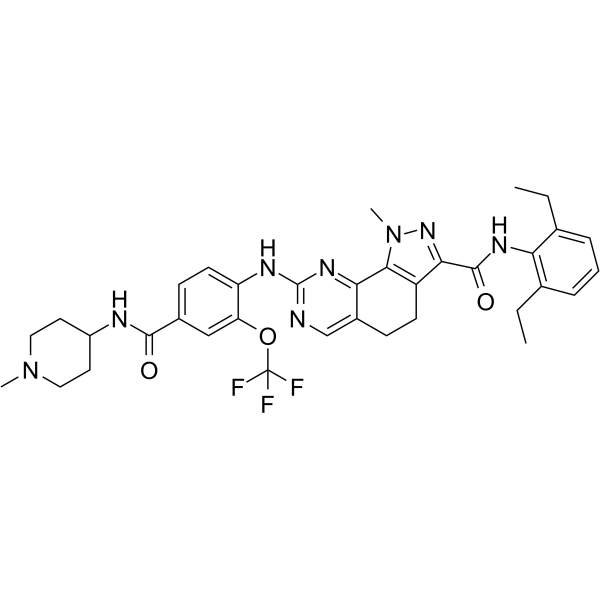
-
- HY-11003
-
|
GW843682
|
Polo-like Kinase (PLK)
|
Cancer
|
|
GW843682X is a selective, ATP-competitive inhibitor of PLK1 and PLK3, with IC50s of 2.2 nM and 9.1 nM, respectively, and is also >100-fold selective against ~30 other kinases.
|
-
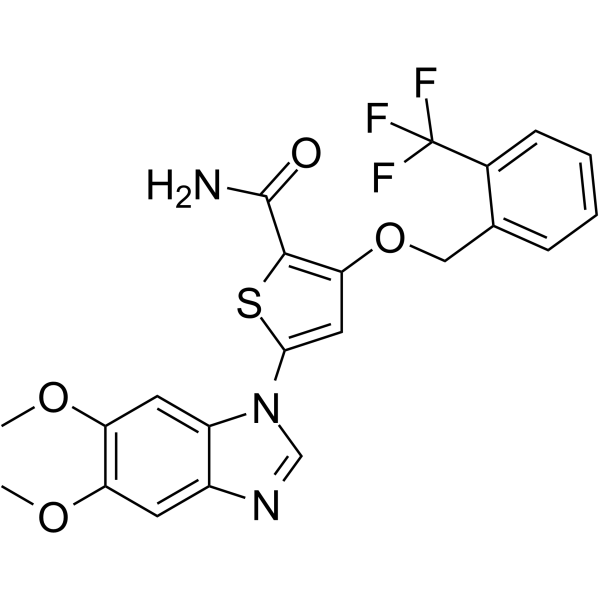
-
- HY-100844
-
|
|
MAP3K
Apoptosis
|
Cancer
|
|
GS-444217 is a potent, orally available and selective ATP-competitive inhibitor of apoptosis signal-regulating kinase 1 (ASK1) with an IC50 of 2.87 nM .
|
-
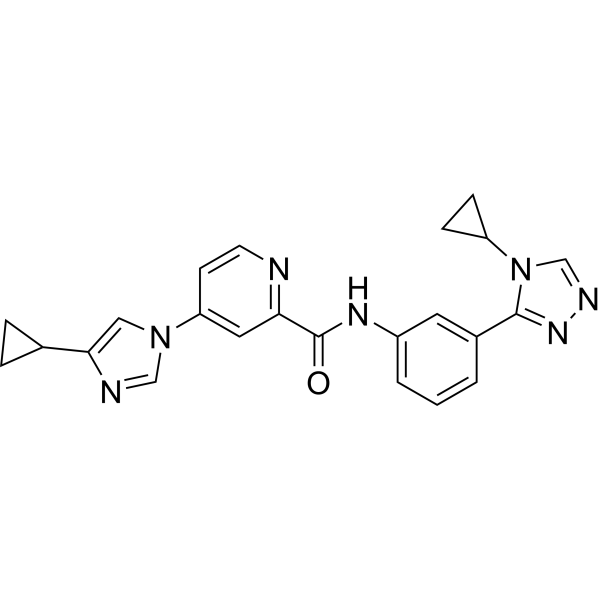
-
- HY-103351
-
|
|
Cathepsin
|
Inflammation/Immunology
|
|
Cathepsin G Inhibitor I (Compound 7) is a potent, selective, reversible, competitive, non-peptidic Cathepsin G inhibitor (IC50 = 53 nM; Ki = 63 nM). Cathepsin G Inhibitor I can be used in research related to immune disorders .
|
-
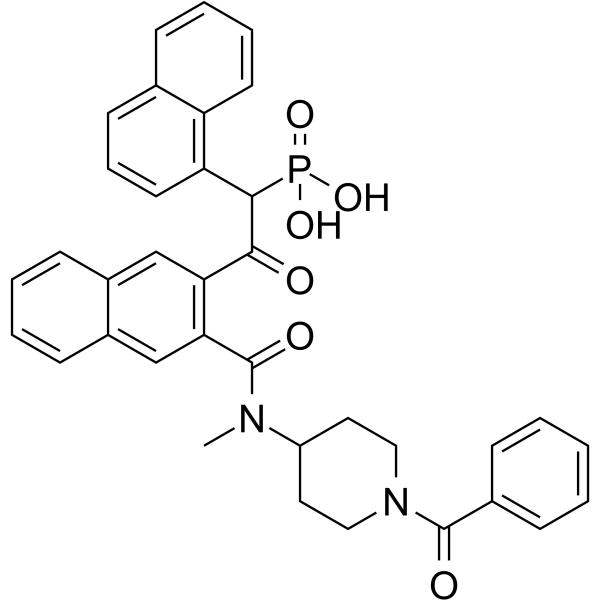
-
- HY-16755
-
|
GSK-2586184; GLPG-0778
|
JAK
|
Inflammation/Immunology
|
|
Solcitinib is an orally active, competitive, potent, selective JAK1 inhibitor, with an IC50 of 9.8 nM, and 11-, 55- and 23-fold selectivity over JAK2, JAK3 and TYK2, respectively; Solcitinib is used in the research of moderate-to-severe plaque-type psoriasis.
|
-
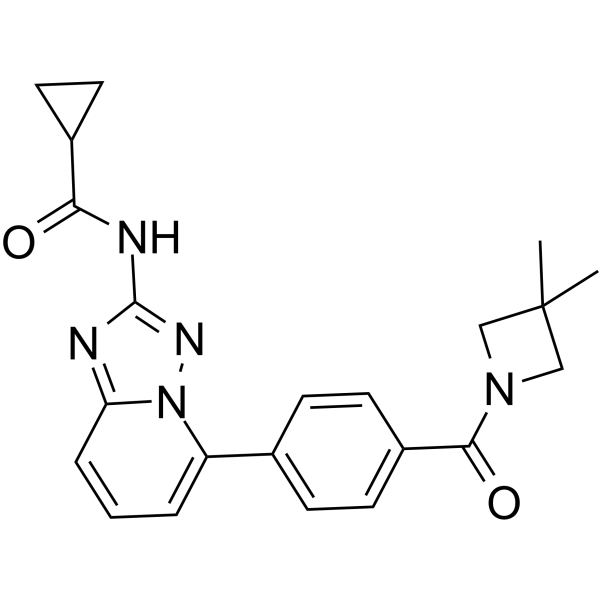
-
- HY-12660
-
|
|
Mps1
Apoptosis
|
Cancer
|
|
MPI-0479605 is a potent and selective ATP-competitive inhibitor of Mps1, with an IC50 of 1.8 nM.
|
-
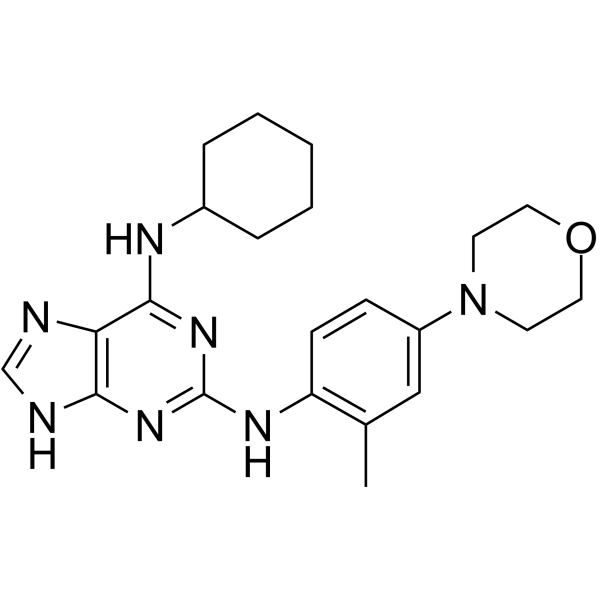
-
- HY-19712
-
-
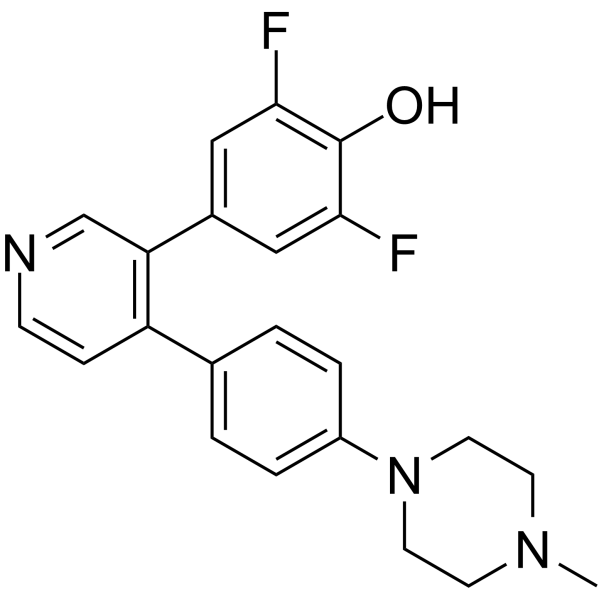
-
- HY-11007
-
GNF-2
3 Publications Verification
|
Bcr-Abl
SARS-CoV
|
Cancer
|
|
GNF-2 is a highly selective, allosteric, non-ATP competitive inhibitor of Bcr-Abl. GNF-2 inhibits Ba/F3.p210 proliferation with an IC50 of 138 nM .
|
-
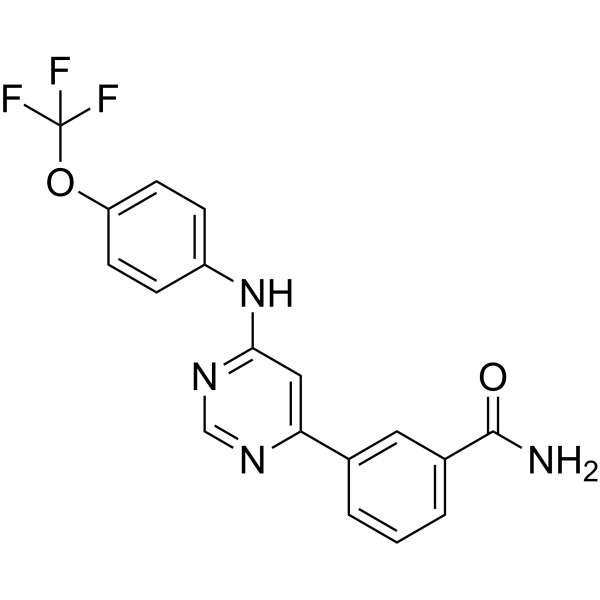
-
- HY-110399
-
-
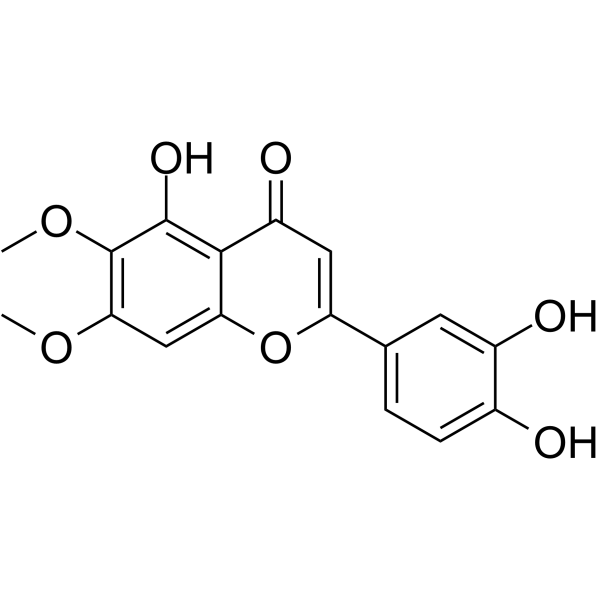
-
- HY-117769
-
|
|
Fatty Acid Synthase (FASN)
|
Metabolic Disease
|
|
GSK837149A is a selective inhibitor of human Fatty Acid Synthase (FASN) targeting the KR domain. GSK837149A has reversible inhibition effect on FASN and selectivity for type I FASN (Ki=30 nM). GSK837149A is also a competitive inhibitor of NADPH and a non-competitive inhibitor of acetoacetyl-CoA. GSK837149A can be used for the research of obesity and breast cancer .
|
-
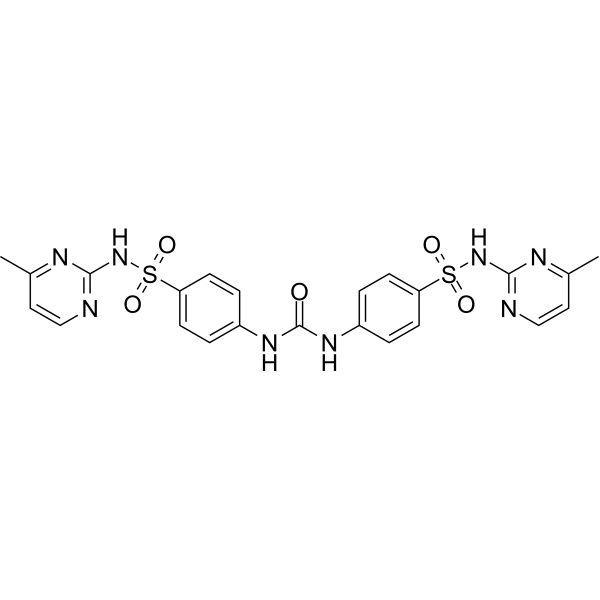
-
- HY-15248
-
|
|
mTOR
Autophagy
|
Cancer
|
|
GDC-0349 is a potent and selective ATP-competitive mTOR inhibitor with a Ki of 3.8 nM. GDC-0349 inhibits of both mTORC1 and mTORC2 complexes.
|
-
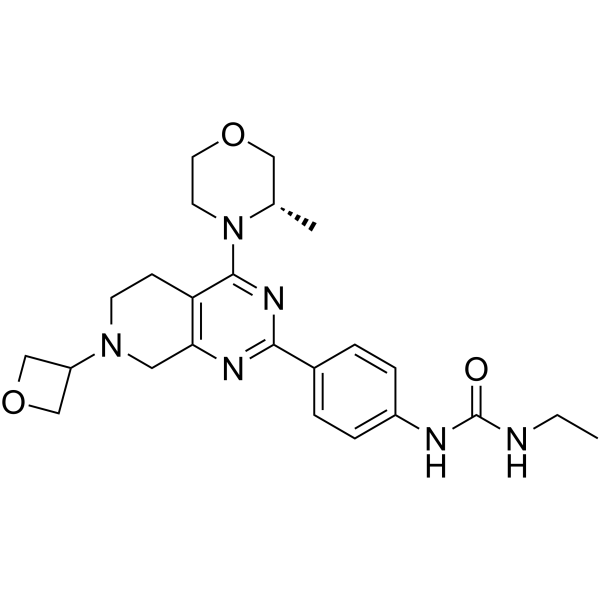
-
- HY-104050
-
M-31850
2 Publications Verification
|
Glycosidase
|
Metabolic Disease
|
|
M-31850 is a potent, selective and competitive β-hexosaminidase (Hex) inhibitor with IC50s of 6.0 μM and 3.1 μM for human HexA and human HexB, respectively. M-31850 also competitively inhibits β-N-acetyl-D-hexosaminidase OfHex2 with a Ki of 2.5 μM .
|
-
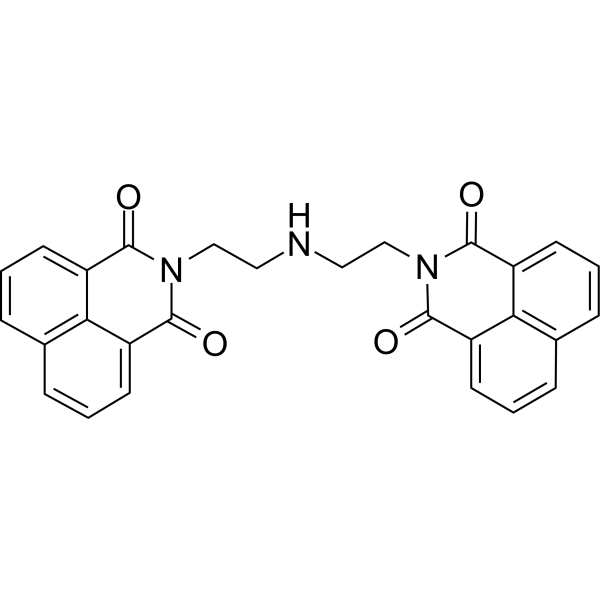
-
- HY-15186A
-
|
GDC-0068 dihydrochloride; RG-7440 dihydrochloride
|
Organoid
Akt
|
Cancer
|
|
Ipatasertib dihydrochloride (GDC-0068 dihydrochloride) is a highly selective and ATP-competitive pan-Akt inhibitor with IC50s of 5, 18 and 8 nM for Akt1, Akt2 and Akt3, respectively.
|
-
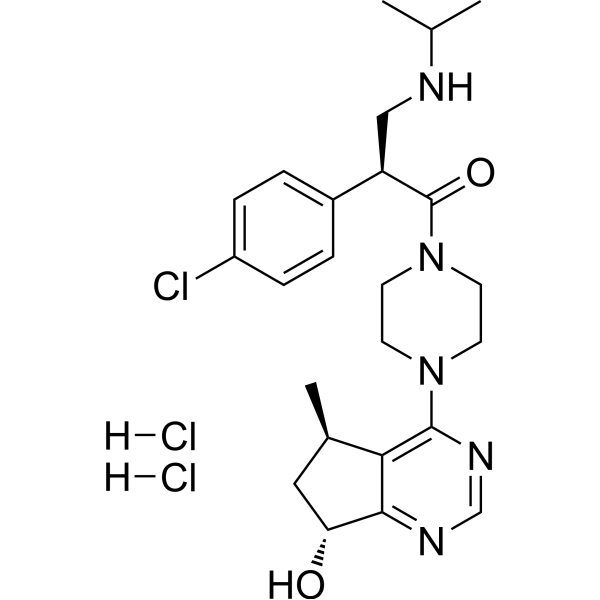
-
- HY-104009A
-
|
|
Histone Methyltransferase
|
Others
|
|
GSK2807 Trifluoroacetate is a potent, selective and SAM-competitive inhibitor of SMYD3, with a Ki of 14 nM and an IC50 of 130 nM .
|
-
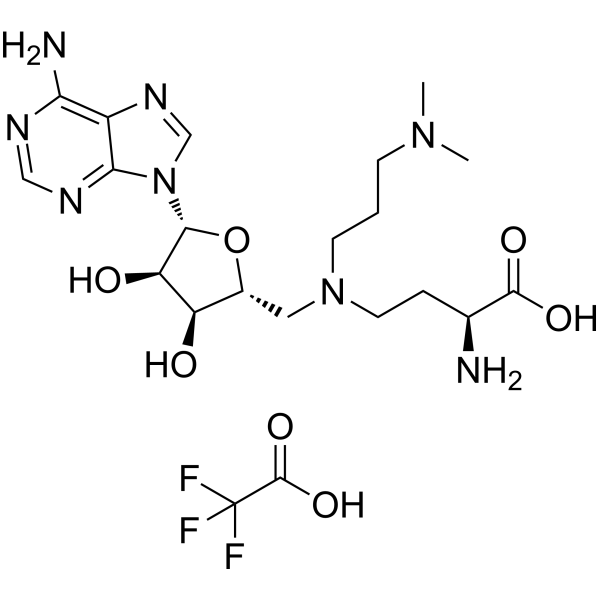
-
- HY-13298
-
|
|
Mps1
|
Cancer
|
|
Mps1-IN-1 is a potent, selective and ATP-competitive Mps1 kinase inhibitor, with an IC50 and a Kd of 367 nM and 27 nM .
|
-
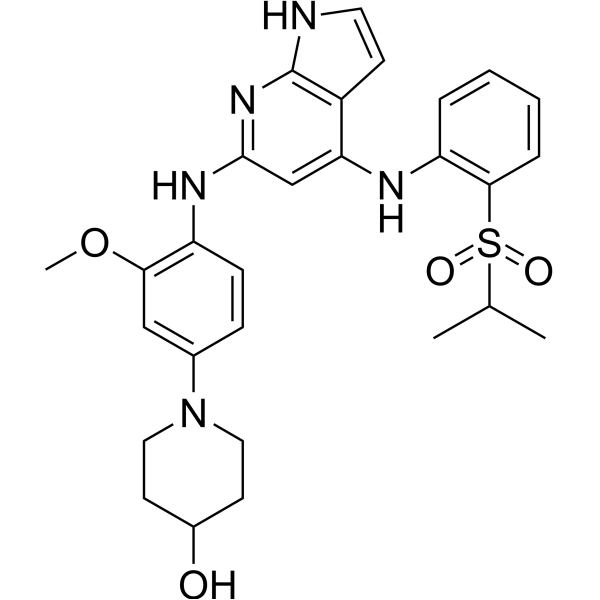
-
- HY-P2271
-
|
|
Apelin Receptor (APJ)
|
Cancer
|
|
MM 54 (compound 5) is a competitive antagonist at APJ, with an IC50 of 93 nM. MM 54 behaves as a potent and selective inhibitor of apelin binding and APLNR activation .
|
-
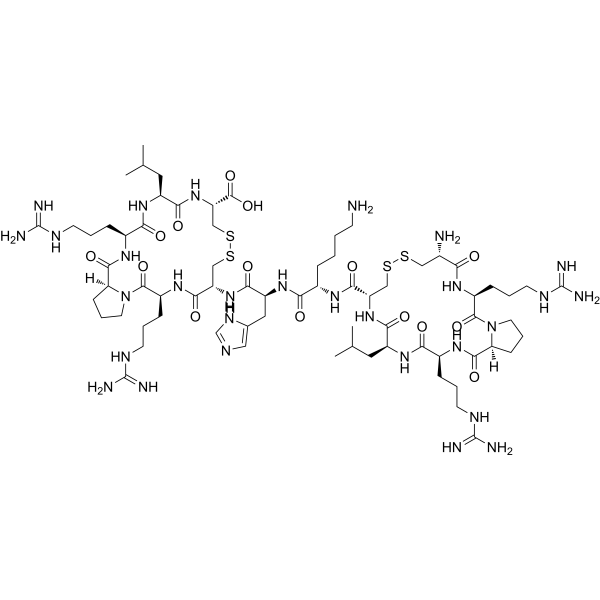
-
- HY-112080
-
|
|
Histone Methyltransferase
|
Cancer
|
|
BAY-6035, a chemical probe, is a potent, selective and substrate-competitive inhibitor of SMYD3. BAY-6035 inhibits methylation of MEKK2 peptide with an IC50 of 88 nM .
|
-
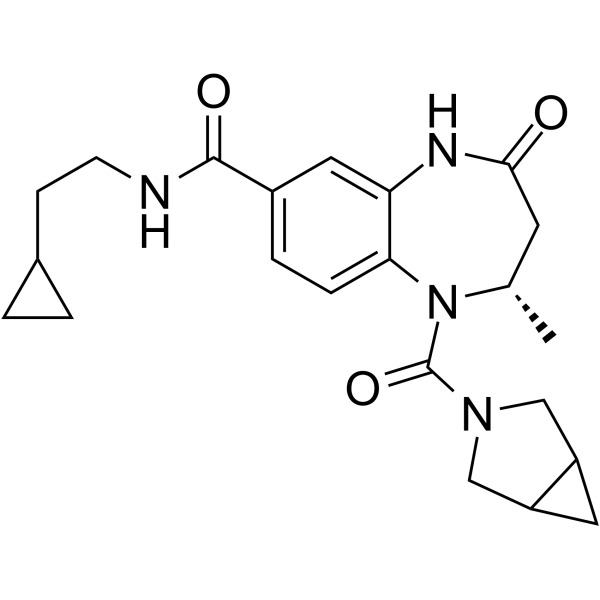
-
- HY-10721
-
|
AKT protein kinase inhibitor
|
Akt
|
Cancer
|
|
PF-AKT400 is a broadly selective, potent, ATP-competitive Akt inhibitor, displays 900-fold greater selectivity for PKBα (IC50=0.5 nM) than PKA (IC50=450 nM).
|
-
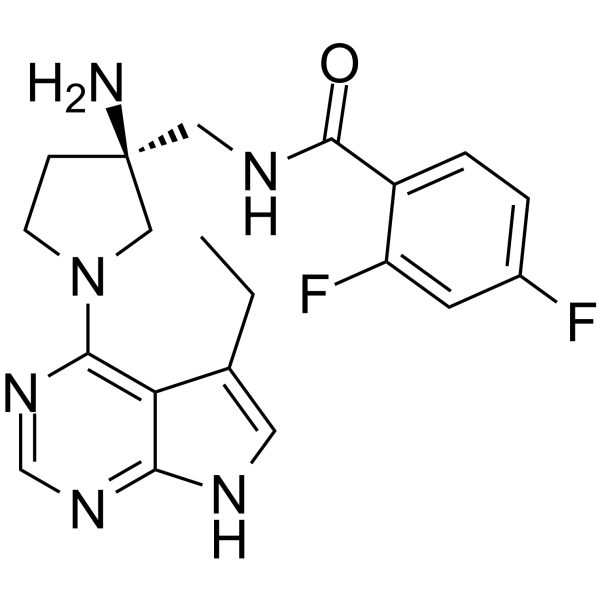
-
- HY-124793
-
|
|
Cyclin G-associated Kinase (GAK)
|
Infection
|
|
GAK inhibitor 49 is a potent, ATP-competitive and highly selective cyclin G associated kinase (GAK) inhibitor with a Ki of 0.54 nM and a cell IC50 of 56 nM. GAK inhibitor 49 also shows binding to RIPK2 .
|
-
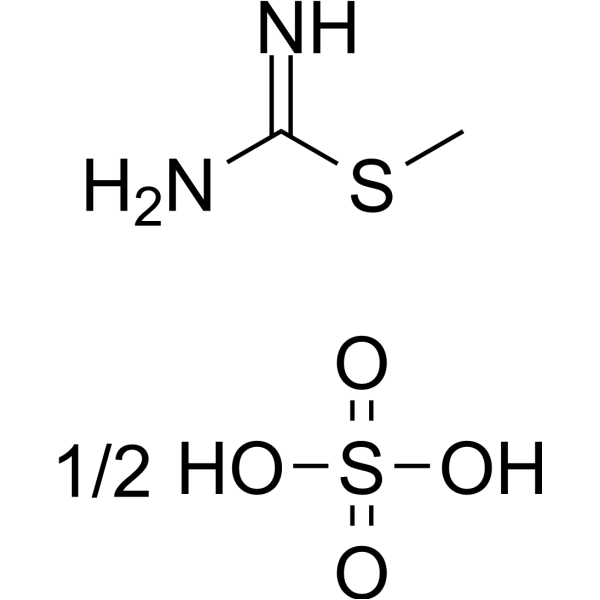
-
- HY-79457
-
|
|
NO Synthase
HSV
|
Infection
Inflammation/Immunology
|
|
S-Methylisothiourea sulfate is a potent, selective and competitive inhibitor of inducible nitric oxide synthase (iNOS). S-Methylisothiourea sulfate exerts beneficial effects in rodent models of septic shock .
|
-
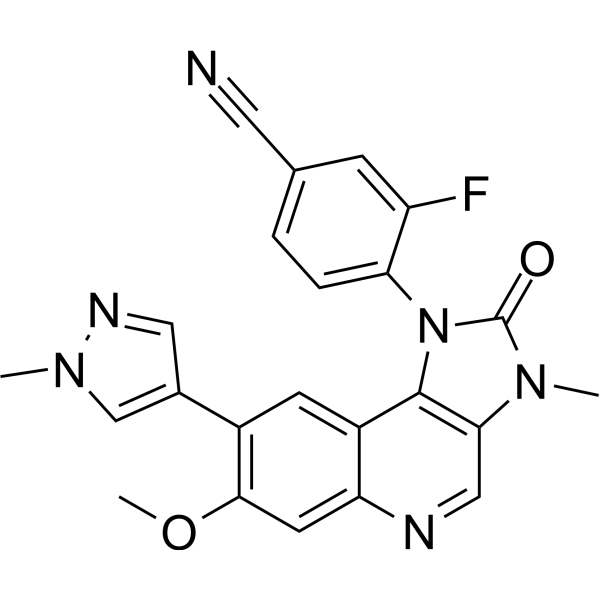
-
- HY-160096
-
|
|
ATM/ATR
|
Cancer
|
|
M3541 is a potent, ATP-competitive and selective ATM inhibitor with an IC50 of 0.25 nM. M3541 shows remarkable selectivity against other protein kinases. M3541 suppresses double-strand breaks (DSB) repair and has antitumor activities .
|
-
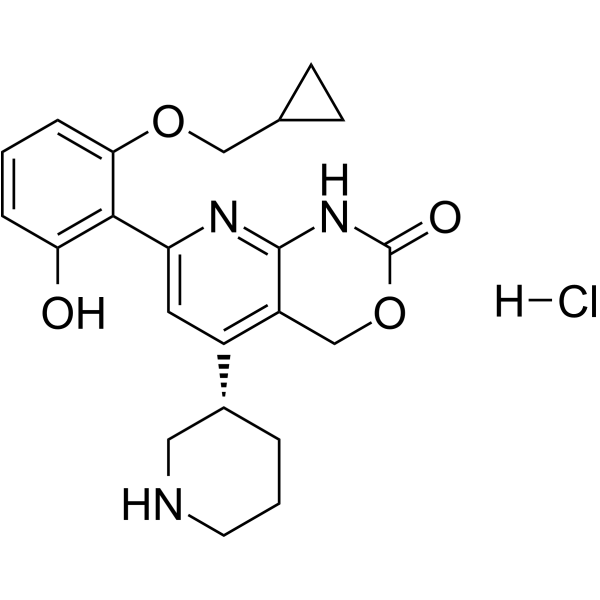
-
- HY-124795
-
|
|
Nucleoside Transporters
|
Others
|
|
FPMINT is a potent, irreversible and non-competitive inhibitor of Equilibrative nucleoside transporters (ENTs), which is more selective to ENT2 than to ENT1. FPMINT plays an important role in uridine uptake .
|
-

-
- HY-50948
-
-
-
- HY-112355
-
|
|
Aurora Kinase
|
Cancer
|
|
Aurora kinase inhibitor-2 is a selective and ATP-competitive Aurora kinase inhibitor with IC50s of 310 nM and 240 nM for Aurora A and Aurora B, respectively .
|
-
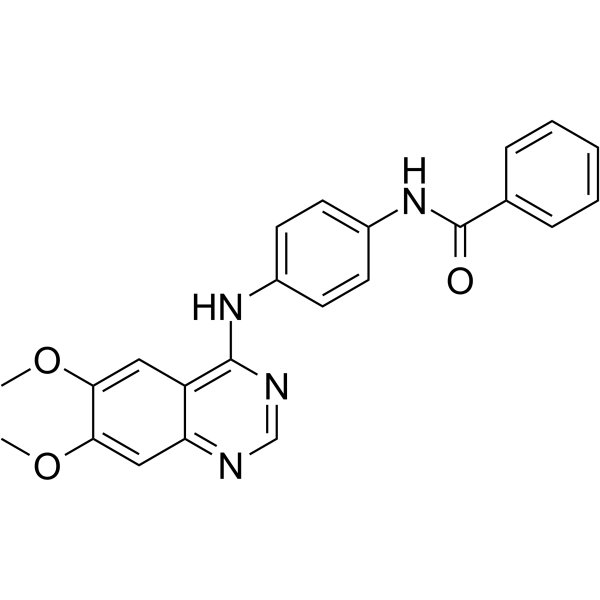
-
- HY-70035
-
|
FXV673
|
Factor Xa
|
Cardiovascular Disease
|
|
Otamixaban(FXV673) is a potent (Ki = 0.5 nM), selective, rapid acting, competitive and reversible fXa inhibitor that effectively inhibits both free and prothrombinase-bound fXa.
|
-
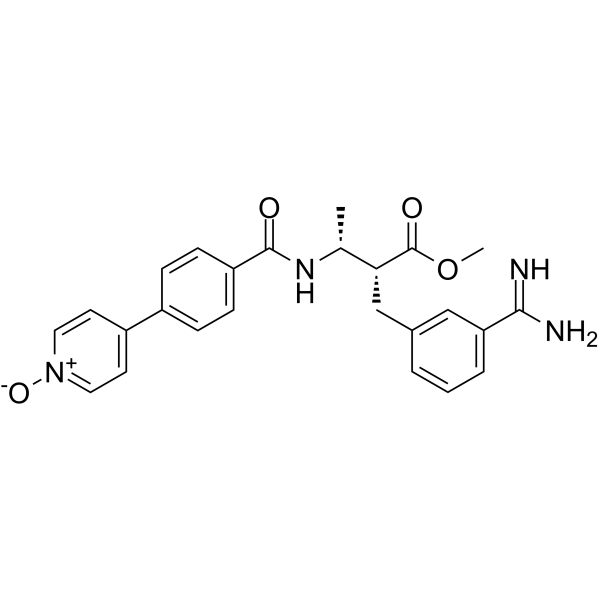
-
- HY-108643
-
|
|
MAPKAPK2 (MK2)
|
Cancer
|
|
CMPD1 is a selective and non-ATP-competitive p38 MAPK-mediated MK2 phosphorylation inhibitor with apparent Ki (Ki app) of 330 nM .
|
-
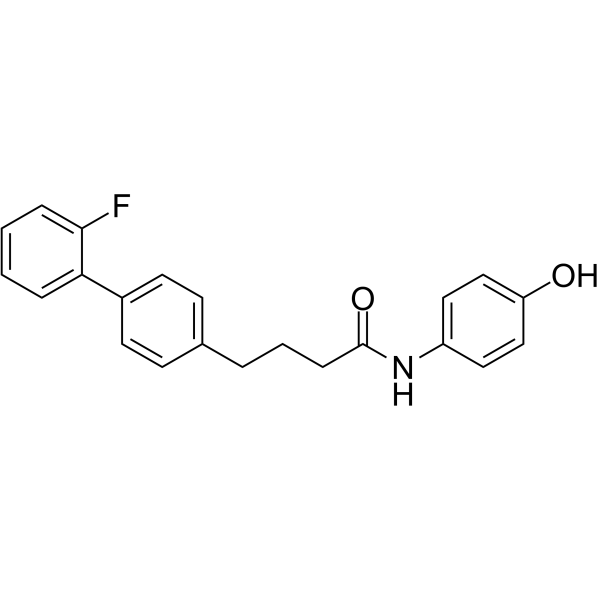
- HY-15687
-
|
|
ROCK
|
Metabolic Disease
|
|
SAR407899 hydrochloride is a selective, potent and ATP-competitive ROCK inhibitor, with an IC50 of 135 nM for ROCK-2, and Kis of 36 nM and 41 nM for human and rat ROCK-2, respectively.
|
-
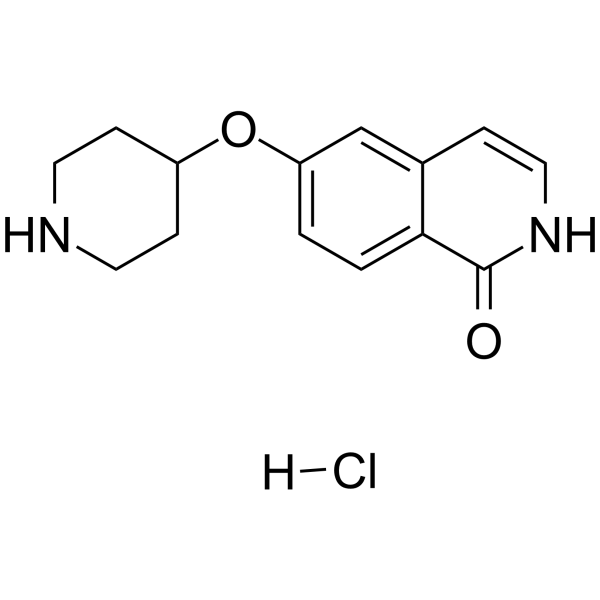
- HY-15159
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
|
Cyclapolin 9 is a potent, selective and ATP-competitive polo-like kinase 1 (PLK1) inhibitor with an IC50 of 500 nM. Cyclapolin 9 is inactive against other kinases .
|
-
- HY-100846
-
JQEZ5
3 Publications Verification
|
Histone Methyltransferase
|
Cancer
|
|
JQEZ5 is a potent and selective EZH2 lysine methyltransferase inhibitor. JQEZ5 SAM-competitively inhibits polycomb repressive complex 2 (PRC2) with an IC50 of 80 nM. JQEZ5 has anti-tumor effects .
|
-
- HY-169884
-
|
|
Aldehyde Dehydrogenase (ALDH)
|
Cancer
|
|
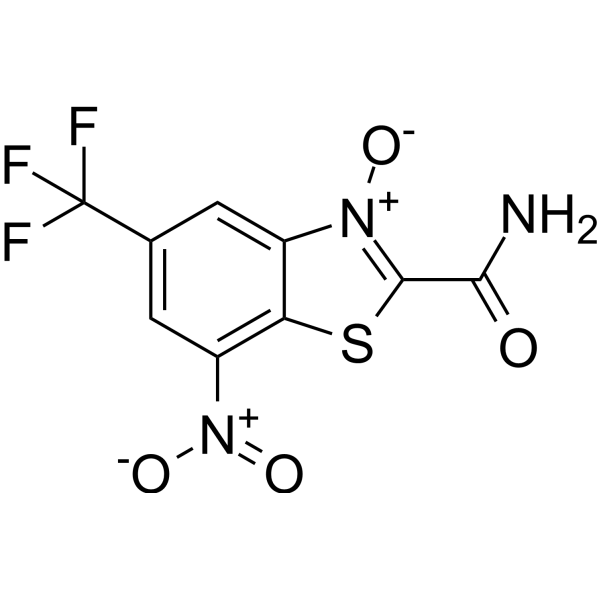
MCI-INI-3 is a selective competitive inhibitor of human ALDH1A3 (with a Ki value of 0.55 μM for ALDH1A3 and a Ki value of 78.2 μM for ALDH1A1). MCIINI-3 inhibits the biosynthesis of retinoic acid and reduces the viability of GSC-83 and GSC-326 glioblastoma cells .
|
-

- HY-14715
-
-
- HY-16985S
-
|
ODM-201-d4; BAY-1841788-d4
|
Isotope-Labeled Compounds
Androgen Receptor
|
Cancer
|
|
Darolutamide-d4 (ODM-201-d4) is deuterium labeled Darolutamide (HY-16985). Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-

- HY-19797
-
|
|
p97
|
Cancer
|
|
ML241 is a potent, selective and competitive p97 ATPase inhibitor with an IC50 of 0.11 μM. ML241 can be used for the research of cancer .
|
-
- HY-111184
-
|
|
PI3K
|
Cancer
|
|
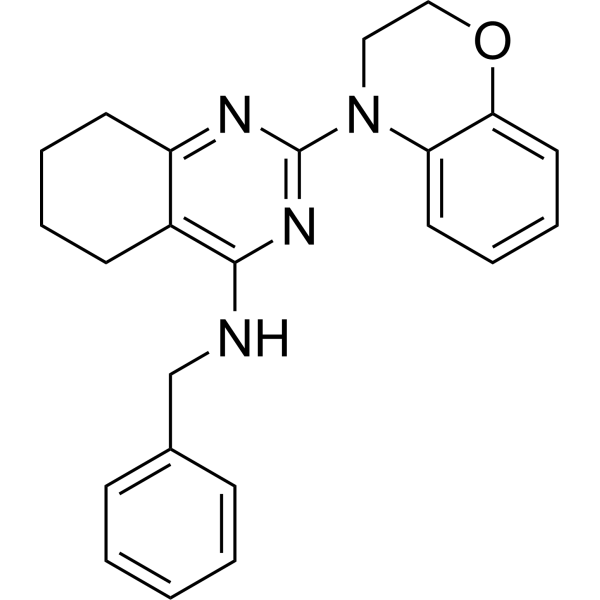
PIK-108 is a non-ATP competitive, allosteric p110β/p110δ selective inhibitor .
|
-
- HY-13450
-
|
|
JAK
EGFR
|
Inflammation/Immunology
|
|
ZM 449829 is a potent, selective and ATP competitive inhibitor of JAK3, with a pIC50 of 6.8. ZM 449829 will be useful pharmacological tools for the investigation of the JAK3 .
|
-

- HY-124230
-
|
|
15-PGDH
|
Metabolic Disease
|
|
15-PGDH-IN-3 (Compound 61) is a selective competitive inhibitor of 15-hydroxyprostaglandin dehydrogenase (15-PGDH) .
|
-
- HY-16985R
-
|
ODM-201 (Standard); BAY-1841788 (Standard)
|
Reference Standards
Androgen Receptor
|
Cancer
|
|
Darolutamide (Standard) is the analytical standard of Darolutamide (HY-16985). This product is intended for research and analytical applications. Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-

- HY-101169
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
Tetrindole mesylate is a selective inhibitor of monoamine oxidase A (MAO A). Tetrindole mesylate inhibits rat brain mitochondrial MAO A in a competitive manner with a Ki value of 0.4 μM and inhibits MAO B with a Ki of 110 μM. Tetrindole mesylate has antidepressant activity .
|
-
- HY-124793A
-
|
|
Cyclin G-associated Kinase (GAK)
|
Infection
|
|
GAK inhibitor 49 hydrochloride is a potent, ATP-competitive and highly selective cyclin G associated kinase (GAK) inhibitor with a Ki of 0.54 nM and a cell IC50 of 56 nM. GAK inhibitor 49 hydrochloride also shows binding to RIPK2 .
|
-
- HY-101169A
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
Tetrindole hydrochloride is a selective inhibitor of monoamine oxidase A (MAO A). Tetrindole hydrochloride inhibits rat brain mitochondrial MAO A in a competitive manner with a Ki value of 0.4 μM and inhibits MAO B with a Ki of 110 μM. Tetrindole hydrochloride has antidepressant activity .
|
-

- HY-119611A
-
-
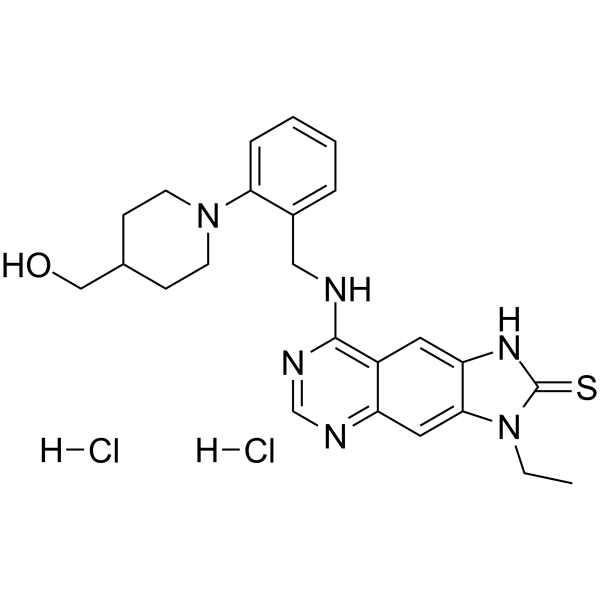
- HY-103384
-
|
|
Casein Kinase
|
Cancer
|
|
TMCB is a selective, ATP-competitive CK2 (casein kinase II) inhibitor with distinct Ki values of 83 nM and 21 nM for the two different catalytic CK2 subunits α and α', respectively .
|
-
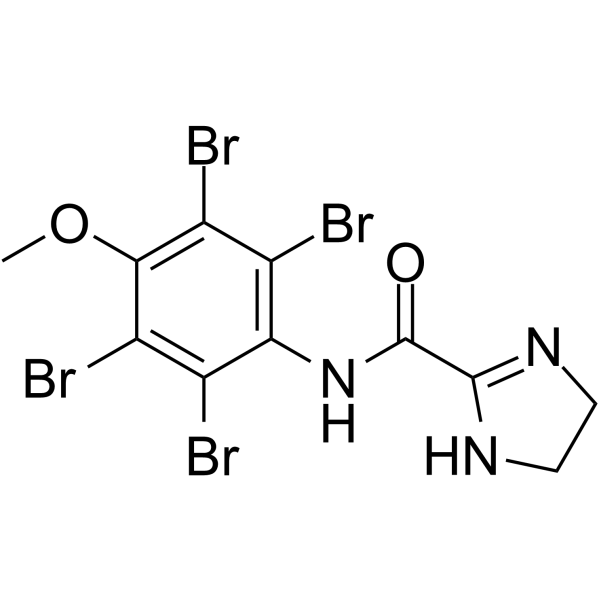
- HY-122592
-
|
|
Factor Xa
|
Cardiovascular Disease
|
|
Zifaxaban is an orally active, competitively and selective Factor Xa (FXa) inhibitor with an IC50 of 11.1 nM for human FXa. Zifaxaban shows >10000-fold greater selectivity than other serine proteases. Zifaxaban can be used for the arterial and venous thrombosis research .
|
-
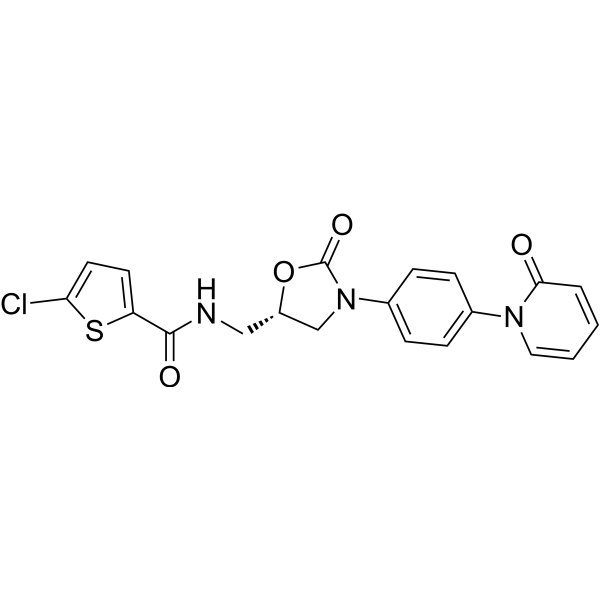
- HY-16194
-
-
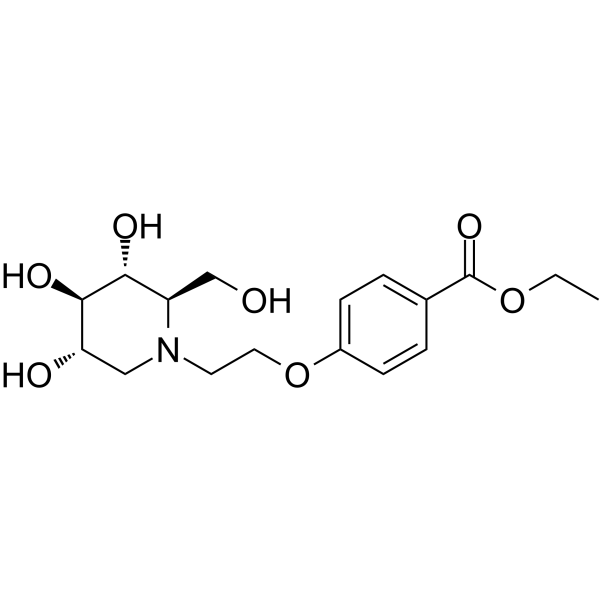
- HY-110331
-
|
|
Checkpoint Kinase (Chk)
|
Cancer
|
|
CCT241533 dihydrochloride is a potent and selective ATP competitive inhibitor of CHK2 with an IC50 of 3 nM and Ki of 1.16 nM .
|
-
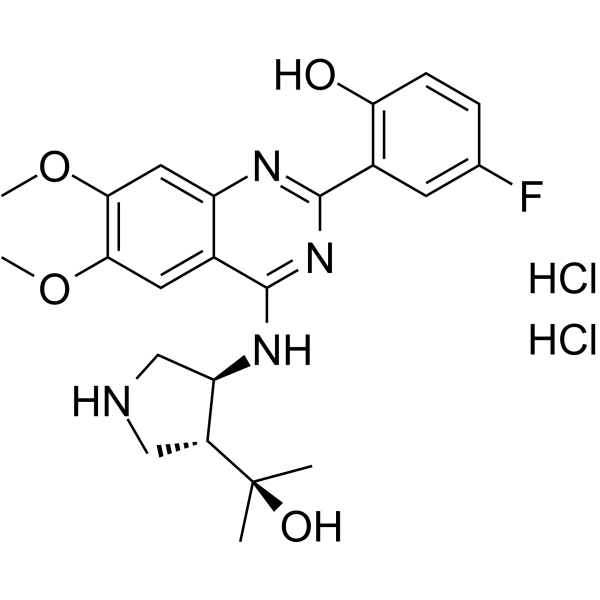
- HY-W042648
-
|
|
Casein Kinase
|
Others
|
|
4,5,6,7-Tetrabromobenzimidazole is a selective and ATP competitive CK2 (casein kinase 2) inhibitor .
|
-
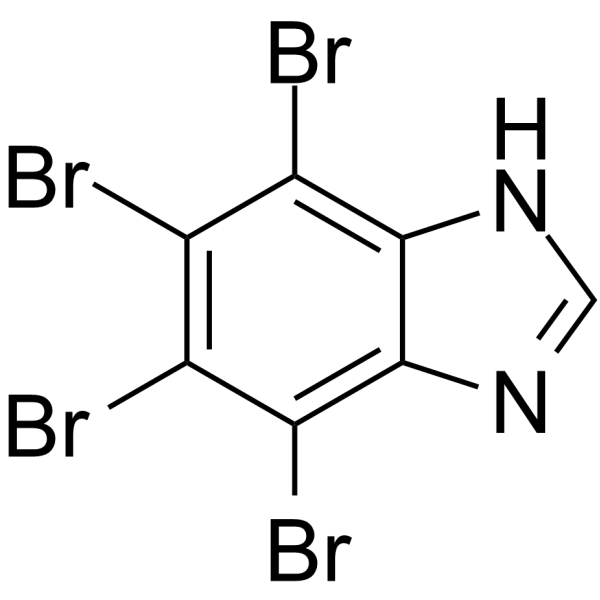
- HY-120279A
-
|
|
Polo-like Kinase (PLK)
|
Cancer
|
|
CFI-400437 is an indolinone-derived, ATP-competitive kinase inhibitor with high selectivity for PLK4 (IC50 of 0.6 nM) .
|
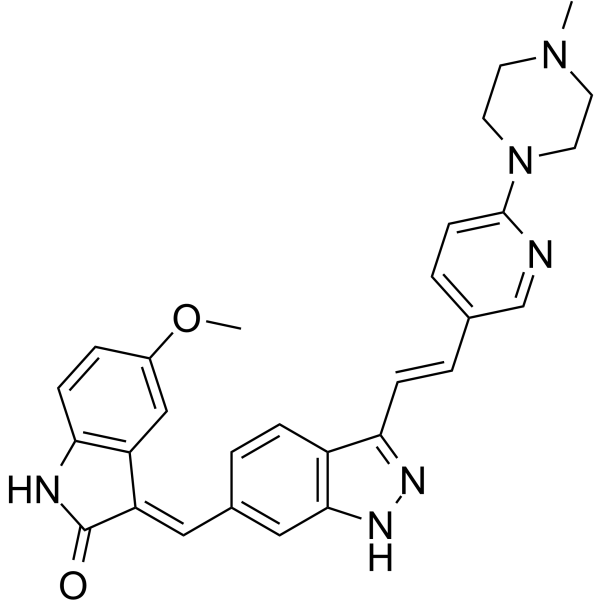
-
- HY-132907
-
-
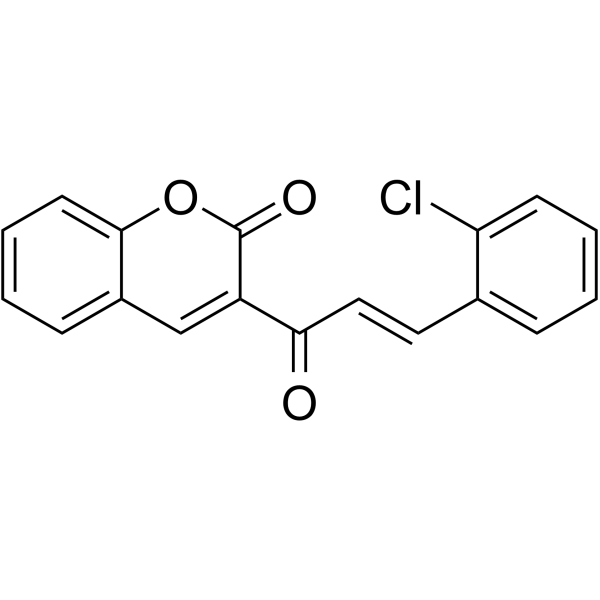
- HY-15656S
-
-
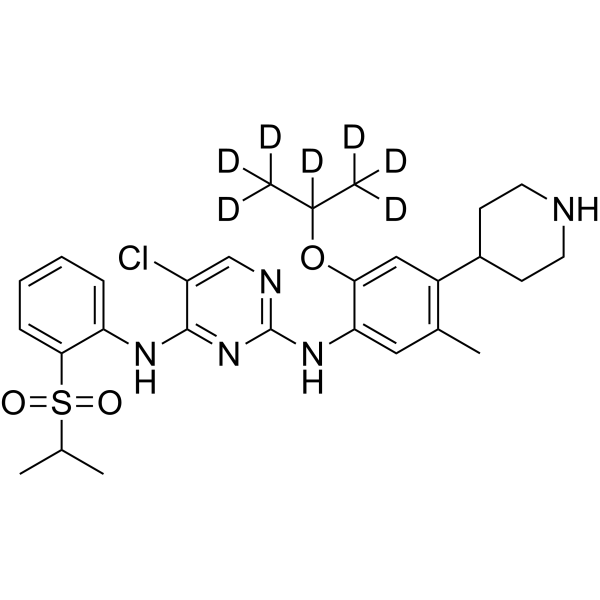
- HY-133117A
-
|
|
IKK
|
Cancer
|
|
(Rac)-BAY-985 (Compound Example 100.01) is a potent, ATP-competitive and selective TBK1 inhibitor with an IC50 of 1.5 nM. Antitumor efficacy .
|
-
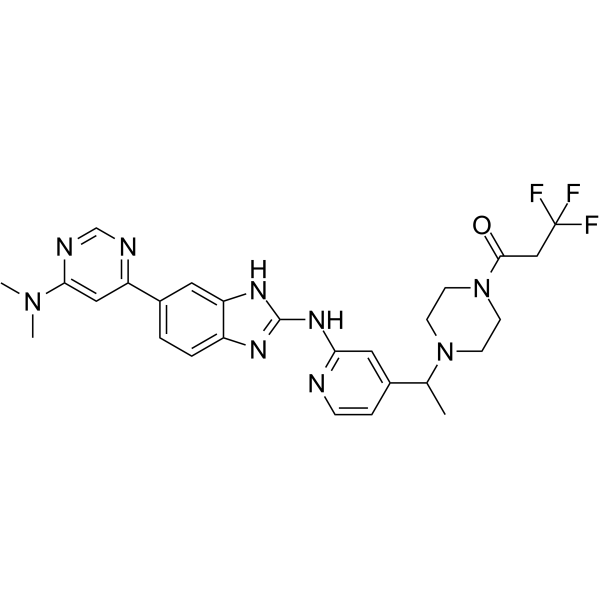
- HY-106009
-
|
|
Checkpoint Kinase (Chk)
|
Cancer
|
|
VRX0466617 is a selective and ATP-competitive Chk2 inhibitor with an IC50 of 120 nM and a Ki of 11 nM. VRX0466617 does not inhibit the related Chk1 activity. VRX0466617 can be used in the study of cancer.
|
-
- HY-119611
-
-
- HY-50949
-
-
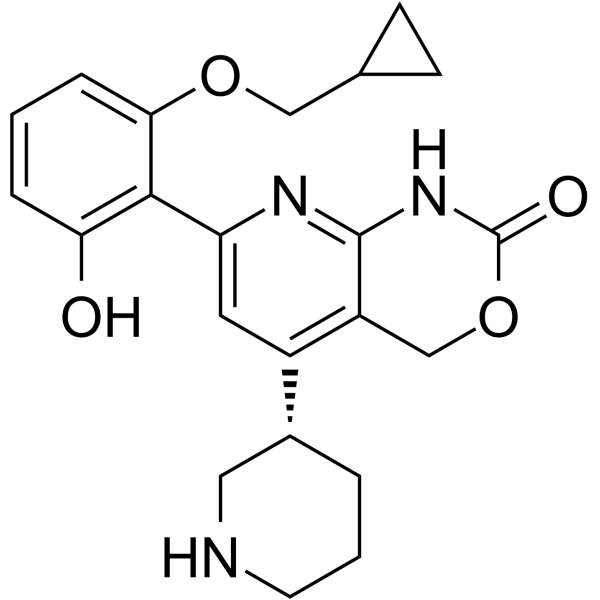
- HY-16015A
-
|
ABC294640 hydrochloride
|
SphK
|
Cancer
|
|
Opaganib (ABC294640) hydrochloride is a selective, competitive sphingosine kinase 2 (SK2) inhibitor with Ki of 9.8 μM.
|
-

- HY-112333
-
-

- HY-12042A
-
|
AS703026 hydrochloride; MSC1936369B hydrochloride
|
MEK
|
Cancer
|
|
Pimasertib hydrochloride is a highly selective, ATP non-competitive allosteric orally available MEK1/2 inhibitor .
|
-

- HY-112081A
-
|
|
DNA/RNA Synthesis
|
Others
|
|
BAY-707 acetate is a highly potent and selective substrate-competitive inhibitor of MTH1 with superior cellular target engagement and pharmacokinetic properties.
|
-

- HY-159741
-
|
FCE 24928
|
Cytochrome P450
|
Cancer
|
|
Minamestane (FCE 24928) is a selective and competitive aromatase inhibitor with an IC50 value of 45.7 nM. Minamestane is promising for research of postmenopausal breast cancer .
|
-

- HY-128126
-
-

- HY-116097
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
PSB-1491 is a selective and competitive monoamine oxidase B (MAO-B) inhibitor with an IC50 of 0.386 nM for hMAO-B. PSB-1491 shows >25000-fold selective versus MAO-A .
|
-
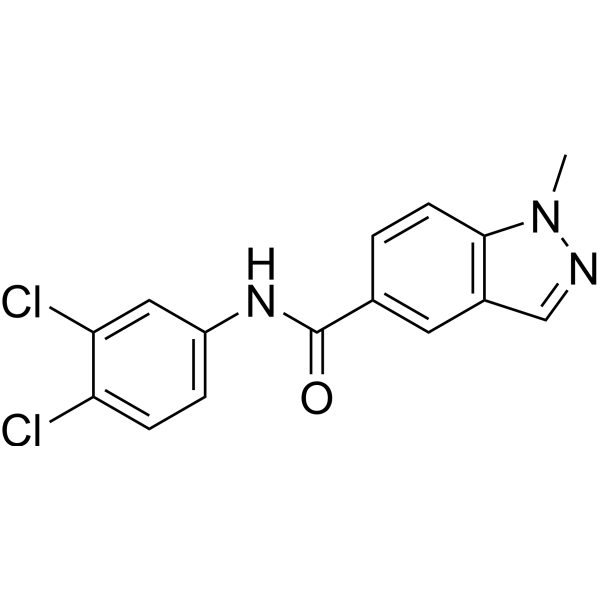
- HY-76409
-
|
FXV673 hydrochloride
|
Factor Xa
|
Inflammation/Immunology
|
|
Otamixaban (FXV673) is a potent, selective, rapid-acting, competitive, and reversible fXa inhibitor (Ki=0.5 nM) that effectively inhibits both free and prothrombinase-bound fXa .
|
-
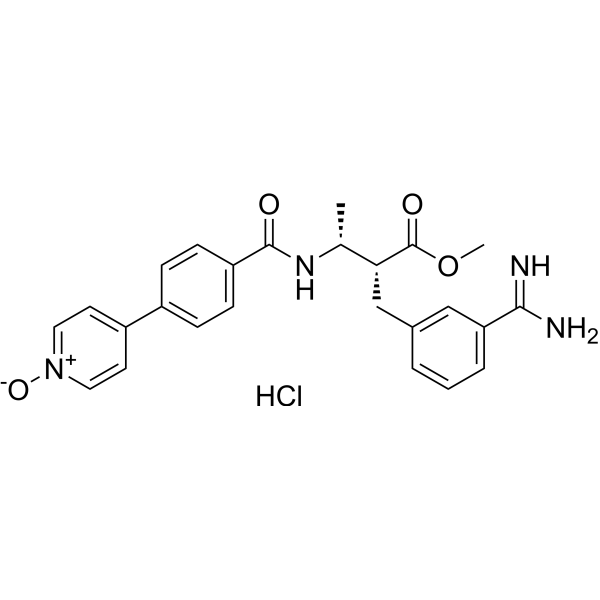
- HY-104009
-
|
|
Histone Methyltransferase
|
Cancer
|
|
GSK2807 is a potent and selective SAM-competitive inhibitor of SMYD3, exhibiting a Ki value of 14 nM, and it may be useful for cancer treatment by preventing the methylation of MEKK2.
|
-

- HY-B0140R
-
|
|
Reference Standards
Phosphodiesterase (PDE)
Adenosine Receptor
|
Inflammation/Immunology
Cancer
|
|
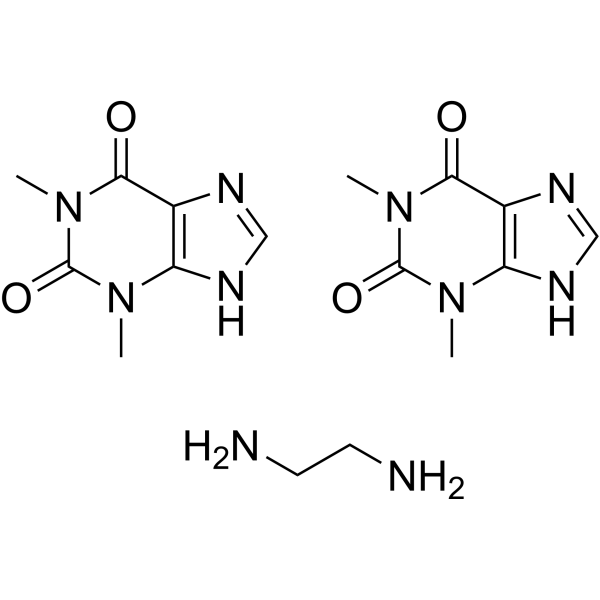
Aminophylline (Standard) is the analytical standard of Aminophylline. This product is intended for research and analytical applications. Aminophylline is a competitive and non-selective phosphodiesterase (PDE) inhibitor. Aminophylline is a competitive adenosine receptor antagonist. Aminophylline has apulmonary vasodilator action as well as a bronchodilator action and has the potential for asthma research .
|
-

- HY-50949A
-
|
|
IKK
|
Inflammation/Immunology
|
|
Bay 65-1942 R form is the less active R-form of Bay 65-1942. Bay 65-1942 is an ATP-competitive and selective IKKβ inhibitor.
|
-
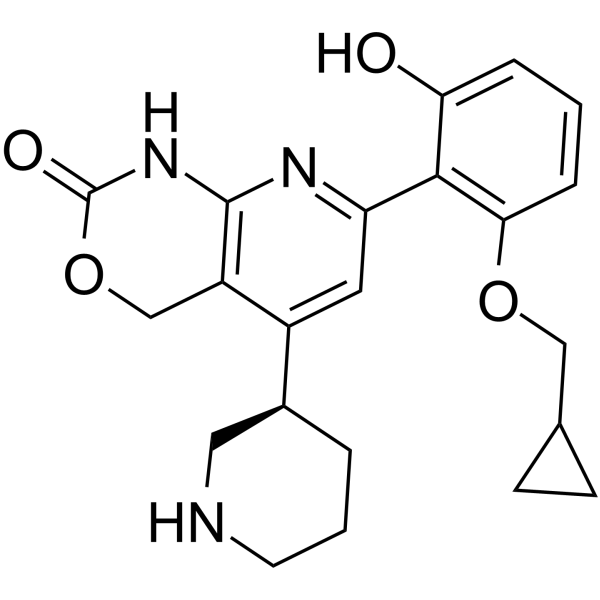
- HY-149425
-
|
|
Sirtuin
|
Inflammation/Immunology
Cancer
|
|
SIRT5 Inhibitor 6 is a potent, substrate-competitive and selective SIRT5 inhibitor with an IC50 of 3.0 μM. SIRT5 Inhibitor6 has a therapeutic potential against septic AKI in vivo .
|
-
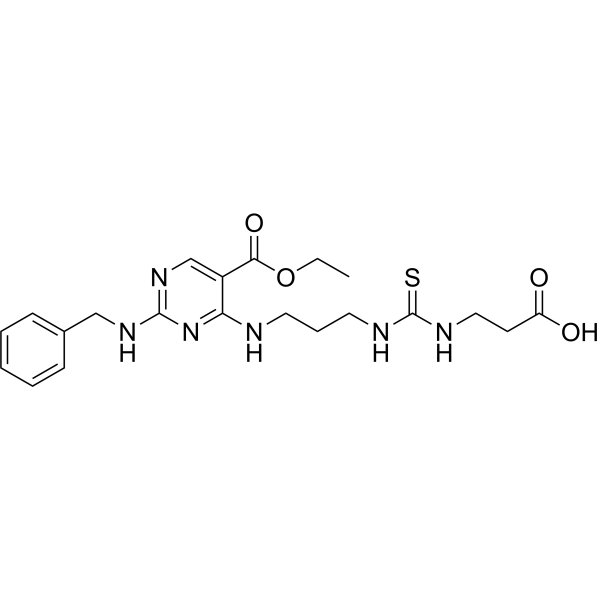
- HY-146154
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-58 (Compound 4a) is a potent, ATP-competitive, and selective EGFR inhibitor. EGFR-IN-58 shows potent cytotoxicity against melanoma, colon, and blood cancers .
|
-
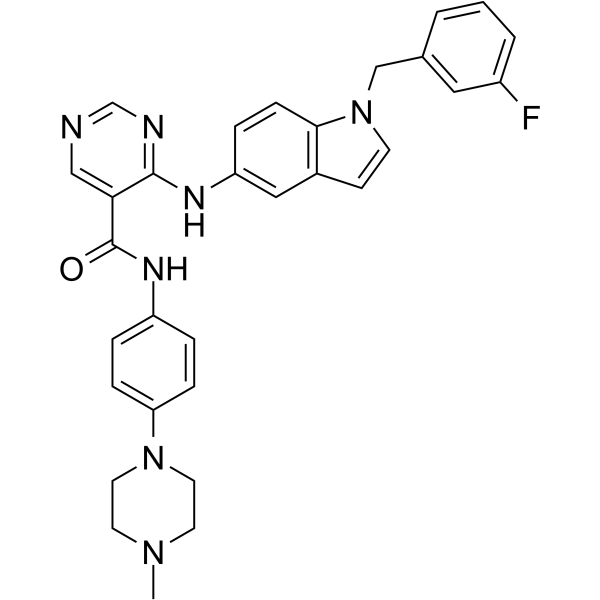
- HY-110399R
-
|
|
Lipoxygenase
|
Neurological Disease
|
|
Cirsiliol (Standard) is the analytical standard of Cirsiliol. This product is intended for research and analytical applications. Cirsiliol is a potent and selective 5-lipoxygenase inhibitor and a competitive low affinity benzodiazepine receptor ligand.
|
-

- HY-120650
-
|
|
FAAH
|
Infection
|
|
CAY10435 is a β-ketooxazapyridine, selective FAAH inhibitor with antimicrobial activity. CAY10435 binds non-competitively to the FAAH of Dictyostelium discoideum with a Kd value of 0.57 nM .
|
-
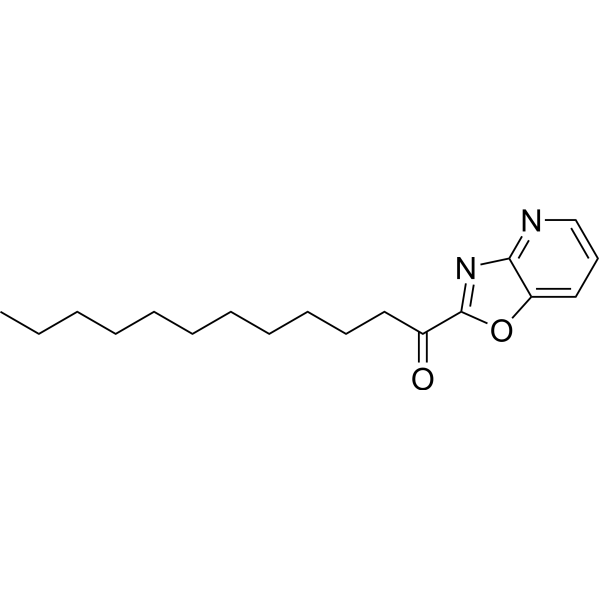
- HY-103366
-
|
|
Checkpoint Kinase (Chk)
|
Cancer
|
|
NSC 109555 ditosylate is a potent, selective, ATP-competitive checkpoint kinase 2 (Chk2) inhibitor with an IC50 of 240 nM. NSC 109555 ditosylate can be used for the research of cancer .
|
-
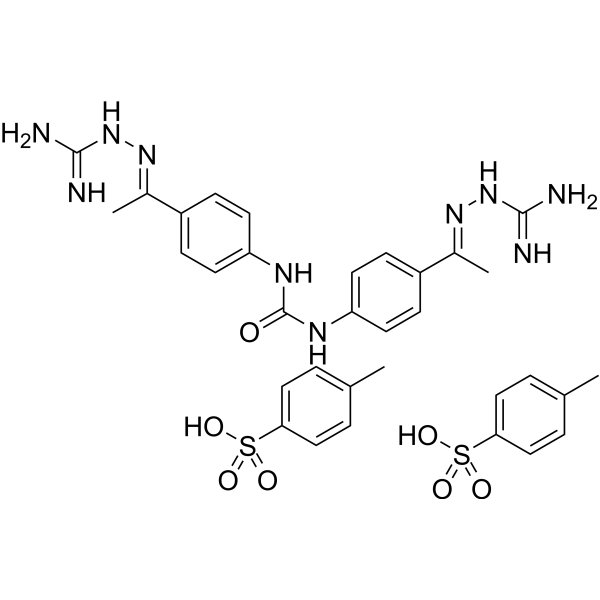
- HY-13303
-
|
|
MEK
|
Cancer
|
|
RO 4927350 is a potent and selective non-ATP-competitive MEK1/2 inhibitor. RO 4927350 exhibits significant antitumor efficacy in a broad spectrum of tumor models .
|
-

- HY-16015R
-
|
ABC294640 (Standard)
|
SphK
Reference Standards
|
Cancer
|
|
Opaganib (Standard) is the analytical standard of Opaganib. This product is intended for research and analytical applications. Opaganib (ABC294640) is a selective, competitive sphingosine kinase 2 (SK2) inhibitor with Ki of 9.8 μM.
|
-

- HY-117923
-
|
|
mTOR
PI3K
|
Cancer
|
|
PF-06465603 is a highly potent and selective ATP-competitive kinase inhibitor and a class 1 PI3K and mTOR inhibitor. PF-06465603 is a metabolite of PF-04691502 with a terminal carboxylic acid structure .
|
-

- HY-121908
-
|
|
ATP Synthase
|
Cancer
|
|
FCPT, an ATP competitive inhibitor, induces a tight-binding of kinesin-5 onto microtubules and induced loss of microtubules selectively at the poles of Xenopus extract spindles without altering microtubule dynamics .
|
-
- HY-111286
-
-

- HY-129444
-
|
|
Monoamine Oxidase
|
Cancer
|
|
MD 780236 free base is a substrate and selective inhibitor of monoamine oxidase B (MAO-B), competitive with phenylethylamine and 5-hydroxytryptamine (5-HT) .
|
-

- HY-183872
-
|
|
Fungal
N-myristoyltransferase
|
Infection
|
|
FTR1335 is an Antifungal agent as well as a selective, substrate peptide-competitive, and myristoyl-CoA non-competitive inhibitor of N-myristoyltransferase CaNmt, with an IC50 of 0.49 nM against Candida albicans CaNmt. FTR1335 exhibits fungicidal activity against Candida albicans and inhibits the growth of Candida tropicalis. FTR1335 can be used in research related to Candida albicans infections .
|
-

- HY-156350
-
|
|
DAPK
|
Cancer
|
|
SGC-STK17B-1, a chemical probe, is an ATP-competitive and selective STK17B (a member of DAPK family) inhibitor (IC50: 34 nM, KD: 5.6 nM) .
|
-
- HY-12493A
-
|
|
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
LY-2584702 tosylate salt is a selective ATP competitive inhibitor of p70S6K with an IC50 of 4 nM. In S6K1 enzyme assay, the IC50 of LY-2584702 is 2 nM.
|
-
- HY-151115
-
|
|
Smo
|
Inflammation/Immunology
Cancer
|
|
JNJ-1289 is a potent, selective, competitive and allosteric human spermine oxidase (hSMOX) inhibitor (IC50: 50 nM). JNJ-1289 can be used in the research of polyamine catabolism, inflammation and cancers .
|
-
- HY-101190
-
-
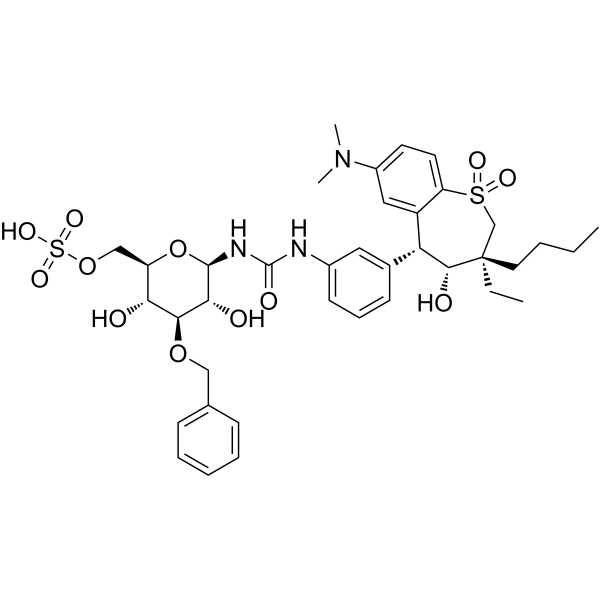
- HY-13217
-
|
GBR-12909 dihydrochloride; I893 dihydrochloride
|
Dopamine Transporter
|
Neurological Disease
|
|
Vanoxerine dihydrochloride (GBR-12909 dihydrochloride) is a competitive, potent, and highly selective dopamine reuptake inhibitor (Ki=1 nM). Vanoxerine dihydrochloride (GBR-12909 dihydrochloride) binds to the target site on the dopamine transporter (DAT) .
|
-
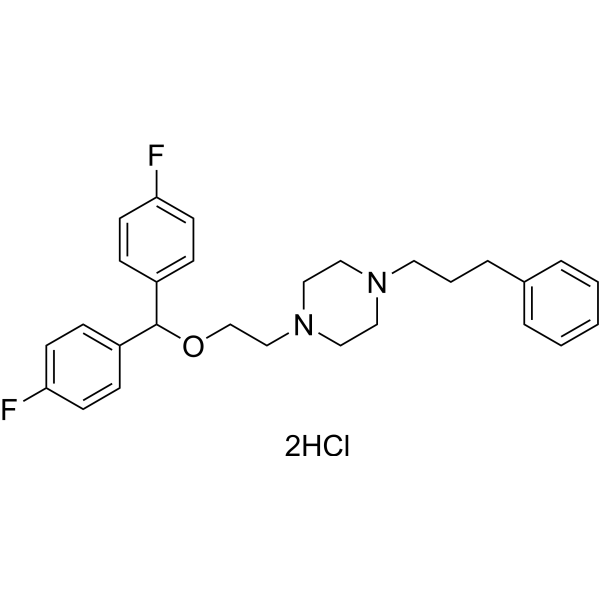
- HY-13217A
-
|
GBR 12909; I893
|
Dopamine Transporter
|
Neurological Disease
|
|
Vanoxerine (GBR-12909) is a competitive, potent, and highly selective dopamine reuptake inhibitor (Ki=1 nM). Vanoxerine (GBR-12909) binds to the target site on the dopamine transporter (DAT) .
|
-
- HY-12853
-
|
|
Environmental Pollutants
Reactive Oxygen Species (ROS)
|
Others
|
|
Mesotrione is a herbicide belongs to the benzoylcyclohexanedione family. Mesotrione is a potent and competitive and reversible inhibitor of HPPD enzyme. Mesotrione is selective to maize due to rapid metabolism and relative high tolerance by the susceptible crop plant .
|
-
- HY-12493B
-
|
|
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
LY-2584702 hydrochloride is a selective ATP competitive inhibitor of p70S6K with an IC50 of 4 nM. In S6K1 enzyme assay, the IC50 of LY-2584702 is 2 nM.
|
-
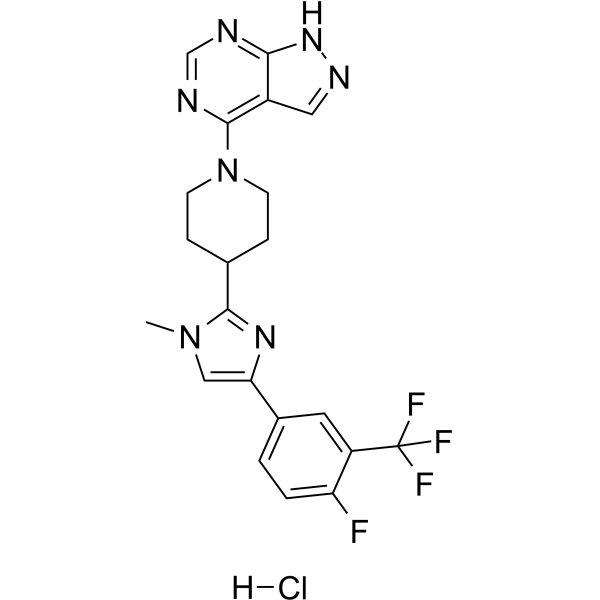
- HY-16082
-
AZD7545
4 Publications Verification
|
PDHK
|
Metabolic Disease
|
|
AZD7545 is a potent, competitive, selective PDHK2 (pyruvate dehydrogenase kinase 2) inhibitor with IC50s of 36.8 nM, 6.4 nM for PDHK1 and PDHK2, respectively .
|
-
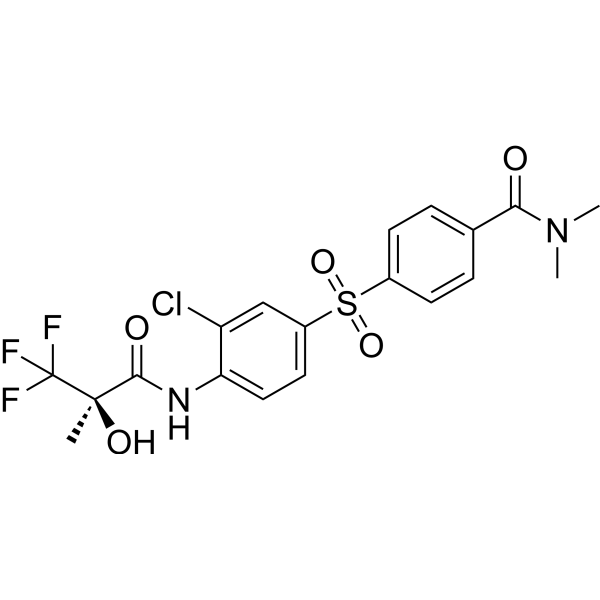
- HY-115732
-
|
|
PKA
|
Inflammation/Immunology
|
|
PKA-IN-1 is a potent and selective cyclic AMP-dependent protein kinase (PKA) catalytic subunit (cAK) inhibitor with an IC50 of 0.03 μM. PKA-IN-1 inhibits cAK in a fashion that is competitive with respect to ATP as substrate .
|
-
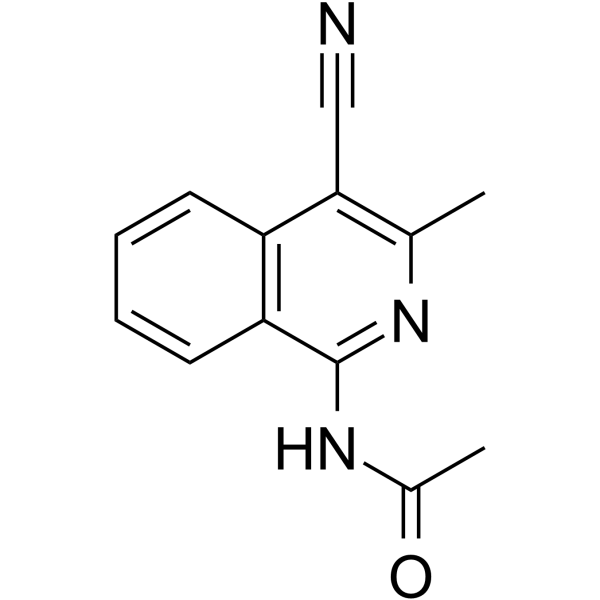
- HY-103316A
-
Ned 19
3 Publications Verification
|
Calcium Channel
|
Cardiovascular Disease
Cancer
|
|
Ned 19 is a selective membrane-permeant non competitive NAADP antagonist and inhibits NAADP-mediated Ca 2+ signaling, with an IC50 of 65 nM . Ned 19 strongly inhibits tumor growth and vascularization as well as lung metastases in mice .
|
-
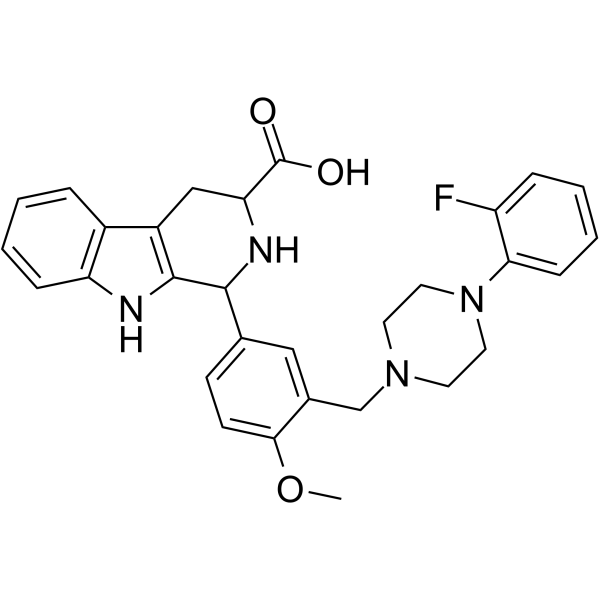
- HY-10787
-
|
H 376/95
|
Thrombin
|
Cardiovascular Disease
|
|
Ximelagatran (H 376/95) is an orally active thrombin inhibitor that selectively and competitively inhibits both free and clot-bound thrombin. Ximelagatran is an anticoagulant agent with a rapid onset of anticoagulant effect, predictable, dose-dependent pharmcokinetics and pharmacodynamics .
|
-
- HY-11087
-
|
SD-06
|
p38 MAPK
Autophagy
|
Inflammation/Immunology
|
|
SD 0006 (SD-06) is an orally active, selective, ATP-competitive and potent diaryl pyrazole inhibitor of p38α MAP kinase, with an IC50 of 110 nM for p38α .
|
-
- HY-100888
-
|
TAK-931
|
CDK
|
Cancer
|
|
Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM. Simurosertib has anti-cancer activity .
|
-
- HY-12774
-
IC261
4 Publications Verification
|
Casein Kinase
Apoptosis
|
Cancer
|
|
IC261 is a selective, ATP-competitive CK1 inhibitor, with IC50s of 1 μM, 1 μM, 16 μM for Ckiδ, Ckiε and Ckiα1, respectively.
|
-
- HY-13994
-
|
|
Mps1
Polo-like Kinase (PLK)
|
Cancer
|
|
Mps1-IN-2 is a potent, selective and ATP-competitive dual Mps1/Plk1 inhibitor, with an IC50 and a Kd of 145 nM and 12 nM for Mps1 and a Kd of 61 nM for Plk1.
|
-
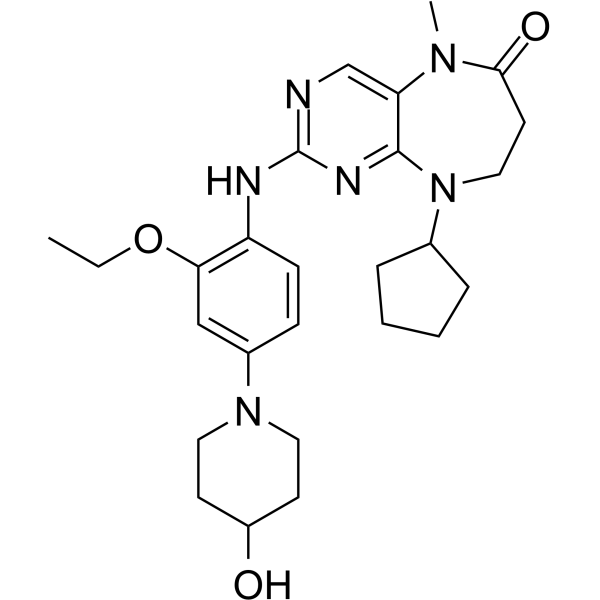
- HY-59090
-
-
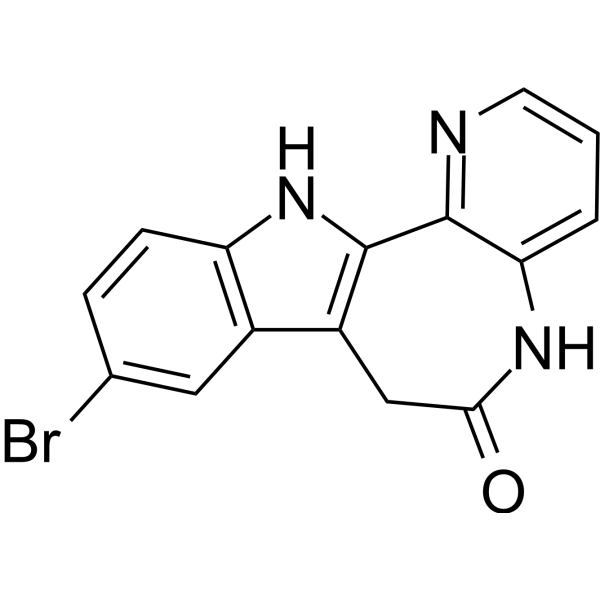
- HY-W015514
-
|
|
JAK
|
Neurological Disease
|
|
N-(3-Aminopropyl)cyclohexylamine, a cyclohexylamine derivative, acts as a selective and competitive inhibitor of spermine synthase. N-(3-Aminopropyl)cyclohexylamine can be used for the research of neurological diseases .
|
-
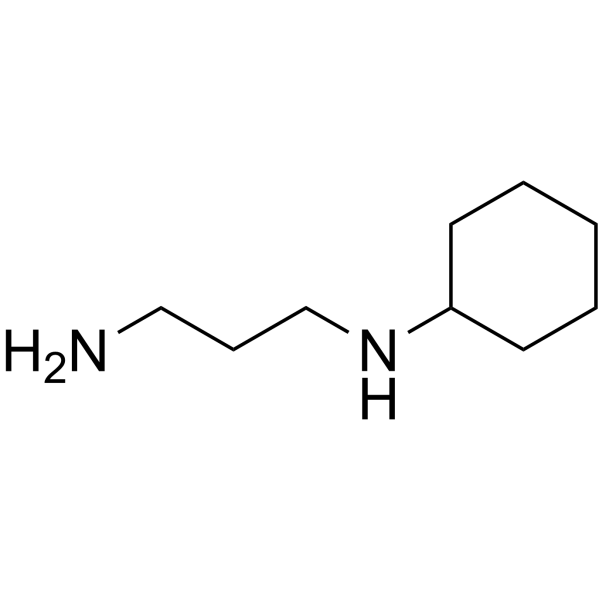
- HY-12012
-
-
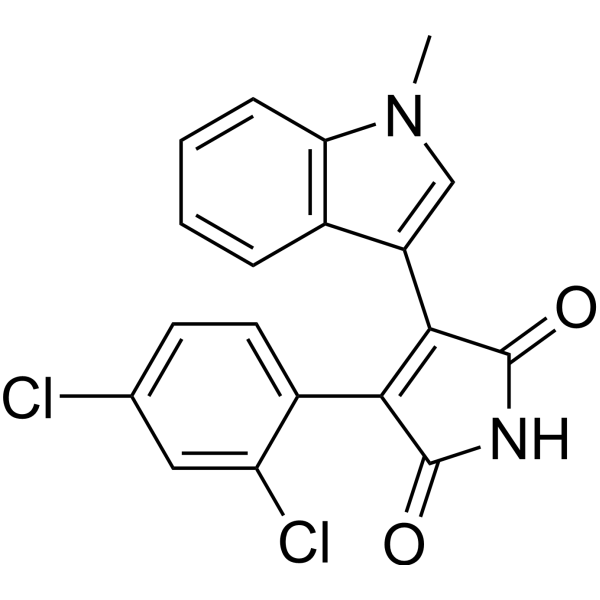
- HY-10963
-
|
CYT387 mesylate
|
JAK
Autophagy
|
Cancer
|
|
Momelotinib mesylate (CYT387 mesylate) is an ATP-competitive inhibitor of JAK1/JAK2 with IC50 of 11 nM/18 nM, appr 10-fold selectivity versus JAK3.
|
-
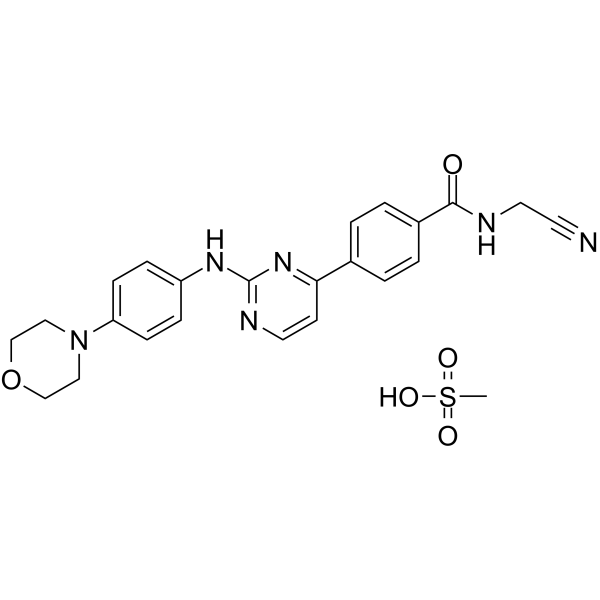
- HY-118047
-
-
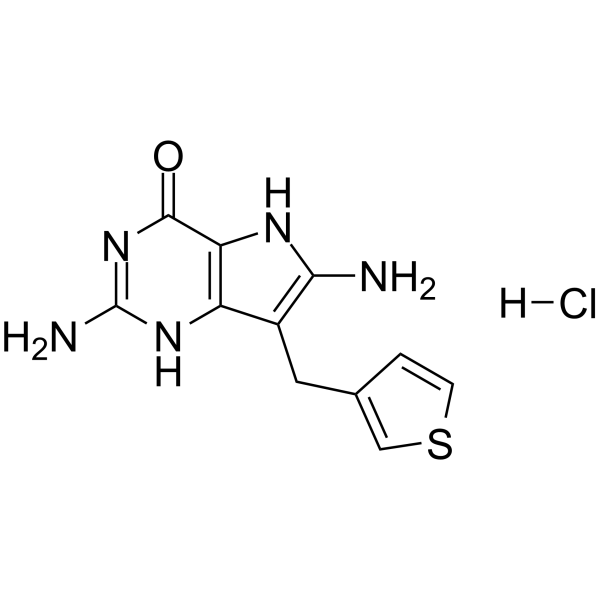
- HY-N10488
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
BChE-IN-11 (compound 10) is a potent, selective and non-competitive BChE (butyrylcholinesterase) inhibitor, with an IC50 of 2.1 μM. BChE-IN-11 can be used for Alzheimer's disease (AD) research .
|
-
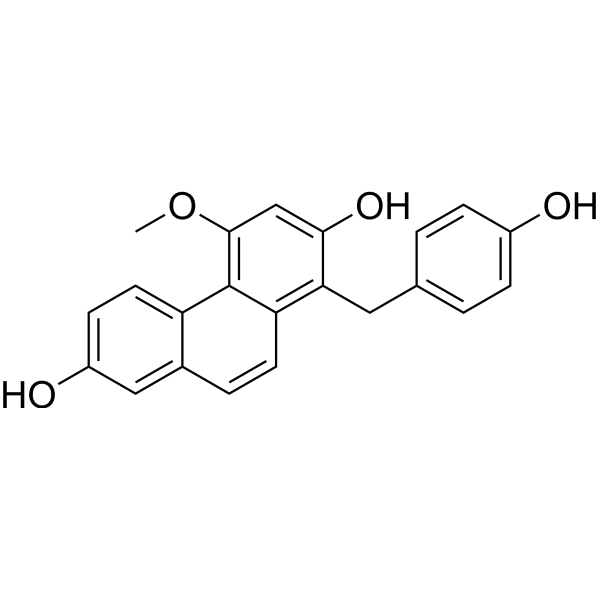
- HY-126015
-
P053
1 Publications Verification
|
Acyltransferase
|
Metabolic Disease
|
|
P053 is a potent, non-competitive and selective ceramide synthase 1 (CerS1) inhibitor wirh an IC50 of 0.5 μM. P053 acts as an endogenous inhibitor of mitochondrial fatty acid oxidation in muscle. Whole-body adiposity regulator .
|
-
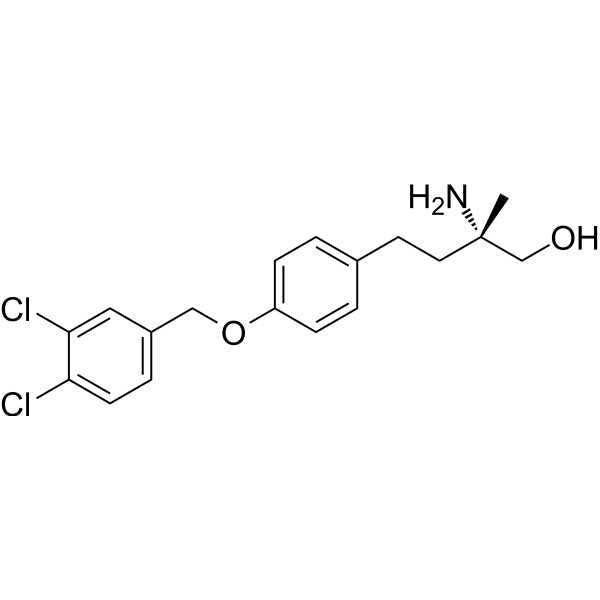
- HY-12832
-
-
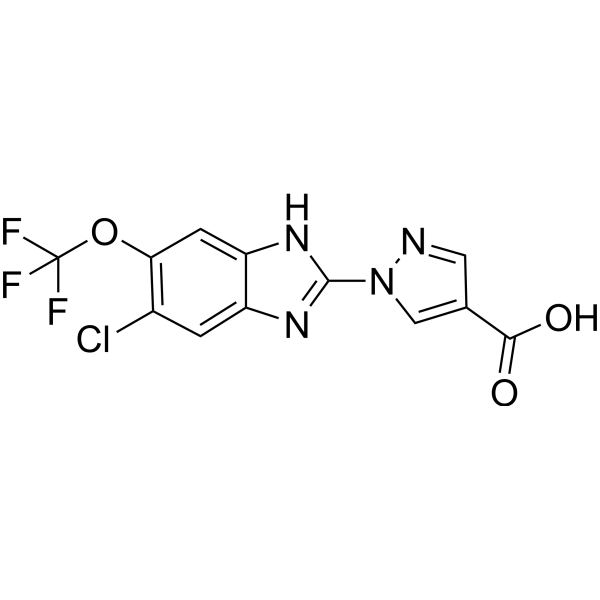
- HY-122011
-
|
|
ROCK
SGK
PKA
PKC
|
Inflammation/Immunology
|
|
PF-4950834 is a potent, selective, orally bioavailable, ATP-competitive rho kinase inhibitor with IC50 values of 8.35 nM and 33.12 nM against ROCK2 and ROCK1, respectively. PF-4950834 inhibits neutrophil migration .
|
-
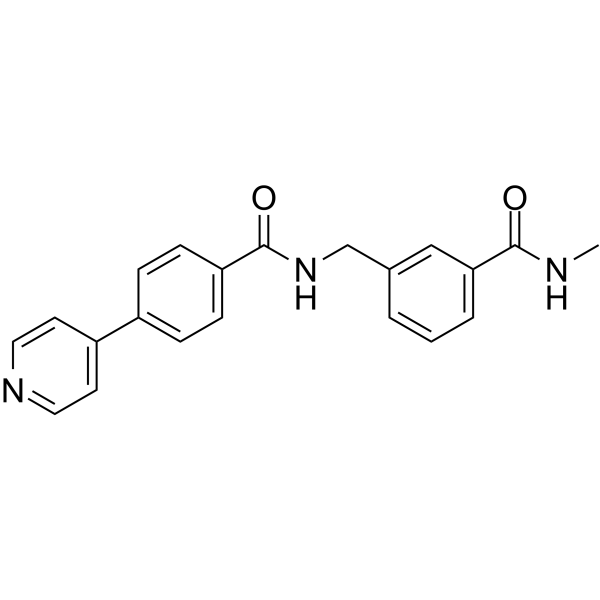
- HY-13418A
-
Dorsomorphin
Maximum Cited Publications
754 Publications Verification
Compound C; BML-275
|
Organoid
AMPK
TGF-β Receptor
Autophagy
|
Cancer
|
|
Dorsomorphin (Compound C) is a selective and ATP-competitive AMPK inhibitor (Ki=109 nM in the absence of AMP). Dorsomorphin (BML-275) selectively inhibits BMP type I receptors ALK2, ALK3, and ALK6. Dorsomorphin can reverse autophagy activation and anti-inflammatory effect of Urolithin A (HY-100599) .
|
-
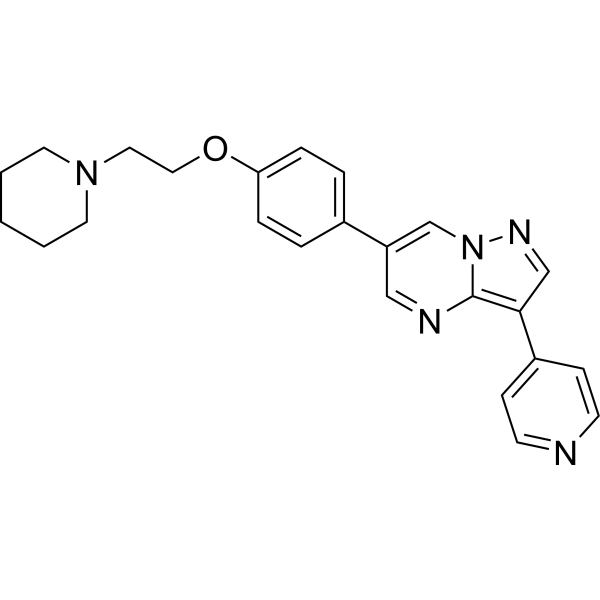
- HY-100616
-
|
cis-1-Aminocyclobutane-1,3-dicarboxylic acid
|
Sodium Channel
|
Neurological Disease
|
|
cis-ACBD is a potent and selective inhibitor of the high-affinity, Na +-dependent plasma membrane glutamate transporter. cis-ACBD is a glutamate reuptake inhibitor. cis-ACBD also acts as linear competitive inhibitor of the uptake of D-[3H]aspartate .
|
-
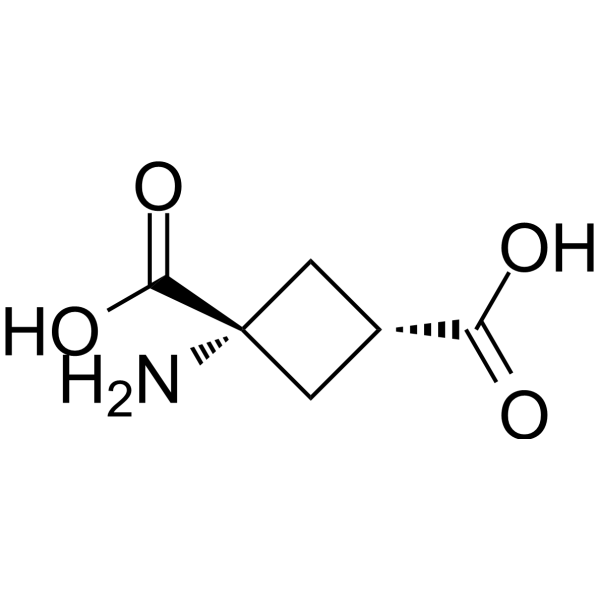
- HY-122060
-
|
|
NO Synthase
|
Metabolic Disease
|
|
BYK 191023 is a selective and L-arginine competitive inducible nitric-oxide synthase inhibitor with an IC50 value of 86 nM, 17 µM, 162 µM for inducible (iNOS), neuronal (nNOS), and endothelial (eNOS) NO synthases respectively .
|
-

- HY-12866
-
|
LOXO-101; ARRY-470
|
Trk Receptor
Apoptosis
|
Cancer
|
|
Larotrectinib (LOXO-101) is an ATP-competitive oral, selective inhibitor of the tropomyosin-related kinase (TRK) family receptors, with low nanomolar 50% inhibitory concentrations against all three isoforms (TRKA, B, and C).
|
-
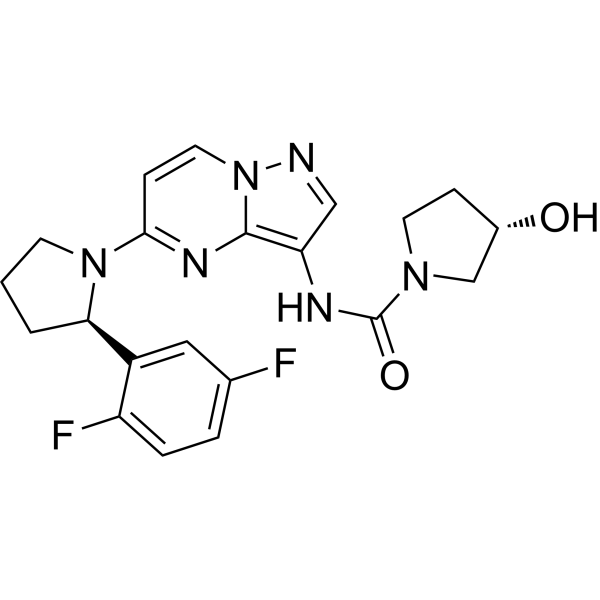
- HY-171962A
-
|
|
Phospholipase
GHSR
|
Neurological Disease
|
|
KARI 201 hydrochloride is a selective, brain penetrant pand competitive acid sphingomyelinase (ASM) inhibitor with an IC50 of 338.3?nM. KARI 201 hydrochloride is a ghrelin receptor agonist. KARI 201 hydrochloride improves neuropathological features of Alzheimer's disease .
|
-

- HY-12493
-
|
|
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
LY-2584702 free base is a selective ATP competitive inhibitor of p70S6K with an IC50 of 4 nM. In S6K1 enzyme assay, the IC50 of LY-2584702 is 2 nM.
|
-
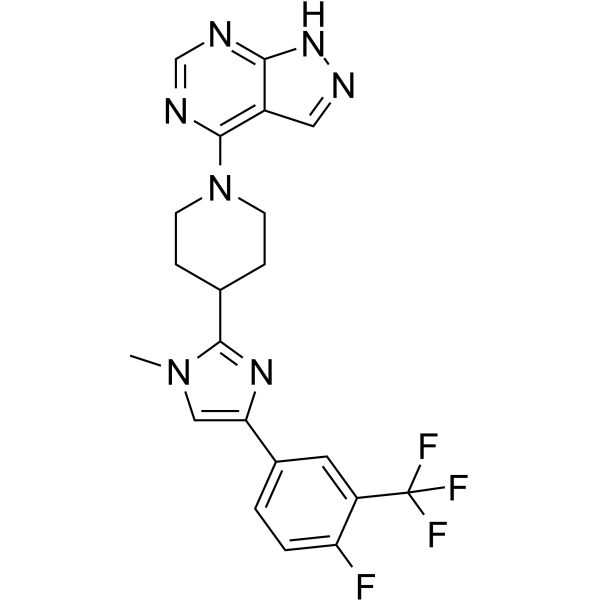
- HY-112668
-
|
SP2086 phosphate
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Retagliptin phosphate (SP2086 phosphate) is a selective, competitive and orally active dipeptidyl peptidase-4 (DPP-4) inhibitor. Retagliptin phosphate can be used for type 2 diabetes mellitus (T2DM) research .
|
-
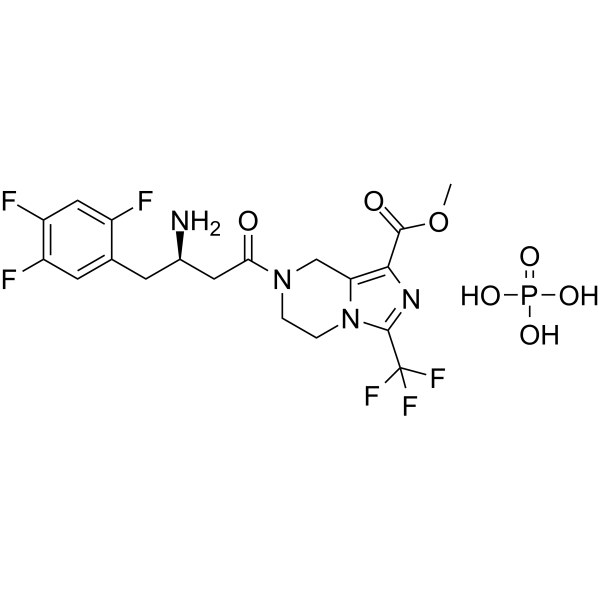
- HY-14691
-
|
BAY 869766; RDEA119
|
MEK
|
Cancer
|
|
Refametinib (BAY 869766; RDEA119) is an orally available, potent, non-ATP-competitive, selective, allosteric MEK1/MEK2 inhibitor with IC50s of 19 nM and 47 nM, respectively.
|
-
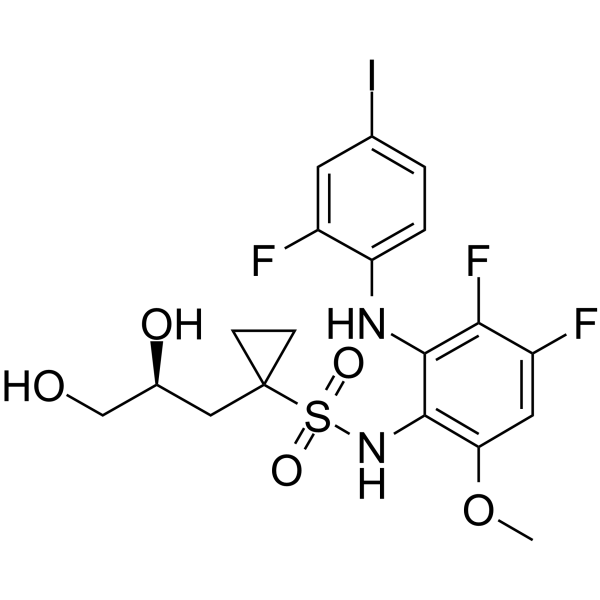
- HY-10515
-
|
|
PDK-1
|
Cancer
|
|
BX-320 is a selective, ATP-competitive, orally acitive, and direct PDK1 inhibitor with an IC50 of 30 nM in a direct kinase assay format. BX-320 also induces apoptosis. Anticancer effect .
|
-
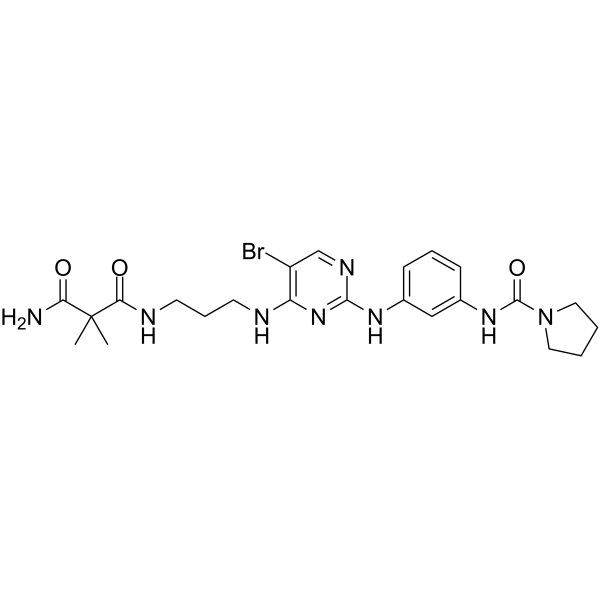
- HY-112668A
-
|
SP2086
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Retagliptin (SP2086) is a selective, competitive and orally active dipeptidyl peptidase-4 (DPP-4) inhibitor. Retagliptin can be used for type 2 diabetes mellitus (T2DM) research .
|
-
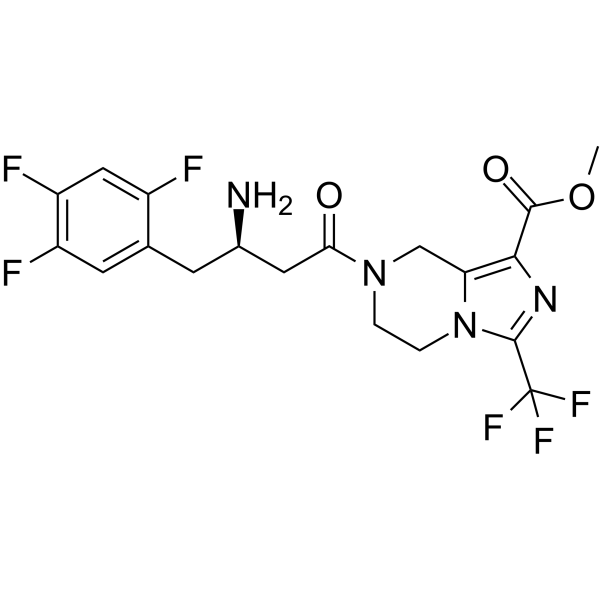
- HY-171962
-
|
|
Phospholipase
GHSR
|
Neurological Disease
|
|
KARI 201 is a selective, brain penetrant pand competitive acid sphingomyelinase (ASM) inhibitor with an IC50 of 338.3?nM. KARI 201 is a ghrelin receptor agonist. KARI 201 improves neuropathological features of Alzheimer's disease .
|
-

- HY-16446
-
|
|
c-Met/HGFR
PI3K
Akt
MEK
Apoptosis
|
Cancer
|
|
SAR125844 is a potent, selective, and ATP-competitive MET kinase inhibitor with the value of IC50 is 4.2 nM and Ki is 2.8 nM. SAR125844 has antitumor activity and can be used for the research of cancer .
|
-
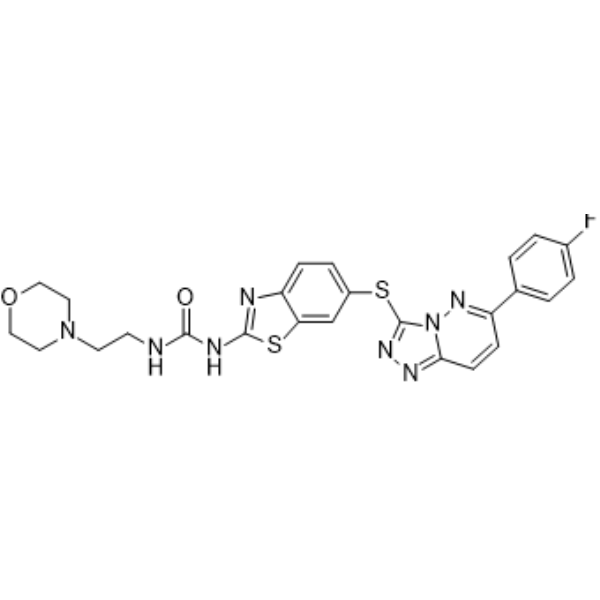
- HY-101053R
-
|
Src Kinase inhibitor 1 (Standard); Src-l1 (Standard)
|
Src
Reference Standards
|
Cancer
|
|
Src Inhibitor 1 (Standard) is the analytical standard of Src Inhibitor 1 (HY-101053). This product is intended for research and analytical applications. Src Inhibitor 1 is a potent, ATP-competitive and selective dual site Src tyrosine kinase inhibitor with IC50 values of 44 nM for Src and 88nM for Lck.
|
-

- HY-10917
-
|
|
c-Fms
|
Inflammation/Immunology
Cancer
|
|
GW2580 is an orally bioavailable and selective inhibitor of c-Fms kinase which completely inhibits human cFMS kinase in vitro at 0.06 μM. GW2580 acts as a competitive inhibitor of ATP binding to the cFMS kinase and inhibits colony-stimulating-factor-1 signaling .
|
-
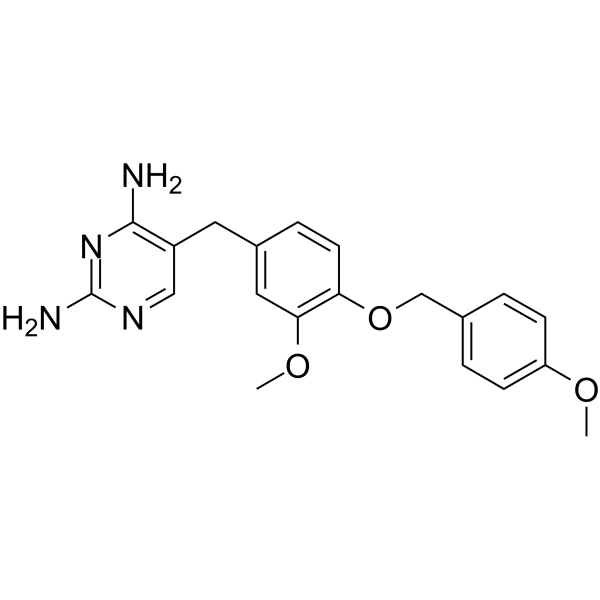
- HY-18086
-
|
SC 204330
|
Pim
|
Cancer
|
|
TCS PIM-1 1 (SC 204330) is a potent, selective and ATP-competitive Pim-1 kianse inhibitor with an IC50 of 50 nM, displays good selectivity over Pim-2 and MEK1/MEK2 (IC50s >20000 nM) .
|
-
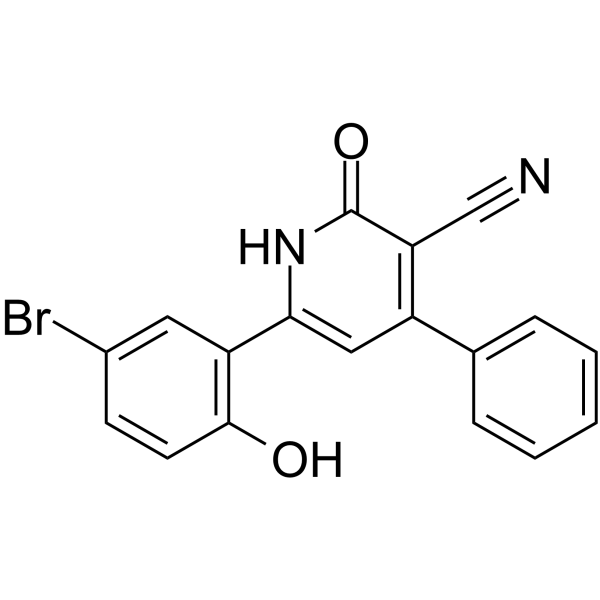
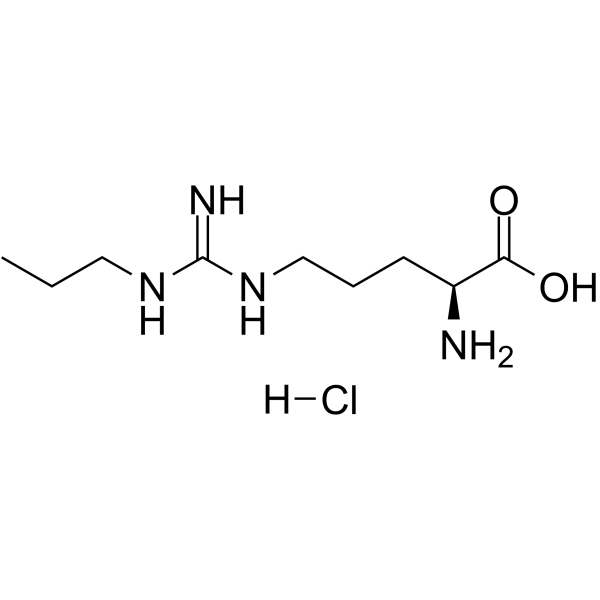
- HY-102062A
-
|
N-omega-Propyl-L-arginine hydrochloride
|
NO Synthase
|
Neurological Disease
|
|
Nω-Propyl-L-arginine (N-omega-Propyl-L-arginine) hydrochloride is a potent, competitive, and highly selective inhibitor of neuronal nitric oxide synthase (nNOS), with a Ki of 57 nM. Nω-Propyl-L-arginine hydrochloride displays a 149-fold selectivity for nNOS over endothelial NOS (eNOS) .
|
-
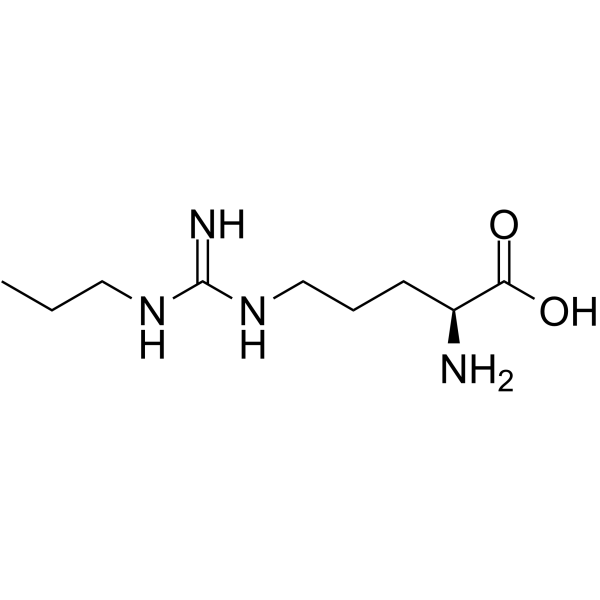
- HY-102062
-
|
N-omega-Propyl-L-arginine
|
NO Synthase
|
Neurological Disease
|
|
Nω-Propyl-L-arginine (N-omega-Propyl-L-arginine) is a potent, competitive, and highly selective inhibitor of neuronal nitric oxide synthase (nNOS), with a Ki of 57 nM. Nω-Propyl-L-arginine displays a 149-fold selectivity for nNOS over endothelial NOS (eNOS) .
|
-
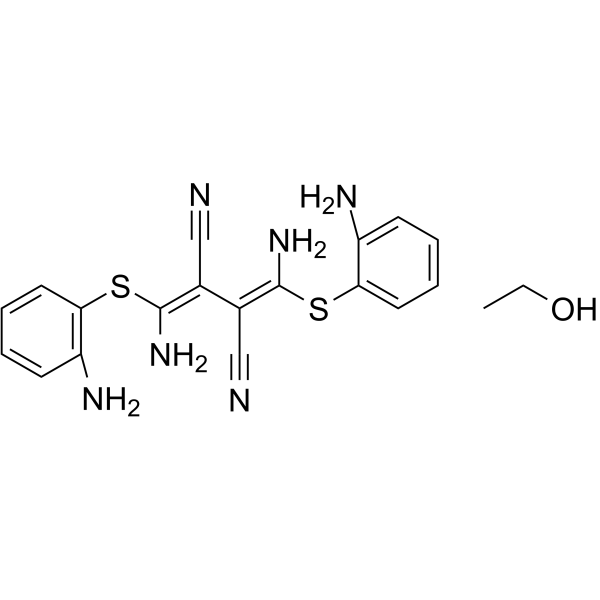
- HY-12031
-
|
|
MEK
Autophagy
Mitophagy
Influenza Virus
|
Cancer
|
|
U0126 (U0126-EtOH) is a potent, non-ATP competitive and selective MEK1 and MEK2 inhibitor, with IC50s of 72 nM and 58 nM, respectively. U0126 is an autophagy and mitophagy inhibitor .
|
-
- HY-125017
-
|
PLB-1001; CBT-101; Vebreltinib
|
c-Met/HGFR
|
Cancer
|
|
Bozitinib (PLB-1001) is a highly selective c-MET kinase inhibitor with blood-brain barrier permeability. Bozitinib (PLB-1001) is a ATP-competitive small-molecule inhibitor, binds to the conventional ATP-binding pocket of the tyrosine kinase superfamily .
|
-
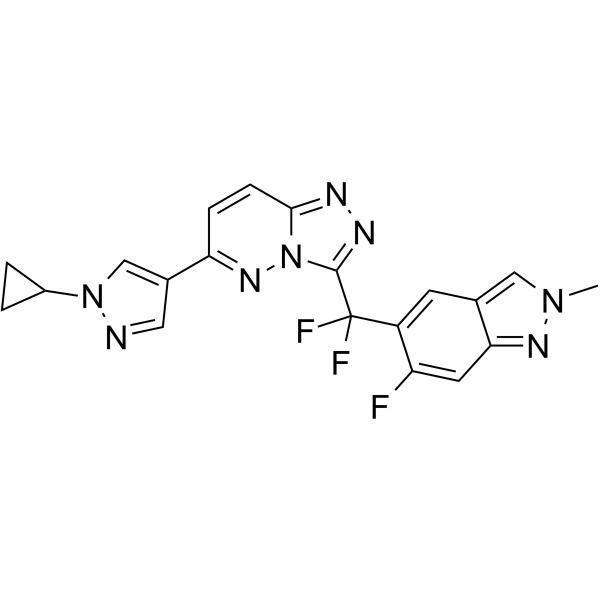
- HY-P1178A
-
|
|
Trk Receptor
|
Neurological Disease
|
|
Cyclotraxin B TFA is a BBB-penetrable and selective TrkB inhibitor. Cyclotraxin B TFA inhibits BDNF-induced TrkB activity in a non-competitive manner, with an IC50 of 0.30 nM. Cyclotraxin B TFA has analgesic and anxiolytic effects .
|
-
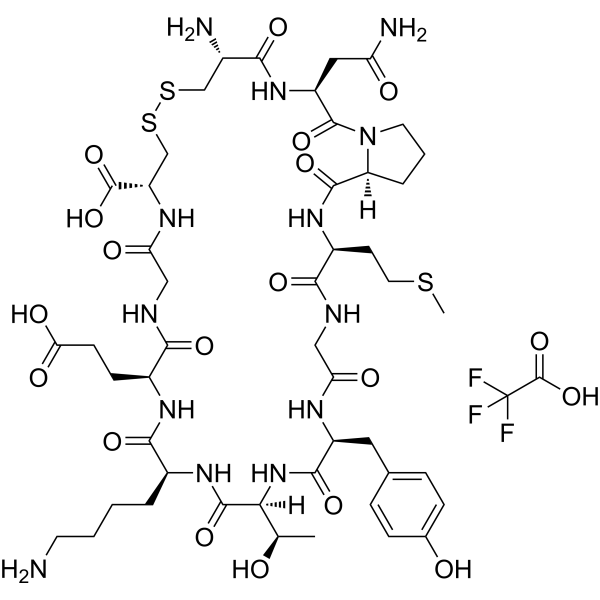
- HY-136270
-
|
VX-803; M4344; ATR inhibitor 2
|
ATM/ATR
|
Cancer
|
|
Gartisertib (VX-803) is an ATP-competitive, orally active, and selective ATR inhibitor, with a Ki of <150 pM. Gartisertib potently inhibits ATR-driven phosphorylated checkpoint kinase-1 (Chk1) phosphorylation with an IC50 of 8 nM. Antitumor activity .
|
-
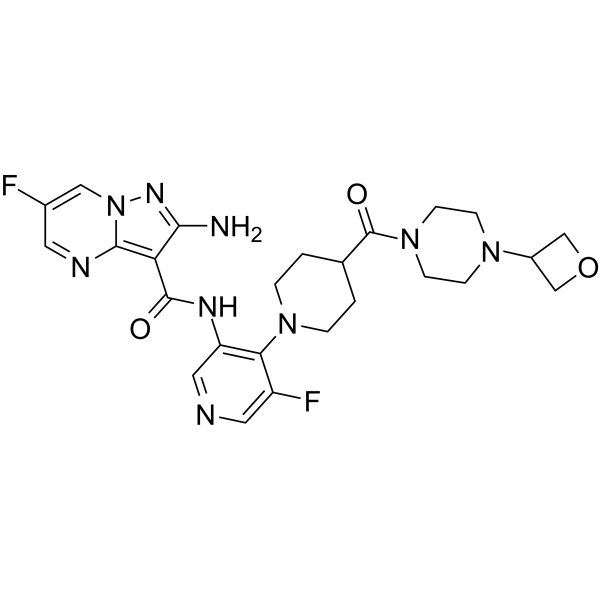
- HY-18963
-
|
RG-14355
|
EGFR
|
Cancer
|
|
Lavendustin A (RG-14355) is a potent, selective and ATP-competitive inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase, with an IC50 of 11 nM. Lavendustin A does not inhibit protein kinase A or C. Lavendustin A can suppress VEGF-induced angiogenesis .
|
-
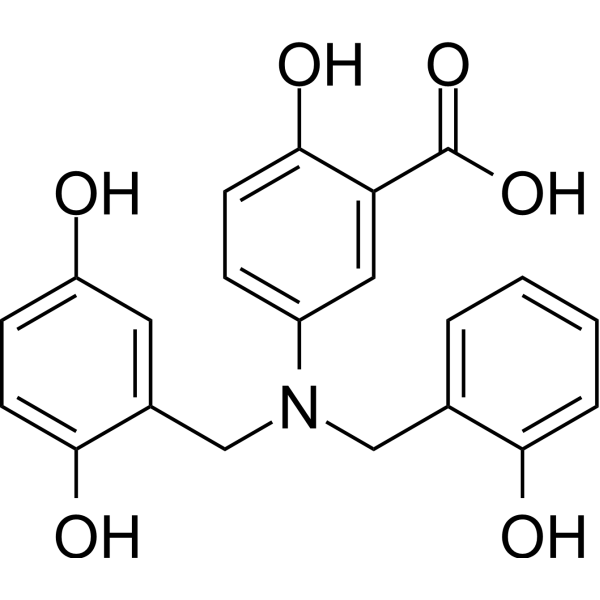
- HY-13531
-
|
|
PI3K
|
Cancer
|
|
AS-604850 is a potent, selective and ATP-competitive PI3Kγ inhibitor with an IC50 value of 0.25 μM and a Ki value of 0.18 μM. AS-604850 shows isoform selective inhibitor of PI3Kγ with over 30-fold selectivity for PI3Kδ and β, and 18-fold selectivity over PI3Kα, respectively .
|
-
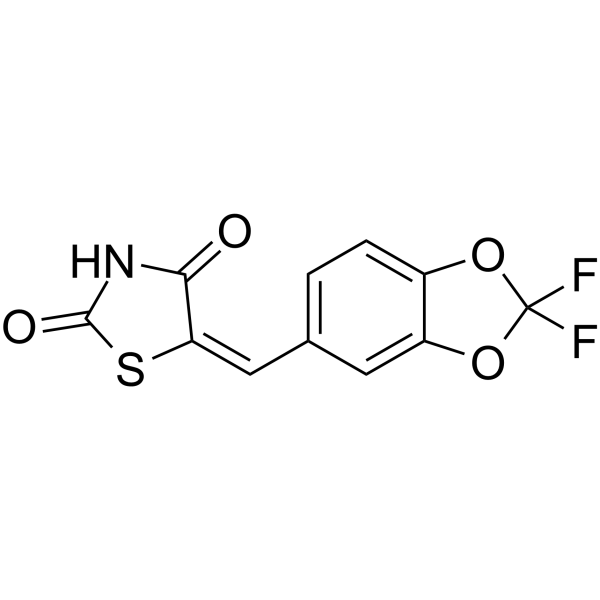
- HY-112136
-
|
CP 43
|
MAP3K
Mitosis
|
Cancer
|
|
TAO Kinase inhibitor 1 (compound 43) is a selective, ATP-competitive thousand-and-one amino acid kinases (TAOK) inhibitor with IC50s of 11 to 15 nM for TAOK1 and 2, respectively. TAO Kinase inhibitor 1 delays mitosis and induces mitotic cell death .
|
-
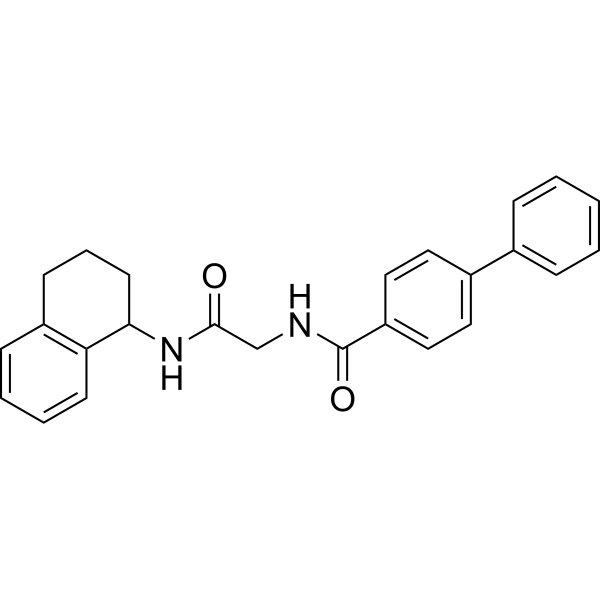
- HY-14721
-
|
EMD-1214063
|
c-Met/HGFR
Autophagy
|
Cancer
|
|
Tepotinib (EMD-1214063) is an orally active and highly selective, reversible, ATP-competitive c-Met inhibitor with an IC50 of 3 nM, >200-fold selective for c-Met than IRAK4, TrkA, Axl, IRAK1, and Mer. Tepotinib inhibits c-Met phosphorylation and induces autophagy. Tepotinib has antitumor effects .
|
-
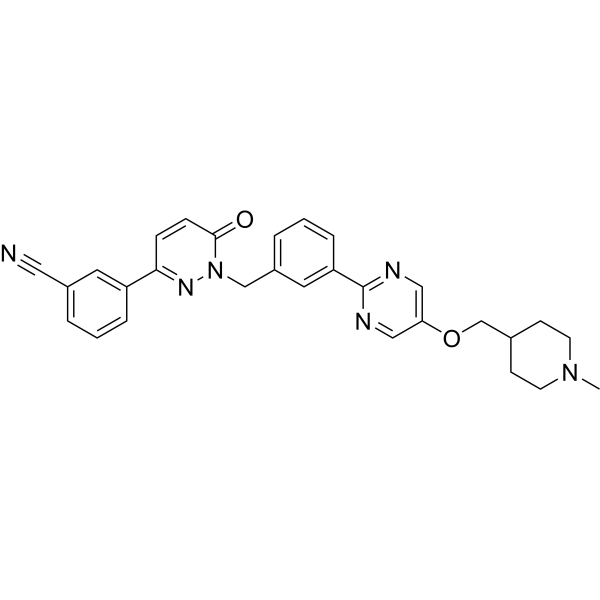
- HY-10335
-
|
|
VEGFR
FGFR
|
Cardiovascular Disease
Inflammation/Immunology
|
|
BMS-645737 is an orally active, selective FGF receptor-1 and VEGF receptor-2 inhibitor. BMS-645737 selectively and competitively inhibits both VEGFR-2 and FGFR-1 tyrosine kinases. BMS-645737 has anti-angiogenic activity. BMS-645737 induces lesions in the incisor teeth .
|
-

- HY-15237
-
|
SL0101
|
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
SL 0101-1 (SL0101), a kaempferol glycoside, isolated from the tropical plant F. refracta, is a cell-permeable, selective, reversible, ATP-competitive p90 Ribosomal S6 Kinase (RSK) inhibitor, with an IC50 of 89 nM . SL 0101-1 (SL0101) is a selective RSK1/2 inhibitor, with a Ki of 1 μM .
|
-
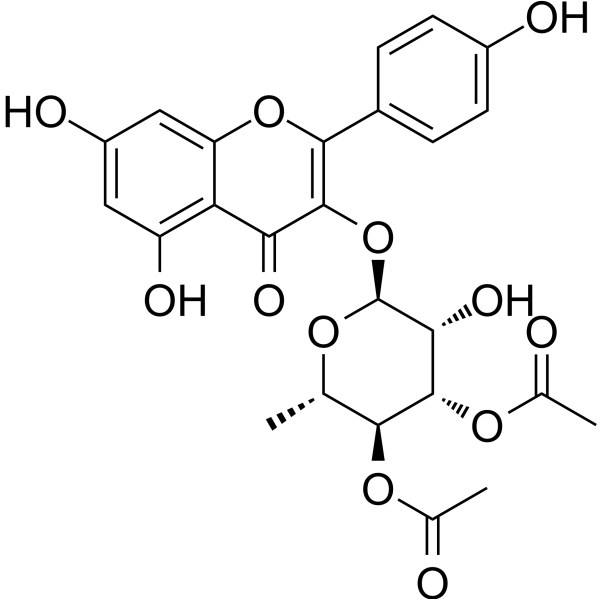
- HY-15959
-
|
Volitinib; HMPL-504; AZD-6094
|
c-Met/HGFR
|
Cancer
|
|
Savolitinib (AZD-6094) is a potent, highly selective, and orally bioavailable c-Met inhibitor with IC50 s of 5 nM and 3 nM for c-Met and p-Met, respectively. Savolitinib (AZD-6094) selectively binds to and inhibits the activation of c-Met in an ATP-competitive manner, and disrupts c-Met signal transduction pathways. Antineoplastic activity .
|
-
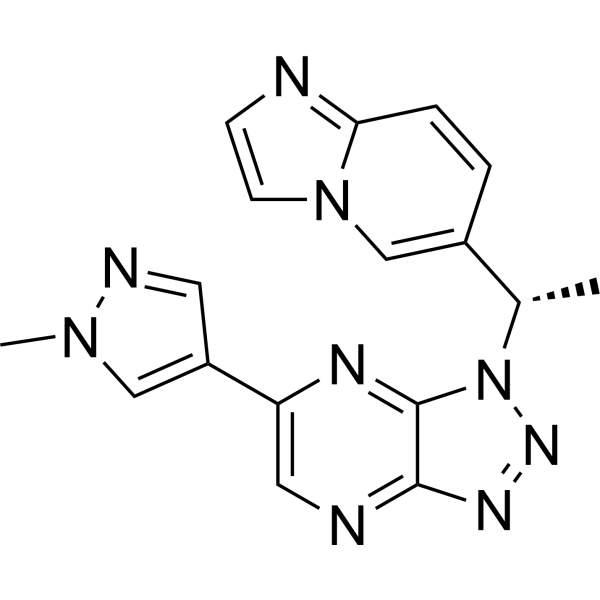
- HY-14721A
-
|
EMD-1214063 hydrochloride
|
c-Met/HGFR
Autophagy
|
Cancer
|
|
Tepotinib (EMD-1214063) hydrochloride is an orally active and highly selective, reversible, ATP-competitive c-Met inhibitor with an IC50 of 3 nM, >200-fold selective for c-Met than IRAK4, TrkA, Axl, IRAK1, and Mer. Tepotinib hydrochloride inhibits c-Met phosphorylation and induces autophagy. Tepotinib hydrochloride has antitumor effects .
|
-

- HY-15272
-
|
|
mTOR
|
Inflammation/Immunology
Cancer
|
|
WAY-600 is a potent, ATP-competitive, and selective mTOR inhibitor with an IC50 of 9 nM for recombinant mTOR enzyme. WAY-600 blocks mTOR complex 1/2 (mTORC1/2) assemble and activation.
|
-
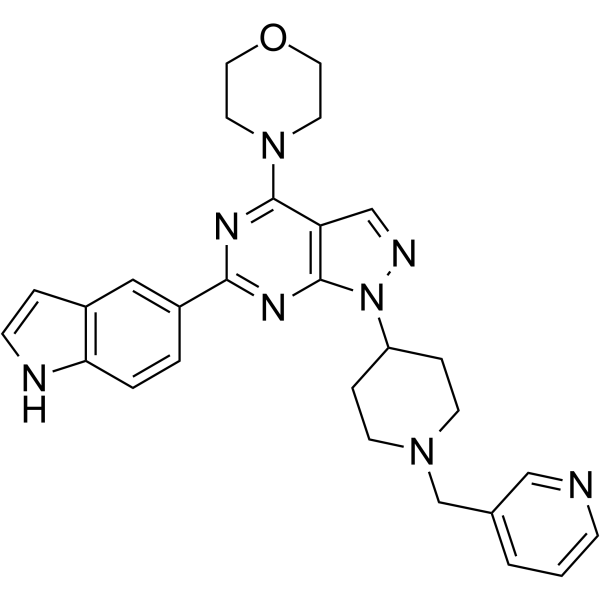
- HY-10716A
-
|
|
GlyT
|
Neurological Disease
|
|
PF-03463275 is a centrally penetrant, orally available, selective, and competitive GlyT1 (glycine transporter-1) reversible inhibitor, with a Ki of 11.6 nM. PF-03463275 has the potential for Schizophrenia research .
|
-
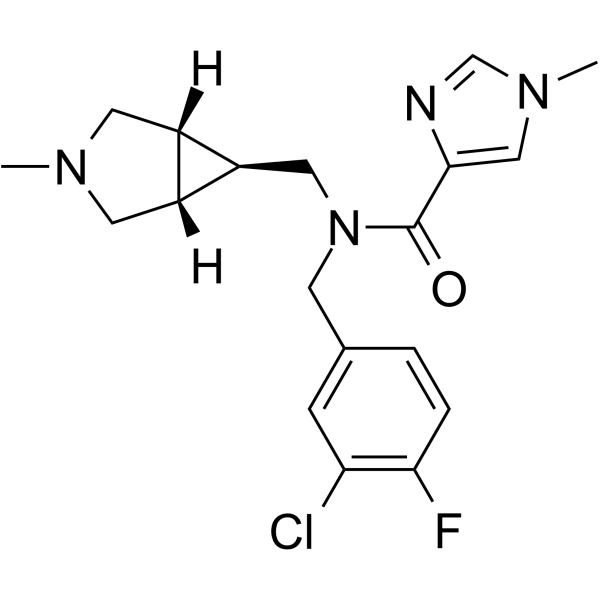
- HY-100714A
-
|
D-APV; D-2-Amino-5-phosphonovaleric acid
|
iGluR
|
Neurological Disease
|
|
D-AP5 (D-APV) is a selective and competitive NMDA receptor antagonist with a Kd of 1.4 μM. D-AP5 (D-APV) inhibits the glutamate binding site of NMDA receptors .
|
-
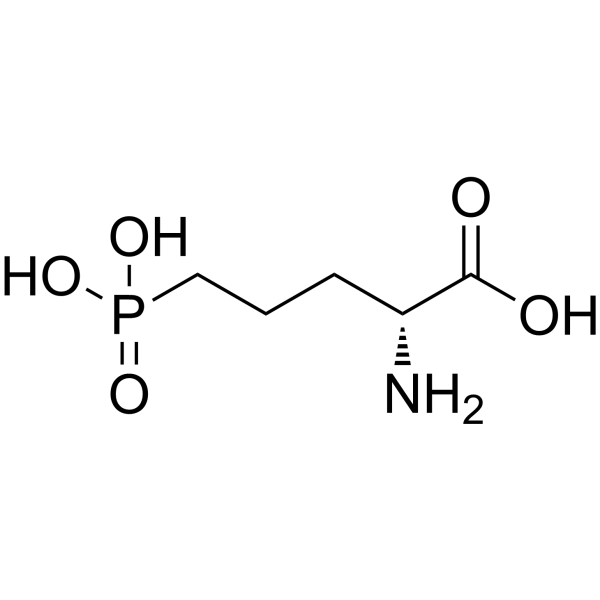
- HY-10716
-
|
|
GlyT
|
Neurological Disease
|
|
PF-03463275 hydrochloride is a centrally penetrant, orally available, selective, and competitive GlyT1 (glycine transporter-1) reversible inhibitor, with a Ki of 11.6 nM. PF-03463275 hydrochloride has the potential for Schizophrenia research .
|
-

- HY-14117
-
-
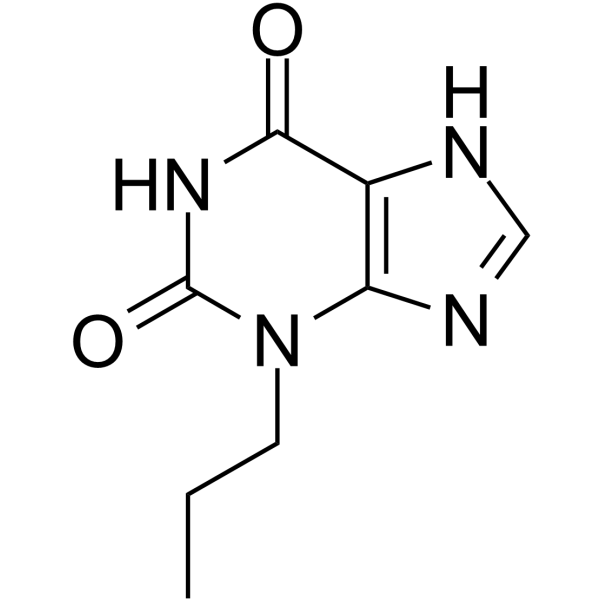
- HY-13634B
-
-
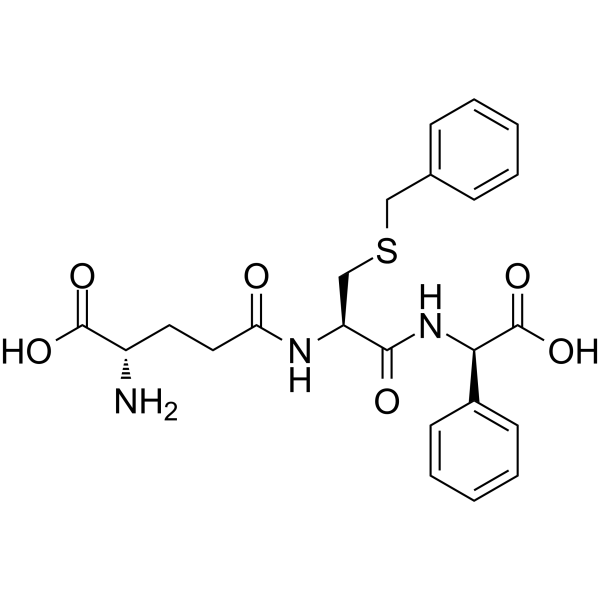
- HY-18780
-
RR6
3 Publications Verification
|
Pantetheinase
|
Cancer
|
|
RR6 is a potent, selective, reversible, competitive and orally active vanin inhibitor with an IC50 of 540 nM for recombinant vanin-1. RR6 also potently inhibits human, bovine and rat serum pantetheinase with IC50 values of 40 nM, 41 nM and 87 nM, respectively .
|
-
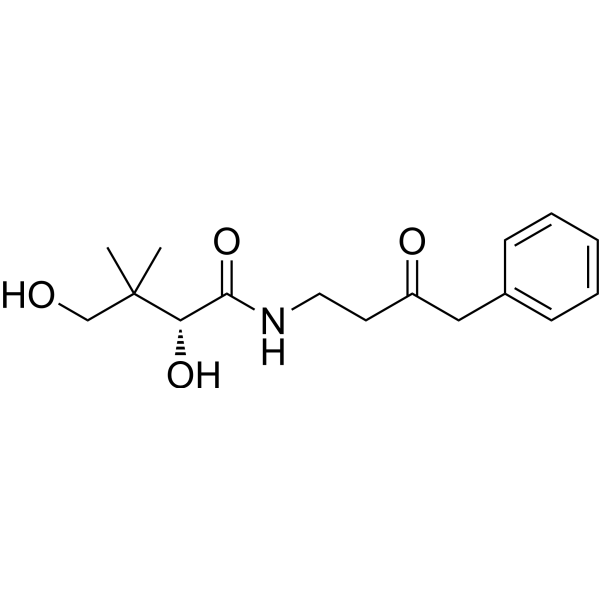
- HY-50846
-
|
|
ERK
|
Cancer
|
|
SCH772984 is a highly selective and ATP-competitive ERK inhibitor, with IC50s of 4 and 1 nM for ERK1 and ERK2, respectively. SCH772984 has antitumor activity in MAPK inhibitor-na?ve and MAPK inhibitor-resistant cells containing BRAF or RAS mutations .
|
-
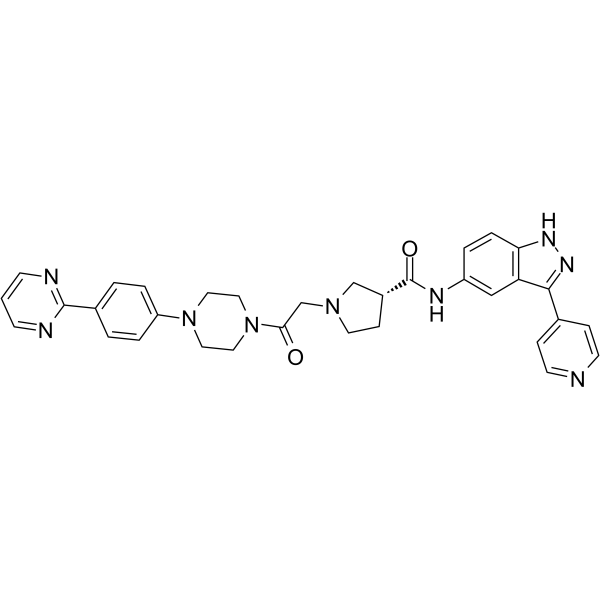
- HY-170798
-
|
|
Monoamine Oxidase
SOD
Glutathione Peroxidase
|
Neurological Disease
|
|
Monoamine Oxidase B inhibitor 6 (Compound BT5) is a BBB-penetrable, highly selective, reversible and competitive MAO-B inhibitor, with an IC50 of 0.11 μM. Monoamine Oxidase B inhibitor 6 has antioxidant and neuroprotective effects and can be used in the research of neurodegenerative diseases .
|
-

- HY-162109
-
|
|
Thrombin
|
Cardiovascular Disease
|
|
Thrombin inhibitor 11 (Compound 46) is an orally active, competitive and selective α-Thrombin inhibitor, with a Ki value of 65 nM against h-αThrombin and a Ki value of 10.3 nM against rat-derived α-thrombin. Thrombin inhibitor 11 can be used for the research of thrombotic diseases .
|
-
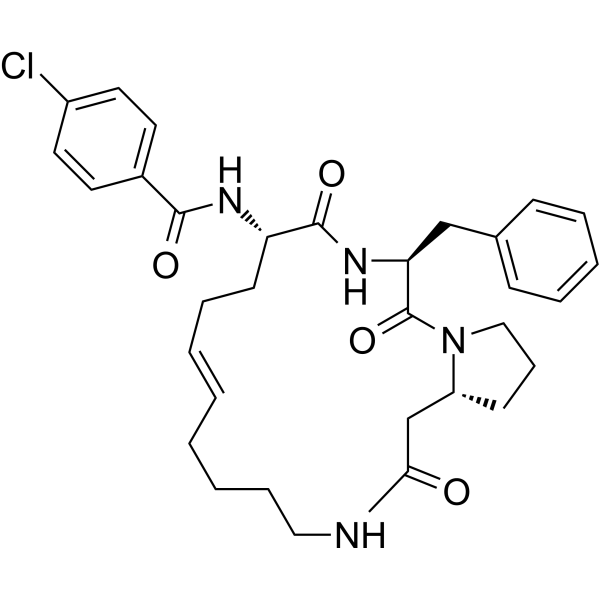
- HY-112390C
-
|
|
5-HT Receptor
Syk
|
Inflammation/Immunology
|
|
Syk Inhibitor II hydrochloride is a potent, high selective and ATP-competitive Syk inhibitor with an IC50 of 41 nM. Syk Inhibitor II hydrochloride inhibits 5-HT release from RBL-cells with an IC50 of 460 nM. Syk Inhibitor II hydrochloride shows less potent against other kinases and has anti-allergic effect .
|
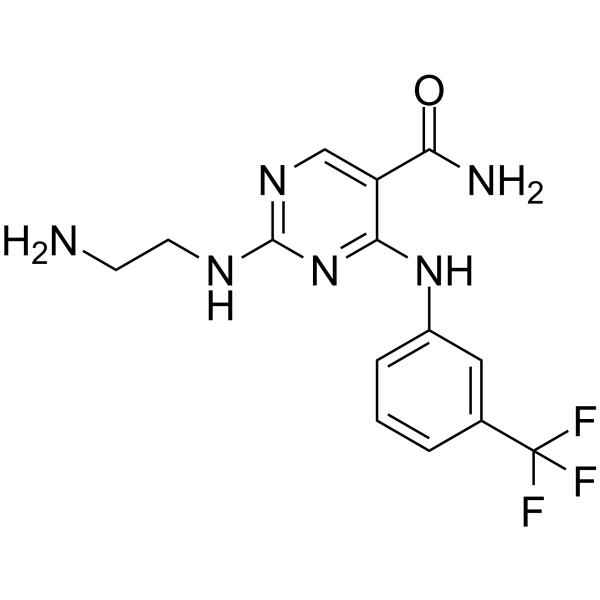
-

- HY-112390A
-
|
|
Syk
5-HT Receptor
|
Inflammation/Immunology
|
|
Syk Inhibitor II is a potent, high selective and ATP-competitive Syk inhibitor with an IC50 of 41 nM. Syk Inhibitor II inhibits 5-HT release from RBL-cells with an IC50 of 460 nM. Syk Inhibitor II shows less potent against other kinases and has anti-allergic effect .
|
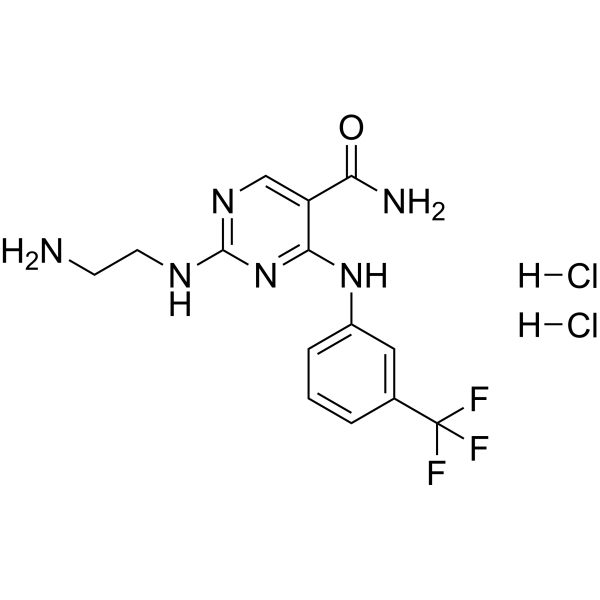
-
- HY-112390
-
|
|
5-HT Receptor
Syk
|
Inflammation/Immunology
|
|
Syk Inhibitor II dihydrochloride is a potent, high selective and ATP-competitive Syk inhibitor with an IC50 of 41 nM. Syk Inhibitor II dihydrochloride inhibits 5-HT release from RBL-cells with an IC50 of 460 nM. Syk Inhibitor II dihydrochloride shows less potent against other kinases and has anti-allergic effect .
|
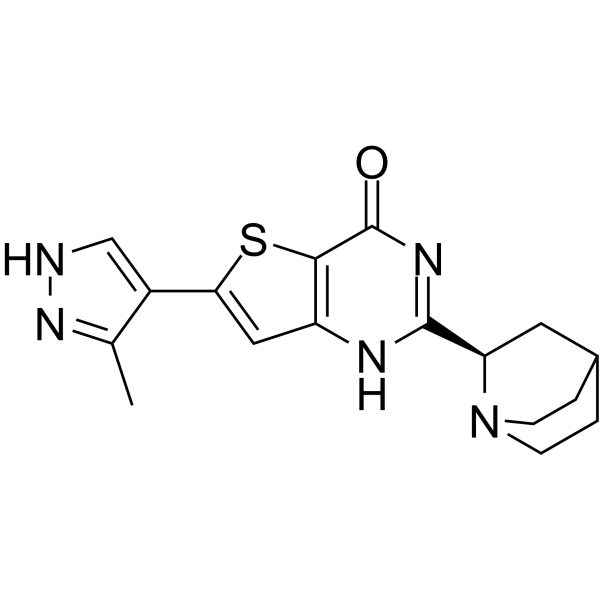
-
- HY-180243
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
AChE-IN-102 (compound C8) is a potent, selective and competitive AChE inhibitor with a Ki of 7.55 nM and an IC50 of 15.25 nM. AChE-IN-102 shows selectivity over BuChE (IC50 = 21.15 nM, b>Ki = 6.17 nM). AChE-IN-102 can be used for Alzheimer’s disease research .
|
-

- HY-N14540
-
|
|
Phosphatase
Akt
|
Cancer
|
|
Glycybenzofuran is a flavonoid. Glycybenzofuran can be isolated from G. uralensis. Glycybenzofuran is a potent, competitive, selective PTP1B inhibitor. Glycybenzofuran shows excellent inhibitory selectivity against PTP1B. Glycybenzofuran increases insulin-stimulated pAkt levels. Glycylbenzofuran can be used in the research of hepatocellular carcinoma .
|
-

- HY-100888A
-
|
(R)-TAK-931
|
CDK
|
Others
|
|
(R)-Simurosertib ((R)-TAK-931) is the (R)-enantiomer of Simurosertib. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM .
|
-
- HY-12219A
-
|
Trodusquemine lactate; Aminosterol-1436 lactate
|
Phosphatase
|
Endocrinology
|
|
MSI-1436 lactate is a selective, non-competitive inhibitor of the enzyme protein-tyrosine phosphatase 1B (PTP1B), with an IC50 of 1 μM, 200-fold preference over TCPTP (IC50 of 224 μM).
|
-
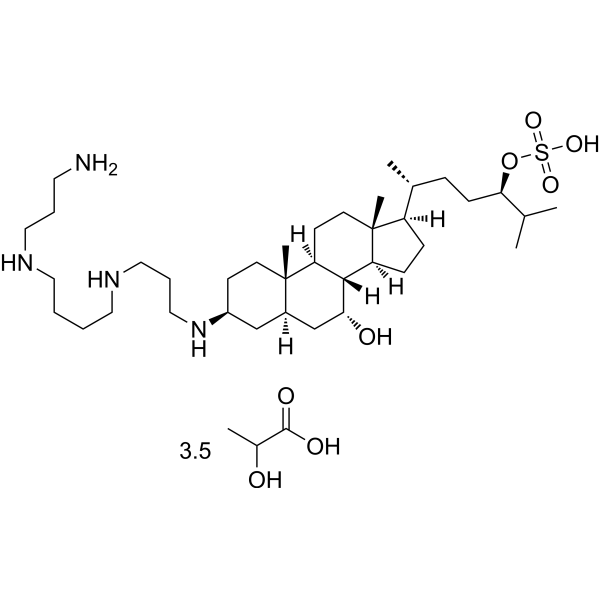
- HY-15260
-
|
BMS-863233
|
CDK
|
Cancer
|
|
XL413 is a potent, selective and ATP competitive inhibitor of Cdc7, with an IC50 of 3.4 nM, and also shows potent effect with IC50s of 215, 42 nM on CK2, PIM1, respectively, and an EC50 of 118 nM on pMCM.
|
-
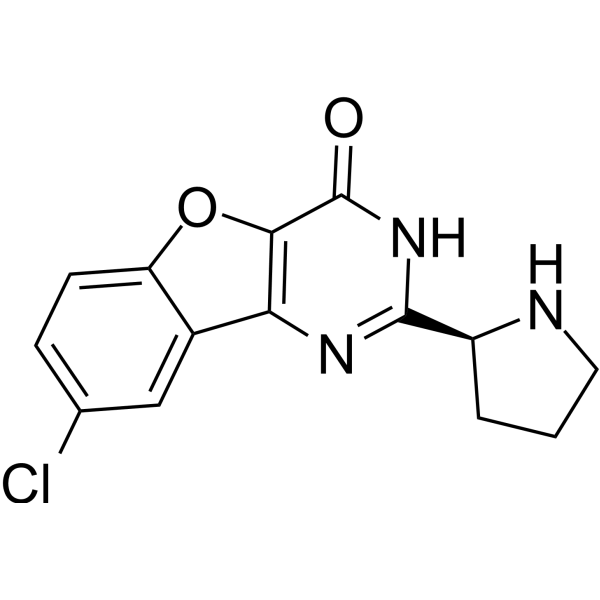
- HY-19794
-
|
|
SphK
|
Cancer
|
|
MP-A08 is a highly selective ATP competitive sphingosine kinase (SPHK1) inhibitor that targets both SphK1 and SphK2 with Ki values of 6.9 ± 0.8 μM and 27 ± 3 μM, respectively.
|
-
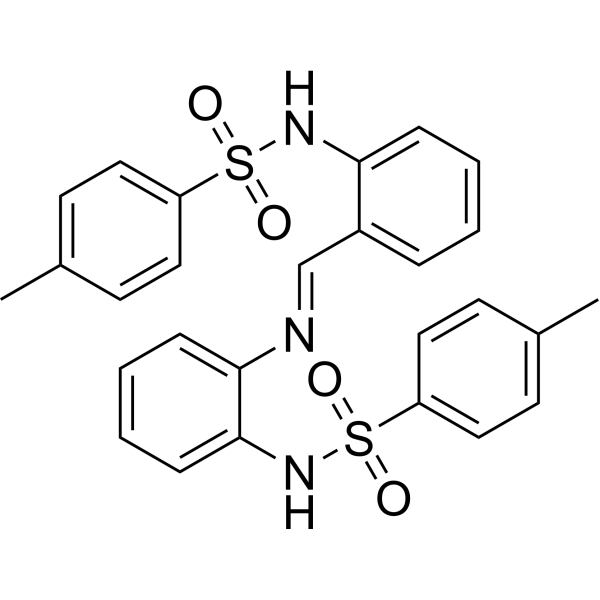
- HY-104009AR
-
|
|
Reference Standards
Histone Methyltransferase
|
Others
|
|
GSK2807 Trifluoroacetate (Standard) is the analytical standard of GSK2807 Trifluoroacetate (HY-104009A). This product is intended for research and analytical applications. GSK2807 Trifluoroacetate is a potent, selective and SAM-competitive inhibitor of SMYD3, with a Ki of 14 nM and an IC50 of 130 nM .
|
-

- HY-15687A
-
|
|
ROCK
|
Metabolic Disease
|
|
SAR407899 is a selective, potent and ATP-competitive ROCK inhibitor, with an IC50 of 135 nM for ROCK-2, and Kis of 36 nM and 41 nM for human and rat ROCK-2, respectively. SAR407899 shows stable inhibition of migrasome formation.
|
-
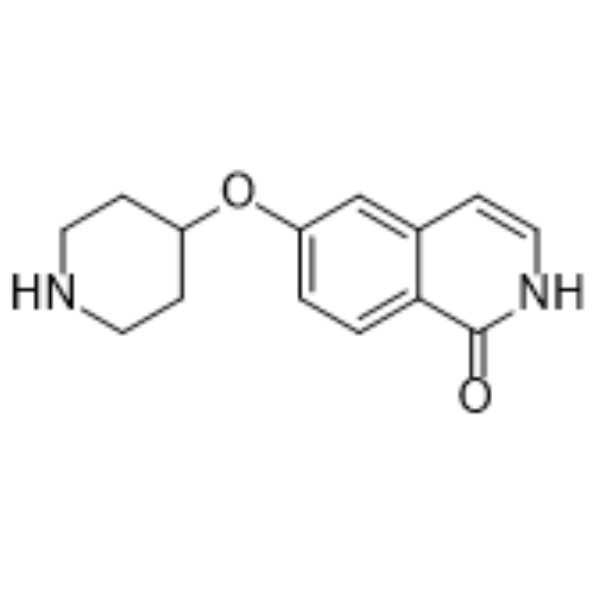
- HY-12122A
-
|
|
NO Synthase
|
Neurological Disease
Inflammation/Immunology
|
|
AR-C102222 hydrochloride is a potent, competitive, orally active and highly selective inducible nitric oxide synthase (iNOS) inhibitor, with an IC50 of 37 nM . AR-C102222 hydrochloride has antinociception and anti-inflammatory activities .
|
-
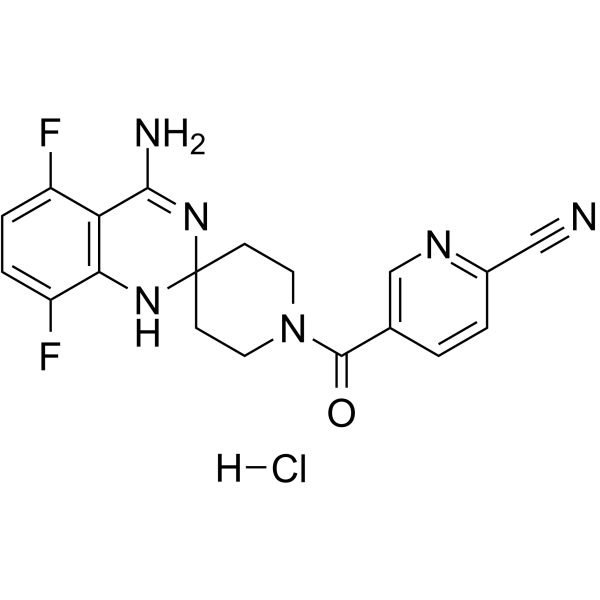
- HY-12219
-
|
Trodusquemine; Aminosterol-1436
|
Phosphatase
|
Endocrinology
|
|
MSI-1436 is a selective, non-competitive inhibitor of the enzyme protein-tyrosine phosphatase 1B (PTP1B), with an IC50 of appr 1 μM, 200-fold preference over TCPTP (IC50, 224 μM).
|
-
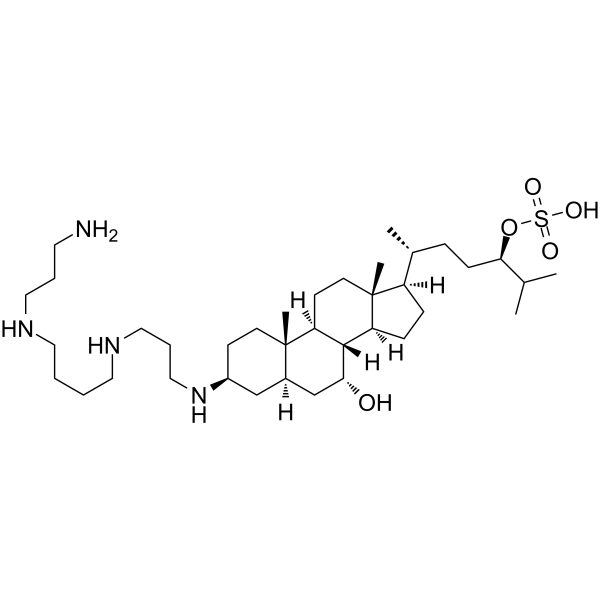
- HY-18990
-
-
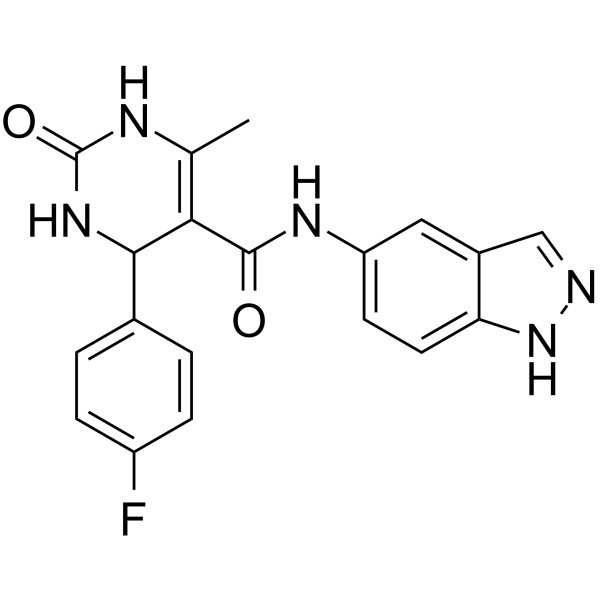
- HY-10195
-
|
LY333531
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
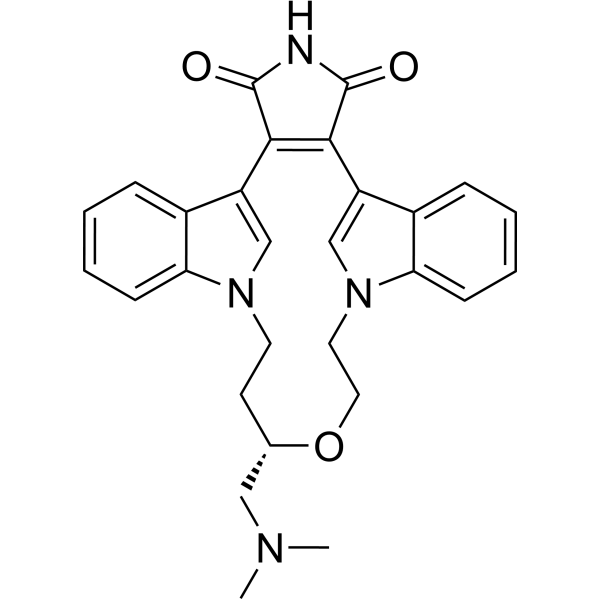
- HY-10195B
-
|
LY333531 hydrochloride
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
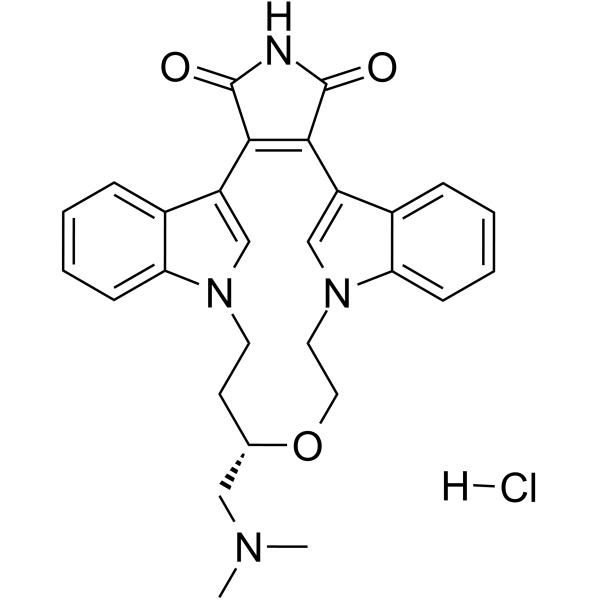
- HY-103021
-
|
|
TGF-β Receptor
|
Inflammation/Immunology
Cancer
|
|
LY3200882 is a potent, highly selective, ATP-competitive and orally active TGF-β receptor type 1 (ALK5) inhibitor with an IC50 of 38.2 nM. LY3200882 inhibits various pro-tumorigenic activities and is also used as an immune modulatory agent .
|
-
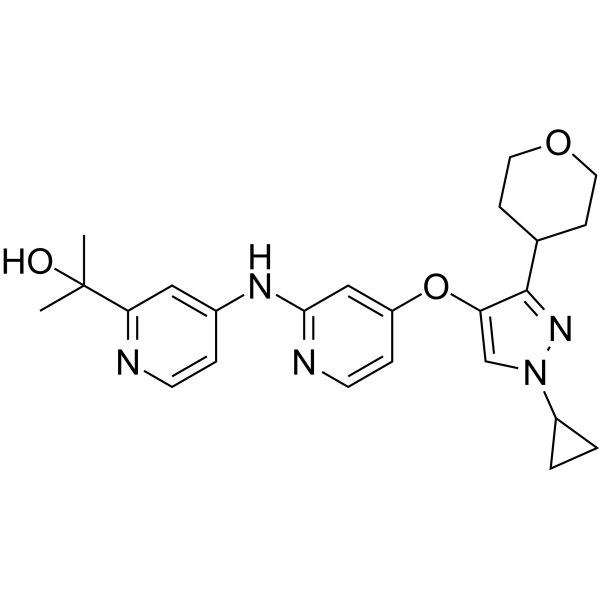
- HY-182481
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
MD-230254 is a reversible, competitive and selective inhibitor inhibitor of MAO-B with an IC50 value of 1.8 nM. MD-230254 can be used for the study of MAO-B-related neurodegenerative diseases including Parkinson’s disease and Alzheimer’s disease .
|
-

- HY-122614
-
S29434
2 Publications Verification
NMDPEF
|
Autophagy
|
Neurological Disease
|
|
S29434 (NMDPEF) is a potent, competitive, selective and cell-permeable inhibitor of quinone reductase 2 (QR2), with IC50s ranging from 5 to 16 nM for human QR2 at different organizational levels, and has good selectivity for QR2 over QR1. S29434 induces autophagy and inhibits QR2-mediated ROS production .
|
-
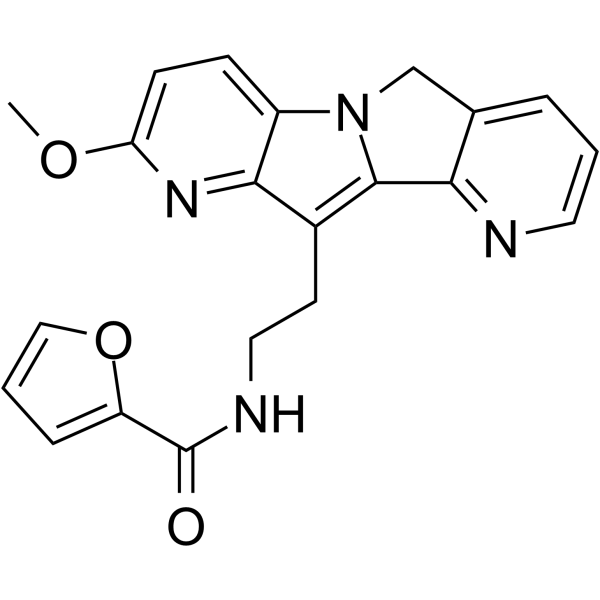
- HY-124366
-
|
|
Phosphatase
|
Infection
Cancer
|
|
Slingshot inhibitor D3 is a potent, selective, reversible and competitive inhibitor of Slingshot. The IC50 value for Slingshot 1 is 3 μM and the Ki value for Slingshot 2 is 3.9 μM. Slingshot inhibitor D3 has similar inhibitory activities toward both Slingshot 1 and Slingshot 2 .
|
-
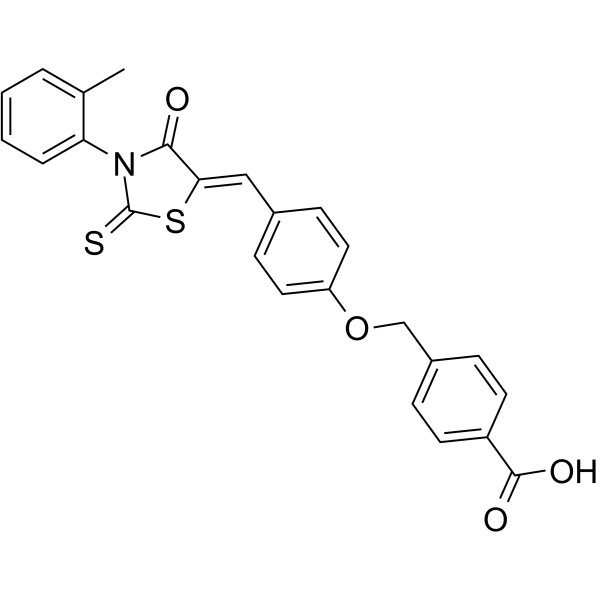
- HY-15658
-
GSK2801
3 Publications Verification
|
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
GSK2801, a chemical probe, is a potent, selective, orally active and cell active acetyl-lysine competitive BAZ2A and BAZ2B bromodomains inhibitor with Kd values of 136 nM and 257 nM, respectively. GSK2801 shows >50-fold selectivity for BAZ2A/B over BRD4 .
|
-
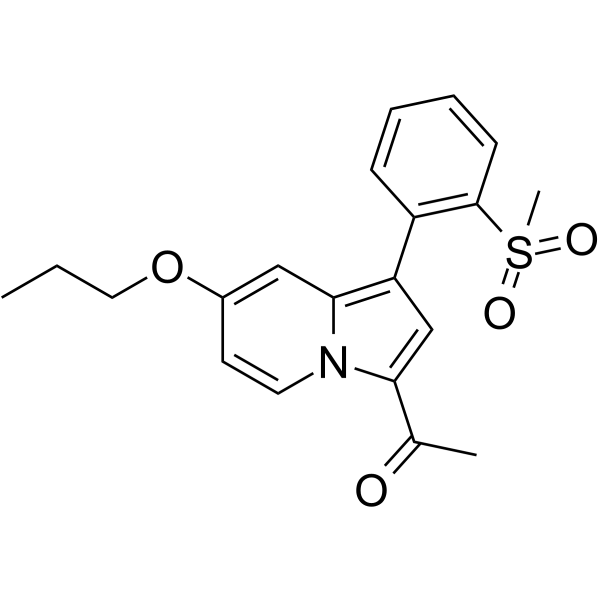
- HY-108540
-
|
2-Amino-2-norbornanecarboxylic acid; LAT1-IN-1
|
Apoptosis
|
Cancer
|
|
BCH (2-Amino-2-norbornanecarboxylic acid) is a selective and competitive inhibitor of large neutral amino acid transporter 1 (LAT1) significantly inhibit cellular uptake of amino acids and mTOR phosphorylation, which induces the suppression of cancer growth and apoptosis .
|
-
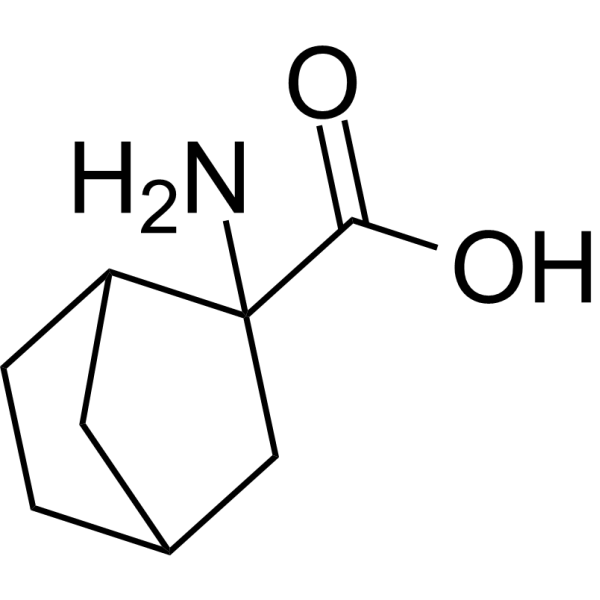
- HY-12275
-
|
|
Flavivirus
Dengue Virus
ERK
Apoptosis
|
Cancer
|
|
FR 180204 is an ATP-competitive and selective ERK inhibitor. FR 180204 inhibits ERK1 and ERK2 with IC50s of 0.51 μM (Ki=0.31 μM) and 0.33 μM (Ki=0.14 μM), respectively .
|
-
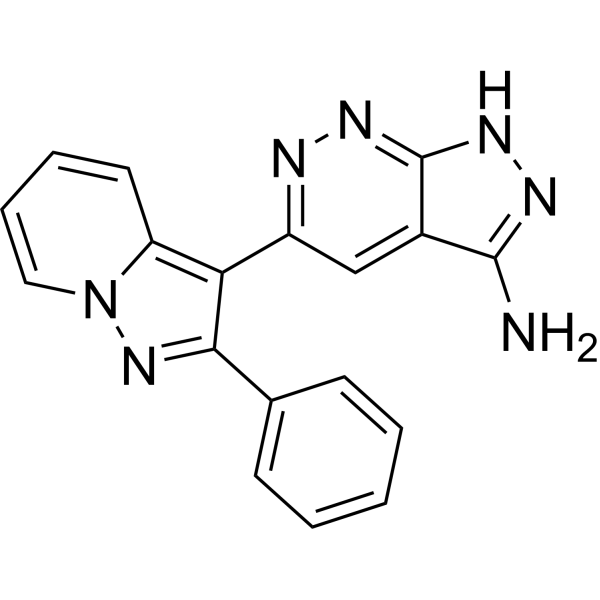
- HY-101920
-
|
|
Autophagy
PI3K
|
Neurological Disease
Cancer
|
|
Autophinib is a potent, selective autophagy inhibitor with IC50s of 90 nM and 40 nM for starvation- and Rapamycin-induced autophagy, respectively. Autophinib is also an ATP competitive Vacuolar Protein Sorting 34 (VPS34) inhibitor with an IC50 of 19 nM. Autophinib inhibits autophagy induced by starvation or Rapamycin by targeting VPS34 .
|
-
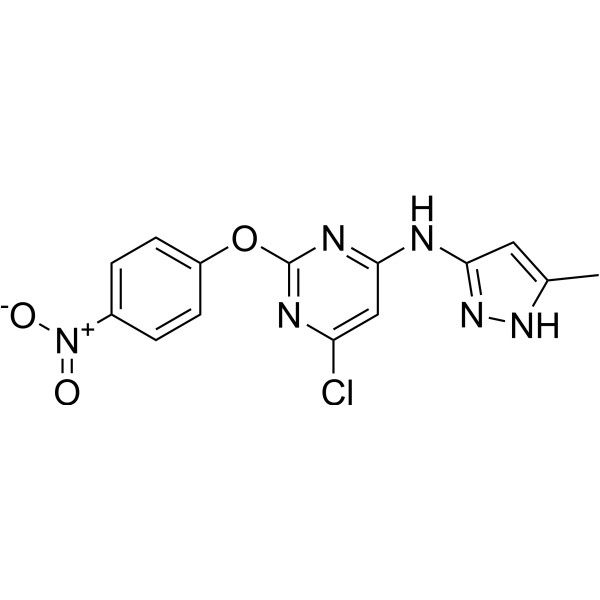
- HY-169422
-
|
IDE275
|
DNA/RNA Synthesis
|
Cancer
|
|
GSK4418959 (IDE275) is a selective, reversible and orally active WRN helicase inhibitor. GSK4418959 shows >10,000-fold selectivity over other helicases. GSK4418959 inhibits ATPase and DNA unwinding functions in an ATP-competitive manner. GSK4418959 can be used for the study of microsatellite instability-high (MSI-H) cancer, such as colorectal cancer (CRC) and endometrial cancer (EC) .
|
-

- HY-12037A
-
|
ON-01910
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib (ON-01910) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3 kinase/Akt pathway, promots the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
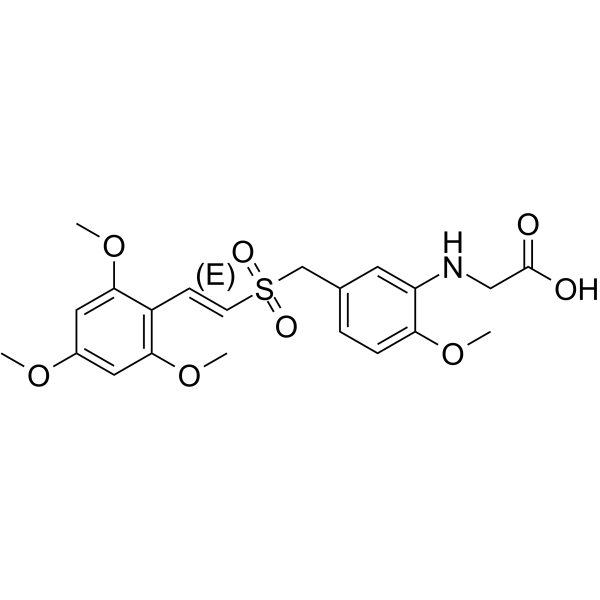
- HY-13241
-
|
LY2228820 dimesylate
|
p38 MAPK
Autophagy
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
Ralimetinib dimesylate (LY2228820 dimesylate) is a selective, ATP-competitive inhibitor of p38 MAPK α/β with IC50s of 5.3 and 3.2 nM, respectively. Ralimetinib (LY2228820) selectively inhibits phosphorylation of MK2 (Thr334), with no effect on phosphorylation of p38a MAPK, JNK, ERK1/2, c-Jun, ATF2, or c-Myc.
|
-
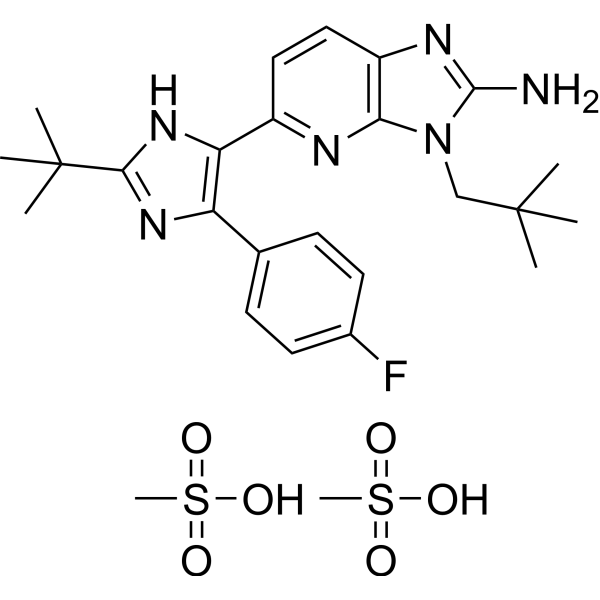
- HY-112390B
-
|
|
Syk
5-HT Receptor
|
Inflammation/Immunology
|
|
Syk Inhibitor II dihydrochloride dihydrate is a potent, high selective and ATP-competitive Syk inhibitor with an IC50 of 41 nM. Syk Inhibitor II dihydrochloride dihydrate inhibits 5-HT release from RBL-cells with an IC50 of 460 nM. Syk Inhibitor II dihydrochloride dihydrate shows less potent against other kinases and has anti-allergic effect .
|
-
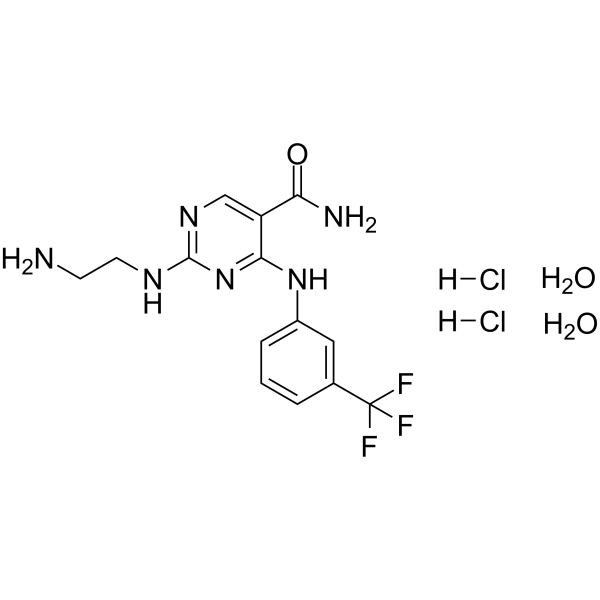
- HY-50706R
-
|
AZD6244 (Standard); ARRY-142886 (Standard)
|
MEK
Apoptosis
Reference Standards
|
Cancer
|
|
Selumetinib (Standard) is the analytical standard of Selumetinib. This product is intended for research and analytical applications. Selumetinib (AZD6244) is selective, non-ATP-competitive oral MEK1/2 inhibitor, with an IC50 of 14 nM for MEK1. Selumetinib (AZD6244) inhibits ERK1/2 phosphorylation.
|
-

- HY-10474R
-
|
PP 242 (Standard)
|
Reference Standards
mTOR
Autophagy
Mitophagy
Apoptosis
|
Cancer
|
|
Torkinib (Standard) is the analytical standard of Torkinib. This product is intended for research and analytical applications. Torkinib (PP 242) is a selective and ATP-competitive mTOR inhibitor with an IC50 of 8 nM . PP242 inhibits both mTORC1 and mTORC2 with IC50s of 30 nM and 58 nM, respectively .
|
-

- HY-13418
-
|
Compound C dihydrochloride; BML-275 dihydrochloride
|
Organoid
AMPK
TGF-β Receptor
Autophagy
|
Cancer
|
|
Dorsomorphin (Compound C) dihydrochloride is a potent, selective and ATP-competitive AMPK inhibitor, with a Ki of 109 nM. Dorsomorphin dihydrochloride inhibits BMP pathway by targeting the type I receptors ALK2, ALK3, and ALK6. Dorsomorphin dihydrochloride can reverse autophagy activation and anti-inflammatory effect of Urolithin A (HY-100599).
|
-
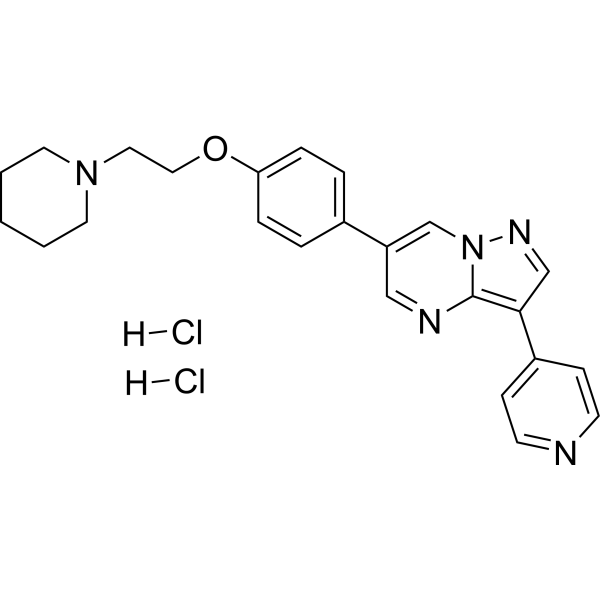
- HY-122369
-
|
|
Carboxypeptidase
|
Metabolic Disease
|
|
Histargin is a selective carboxypeptidase B inhibitor with an IC50 of 17 μg/mL and a Ki of 30 μM. Histargin exerts competitive inhibition with substrate, with inhibitory activity abolished by metal cations. Histargin shows no significant inhibitory activity against carboxypeptidase A, aminopeptidase A, or aminopeptidase B .
|
-

- HY-122661
-
|
MPH
|
PARP
Apoptosis
|
Cancer
|
|
Mefuparib hydrochloride (MPH) is an orally active, substrate-competitive and selective PARP1/2 inhibitor with IC50s of 3.2 nM and 1.9 nM, respectively. Mefuparib hydrochloride induces apoptosis and possesses prominent anticancer activity in vitro and in vivo .
|
-
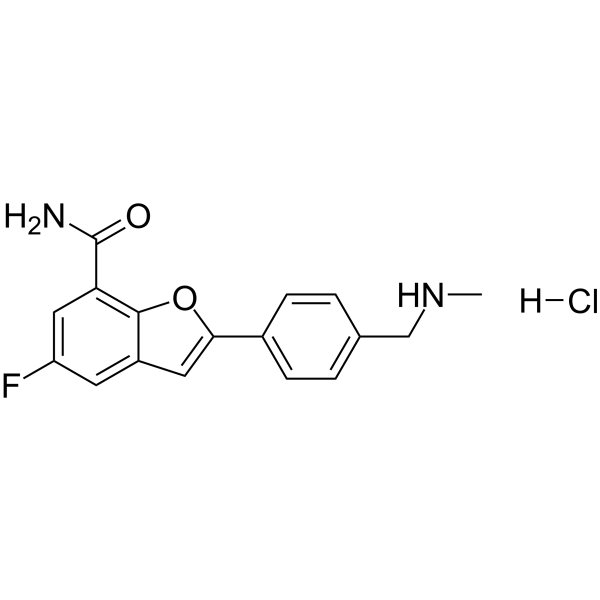
- HY-10962
-
|
CYT387 sulfate salt
|
JAK
Autophagy
Apoptosis
|
Cancer
|
|
Momelotinib sulfate (CYT387 sulfate salt) is an ATP-competitive inhibitor of JAK1/JAK2 with IC50 of 11 nM/18 nM, 10-fold selectivity versus JAK3 (IC50=155 nM).
|
-
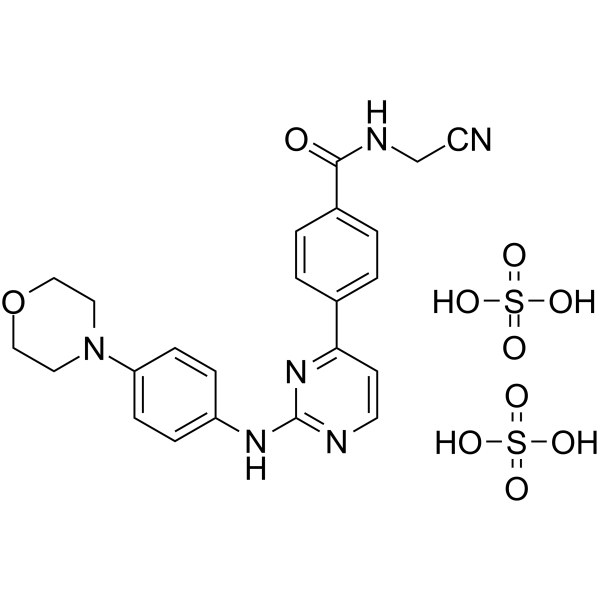
- HY-130199
-
|
Parellic acid
|
Others
|
Metabolic Disease
|
|
Psoromic acid is a potent and selective RabGGTase (Rab geranylgeranyl transferase) inhibitor with an IC50 value of 1.3 µM. Psoromic acid is an antioxidative agent. Psoromic acid exhibits a competitive type of HMGR inhibition and mixed type of ACE (angiotensin converting enzyme) inhibition .
|
-
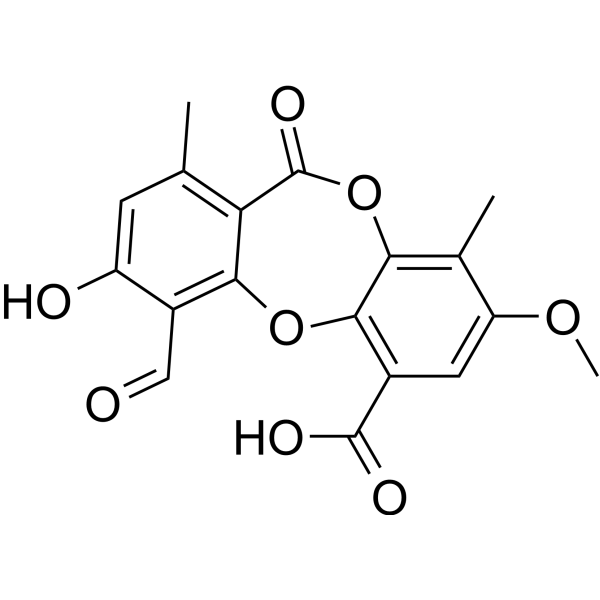
- HY-110150
-
|
|
PI4P5K
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
UNC3230 is a potent, selective and ATP-competitive PIP5K1C inhibitor with an IC50 of ~41 nM. UNC3230 also inhibits PIP4K2C and does not inhibit any of the other lipid kinases that regulate phosphoinositide levels. UNC3230 has antinociceptive and anticancer effects .
|
-
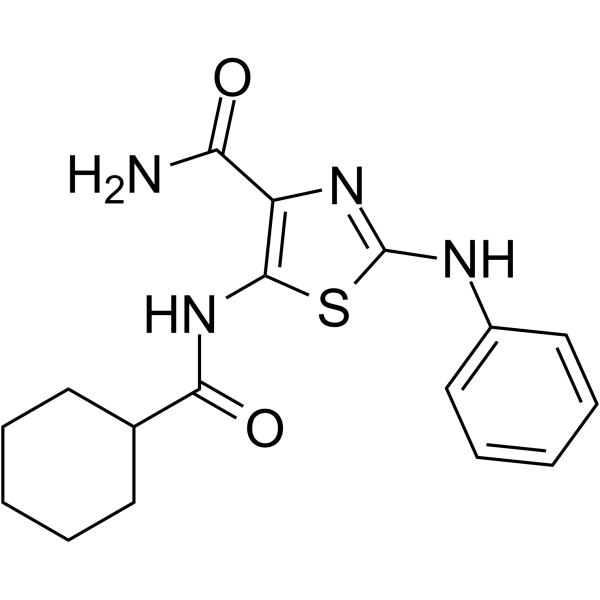
- HY-100513
-
|
|
DNA/RNA Synthesis
Apoptosis
Antibiotic
|
Cancer
|
|
(±)-Dehydroaltenusin, an antibiotic, is a selective eukaryotic DNA polymerase α (pol α) inhibitor with an IC50 of 0.68 μM. (±)-Dehydroaltenusin can be isolated from fungus Alternaria tenuis. (±)-Dehydroaltenusin competitively inhibits the DNA template primer (Ki: 0.23 μM) and non-competitively suppresses the 2'-deoxyribonucleoside 5'-triphosphate substrate (Ki: 0.18 μM). (±)-Dehydroaltenusin induces the cancer cell S-phase cycle arrest and apoptosis. (±)-Dehydroaltenusin can be used for cancers like human adenocarcinoma research .
|
-

- HY-104050R
-
|
|
Glycosidase
Reference Standards
|
Metabolic Disease
|
|
M-31850 (Standard) is the analytical standard of M-31850 (HY-104050). This product is intended for research and analytical applications. M-31850 is a potent, selective and competitive β-hexosaminidase (Hex) inhibitor with IC50s of 6.0 μM and 3.1 μM for human HexA and human HexB, respectively. M-31850 also competitively inhibits β-N-acetyl-D-hexosaminidase OfHex2 with a Ki of 2.5 μM .
|
-

- HY-50706
-
-
- HY-10253
-
|
Tyrphostin AG 1024
|
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
AG1024 (Tyrphostin AG 1024) is a reversible, competitive and selective IGF-1R inhibitor with an IC50 of 7 μM. AG1024 inhibits phosphorylation of IR (IC50=57 μM). AG1024 induces apoptosis and has anti-cancer activity .
|
-
- HY-136532
-
ZT-1a
3 Publications Verification
|
NKCC
|
Neurological Disease
|
|
ZT-1a is a potent, non-ATP-competitive and selective SPAK inhibitor. ZT-1a inhibits SPAK activity with IC50s of 44.3, 35.0, 46.7 μM at ATP concentrations of 0.01, 0.1 and 1 mM, respectively .
|
-
- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-
- HY-161913
-
|
|
Dopamine Transporter
|
Neurological Disease
|
|
AC-4-248 is an atypical non-competitive dopamine transporter (DAT) inhibitor and reduces the potency of cocaine to inhibit DAT. AC-4-248 is a non-selective inhibitor of SLC6 transporters with IC50 values for hDAT, hSERT, and hNET of 45.5 μM, 96.2 μM, and 250 μM, respectively .
|
-

- HY-103351R
-
|
|
Reference Standards
Cathepsin
|
Inflammation/Immunology
|
|
Cathepsin G Inhibitor I (Standard) is the analytical standard of Cathepsin G Inhibitor I (HY-103351). This product is intended for research and analytical applications. Cathepsin G Inhibitor I (compound 7) is an effective, selective, reversible, competitive, non-peptide cathepsin G inhibitor (IC50=53 nM; Ki=63 nM) .
|
-

- HY-N6057
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
Obtusin, isolated from Cassia obtusifolia Linn seed, is a highly selective and competitive human monoamine oxidase-A (hMAO-A) inhibitor with an IC50 of 11.12 μM and a Ki of 6.15 μM. Obtusin plays a preventive role in neurodegenerative diseases, especially anxiety and depression .
|
-
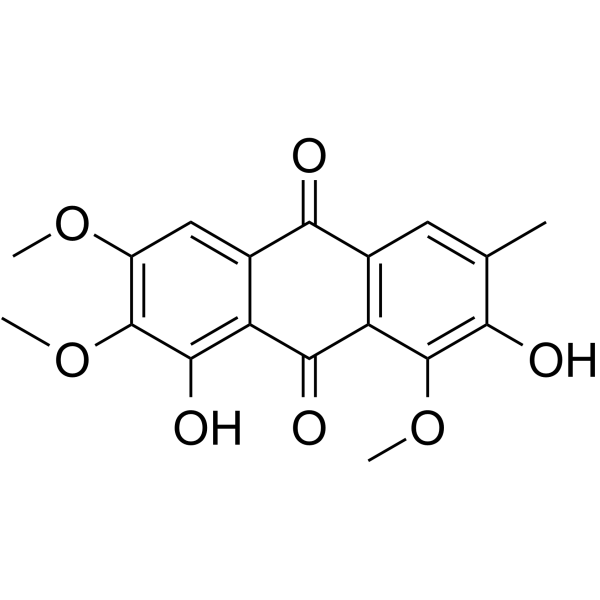
- HY-15269
-
|
|
mTOR
Akt
PI3K
|
Cancer
|
|
PP30 is a potent, selective, ATP-competitive mTOR inhibitor with an IC50 of 80 nM. PP30 blocks insulin-stimulated phosphorylation of Akt at residues S473 and T308, preventing the full activation of Akt. PP30 is applicable for cancer research .
|
-
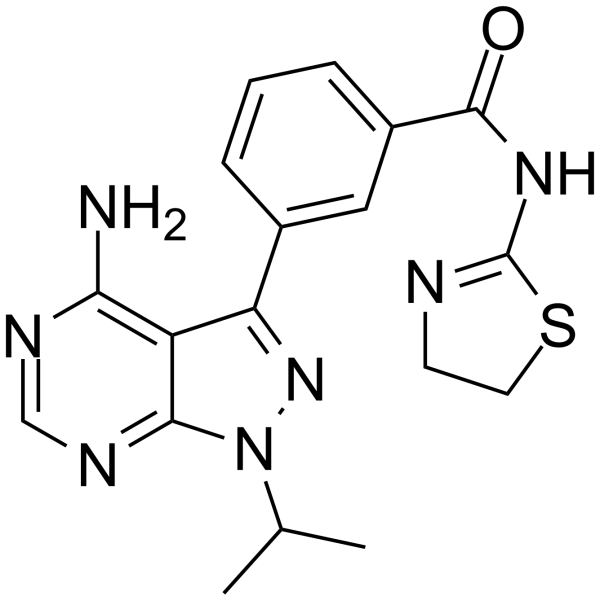
- HY-150076
-
BLU2864
2 Publications Verification
|
PKA
|
Metabolic Disease
Cancer
|
|
BLU2864 is an orally active, highly selective, ATP-competitive PRKACA inhibitor (IC50=0.3 nM). BLU2864 shows anti-tumor activity. BLU2864 can be used in cancer and polycystic kidney disease research .
|
-
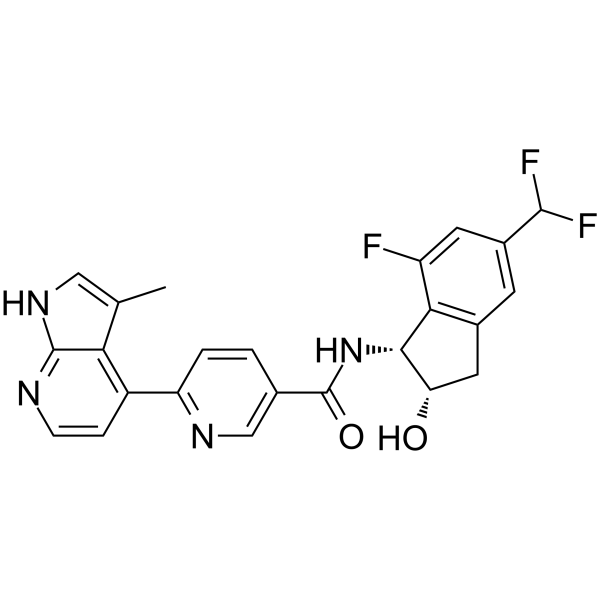
- HY-W371164
-
-
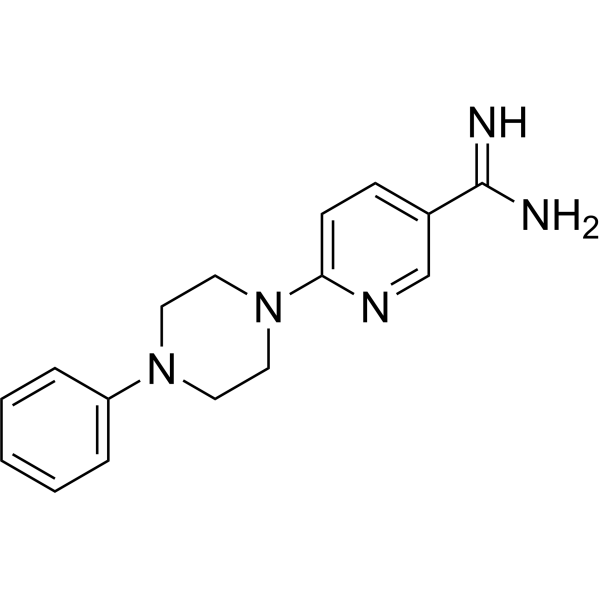
- HY-15260A
-
|
BMS-863233 monohydrochloride
|
CDK
|
Cancer
|
|
XL413 (BMS-863233) hydrochloride is a potent, selective and ATP competitive inhibitor of Cdc7, with an IC50 of 3.4 nM, and also shows potent effect with IC50s of 215, 42 nM on CK2, PIM1, respectively, and an EC50 of 118 nM on pMCM.
|
-
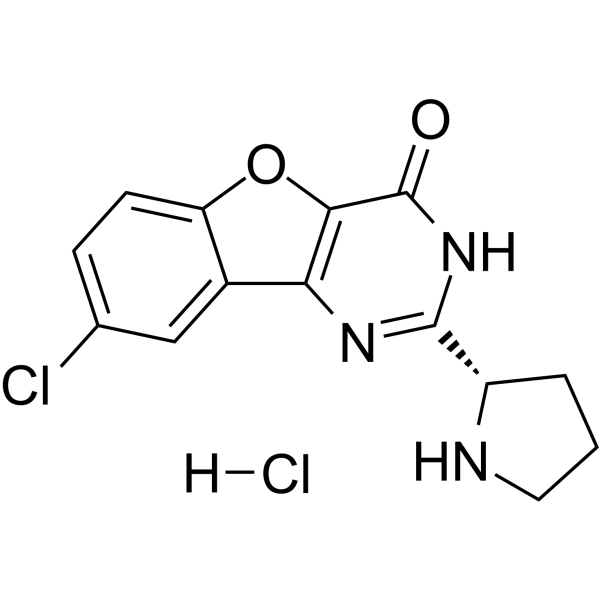
- HY-13335
-
|
|
PKC
Apoptosis
|
Cancer
|
|
PKCβ inhibitor 1 is a potent, ATP-competitive, and selective PKCβ inhibitor with IC50s of 21 and 5 nM for human PKCβ1 and PKCβ2, respectively. PKCβ inhibitor 1 exhibits selectivity of more than 60-fold in favor of PKCβ2 relative to other PKC isozymes (PKCα, PKCγ, and PKCε) .
|
-
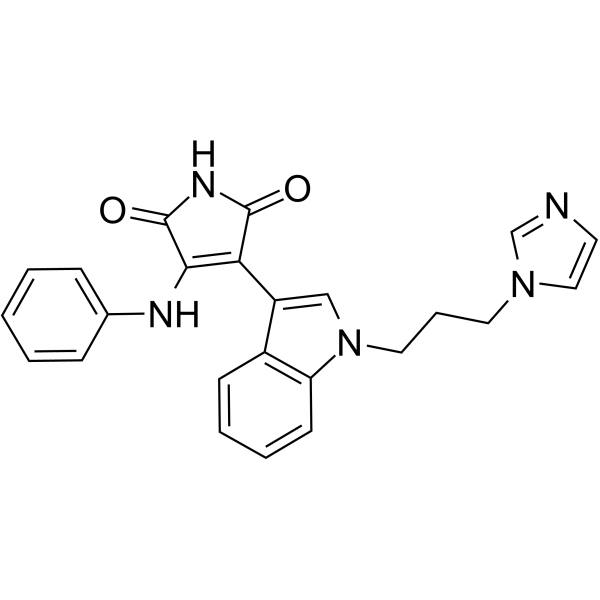
- HY-177936
-
|
|
NEDD8-activating Enzyme
|
Cancer
|
|
NAE-IN-3 (compound 1) is a potent, selective and non-covalent competitive NEDD8-activating enzyme (NAE) inhibitor (IC50 = 0.8 μM). NAE-IN-3 inhibits NAE by blocking the ATP-binding domain. NAE-IN-3 exhibits selectivity over analogous E1 enzymes UAE and SAE. NAE-IN-3 can be used for cancer research .
|
-

- HY-179266
-
|
|
Fat Mass and Obesity-associated Protein (FTO)
|
Cancer
|
|
FTO-IN-15 (Compound 8a) is a potent and selective dual-competitive FTO inhibitor with an IC50 of 43.7 nM, showing high selectivity over ALKBH3 and ALKBH5. FTO-IN-15 substantially inhibits FTO demethylation by simultaneously occupying the substrate and 2-oxoglutarate (2-OG) pockets. FTO-IN-15 can be used for the research of acute myeloid leukemia (AML) .
|
-

- HY-10403
-
-
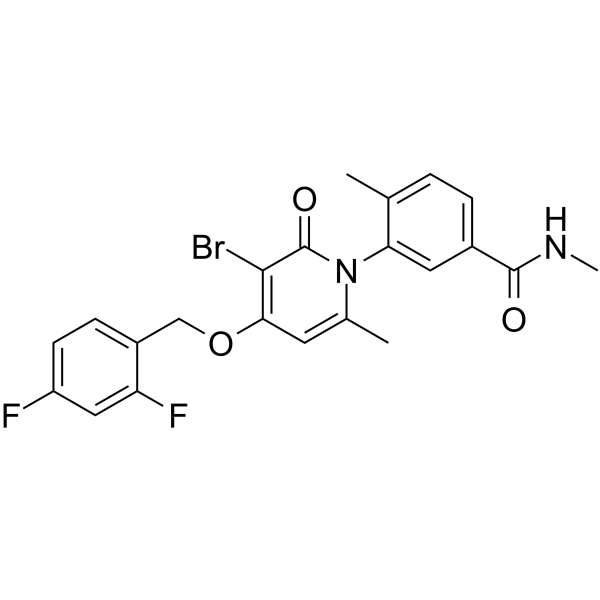
- HY-11005
-
|
|
PDK-1
Apoptosis
|
Cancer
|
|
BX-912 is a direct, selective, and ATP-competitive PDK1 inhibitor (IC50=26 nM). BX-912 blocks PDK1/Akt signaling in tumor cells and inhibits the anchorage-dependent growth of a variety of tumor cell lines in culture or induces apoptosis .
|
-
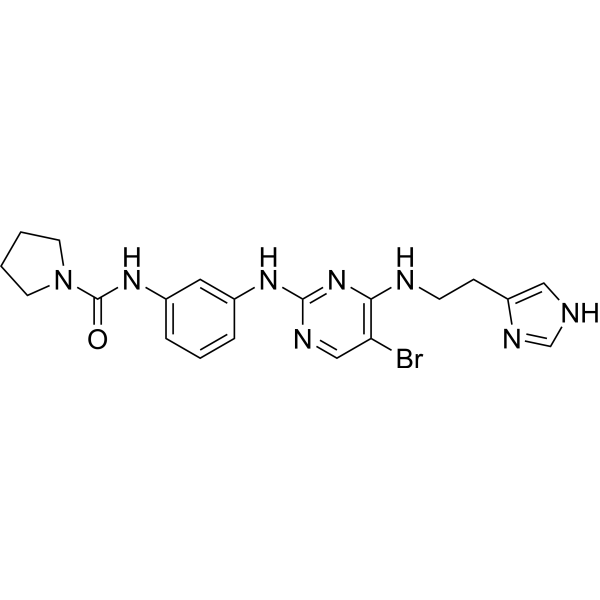
- HY-50706AR
-
|
AZD6244 sulfate (Standard); ARRY-142886 sulfate (Standard)
|
MEK
Apoptosis
Reference Standards
|
Cancer
|
|
Selumetinib (sulfate) (Standard) is the analytical standard of Selumetinib (sulfate). This product is intended for research and analytical applications. Selumetinib (AZD6244) is selective, non-ATP-competitive oral MEK1/2 inhibitor, with an IC50 of 14 nM for MEK1. Selumetinib (AZD6244) inhibits ERK1/2 phosphorylation.
|
-

- HY-160564
-
|
|
EGFR
|
Cancer
|
|
ZNL-0056 is an orally active ATP-competitive inhibitor that targets both the Cys797 and Cys775 in the ATP binding site of EGFR. ZNL-0056 selectively inhibits EGFR and its downstream signaling in H3255 cells. ZNL-0056 can be used for the research of cancer .
|
-
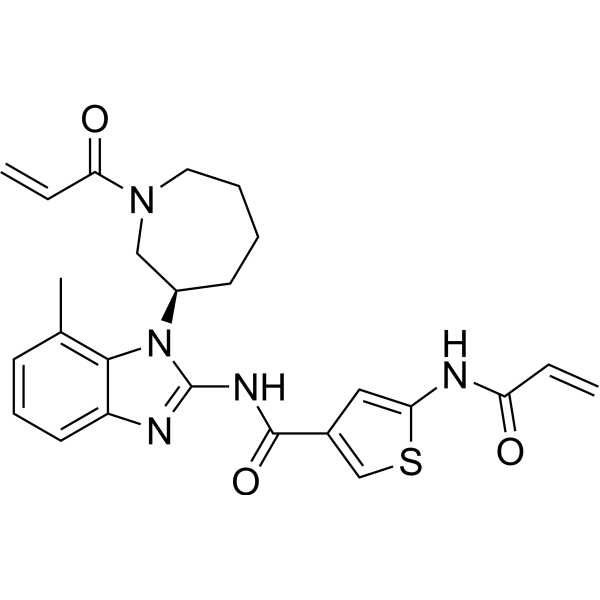
- HY-W484263
-
|
|
NO Synthase
|
Neurological Disease
|
|
hnNOS-IN-3 (compound 39) is a selective nNOS inhibitor, with a Ki of 0.32 μM. The nNOS binding of hnNOS-IN-3 is competitive with L-arginine. The selectivity of hnNOS-IN-3 for nNOS versus iNOS (Ki=37 μM) and eNOS (Ki=9.4 μM) is 115-fold and 29-fold, respectively .
|
-
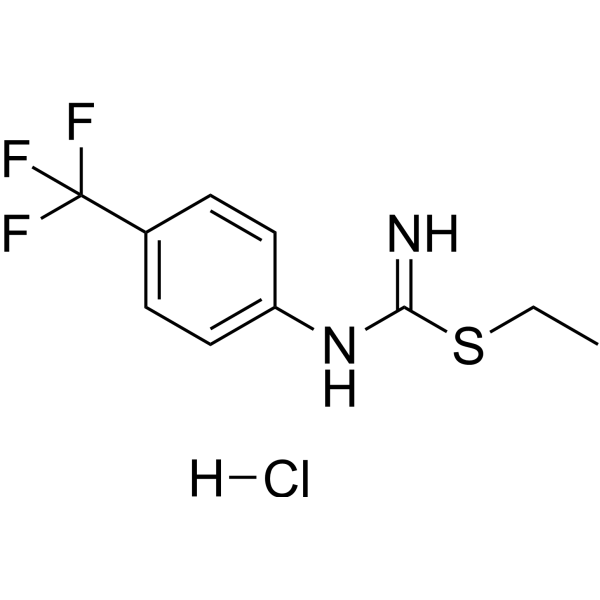
- HY-122805
-
|
|
Ser/Thr Protease
|
Neurological Disease
|
|
PF-794 is a potent, selective and ATP-competitive Traf2- and Nck-interacting kinase (TNIK) inhibitor with an IC50 of 39 nM. PF-794 shows selective for the TNIK family. PF-794 reduces endogenous p120-catenin phosphorylation in cells. PF-794 can be used for teh study of psychiatric disorders .
|
-

- HY-15281
-
|
|
mTOR
|
Cancer
|
|
QL-IX-55 is a selective ATP-competitive inhibitor of mTORC1/2 with IC50s of 50/50/10-50 nM for Human mTORC1/Yeast mTORC1/Yeast mTORC2, respectively.
|
-
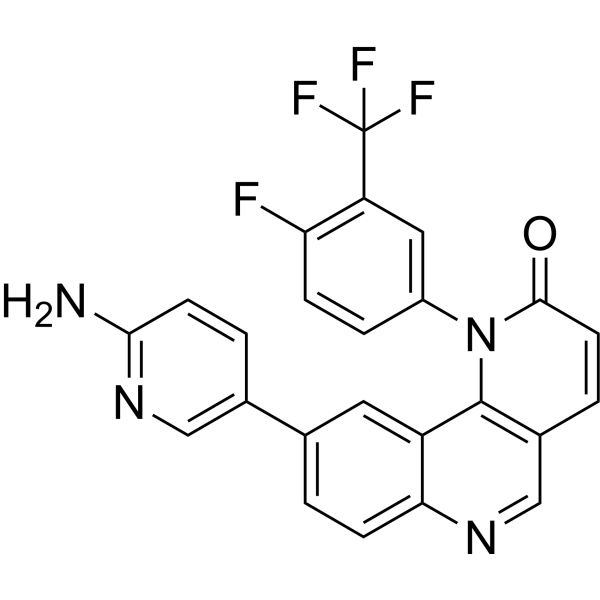
- HY-111294
-
|
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
ASP4000 hydrochloride is a potent, competitive, selective, orally active DPP4 inhibitor with an IC50 value of 2.25 nM against human recombinant DPP4. ASP4000 hydrochloride shows antihyperglycemic activity. ASP4000 hydrochloride can be used in the research of type 2 diabetes .
|
-

- HY-10285A
-
|
BMS-477118 hydrate
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin hydrate (BMS-477118 hydrate) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin hydrate has the peotential for type 2 diabetes mellitus research .
|
-
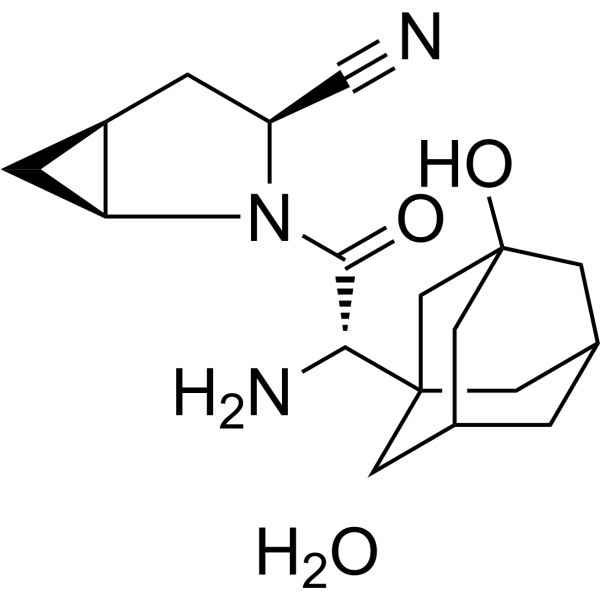
- HY-108241A
-
|
|
Glycosidase
|
Neurological Disease
|
|
Galacto-PUGNAc is a selective competitive inhibitor of lysosomal β-hexosaminidases (HEX), with Ki values of 51 and 18 nM, for hHEXA and hHEXB, respectively. Galacto-PUGNAc elevates GM2 ganglioside levels in neuroblastoma cells. Galacto-PUGNAc can be used for the research of neurodegenerative disorders .
|
-

- HY-170546
-
|
|
Haspin Kinase
|
Cancer
|
|
MU1920 is an ATP-competitive, selective inhibitor for haspin with an IC50 of 6 nM. MU1920 exhibits good pharmacokinetic properties and metabolic stability in mouse plasma and microsomes without obvious anticancer effects, which can be used for development of chemical probes .
|
-

- HY-116605
-
|
|
Histone Methyltransferase
|
Cancer
|
|
GSK926 (compound 3) is a selective histone lysine methyltransferase EZH2 inhibitor (IC50=0.02 μM; Ki=7.9 nM), with SAM-competitive and cell-active properties. GSK926 can be used in cancer research .
|
-

- HY-12017
-
|
|
c-Met/HGFR
|
Cancer
|
|
PF-04217903 is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
|
-
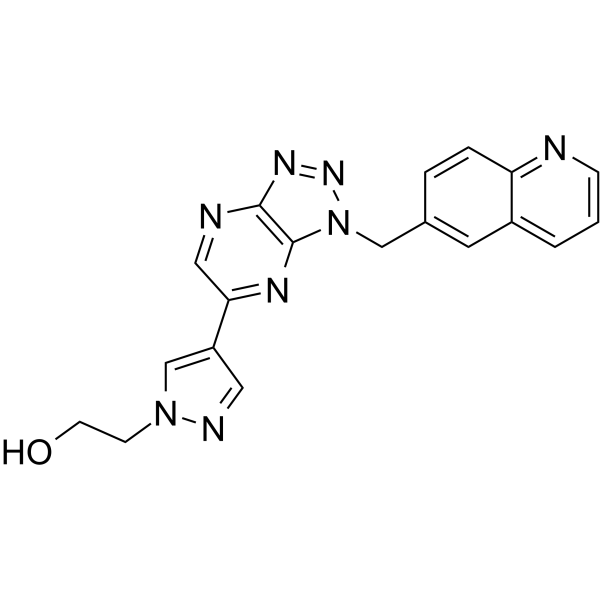
- HY-145940
-
|
BRD3727
|
Casein Kinase
|
Cancer
|
|
BAY-204 (BRD3727) is a potent, ATP-competitive, and selective CSNK1α inhibitor (IC50 = 2 nM at 10 μM ATP; 12 nM at 1 mM ATP). BAY-204 can be used for the study of acute myeloid leukemia (AML) .
|
-
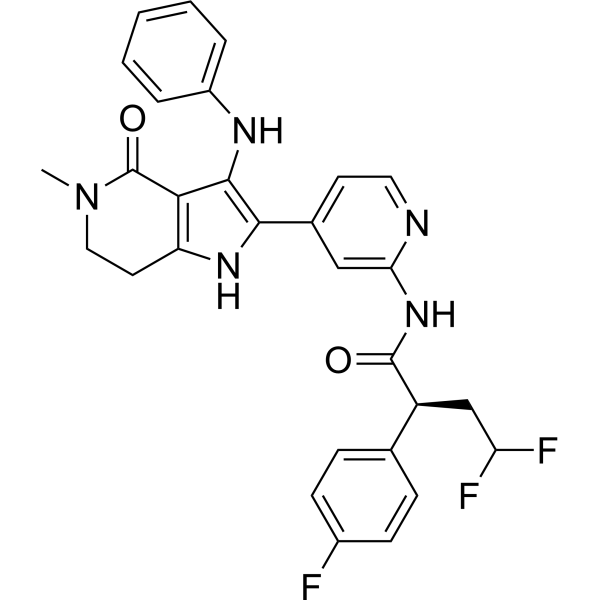
- HY-111055
-
|
|
GSK-3
|
Neurological Disease
|
|
BIP-135 is a potent and selective ATP-competitive GSK-3 inhibitor, with IC50s of 16 nM and 21 nM for GSK-3α and GSK-3β, respectively. BIP 135 exhibits neuroprotective effect .
|
-
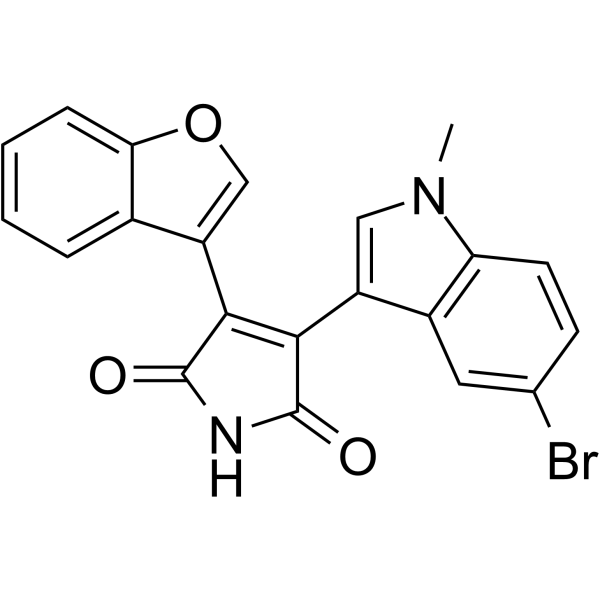
- HY-12017A
-
|
|
c-Met/HGFR
|
Cancer
|
|
PF-04217903 mesylate is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 mesylate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
|
-
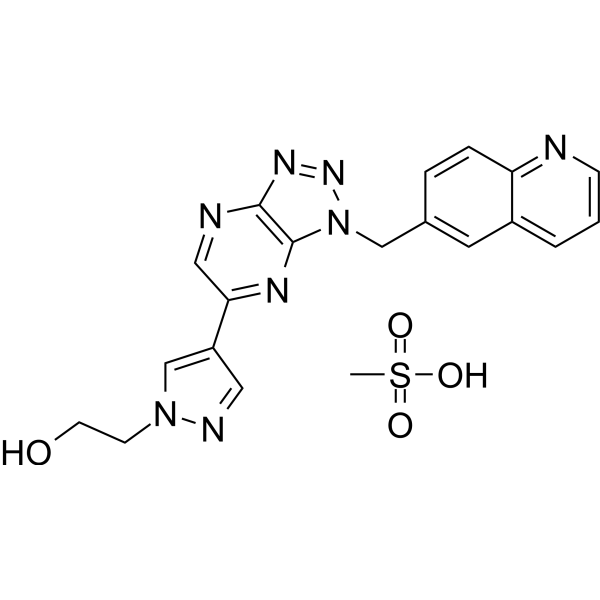
- HY-10285
-
|
BMS-477118
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-
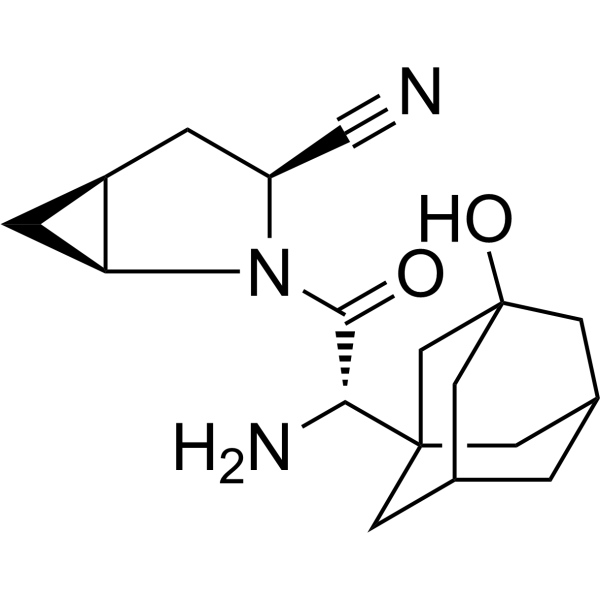
- HY-15727
-
|
GSK2110183; LAE002
|
Akt
PKC
ROCK
|
Cancer
|
|
Afuresertib (GSK2110183) is an orally bioavailable, selective, ATP-competitive and potent pan-Akt kinase inhibitor with Kis of 0.08/2/2.6 nM for Akt1/Akt2/Akt3, respectively .
|
-
- HY-15727A
-
|
GSK2110183 hydrochloride; LAE002 hydrochloride
|
Akt
PKC
ROCK
|
Cancer
|
|
Afuresertib hydrochloride (GSK 2110183 hydrochloride) is an orally bioavailable, selective, ATP-competitive and potent pan-Akt kinase inhibitor with Kis of 0.08/2/2.6 nM for Akt1/Akt2/Akt3 respectively .
|
-
- HY-16448
-
|
BMS-477118 hydrochloride
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin hydrochloride (BMS-477118 hydrochloride) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin hydrochloride has the peotential for type 2 diabetes mellitus research .
|
-
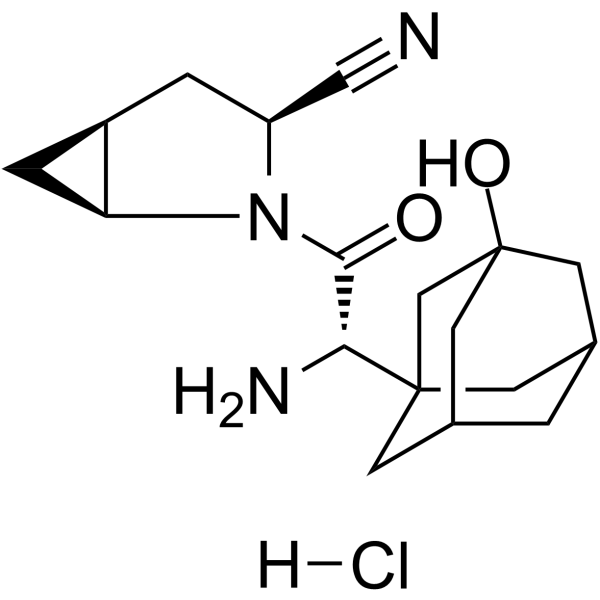
- HY-108657
-
|
|
P2Y Receptor
|
Neurological Disease
|
|
MRS2279 is a selective and high affinity P2Y1 receptor antagonist, with a Ki of 2.5 nM and an IC50 of 51.6 nM. MRS2279 competitively inhibits ADP-promoted platelet aggregation with an apparent affnity (pKB=8.05) .
|
-
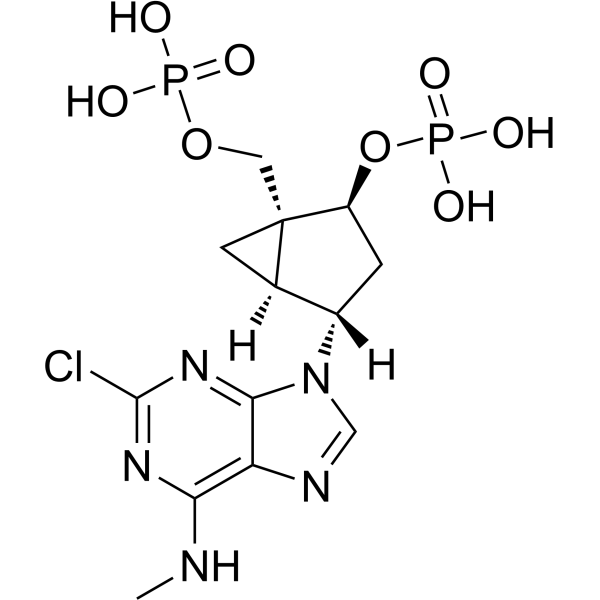
- HY-12866A
-
|
LOXO-101 sulfate; ARRY-470 sulfate
|
Trk Receptor
Apoptosis
|
Cancer
|
|
Larotrectinib sulfate (LOXO-101 sulfate; ARRY-470 sulfate) is an ATP-competitive oral, selective inhibitor of the tropomyosin-related kinase (TRK) family receptors, with low nanomolar 50% inhibitory concentrations against all three isoforms (TRKA, B, and C).
|
-
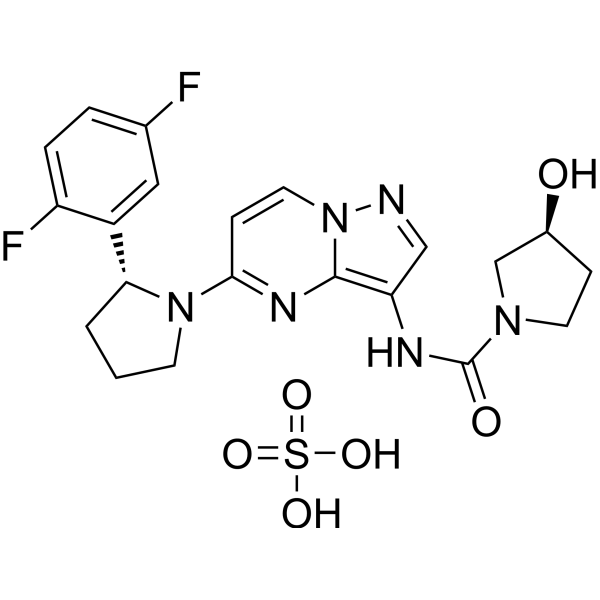
- HY-103353
-
|
|
Cathepsin
Parasite
|
Infection
Cancer
|
|
SID 26681509 is a potent, reversible, competitive, and selective inhibitor of human cathepsin L with an IC50 of 56 nM. SID 26681509 inhibits in vitro propagation of malaria parasite Plasmodium falciparum and inhibits Leishmania major with IC50s of 15.4 μM and 12.5 μM, respectively. SID 26681509 shows no inhibitory activity against cathepsin G .
|
-
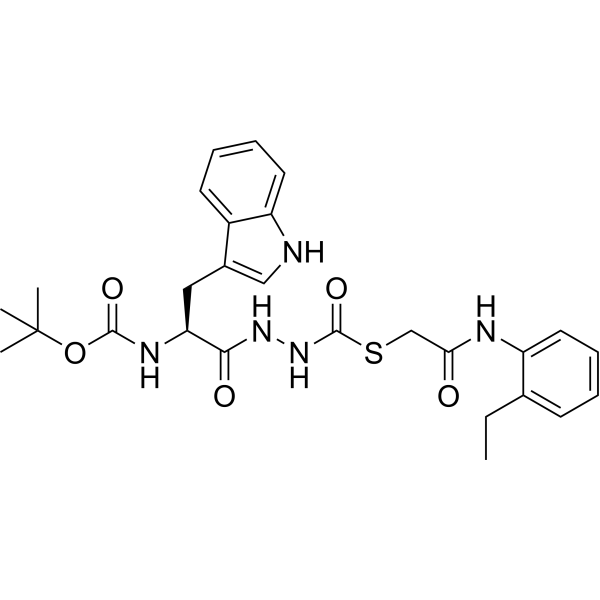
- HY-182434
-
|
|
Phosphatase
FAP
|
Inflammation/Immunology
|
|
CL 118326 is a potent, selective, competitive inhibitor of mammalian pancreatic phospholipase A2 (PLA2) and a weak antagonist of platelet-activating factor receptor (PAF receptor). CL 118326 competitively inhibits mammalian pancreatic PLA2 (porcine: IC50 = 1.55 μg/mL), and shows no activity against snake venom or bee venom PLA2. CL 118326 inhibits PAF-induced and thrombin-induced platelet aggregation, as well as the release of leukotriene (LTC4) and histamine from basophil-enriched leukocytes. CL 118326 can be used for research on inflammation and allergic reactions .
|
-

- HY-14721R
-
|
EMD-1214063 (Standard)
|
Reference Standards
c-Met/HGFR
Autophagy
|
Cancer
|
|
Tepotinib (Standard) is the analytical standard of Tepotinib. This product is intended for research and analytical applications. Tepotinib (EMD-1214063) is an orally active and highly selective, reversible, ATP-competitive c-Met inhibitor with an IC50 of 3 nM, >200-fold selective for c-Met than IRAK4, TrkA, Axl, IRAK1, and Mer. Tepotinib inhibits c-Met phosphorylation. Tepotinib has antitumor effects .
|
-

- HY-103265B
-
|
FPL 67156 trisodium hydrate
|
NTPDase
|
Cardiovascular Disease
Inflammation/Immunology
|
|
ARL67156 (FPL 67156) trisodium hydrate is a selective small is a selective samll molecular inhibitor, targeting to ecto-ATPase, CD39, and CD73. ARL67156 trisodium hydrate is also a competitive inhibitor of NTPDase1 (CD39), NTPDase3 and NPP1, with Kis of 11, 18 and 12 μM, respectively. ARL67156 trisodium hydrate can be used in the research of disease like calcific aortic valve disease, asthma .
|
-
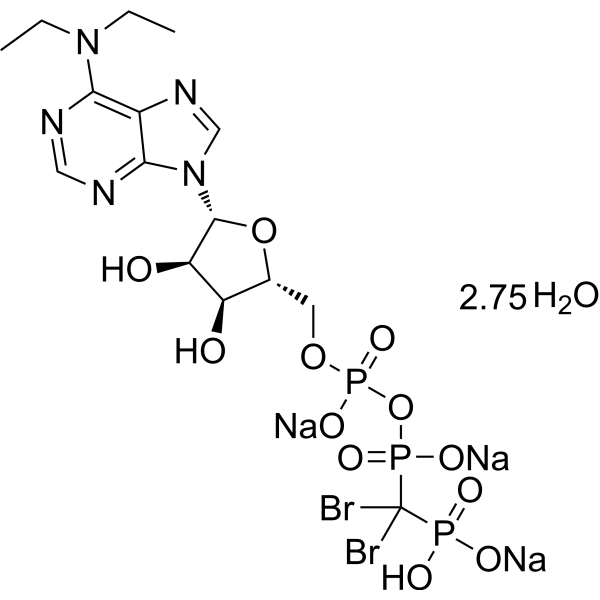
- HY-103265D
-
|
FPL 67156 triethylamine
|
NTPDase
|
Cardiovascular Disease
Inflammation/Immunology
|
|
ARL67156 (FPL 67156) triethylamine is a selective small is a selective samll molecular inhibitor, targeting to ecto-ATPase, CD39, and CD73. ARL67156 triethylamine is also a competitive inhibitor of NTPDase1 (CD39), NTPDase3 and NPP1, with Kis of 11, 18 and 12 μM, respectively. ARL67156 triethylamine can be used in the research of disease like calcific aortic valve disease, asthma .
|
-
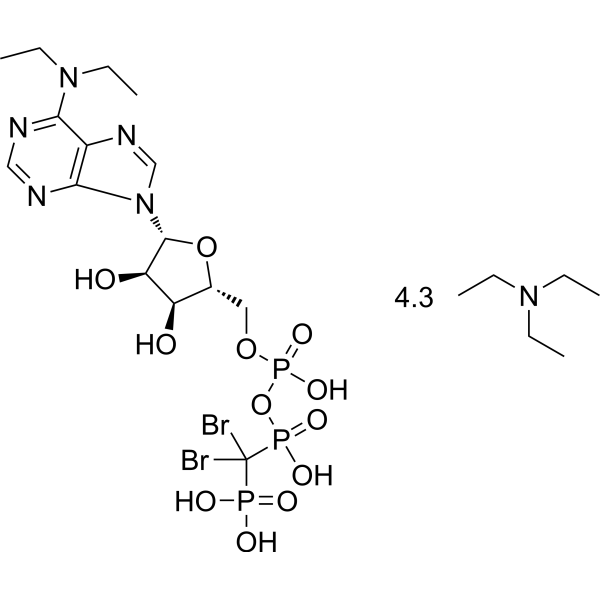
- HY-12037
-
|
ON-01910 sodium
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib sodium (ON-01910 sodium) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3K/Akt pathway, promotes the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib sodium is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
- HY-107597
-
Halicin
2 Publications Verification
SU3327
|
JNK
|
Metabolic Disease
Inflammation/Immunology
|
|
Halicin (SU3327) is a potent, selective and substrate-competitive JNK inhibitor with an IC50 of 0.7 μM. Halicin also inhibits protein-protein interactions between JNK and JNK Interacting Protein (JIP) with an IC50 of 239 nM. Halicin shows less active against p38α and Akt kinase .
|
-
- HY-123206
-
|
|
5-HT Receptor
|
Inflammation/Immunology
|
|
R-96544 (free base) is a potent, selective and competitive 5-HT2 receptor antagonist. R-96544 (free base) inhibits platelet aggregation induced by serotonin, and inhibits 5-HT2A receptor-mediated contraction of guinea pig trachea .
|
-
- HY-B1558A
-
|
MCI-2016
|
Monoamine Oxidase
|
Neurological Disease
|
|
Bifemelane hydrochloride (MCI-2016) is a potent, selective and competitive inhibitor of monoamine oxidase A (MAO-A), with a Ki of 4.20 μM. Bifemelane hydrochloride also inhibits MAO-B noncompetitively with a Ki of 46.0 μM. Bifemelane hydrochloride has a potent antidepressant activity and can be used for the research of cognitive and emotional disturbances related to cerebrovascular disease .
|
-
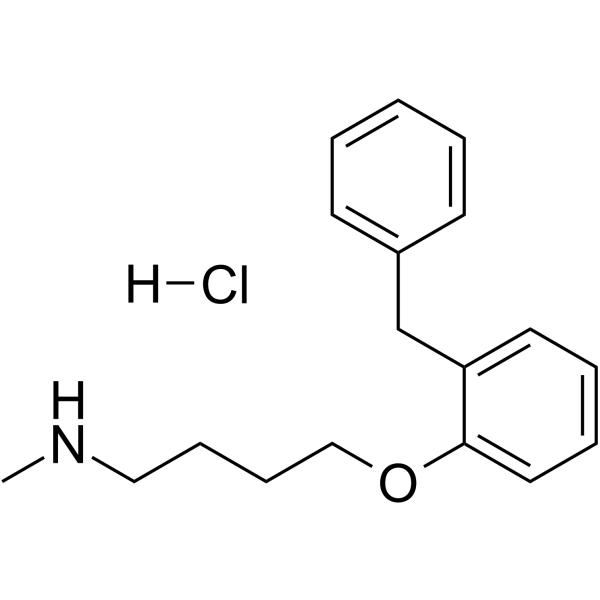
- HY-148757
-
|
|
CaMK
|
Metabolic Disease
|
|
TIM-063 is a selective and cell-permeable CaMKK inhibitor, ATP competitive inhibitor, can directly target the catalytic domain of CaMKK, with the Ki values of 0.35 μM and 0.2 μM for CaMKKα and CaMKKβ, respectively, the IC50 values are 0.63 μM and 0.96 μM, respectively .
|
-
- HY-15141
-
Staurosporine
Maximum Cited Publications
207 Publications Verification
Antibiotic AM-2282; STS; AM-2282
|
PKC
PKA
Apoptosis
Bacterial
Fungal
Antibiotic
|
Infection
Cancer
|
|
Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases with IC50s of 6 nM, 15 nM, 2 nM, and 3 nM for PKC, PKA, c-Fgr, and Phosphorylase kinase respectively. Staurosporine also inhibits TAOK2 with an IC50 of 3 μM. Staurosporine is an apoptosis inducer .
|
-
- HY-18953
-
|
|
mTOR
|
Cancer
|
|
mTOR inhibitor-23 (compound DHM25) is a selective, competitive, irreversible and covalent inhibitor of mTOR. mTOR inhibitor-23 has the mechanism of inhibition occurs mainly through its capacity to covalently interact with a nucleophilic amino acid inside the ATP pocket. mTOR inhibitor-23 exerts potent antitumor activity against triple-negative breast tumor cell lines .
|
-

- HY-10512S
-
-
- HY-184028
-
-

- HY-10423
-
|
ASP7486
|
mTOR
Autophagy
|
Cancer
|
|
OSI-027 (ASP7486) is a potent, selective, orally active and ATP-competitive mTOR kinase activity inhibitor with an IC50 of 4 nM. OSI-027 targets both mTORC1 and mTORC2 with IC50s of 22 nM and 65 nM, respectively .
|
-
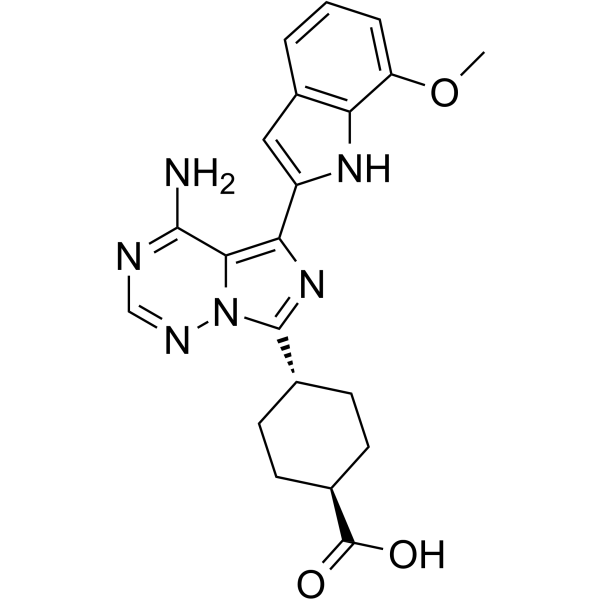
- HY-P1694
-
|
|
Bradykinin Receptor
|
Cardiovascular Disease
|
|
B4148 is a selective competitive bradykinin (BK) antagonist that significantly inhibits BK-induced hypotension in rats. In a rat model of endotoxin shock induced by Escherichia coli lipopolysaccharide, B4148 significantly attenuated the decrease in mean arterial blood pressure compared with the control group .
|
-

- HY-10254G
-
|
PD0325901; PD325901
|
MEK
|
Cancer
|
|
Mirdametinib (PD0325901) (GMP) is Mirdametinib (HY-10254) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Mirdametinib is an orally active, selective and non-ATP-competitive MEK inhibitor .
|
-
- HY-111388
-
|
|
CDK
|
Cancer
|
|
SEL120-34A is a potent, selective, orally available, ATP-competitive CDK8 inhibitor, with IC50s of 4.4 nM and 10.4 nM for CDK8/CycC and CDK19/CycC, respectively, with antitumor activity.
|
-
- HY-101964
-
SPI-112
1 Publications Verification
|
SHP2
Phosphatase
|
Cancer
|
|
SPI-112 is a potent, selective and competitive SHP2 (PTPN11) inhibitor with IC50s of 1 μM, 18.3 μM and 14.5 μM for SHP2, protein tyrosine phosphatase (PTP) and PTP1B, respectively .
|
-
- HY-P1293
-
|
|
iGluR
|
Neurological Disease
|
|
Conantokin G, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G has neuroprotective properties .
|
-
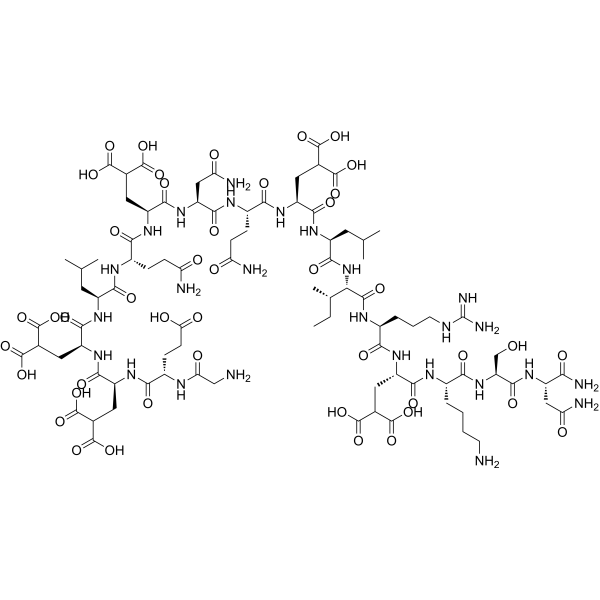
- HY-111388B
-
|
SEL120-34A hydrochloride
|
CDK
|
Cancer
|
|
SEL120-34A hydrochloride is a potent, selective, orally available, ATP-competitive CDK8 inhibitor, with IC50s of 4.4 nM and 10.4 nM for CDK8/CycC and CDK19/CycC, respectively, with antitumor activity.
|
-
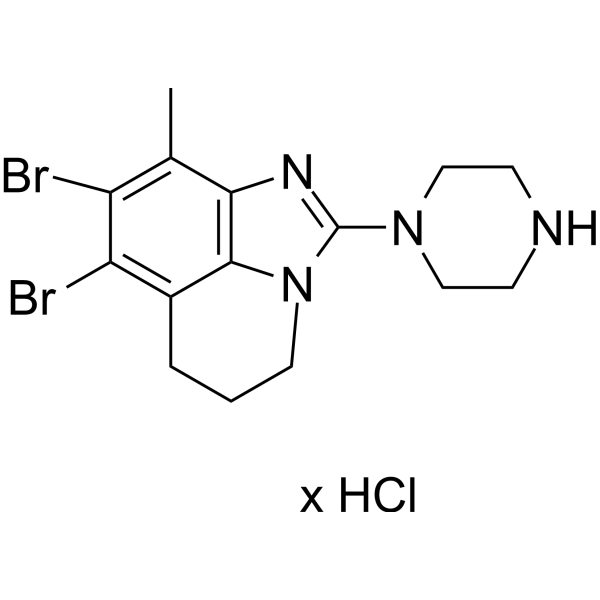
- HY-B0418AR
-
|
R-18553 hydrochloride (Standard)
|
Reference Standards
Opioid Receptor
Autophagy
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Loperamide (hydrochloride) (Standard) is the analytical standard of Loperamide (hydrochloride). This product is intended for research and analytical applications. Loperamide (hydrochloride) (R-18553 (hydrochloride)) is an opioid receptor agonist . Loperamide hydrochloride is a selective and competitive human intestinal carboxylesterases (hiCE) inhibitor. Loperamide hydrochloride has anti-diarrheal effect .
|
-
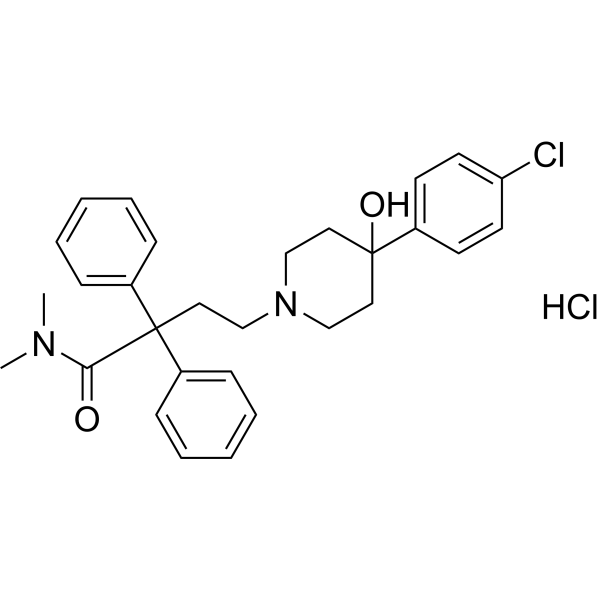
- HY-108643R
-
|
|
Reference Standards
MAPKAPK2 (MK2)
|
Cancer
|
|
CMPD1 (Standard) is the analytical standard of CMPD1 (HY-108643). This product is intended for research and analytical applications. CMPD1 is a selective and non-ATP-competitive p38 MAPK-mediated MK2 phosphorylation inhibitor with apparent Ki (Kiapp) of 330 nM .
|
-

- HY-50672
-
|
|
Kinesin
Apoptosis
Lipoxygenase
|
Cancer
|
|
MK-0731 is a selective, non-competitive and allosteric kinesin spindle protein (KSP) inhibitor with an IC50 of 2.2 nM and a pKa of 7.6. MK-0731 is >20,000 fold selectivity against other kinesins. MK-0731 induces mitotic arrest and induces apoptosis in tumors. MK-0731 provides significant antitumor efficacy .
|
-
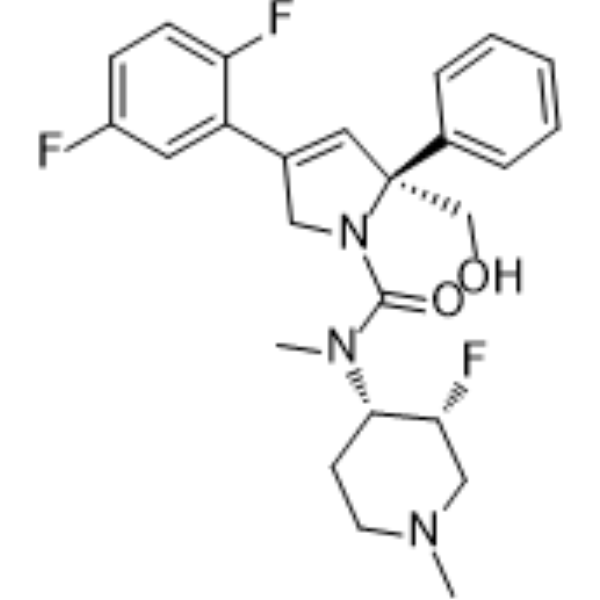
- HY-116000
-
|
Gumarontinib; SCC244
|
c-Met/HGFR
|
Cancer
|
|
Glumetinib (SCC244) is a highly selective, orally bioavailable, ATP-competitive c-Met inhibitor with an IC50 of 0.42 nM. Glumetinib has greater than 2400-fold selectivity for c-Met over those 312 kinases evaluated, including the c-Met family member RON and highly homologous kinases Axl, Mer, TyrO3. Antitumor activity .
|
-
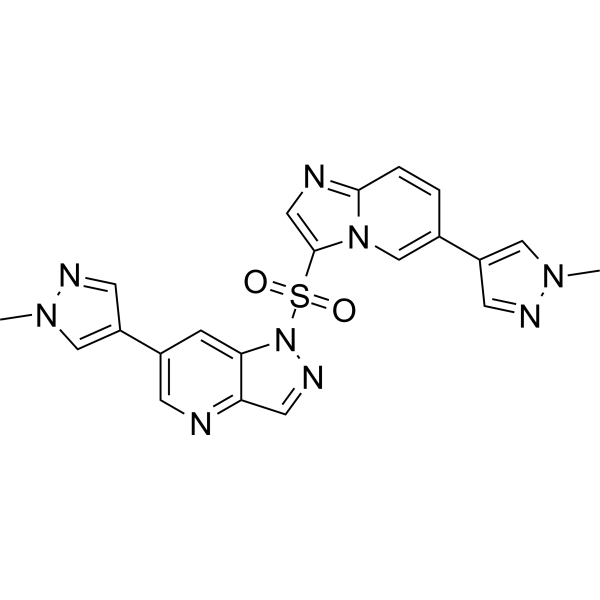
- HY-171805
-
|
|
CD73
|
Cancer
|
|
PSB-0963 is a selective and competitive ecto-5'-nucleotidase (eN/CD73) inhibitor with a Ki of 150 nM for rat ecto-5'-nucleotidase. PSB-0963 shows high selectivity over other ectonucleotidases (NTPDases 1-3) and P2Y receptors. PSB-0963 can be used for the study of cancer .
|
-

- HY-176727
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
WRN inhibitor 19 (Compound 40) is a WRN helicase inhibitor (IC50: 3.7 nM). WRN inhibitor 19 exhibits selective antiproliferative activity in WRN-dependent cancer cells. WRN inhibitor 19 inhibits WRN helicase function by competitively binding to the ATP site and induces DNA damage and cell cycle arrest. WRN inhibitor 19 can be used in the study of WRN-dependent cancers .
|
-

- HY-10195BS
-
|
|
Isotope-Labeled Compounds
PKC
|
Metabolic Disease
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
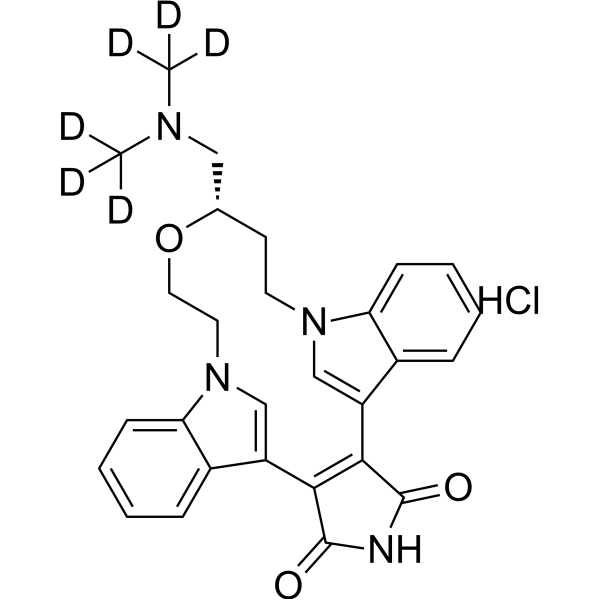
- HY-50706A
-
-
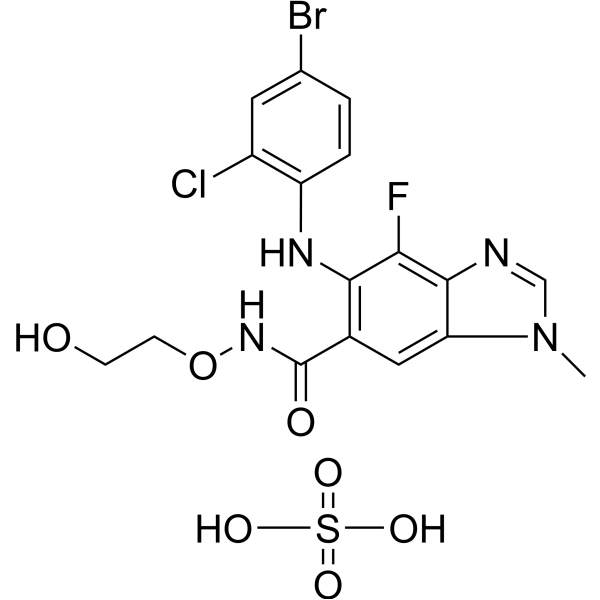
- HY-10279
-
|
|
Factor Xa
|
Cardiovascular Disease
|
|
YM-60828 hydrochloride is an orally active, selective and competitive factor Xa inhibitor with a Ki of 1.3 nM and an IC50 of 2.3 nM. YM-60828 hydrochloride inhibits thrombus formation and platelet aggregation. YM-60828 hydrochloride can be used for the research of venous thrombosis, arterial thrombosis, and thromboembolic disorders .
|
-

- HY-10044
-
WYE-132
4 Publications Verification
WYE-125132
|
mTOR
Apoptosis
|
Cancer
|
|
WYE-132 (WYE-125132) is a highly potent, ATP-competitive, and specific mTOR kinase inhibitor (IC50: 0.19±0.07 nM; >5,000-fold selective versus PI3Ks). WYE-132 (WYE-125132) inhibits mTORC1 and mTORC2.
|
-
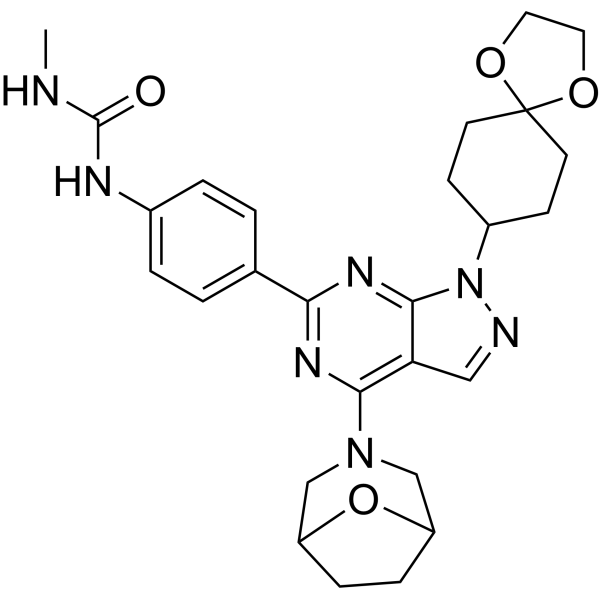
- HY-15656A
-
-
- HY-124817
-
|
|
HSP
|
Inflammation/Immunology
|
|
Col003 is a selective and potent inhibitor of Hsp47 and competitively binds to the collagen binding site on Hsp47 (IC50=1.8 μM). Col003 discourages the interaction of Hsp47 with collagen and inhibits collagen secretion by destabilizing the collagen triple helix. Col003 can be used for the investigation of fibrosis
|
-
- HY-15656B
-
-

- HY-15945
-
|
JRF 12
|
p97
Autophagy
Apoptosis
|
Cancer
|
|
DBeQ is a selective, potent, reversible, and ATP-competitive p97 inhibitor, with an IC50 value of 1.5 μM and 1.6 μM for p97(wt) and p97(C522A), respectively; DBeQ also inhibits Vps4 with an IC50 of 11.5 μM.
|
-
- HY-15656
-
-
- HY-133117
-
BAY-985
4 Publications Verification
|
IKK
|
Cancer
|
|
BAY-985, a chemical probe, is a highly potent, orally active and selective ATP-competitive dual inhibitor of TBK1 and IKKε with IC50s of 2/30 and 2 nM for TBK1 (low/high ATP) and IKKε, respectively. Antitumor efficacy .
|
-
- HY-108657A
-
|
|
P2Y Receptor
|
Neurological Disease
|
|
MRS2279 diammonium is a selective and high affinity P2Y1 receptor antagonist, with a Ki value of 2.5 nM and an IC50 value of 51.6 nM. MRS2279 diammonium competitively inhibits ADP-promoted platelet aggregation with an pKb value of 8.05 .
|
-
- HY-148514
-
|
|
PDGFR
|
Cancer
|
|
CT52923 is a selective, orally active platelet-derived growth factor receptor (PDGFR) antagonist. CT52923 also is an ATP-competitive inhibitor. CT52923 can be used for the research variety of pathological diseases, including atherosclerosis, glomerulonephritis, liver cirrhosis, pulmonary fibrosis, and cancer .
|
-
- HY-13204
-
|
KL 373 hydrochloride
|
mAChR
|
Neurological Disease
|
|
Biperiden (KL 373) hydrochloride is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden hydrochloride has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-
- HY-12853R
-
|
|
Reference Standards
Reactive Oxygen Species (ROS)
|
Others
|
|
Mesotrione (Standard) is the analytical standard of Mesotrione. This product is intended for research and analytical applications. Mesotrione is a herbicide belongs to the benzoylcyclohexanedione family. Mesotrione is a potent and competitive and reversible inhibitor of HPPD enzyme. Mesotrione is selective to maize due to rapid metabolism and relative high tolerance by the susceptible crop plant .
|
-

- HY-12335
-
|
|
Histone Methyltransferase
|
Inflammation/Immunology
Cancer
|
|
UNC0379 is a selective, substrate-competitive inhibitor of lysine methyltransferase SETD8 (KMT5A) with an IC50 of 7.3 μM, KD value of 18.3 μM. UNC0379 can be used in the research of inflammation and cancers, such as pulmonary fibrosis, ovarian cancer, neuroblastoma .
|
-
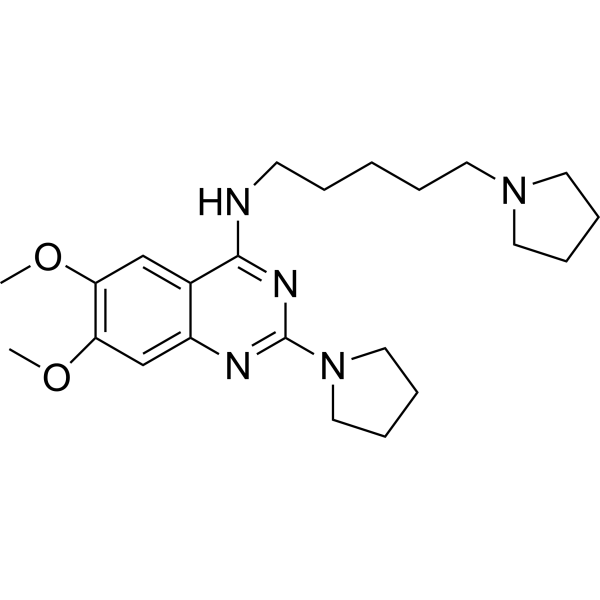
- HY-10195A
-
|
LY333531 mesylate
|
PKC
|
Cardiovascular Disease
Metabolic Disease
|
|
Ruboxistaurin (LY333531) mesylate is an orally active, selective and ATP competitive PKCβ inhibitor with IC50 values of 4.7 and 5.9 nM for PKCβI and PKCβII, respectively. Ruboxistaurin mesylate can be used for the research of eye disorders, heart failure and diabetes .
|
-
- HY-11028
-
|
PF-2413873
|
Progesterone Receptor
|
Endocrinology
|
|
PF-02413873 (PF-2413873) is a potent selective, fully competitive and orally active nonsteroidal progesterone receptor (PR) antagonist, with a Ki of 2.6 nM. PF-02413873 can block progesterone binding and PR nuclear translocation, and inhibit endometrial growth in vivo .
|
-
- HY-13204A
-
|
KL 373
|
mAChR
|
Neurological Disease
|
|
Biperiden (KL 373) is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-
- HY-13217R
-
|
GBR-12909 dihydrochloride (Standard); I893 dihydrochloride (Standard)
|
Dopamine Transporter
Reference Standards
|
Neurological Disease
|
|
Vanoxerine (dihydrochloride) (Standard) is the analytical standard of Vanoxerine (dihydrochloride). This product is intended for research and analytical applications. Vanoxerine dihydrochloride (GBR-12909 dihydrochloride) is a competitive, potent, and highly selective dopamine reuptake inhibitor (Ki=1 nM). Vanoxerine dihydrochloride (GBR-12909 dihydrochloride) binds to the target site on the dopamine transporter (DAT) .
|
-

- HY-12017B
-
|
|
c-Met/HGFR
|
Cancer
|
|
PF-04217903 phenolsulfonate is a potent ATP-competitive c-Met kinase inhibitor with Ki of 4.8 nM for human c-Met. PF-04217903 phenolsulfonate shows more than 1,000-fold selectivity relative to 208 kinases. Antiangiogenic properties .
|
-
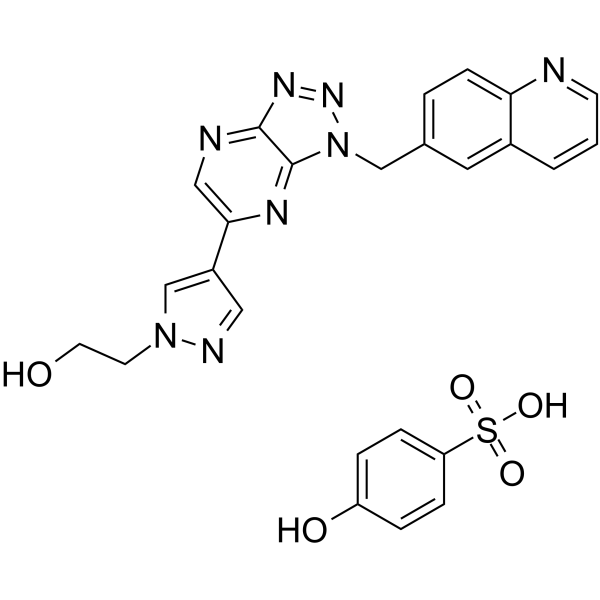
- HY-10917S
-
|
|
Isotope-Labeled Compounds
c-Fms
|
Inflammation/Immunology
Cancer
|
|
GW2580-d6 is the deuterium labeled GW2580. GW2580 is an orally bioavailable and selective inhibitor of c-Fms kinase which completely inhibits human cFMS kinase in vitro at 0.06 μM. GW2580 acts as a competitive inhibitor of ATP binding to the cFMS kinase and inhibits colony-stimulating-factor-1 signaling .
|
-
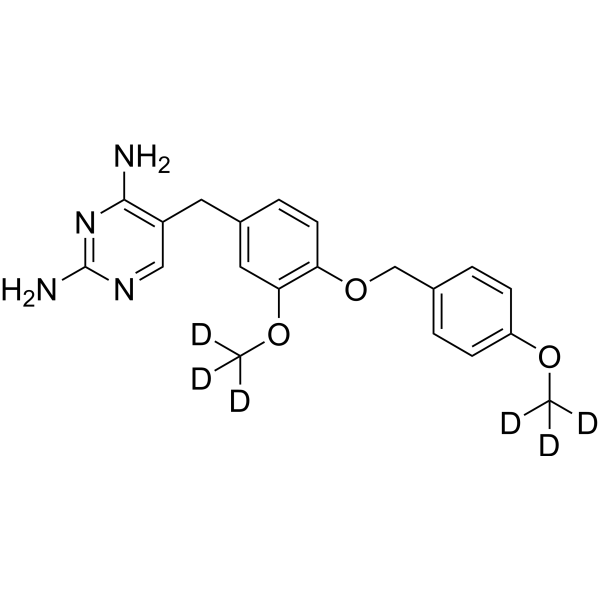
- HY-103353A
-
|
|
Cathepsin
Parasite
|
Infection
Cancer
|
|
SID 26681509 quarterhydrate is a potent, reversible, competitive, and selective inhibitor of human cathepsin L with an IC50 of 56 nM. SID 26681509 quarterhydrate inhibits in vitro propagation of malaria parasite Plasmodium falciparum and inhibits Leishmania major with IC50s of 15.4 μM and 12.5 μM, respectively. SID 26681509 quarterhydrate shows no inhibitory activity against cathepsin G .
|
-
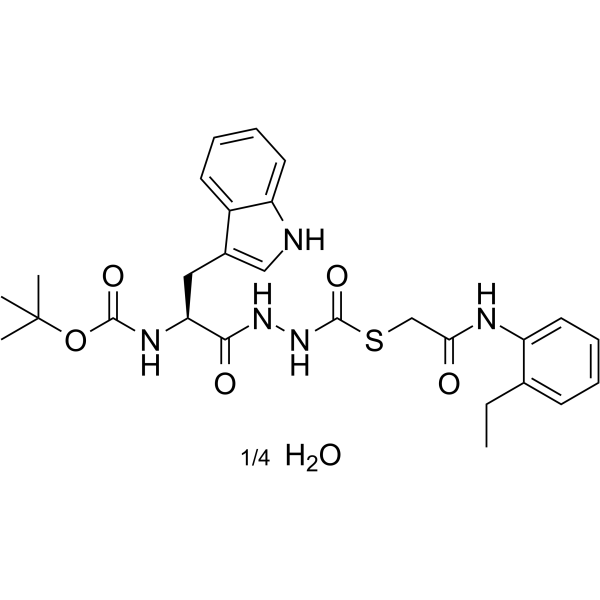
- HY-155098
-
|
|
SHP2
|
Cancer
|
|
CNBCA is a potent, selective, competitive SHP2 enzyme inhibitor, with the IC50 of 0.87 μM. CNBCA binds to full-length SHP2 and inhibits enzyme activity. CNBCA inhibits pAkt and pERK1/2, and the cell growth of BT474 and MDA-MB468 cells. CNBCA can be used for breast cancer study .
|
-
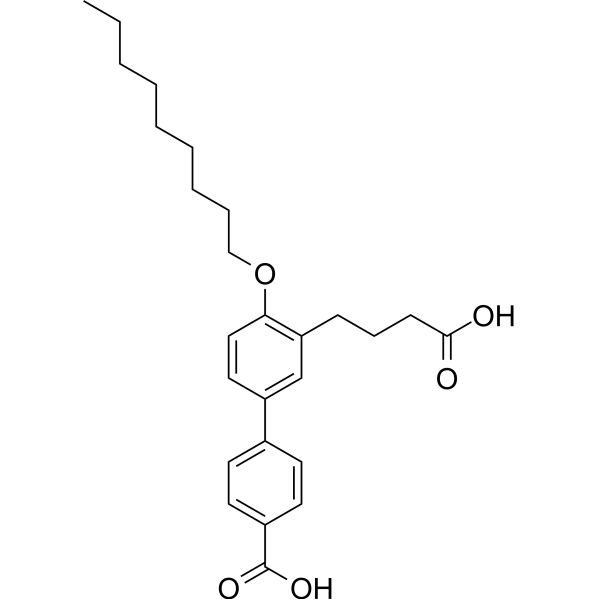
- HY-105711
-
|
|
Lipoxygenase
|
Inflammation/Immunology
|
|
RBx-7796 sodium is a competitive, highly selective, orally active 5-LO (IC50 = 3.5 mM for human 5-LO enzyme) inhibitor. RBx-7796 sodium can effectively inhibit 5-LO activity and LTB4 release. RBx-7796 sodium can significantly inhibit airway inflammation and bronchial constriction .
|
-

- HY-131339
-
SP-96
2 Publications Verification
|
Aurora Kinase
|
Cancer
|
|
SP-96 is a highly potent, selective and non-ATP-competitive Aurora B (IC50=0.316 nM) inhibitor and shows >2000 fold selectivity against FLT3 and KIT. SP-96 shows selective growth inhibition in NCI60 screening, incluing MDA-MD-468 (GI50=107 nM). SP-96 can be used for the research of triple negative breast cancer (TNBC) .
|
-
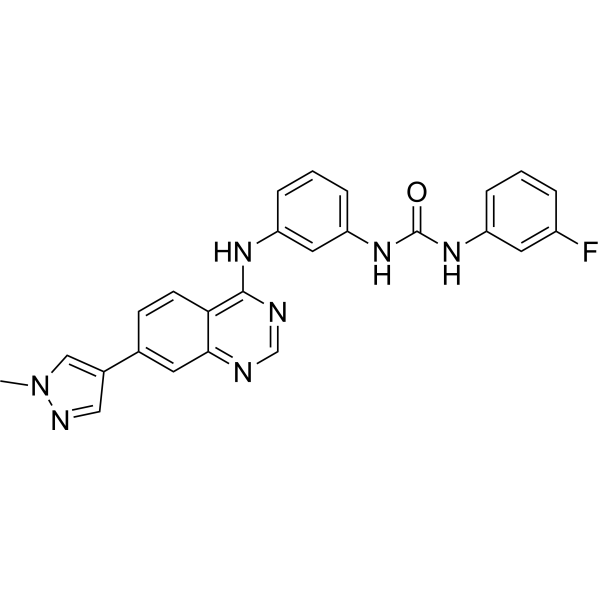
- HY-13260
-
|
|
Akt
Autophagy
Apoptosis
|
Cancer
|
|
CCT128930 is a ATP-competitive and selective inhibitor of AKT (IC50=6 nM for AKT2). CCT128930 has 28-fold selectivity over the closely related PKA kinase (IC50=168 nM) through the targeting of Met282 of AKT (Met173 of PKA-AKT chimera), as well as 20-fold selectivity over p70S6K (IC50=120 nM). Antitumor activity.
|
-
- HY-100414
-
|
BYK61359
|
Proton Pump
|
Metabolic Disease
|
|
Soraprazan (BYK61359) is a selective, reversible K-competitive inhibitor of the H,K-ATPase (Ki=6.4 nM), with an IC50 of 0.19 μM in gastric glands. Soraprazan binds to the H,K-ATPase with a Kd of 28.27 nM. Soraprazan shows immediate inhibition of acid secretion and is more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases .
|
-
- HY-70044G
-
|
GSK-1070916A
|
Apoptosis
Aurora Kinase
|
Cancer
|
|
GSK-1070916 (GMP) is GSK-1070916 (HY-70044) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. GSK-1070916 is a potent and selective ATP-competitive inhibitor of aurora B and aurora C with Kis of 0.38 and 1.5 nM, respectively, and is >250- fold selective over Aurora A .
|
-

- HY-120857
-
|
PD 158294
|
EGFR
|
Others
|
|
BPIQ-II is a linear imidazoloquinazoline that potently inhibits the tyrosine kinase activity of the epidermal growth factor receptor (EGFR; IC50=8 pM). It is selective for EGFR over an assortment of other tyrosine and serine/threonine kinases. Cellular studies indicate that BPIQ-II can enter cells and very selectively shut down EGF-stimulated signal transmission by binding competitively at the ATP site of EGFR.
|
-

- HY-170442
-
|
CDK2-IN-37
|
CDK
|
Cancer
|
|
ECI830 (CDK2-IN-37) is an orally active, ATP-competitive and highly selective CDK2 inhibitor. ECI830 shows high selectivity over other CDK family members. ECI830 can be used for the study of HR+/HER2- breast cancer and CCNE1-amplified tumors (such as ovarian cancer and lung cancer) .
|
-

- HY-139979
-
|
|
Deubiquitinase
|
Cancer
|
|
USP5-IN-1 (compound 64) is a selective competitive inhibitor of USP5 zinc finger ubiquitin binding domain (ZnF-UBD) (KD=2.8 μM). USP5-IN-1 competitively blocks the binding of ubiquitin to ZnF-UBD, inhibits the catalytic activity of USP5, and thus hinders the hydrolysis of ubiquitin chains. USP5-IN-1 can inhibit USP5 cleavage of Lys48-linked diubiquitin substrates in vitro and is a potential USP5 chemical probe and potential inhibitor of USP5-related cancers.
|
-
- HY-114620
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
MAO-B-IN-55 (Compound 5c) is a reversible competitive MAO-B inhibitor with an IC50 of 2.9 nM, and it exhibits approximately 2750-fold higher selectivity for MAO-B over MAO-A. MAO-B-IN-55 can be used for the research of Parkinson's disease .
|
-

- HY-111538
-
|
|
MAGL
|
Cancer
|
|
MAGL-IN-1 is a potent, selective, reversible and competitive inhibitor of MAGL, with an IC50 of 80 nM. MAGL-IN-1 exhibits anti-proliferative effects against human breast, colorectal, and ovarian cancer cells. MAGL-IN-1 blocks MAGL in cell-based as well as in vivo assays .
|
-
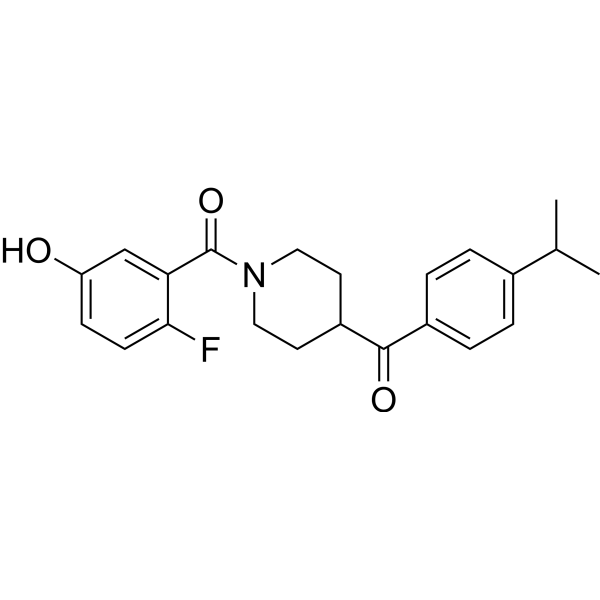
- HY-15779
-
|
|
SphK
Apoptosis
|
Cancer
|
|
K145 is a selective, substrate-competitive and orally active SphK2 inhibitor with an IC50 of 4.3 µM and a Ki of 6.4 µM. K145 is inactive against SphK1 and other protein kinases. K145 induces cell apoptosis and has potently antitumor activity .
|
-
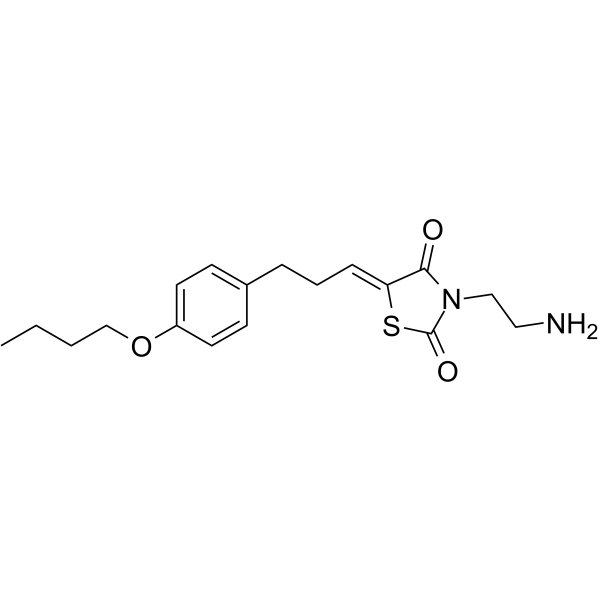
- HY-15779A
-
|
|
SphK
Apoptosis
|
Cancer
|
|
K145 hydrochloride is a selective, substrate-competitive and orally active SphK2 inhibitor with an IC50 of 4.3 μM and a Ki of 6.4 μM. K145 hydrochloride is inactive against SphK1 and other protein kinases. K145 hydrochloride induces cell apoptosis and has potently antitumor activity .
|
-
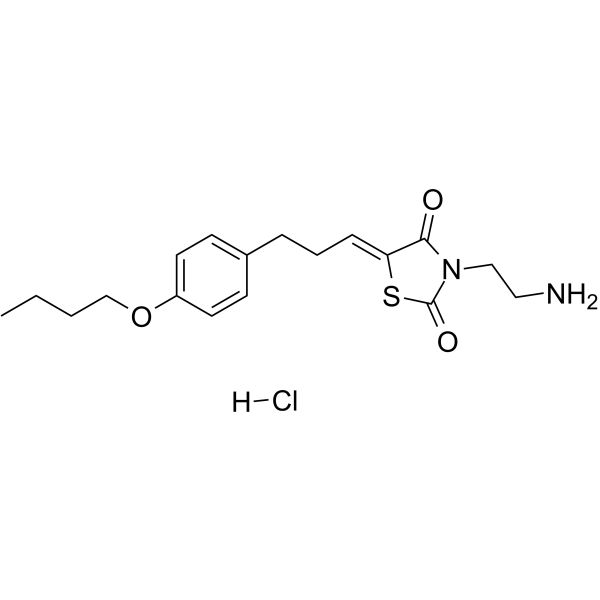
- HY-126752
-
|
|
Reactive Oxygen Species (ROS)
|
Neurological Disease
Metabolic Disease
Inflammation/Immunology
|
|
Ophthalmic acid is a ubiquitous metabolite and glutathione modulator, with a Ki of 0.95 mM for glyoxalase I. Ophthalmic acid competitively inhibits glyoxalase I, glutathione S-transferase, glutaredoxin, glutamate-cysteine ligase, protein disulfide reductase (glutathione), as well as non-selective cation channels. Ophthalmic acid is applicable in diabetes-related research .
|
-
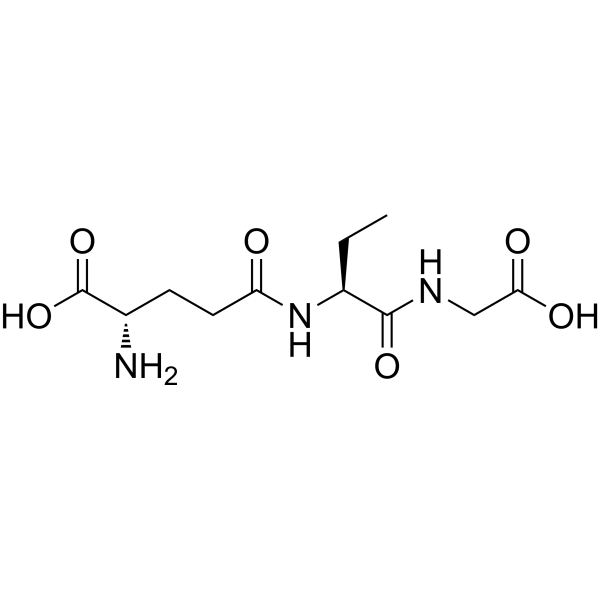
- HY-105911
-
|
|
5-HT Receptor
|
Neurological Disease
|
|
Org 6582 is a competitive, selective, and long-acting 5-HT (Ki = 89 nM) uptake inhibitor. Org 6582’s inhibitory effect on 5-HT uptake can last for more than 48 hours. Org 6582 can be used for research on depressive disorders .
|
-

- HY-136128
-
-
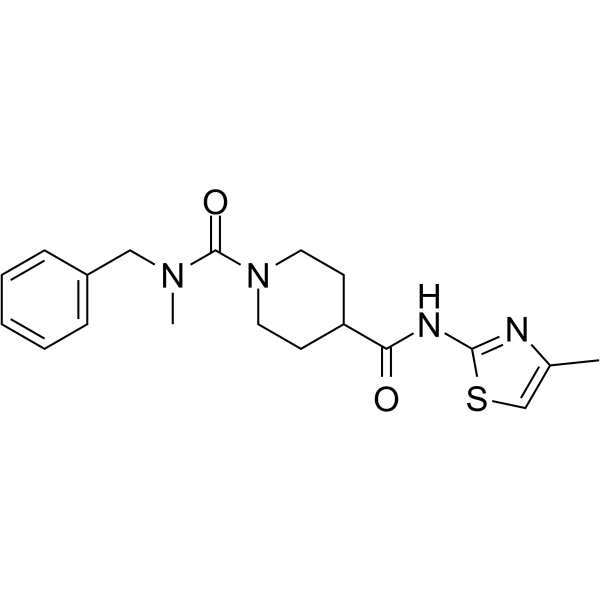
- HY-15663R
-
|
|
PAK
|
Cancer
|
|
IPA-3 (Standard) is the analytical standard of IPA-3. This product is intended for research and analytical applications. IPA-3 is a selective non-ATP competitive PAK1 inhibitor with IC50 of 2.5 μM, and shows no inhibition to group II PAKs (PAKs 4-6).
|
-

- HY-10512R
-
|
AR 0133418 (Standard); GSK 3β inhibitor VIII (Standard); AR 014418 (Standard)
|
Reference Standards
GSK-3
|
Metabolic Disease
Cancer
|
|
AR-A014418 (Standard) is the analytical standard of AR-A014418. This product is intended for research and analytical applications. AR-A014418 is a potent, selective and ATP-competitive GSK3β inhibitor (IC50=104 nM; Ki=38 nM) .
|
-

- HY-15425
-
|
Sphingosine Kinase 1 inhibitor II
|
SphK
LPL Receptor
Apoptosis
Autophagy
|
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
PF-543 (Sphingosine Kinase 1 Inhibitor II) is a potent, selective, reversible and sphingosine-competitive SPHK1 inhibitor with an IC50 of 2 nM and a Ki of 3.6 nM. PF-543 is >100-fold selectivity for SPHK1 over SPHK2. PF-543 is an effective potent inhibitor of sphingosine 1-phosphate (S1P) formation in whole blood with an IC50 of 26.7 nM. PF-543 induces apoptosis, necrosis, and autophagy .
|
-
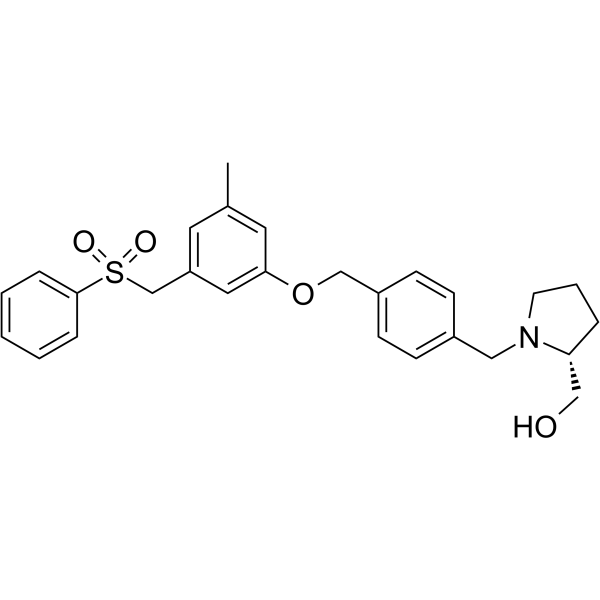
- HY-15425B
-
|
Sphingosine Kinase 1 inhibitor II hydrochloride
|
SphK
LPL Receptor
Apoptosis
Autophagy
|
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
PF-543 hydrochloride (Sphingosine Kinase 1 Inhibitor II hydrochloride) is a potent, selective, reversible and sphingosine-competitive SPHK1 inhibitor with an IC50 of 2 nM and a Ki of 3.6 nM. PF-543 hydrochloride is >100-fold selectivity for SPHK1 over SPHK2. PF-543 hydrochloride is an effective potent inhibitor of sphingosine 1-phosphate (S1P) formation in whole blood with an IC50 of 26.7 nM. PF-543 hydrochloride induces apoptosis, necrosis, and autophagy .
|
-
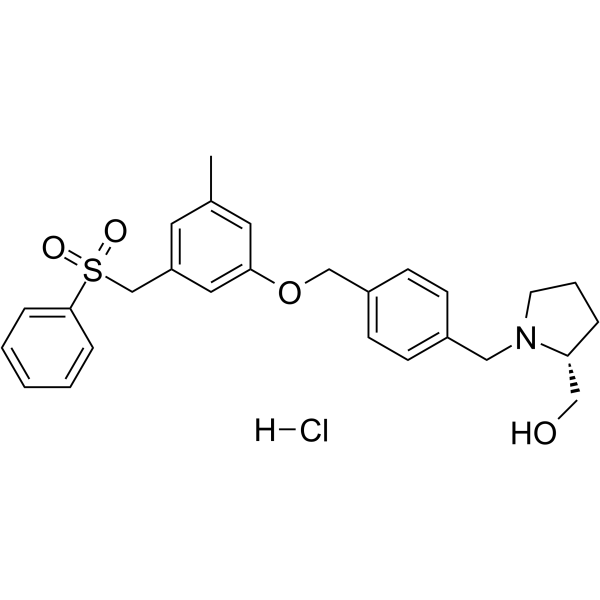
- HY-116919
-
|
|
Lipoxygenase
|
Cardiovascular Disease
|
|
MLS000536924 is a potent and selective inhibitor of human epithelial 15-lipoxygenase-2 with competitive activity. MLS000536924 exhibits more than 50-fold selectivity in inhibiting h15-LOX-2 and can be effectively applied to study its role in atherosclerosis, cystic fibrosis, and ferroptosis. The binding mode of MLS000536924 shows stronger restriction of protein movement than other inhibitors, further verifying its higher biological activity .
|
-

- HY-10787R
-
|
H 376/95 (Standard)
|
Reference Standards
Thrombin
|
Cardiovascular Disease
|
|
Ximelagatran (Standard) is the analytical standard of Ximelagatran (HY-10787). This product is intended for research and analytical applications. Ximelagatran (H 376/95) is an orally active thrombin inhibitor that selectively and competitively inhibits both free and clot-bound thrombin. Ximelagatran is an anticoagulant agent with a rapid onset of anticoagulant effect, predictable, dose-dependent pharmcoKinetics and pharmacodynamics .
|
-

- HY-18963R
-
|
RG-14355 (Standard)
|
Reference Standards
EGFR
|
Cancer
|
|
Lavendustin A (Standard) is the analytical standard of Lavendustin A. This product is intended for research and analytical applications. Lavendustin A (RG-14355) is a potent, selective and ATP-competitive inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase, with an IC50 of 11 nM. Lavendustin A does not inhibit protein kinase A or C. Lavendustin A can suppress VEGF-induced angiogenesis[1][2].
|
-

- HY-101523
-
|
|
CDK
|
Cancer
|
|
Cdc7-IN-1 (Compound 13) is a highly potent, selective and ATP competitive inhibitor of Cdc7 kinase, with an IC50 value of 0.6 nM at 1 mM ATP and with slow off-rate characteristics. Cdc7-IN-1 potently inhibits Cdc7 activity in cancer cells, and effectively induces cell death .
|
-
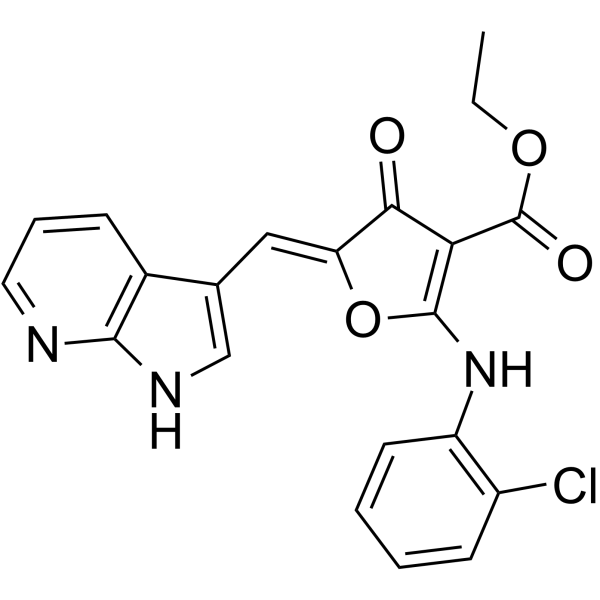
- HY-P10392
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R inhibits competitively the binding of β-catenin to TCF4. aStAx-35R selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-

- HY-12481
-
|
|
PI3K
Autophagy
|
Cancer
|
|
SAR405 is a first-in-class, selective, and ATP-competitive PI3K class III (PIK3C3) isoform Vps34 inhibitor (IC50=1.2 nM; Kd=1.5 nM). SAR405 inhibits autophagy induced either by starvation or by mTOR inhibition. Anticancer activity .
|
-
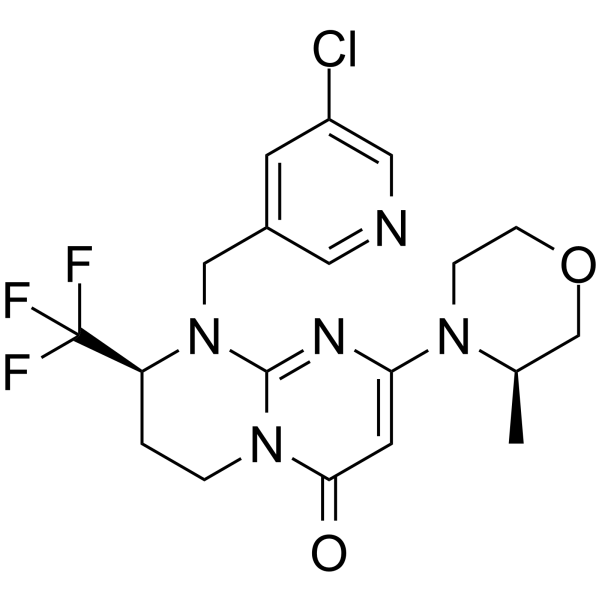
- HY-122203A
-
PCS1055
2 Publications Verification
|
mAChR
Cholinesterase (ChE)
|
Neurological Disease
|
|
PCS1055 is a selective and competitive antagonist for muscarinic M4 receptor with an IC50 of 18.1 nM and a Kd of 5.72 nM. PCS1055 inhibits radioligand [ 3H]-NMS binding to the M4 receptor with a Ki of 6.5 nM. PCS1055 is also an inhibitor for AChE with IC50 of 22 nM and 120 nM for electric eel and human AChE, respectively .
|
-

- HY-10422R
-
|
|
Reference Standards
mTOR
Autophagy
Apoptosis
|
Cancer
|
|
AZD-8055 (Standard) is the analytical standard of AZD-8055 (HY-10422). This product is intended for research and analytical applications. AZD-8055 is a potent, selective, and orally bioavailable ATP-competitive mTOR kinase inhibitor with an IC50 of 0.8 nM. AZD-8055 inhibits both mTORC1 and mTORC2 .
|
-

- HY-15816A
-
|
BVD-523 hydrochloride; VRT752271 hydrochloride
|
ERK
|
Cancer
|
|
Ulixertinib hydrochloride (BVD-523 hydrochloride) is a potent, orally active, highly selective, ATP-competitive and reversible covalent inhibitor of ERK1/2 kinases, with an IC50 of <0.3 nM against ERK2. Ulixertinib hydrochloride inhibits the phosphorylated ERK2 (pERK) and downstream kinase RSK (pRSK) in an A375 melanoma cell line .
|
-
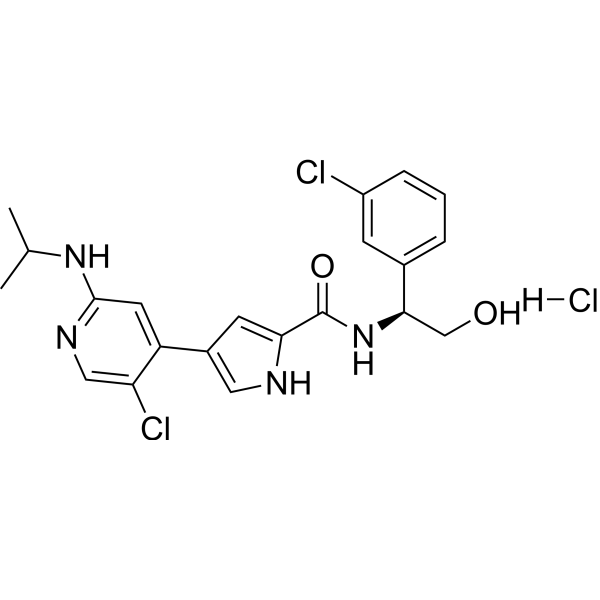
- HY-169422A
-
|
IDE275 (enantiomer)
|
DNA/RNA Synthesis
|
Cancer
|
|
GSK4418959 enantiomer is an enantiomer of GSK4418959 (HY-169422). GSK4418959 (IDE275) is a non-covalent, reversible, selective and orally active WRN helicase inhibitor. GSK4418959 inhibits ATPase and DNA unwinding functions in an ATP-competitive manner. GSK4418959 can be used for the study of microsatellite instability-high (MSI-H) cancer .
|
-

- HY-18732A
-
|
Tilarginine acetate; Methylarginine acetate
|
NO Synthase
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
L-NMMA (Tilarginine) acetate is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA acetate inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA acetate alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA acetate is applicable to research related to nociception, bone cancer pain, and myopia .
|
-
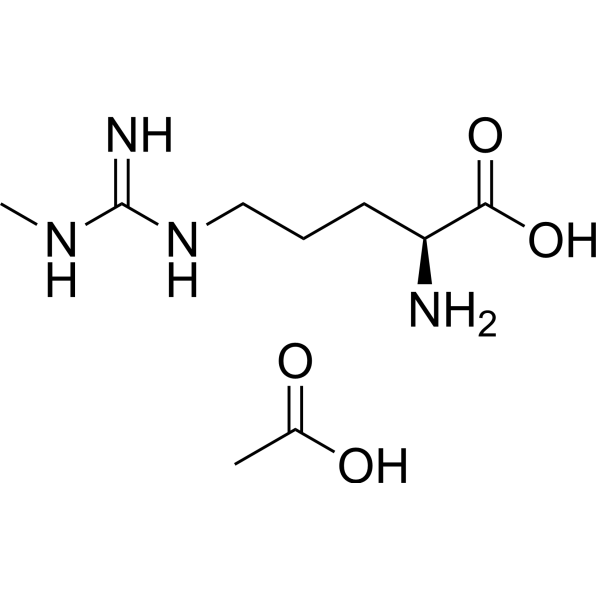
- HY-15901A
-
|
|
Pim
mTOR
|
Cancer
|
|
LGB321 monohydrochloride is a potent, selective, orally active and ATP competitive inhibitor of all three PIM kinases. LGB321 monohydrochloride inhibits proliferation, mTOR-C1 signaling and phosphorylation of BAD in a number of cell lines derived from diverse hematologic malignancies. LGB321 monohydrochloride can be used for the research of hematologic malignancies .
|
-
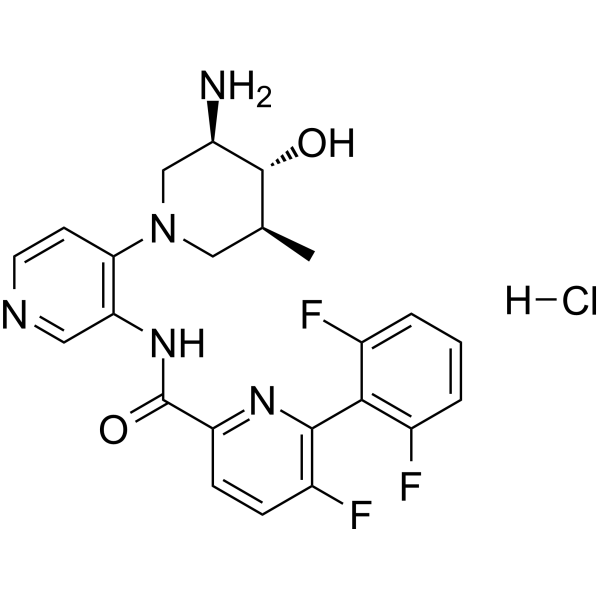
- HY-10281
-
|
|
Factor Xa
|
Cardiovascular Disease
|
|
YM-60828 methanesulfonate is an orally active, selective and competitive factor Xa inhibitor with a Ki of 1.3 nM and an IC50 of 2.3 nM. YM-60828 methanesulfonate inhibits thrombus formation and platelet aggregation. YM-60828 methanesulfonate can be used for the research of venous thrombosis, arterial thrombosis, and thromboembolic disorders .
|
-

- HY-18732C
-
|
Tilarginine hydrochloride; Methylarginine hydrochloride
|
NO Synthase
Endogenous Metabolite
|
Cardiovascular Disease
Cancer
|
|
L-NMMA (Tilarginine) hydrochloride is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA hydrochloride inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA hydrochloride alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA hydrochloride is applicable to research related to nociception, bone cancer pain, and myopia .
|
-

- HY-103442
-
|
DAPH
|
EGFR
Amyloid-β
|
Neurological Disease
Cancer
|
|
CGP52411 (DAPH) is a high selective, potent, orally active and ATP-competitive EGFR inhibitor with an IC50 of 0.3 μM. CGP52411 blocks the toxic influx of Ca 2+ ions into neuronal cells, and dramatic inhibits and reverses the formation of β-amyloid (Aβ42) fibril aggregates associated with Alzheimer's disease .
|
-
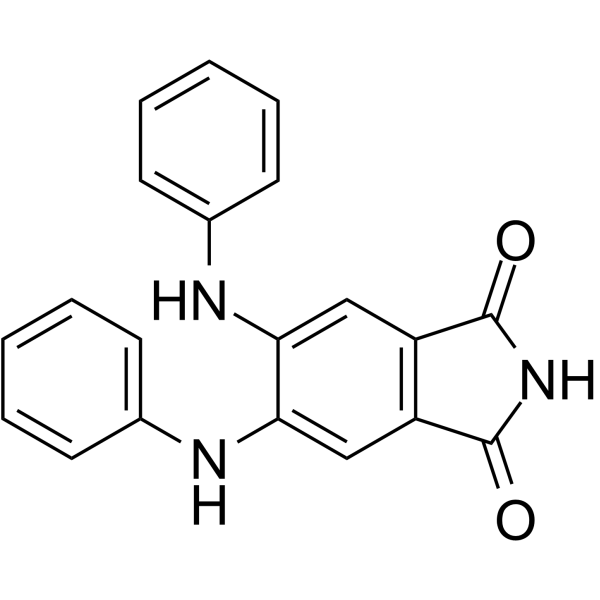
- HY-109184
-
|
AMG 397
|
Bcl-2 Family
|
Cancer
|
|
Murizatoclax (AMG 397) is a potent, selective and orally active inhibitor of myeloid leukemia 1 (MCL-1) inhibitor, with a Ki of 15 pM. Murizatoclax competitive binds to the BH3-binding groove of MCL1 with pro-apoptotic BCL-2 family members. Murizatoclax can be used for the research of cancer .
|
-
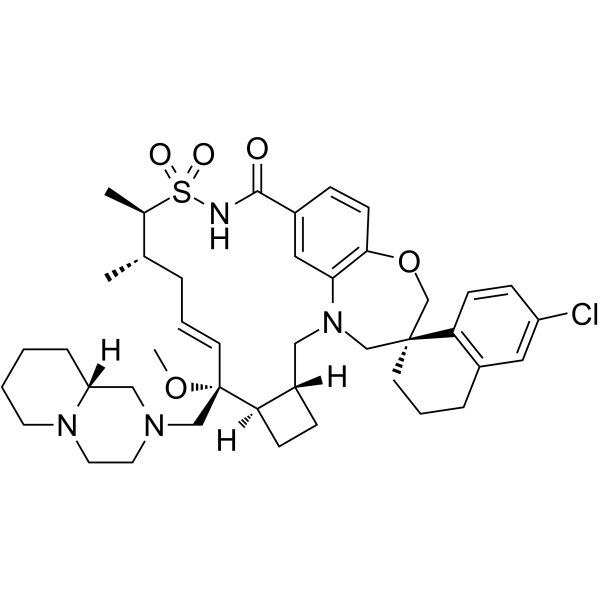
- HY-11007R
-
|
|
Bcr-Abl
SARS-CoV
|
Cancer
|
|
GNF-2 (Standard) is the analytical standard of GNF-2. This product is intended for research and analytical applications. GNF-2 is a highly selective, allosteric, non-ATP competitive inhibitor of Bcr-Abl. GNF-2 inhibits Ba/F3.p210 proliferation with an IC50 of 138 nM .
|
-

- HY-18732
-
|
Tilarginine; Methylarginine
|
NO Synthase
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
L-NMMA (Tilarginine) is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA is applicable to research related to nociception, bone cancer pain, and myopia .
|
-

- HY-103712
-
|
CT7001; ICEC0942
|
CDK
Apoptosis
|
Cancer
|
|
Samuraciclib (CT7001) is a potent, selective, ATP-competitive and orally active CDK7 inhibitor, with an IC50 of 41 nM. Samuraciclib displays 45-, 15-, 230- and 30-fold selectivity over CDK1, CDK2 (IC50 of 578 nM), CDK5 and CDK9, respectively. Samuraciclib inhibits the growth of breast cancer cell lines with GI50 values between 0.2-0.3 μM. Samuraciclib has anti-tumor effects .
|
-
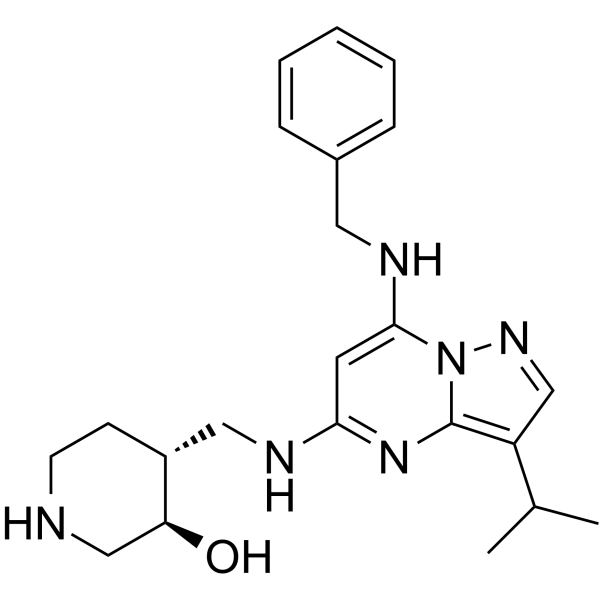
- HY-103712A
-
|
CT7001 hydrochloride; ICEC0942 hydrochloride
|
CDK
Apoptosis
|
Cancer
|
|
Samuraciclib hydrochloride (CT7001 hydrochloride) is a potent, selective, ATP-competitive and orally active CDK7 inhibitor, with an IC50 of 41 nM. Samuraciclib hydrochloride displays 45-, 15-, 230- and 30-fold selectivity over CDK1, CDK2 (IC50 of 578 nM), CDK5 and CDK9, respectively. Samuraciclib hydrochloride inhibits the growth of breast cancer cell lines with GI50 values between 0.2-0.3 µM. Samuraciclib hydrochloride has anti-tumor effects .
|
-
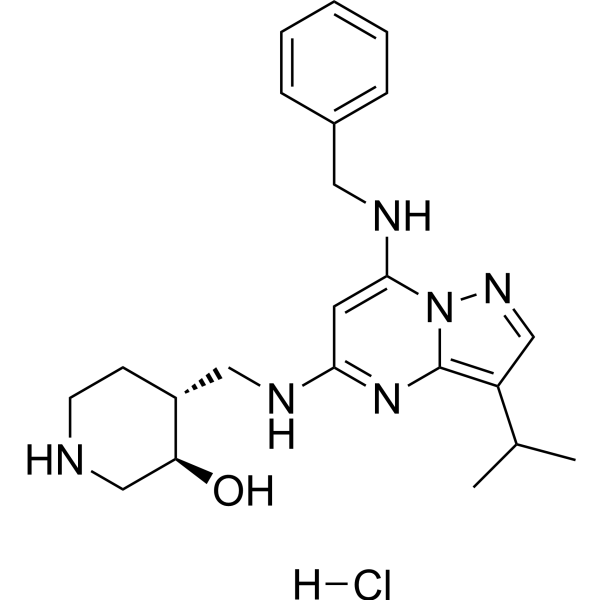
- HY-103712C
-
|
CT7001 hydrochloride hydrate; ICEC0942 hydrochloride hydrate
|
CDK
Apoptosis
|
Cancer
|
|
Samuraciclib (CT7001) hydrochloride hydrate is a potent, selective, ATP-competitive and orally active CDK7 inhibitor, with an IC50 of 41 nM. Samuraciclib hydrochloride hydrate displays 45-, 15-, 230- and 30-fold selectivity over CDK1, CDK2 (IC50 of 578 nM), CDK5 and CDK9, respectively. Samuraciclib hydrochloride hydrate inhibits the growth of breast cancer cell lines with GI50 values between 0.2-0.3 μM. Samuraciclib hydrochloride hydrate has anti-tumor effects .
|
-
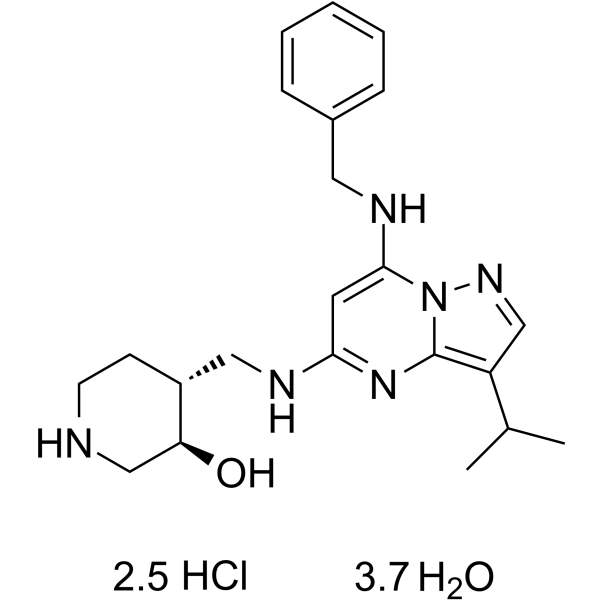
- HY-B0442S
-
|
|
Isotope-Labeled Compounds
Phosphodiesterase (PDE)
Endogenous Metabolite
|
Endocrinology
|
|
Vardenafil-d5 is deuterium labeled Vardenafil. Vardenafil is a selective, orally active, potent inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil shows selectivity over PDE1 (180 nM), PDE6 (11 nM), PDE2, PDE3, and PDE4 (>1000 nM). Vardenafil competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil can be used for the research of erectile dysfunction .
|
-
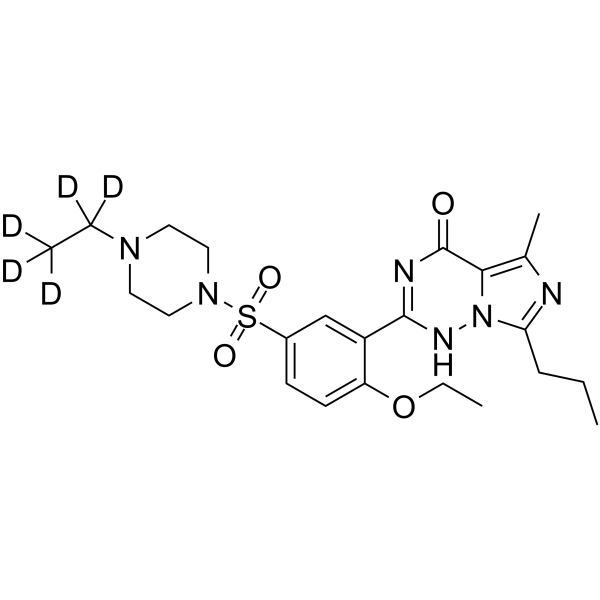
- HY-50703
-
MK-2461
1 Publications Verification
|
c-Met/HGFR
|
Cancer
|
|
MK-2461 is an ATP-competitive, selective and orally active wild-type and mutant c-Met inhibitor (IC50s: 0.4-2.5 nM). MK-2461 also inhibits Ron (IC50 of 7 nM) and Flt1 (IC50 of 10 nM), MK-2461 shows selective for c-MET over other kinases (lC50s = 22-7800 nM). MK-2461 can be used for the study of cancer, such as gastric cancer .
|
-
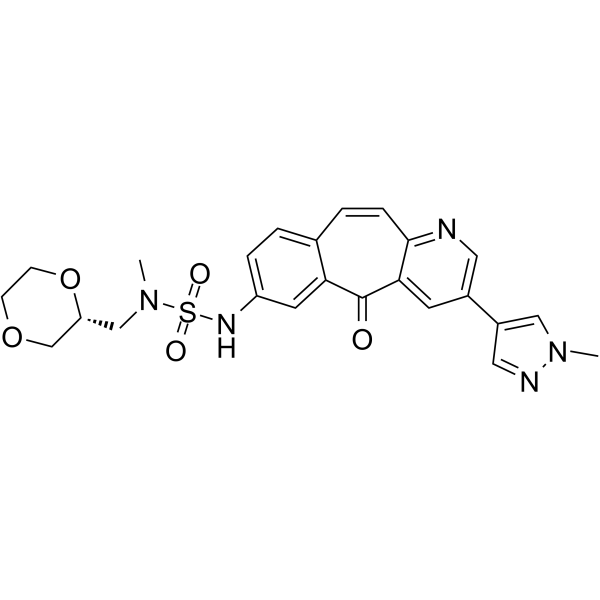
- HY-103712B
-
|
CT7001 hydrochloride dihydrate; ICEC0942 hydrochloride dihydrate
|
CDK
Apoptosis
|
Cancer
|
|
Samuraciclib (CT7001) hydrochloride hydrate is a potent, selective, ATP-competitive and orally active CDK7 inhibitor, with an IC50 of 41 nM. Samuraciclib hydrochloride hydrate displays 45-, 15-, 230- and 30-fold selectivity over CDK1, CDK2 (IC50 of 578 nM), CDK5 and CDK9, respectively. Samuraciclib hydrochloride hydrate inhibits the growth of breast cancer cell lines with GI50 values between 0.2-0.3 μM. Samuraciclib hydrochloride hydrate has anti-tumor effects .
|
-
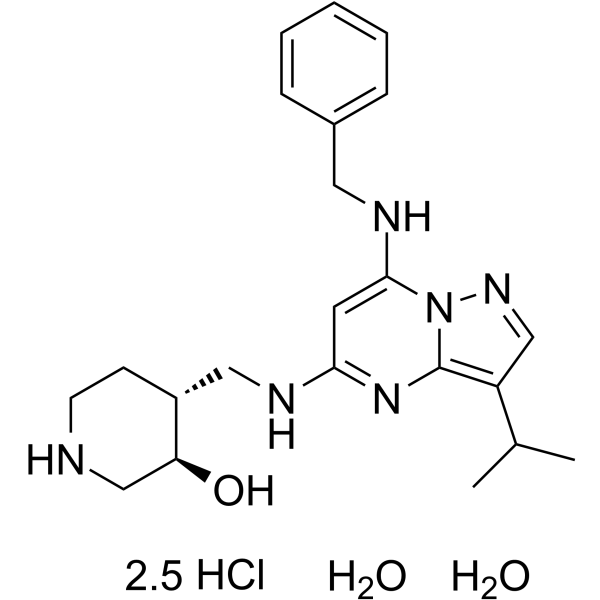
- HY-10917R
-
|
|
c-Fms
|
Inflammation/Immunology
Cancer
|
|
GW2580 (Standard) is the analytical standard of GW2580. This product is intended for research and analytical applications. GW2580 is an orally bioavailable and selective inhibitor of c-Fms kinase which completely inhibits human cFMS kinase in vitro at 0.06 μM. GW2580 acts as a competitive inhibitor of ATP binding to the cFMS kinase and inhibits colony-stimulating-factor-1 signaling .
|
-

- HY-15141G
-
|
Antibiotic AM-2282; STS; AM-2282
|
PKC
|
Infection
Cancer
|
|
Staurosporine (AM-2282) (GMP) is Staurosporine (HY-15141) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases .
|
-
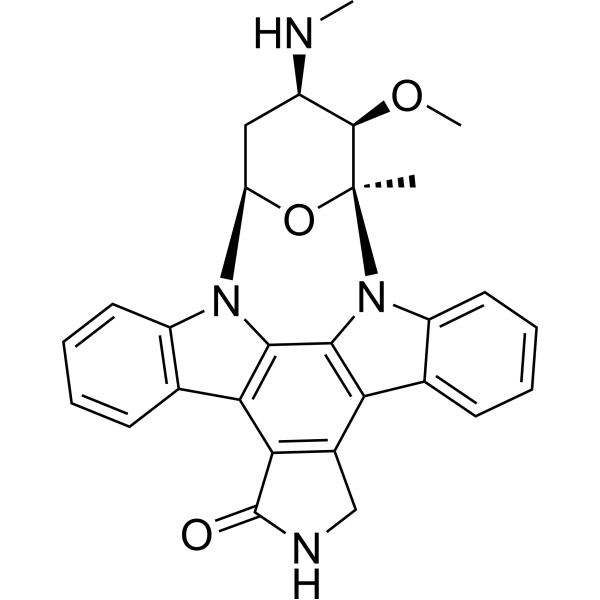
- HY-133130
-
|
|
MAGL
|
Neurological Disease
|
|
JNJ-42226314, a chemical probe, is a competitive, highly selective and reversible non-covalent monoacylglycerol lipase (MAGL) inhibitor. JNJ-42226314 demonstrates dose-dependent enhancement of the major endocannabinoid 2-arachidonoylglycerol (2-AG) as well as efficacy in models of neuropathic and inflammatory pain .
|
-
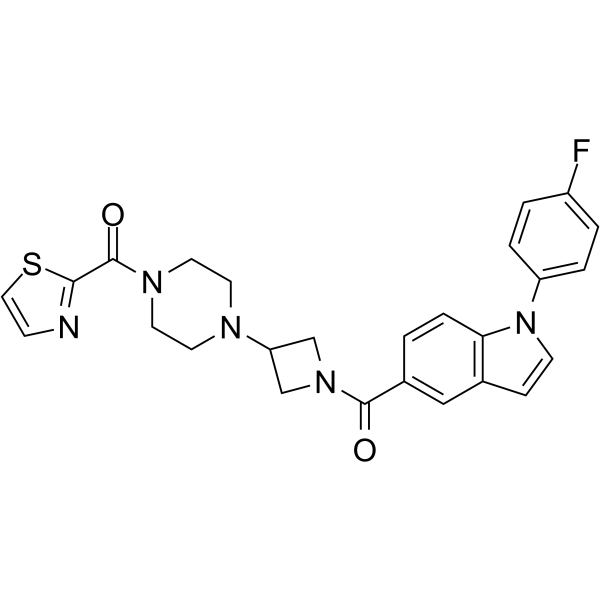
- HY-12335A
-
|
|
Histone Methyltransferase
|
Inflammation/Immunology
Cancer
|
|
UNC0379 TFA is a selective, substrate-competitive inhibitor of lysine methyltransferase SETD8 (KMT5A) with an IC50 of 7.3 μM, KD value of 18.3 μM. UNC0379 TFA can be used in the research of inflammation and cancers, such as pulmonary fibrosis, ovarian cancer, neuroblastoma .
|
-
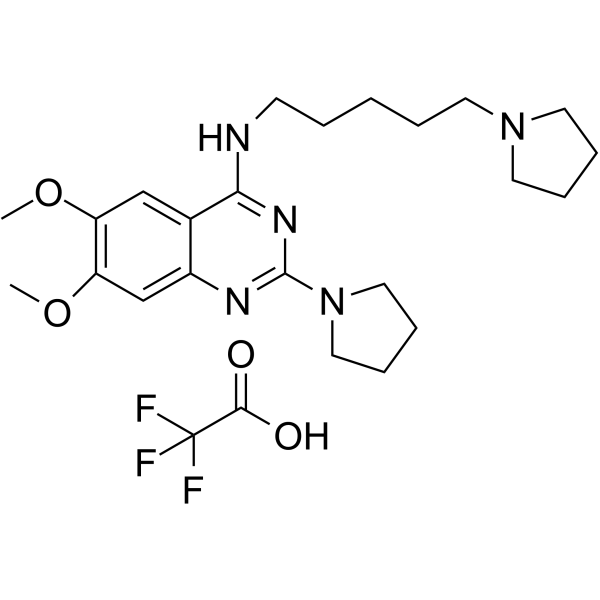
- HY-161029
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
T14-A24 is an orally active, reversible, competitive, and selective AChE inhibitor (Ki=22 nM, IC50=6 nM). T14-A24 has benign BBB penetration, remarkable neuroprotective effect, and safe toxicological profile .
|
-
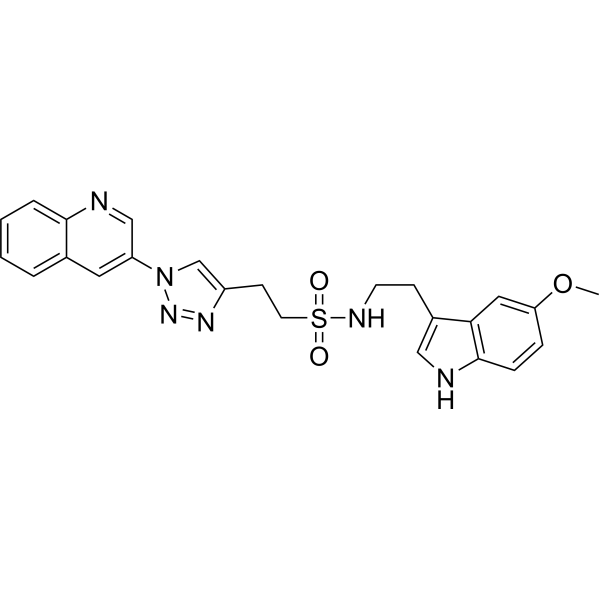
- HY-10262
-
|
|
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
BMS-536924 is an orally active, competitive and selective insulin-like growth factor receptor (IGF-1R) kinase and insulin receptor (IR) inhibitor with IC50s of 100 nM and 73 nM, respectively. BMS-536924 has anti-cancer activity .
|
-
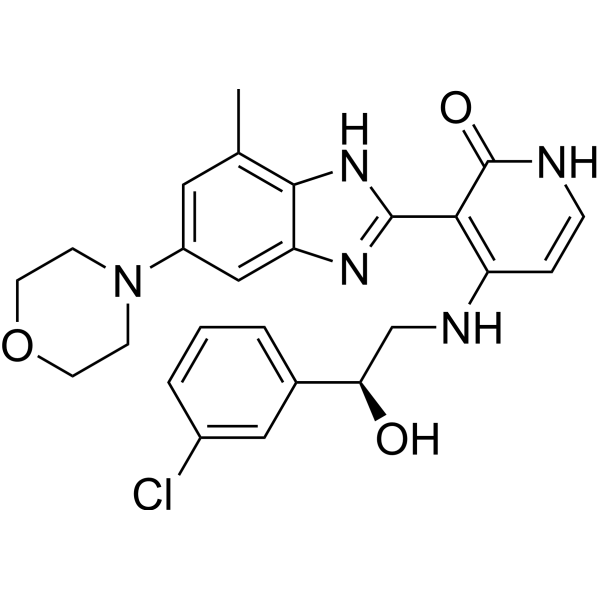
- HY-12866R
-
|
LOXO-101 (Standard); ARRY-470 (Standard)
|
Reference Standards
Trk Receptor
Apoptosis
|
Cancer
|
|
Larotrectinib (Standard) is the analytical standard of Larotrectinib. This product is intended for research and analytical applications. Larotrectinib (LOXO-101) is an ATP-competitive oral, selective inhibitor of the tropomyosin-related kinase (TRK) family receptors, with low nanomolar 50% inhibitory concentrations against all three isoforms (TRKA, B, and C).
|
-

- HY-122629
-
|
|
DAPK
|
Others
|
|
DRAK2-IN-1, compound 16, is a potent, selective and ATP-competitive DRAK2 inhibitor with IC50and Kivalues of 3 nM and 0.26 nM, respectively.
DRAK2-IN-1 also has inbitory effect on DRAK1 (IC50=51 nM) .
|
-
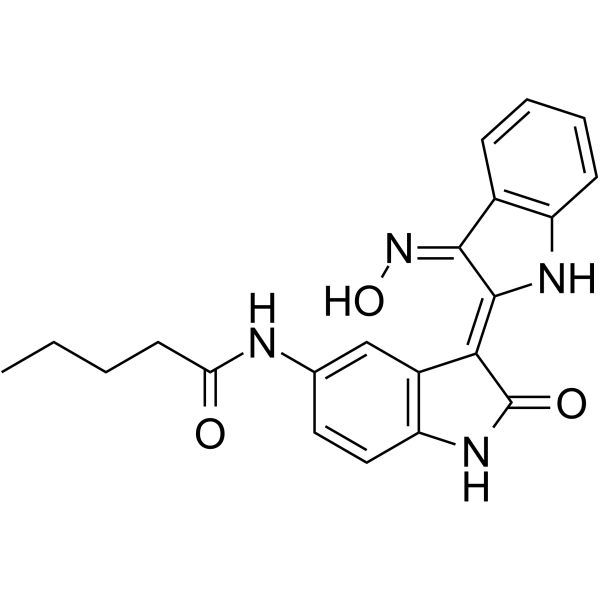
- HY-175750
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
MOMA-341 is a selective Werner RecQ like helicase (WRN) inhibitor. MOMA-341 binds to WRN at cysteine 727 through an allosteric and ATP-competitive binding mechanism. MOMA-341 has antitumor activity and can be used for advanced and metastatic solid tumors research .
|
-

- HY-P1293A
-
|
|
iGluR
|
Neurological Disease
|
|
Conantokin G TFA, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G TFA inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G TFA has neuroprotective properties .
|
-
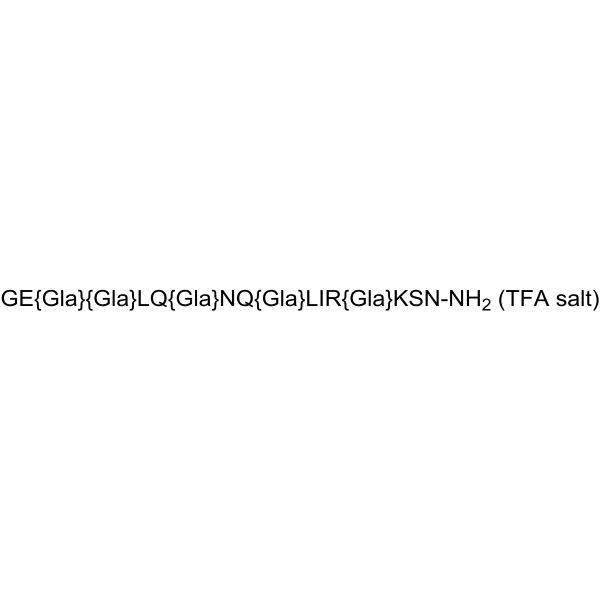
- HY-14691R
-
|
BAY 869766 (Standard); RDEA119 (Standard)
|
MEK
Reference Standards
|
Cancer
|
|
Refametinib (Standard) is the analytical standard of Refametinib. This product is intended for research and analytical applications. Refametinib (BAY 869766; RDEA119) is an orally available, potent, non-ATP-competitive, selective, allosteric MEK1/MEK2 inhibitor with IC50s of 19 nM and 47 nM, respectively.
|
-

- HY-P11019
-
|
|
CXCR
Calcium Channel
|
Cancer
|
|
IS4 is a selective CXCR4 competitive antagonist, with an IC50 of 0.65 nM in THP-1 cells and 38.75 nM in Jurkat cells. IS4 is stable in serum and non-cytotoxic. IS4 competitively binds to CXCR4 with CXCL12, thereby inhibiting CXCL12-induced intracellular Ca 2+ release and cancer cell migration. IS4 can be used in the research on the prevention of metastasis of breast cancer, prostate cancer, leukemia, and other diseases .
|
-

- HY-10409
-
|
TG-101348; SAR 302503
|
JAK
Apoptosis
|
Cancer
|
|
Fedratinib (TG-101348) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-
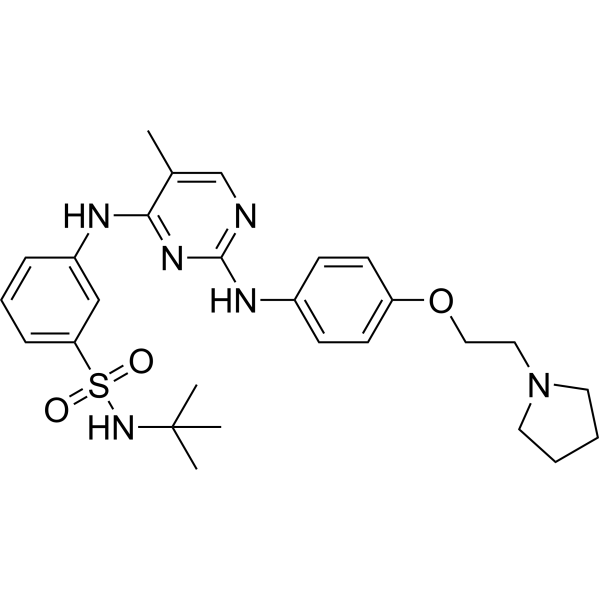
- HY-10409A
-
|
TG-101348 hydrochloride hydrate; SAR 302503 hydrochloride hydrate
|
JAK
Apoptosis
|
Cancer
|
|
Fedratinib hydrochloride hydrate (TG-101348 hydrochloride hydrate) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib hydrochloride hydrate shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib hydrochloride hydrate induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-
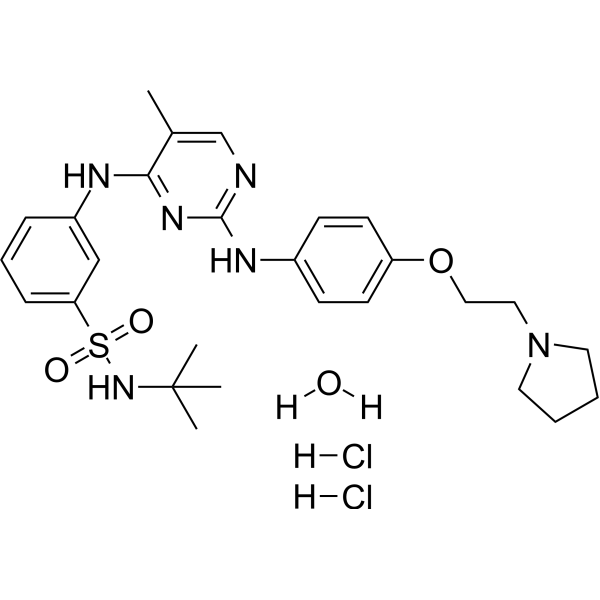
- HY-122203
-
|
|
mAChR
Cholinesterase (ChE)
|
Neurological Disease
|
|
PCS1055 dihydrochloride is a potent, selective and competitive muscarinic M4 receptor antagonist with an IC50 of 18.1 nM and a Kd of 5.72 nM. PCS1055 dihydrochloride inhibits radioligand [ 3H]-NMS binding to the M4 receptor with a Ki of 6.5 nM. PCS1055 dihydrochloride exhibits >100-fold selectivity over M1-, M3-, and M5-receptors and 30-fold selectivity at the M2 receptor. PCS1055 dihydrochloride is also a potent AChE inhibitor with IC50 s of 22 nM and 120 nM for electric eel and human AChE, respectively .
|
-
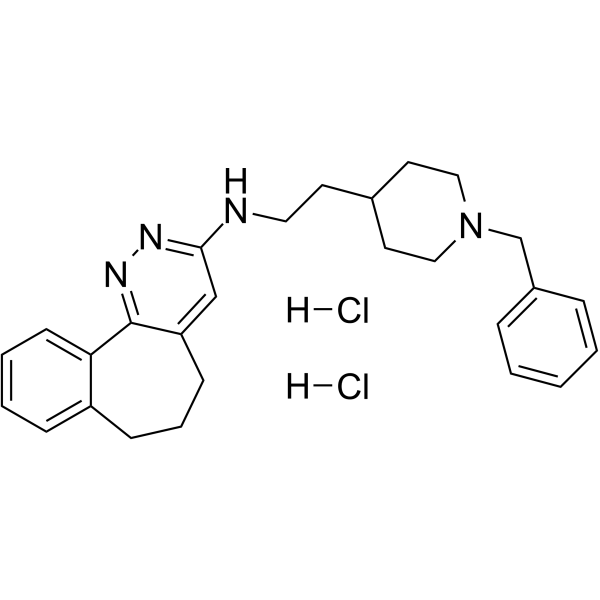
- HY-137506
-
-
- HY-23460A
-
|
4-Ethynyl-L-phenylalanine hydrochloride
|
Tryptophan Hydroxylase
|
Neurological Disease
|
|
p-Ethynylphenylalanine hydrochloride (4-Ethynyl-L-phenylalanine hydrochloride) is a potent, selective, reversible and competitive inhibitor of tryptophan hydroxylase (TPH), with a Ki of 32.6 μM . p-Ethynylphenylalanine (hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-106418A
-
|
|
Adrenergic Receptor
5-HT Receptor
|
Cardiovascular Disease
|
|
SGB-1534 hydrochloride is an orally active, selective and competitive antagonist of the alpha 1-adrenoceptor and the 5-HT2 receptor. SGB-1534 hydrochloride can inhibit vasoconstriction and lower blood pressure. SGB-1534 hydrochloride can be used for the research of cardiovascular disease, such as hypertension .
|
-

- HY-23460
-
|
4-Ethynyl-L-phenylalanine
|
Tryptophan Hydroxylase
|
Neurological Disease
|
|
p-Ethynylphenylalanine (4-Ethynyl-L-phenylalanine) is a potent, selective, reversible and competitive inhibitor of tryptophan hydroxylase (TPH), with a Ki of 32.6 μM . p-Ethynylphenylalanine is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
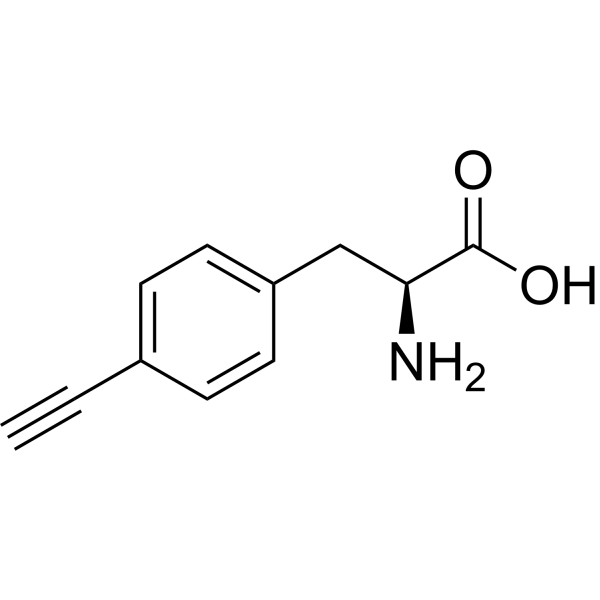
- HY-137472
-
|
|
GSK-3
|
Neurological Disease
|
|
SAR502250 is a potent, selective, ATP competitive, orally active and brain-penetrant inhibitor of GSK3, with an IC50 of 12 nM for human GSK-3β. SAR502250 displays antidepressant-like activity. SAR502250 can be used for the research of Alzheimer’s disease (AD) .
|
-
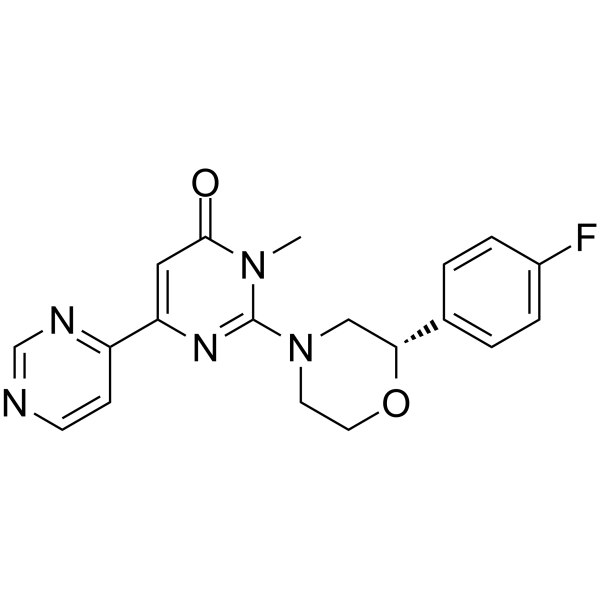
- HY-100844R
-
|
|
MAP3K
Reference Standards
Apoptosis
|
Cancer
|
|
GS-444217 (Standard) is the analytical standard of GS-444217 (HY-100844). This product is intended for research and analytical applications. GS-444217 is a potent, orally available and selective ATP-competitive inhibitor of apoptosis signal-regulating kinase 1 (ASK1) with an IC50 of 2.87 nM .
|
-

- HY-12012R
-
|
|
GSK-3
Autophagy
|
Neurological Disease
Cancer
|
|
SB 216763 (Standard) is the analytical standard of SB 216763. This product is intended for research and analytical applications. SB 216763 is potent, selective and ATP-competitive GSK-3 inhibitor with IC50s of 34.3 nM for both GSK-3α and GSK-3β.
|
-

- HY-10119
-
|
SCH 530348
|
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Vorapaxar (SCH 530348), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar (SCH 530348) inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-
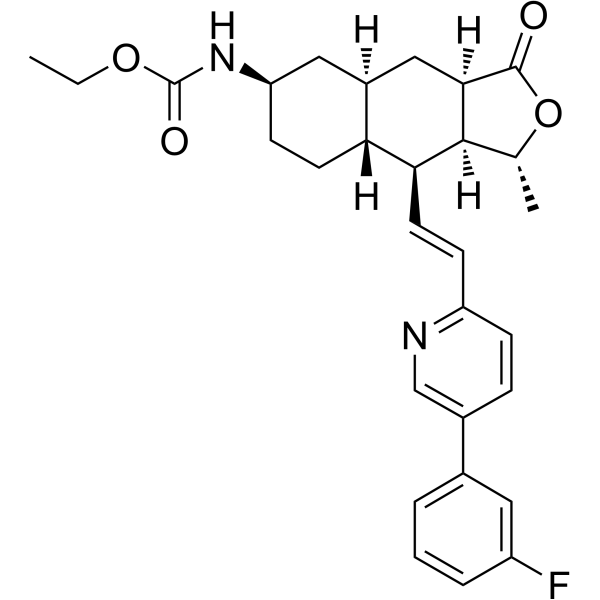
- HY-10228
-
|
AMG 706
|
c-Kit
VEGFR
|
Cancer
|
|
Motesanib (AMG 706) is a potent ATP-competitive inhibitor of VEGFR1/2/3 with IC50s of 2 nM/3 nM/6 nM, respectively, and has similar activity against Kit, and is appr 10-fold more selective for VEGFR than PDGFR and Ret.
|
-
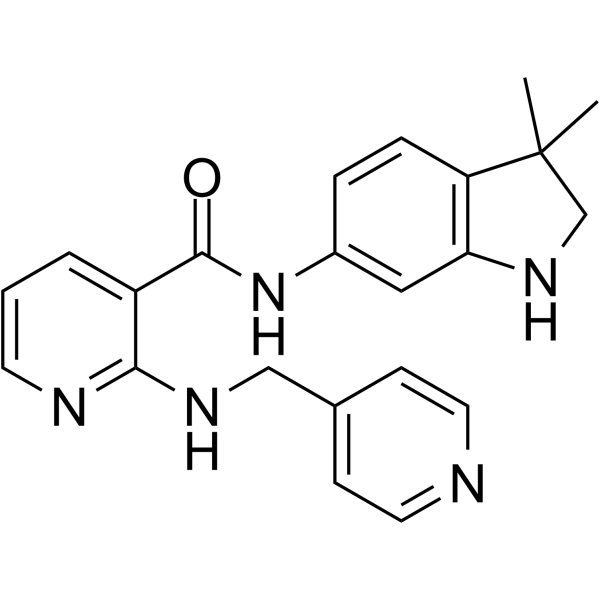
- HY-173055
-
|
|
Lipoxygenase
|
Inflammation/Immunology
|
|
Ferroleuton is a competitive, selective inhibitor for 5-lipoxygenase with an IC50 of 0.21 μM. Ferroleuton exhibits antioxidant activity in DPPH (scavenges 86% DPPH-H at 50 μM), ABTS (EC50=19.42 μM) and FRAP (EC50=3.32 μM) assays .
|
-

- HY-117043
-
|
|
Deubiquitinase
SARS-CoV
|
Infection
|
|
GRL0617 is a selective and competitive noncovalent inhibitor of severe acute respiratory syndrome (SARS-CoV) papain-like protease (PLpro), with an IC50 of 0.6 μM and a Ki value of 0.49 μM. GRL0617 also inhibits SARS-CoV with an EC50 of 14.5 μM. GRL0617 can be used for the research of severe acute respiratory syndrome .
|
-
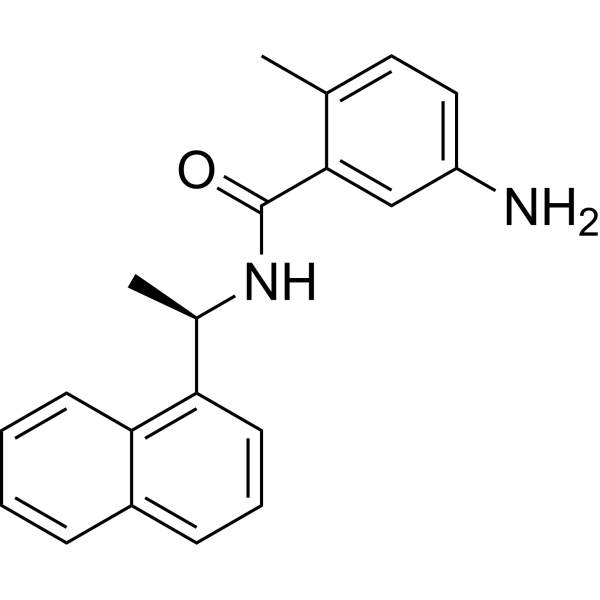
- HY-108540R
-
|
2-Amino-2-norbornanecarboxylic acid (Standard); LAT1-IN-1 (Standard)
|
Apoptosis
Reference Standards
|
Cancer
|
|
BCH (Standard) is the analytical standard of BCH. This product is intended for research and analytical applications. BCH (2-Amino-2-norbornanecarboxylic acid) is a selective and competitive inhibitor of large neutral amino acid transporter 1 (LAT1) significantly inhibit cellular uptake of amino acids and mTOR phosphorylation, which induces the suppression of cancer growth and apoptosis .
|
-

- HY-15847
-
HS38
3 Publications Verification
|
DAPK
|
Cardiovascular Disease
|
|
HS38 is a potent, selective, and ATP-competitive inhibitor of death-associated protein kinase 1 (DAPK1) and zipper-interacting protein kinase (ZIPK, also called DAPK3), with Kds of 300 nM and 280 nM, respectively. HS38 is also a PIM3 inhibitor with an IC50 of 200 nM. HS38 can be used for the research of smooth muscle related disorders .
|
-
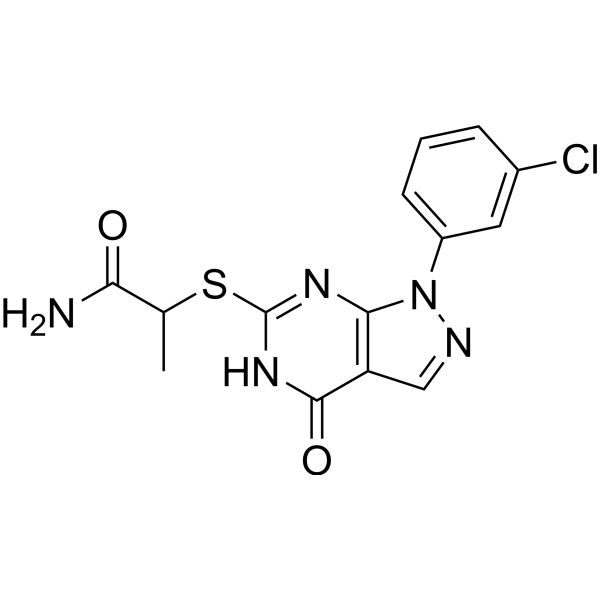
- HY-100846R
-
|
|
Histone Methyltransferase
Reference Standards
|
Cancer
|
|
JQEZ5 (Standard) is the analytical standard of JQEZ5 (HY-100846). This product is intended for research and analytical applications. JQEZ5 is a potent and selective EZH2 lysine methyltransferase inhibitor. JQEZ5 SAM-competitively inhibits polycomb repressive complex 2 (PRC2) with an IC50 of 80 nM. JQEZ5 has anti-tumor effects .
|
-

- HY-181092
-
|
|
PAK
|
Cancer
|
|
PAK4-IN-7 is a selective ATP-competitive inhibitor of p21-activated kinase 4 (PAK4), with an IC50 of 1.88 μM. PAK4-IN-7 inhibits the proliferation of cancer cells. PAK4-IN-7 is applicable for research on tumors such as colorectal cancer and lung cancer .
|
-

- HY-10195R
-
|
LY333531 (Standard)
|
Reference Standards
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (Standard) is the analytical standard of Ruboxistaurin. This product is intended for research and analytical applications. Ruboxistaurin (LY333531) is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin inhibits PKC beta II with an IC50 of 5.9 nM .
|
-

- HY-162675
-
|
|
GSK-3
|
Infection
Neurological Disease
Metabolic Disease
Cancer
|
|
COB-187 is a potent, ATP-competitive and selective inhibitor of GSK-3β. COB-187 inhibits GSK-3 through a reversible and Cysteine (Cys)-199-dependent mechanism. COB-187 inhibits LPS induced cytokine production and SARS-CoV-2 spike protein-induced CXCL10 production .
|
-

- HY-108675
-
|
|
MMP
P2X Receptor
|
Infection
Cancer
|
|
PPNDS tetrasodium is a selective and competitive meprin β inhibitor (IC50: 80 nM, Ki: 8 nM), and also inhibits ADAM10 (IC50: 1.2 μM). PPNDS tetrasodium is also a P2X1 receptor antagonist. PPNDS is an agonist for the ATP receptor of Paramecium. PPNDS tetrasodium potently inhibits polymerases from viruses. PPNDS tetrasodium can be used in the research of infection and cancers .
|
-
- HY-15425A
-
|
Sphingosine Kinase 1 inhibitor II Citrate
|
SphK
LPL Receptor
Apoptosis
Autophagy
|
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
PF-543 Citrate (Sphingosine Kinase 1 Inhibitor II Citrate) is a potent, selective, reversible and sphingosine-competitive SPHK1 inhibitor with an IC50 of 2 nM and a Ki of 3.6 nM. PF-543 Citrate is >100-fold selectivity for SPHK1 over SPHK2. PF-543 Citrate is an effective potent inhibitor of sphingosine 1-phosphate (S1P) formation in whole blood with an IC50 of 26.7 nM. PF-543 Citrate induces apoptosis, necrosis, and autophagy .
|
-
- HY-105327
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
P11149 is a competitive, BBB-penetarated weakly, orally active and selective inhibitor of AChE. P11149 exhibits an IC50 of 1.3 μM for rat BChE/AChE. P11149, a Galanthamine derivative, demonstrates central cholinergic activity, behavioral efficacy and safety. P11149 is used in the study for Alzheimer's disease .
|
-
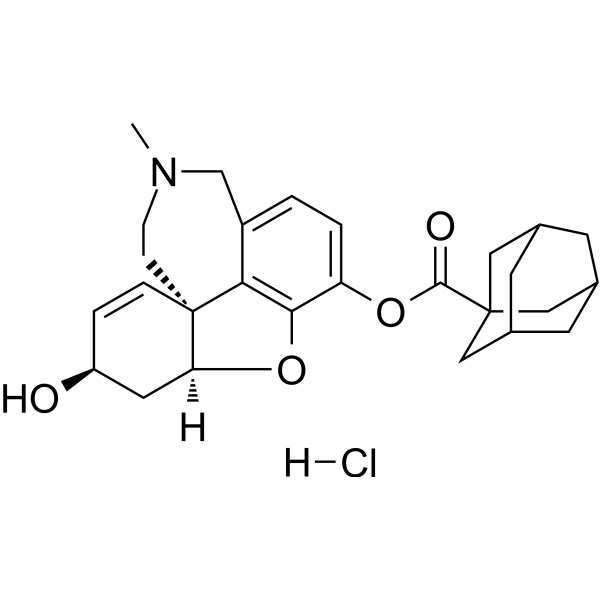
- HY-10014
-
R547
2 Publications Verification
|
CDK
GSK-3
Apoptosis
|
Cancer
|
|
R547 is a potent, selective and orally active ATP-competitive CDK inhibitor, with Kis of 2 nM, 3 nM and 1 nM for CDK1/cyclin B, CDK2/cyclin E and CDK4/cyclin D1, respectively .
|
-
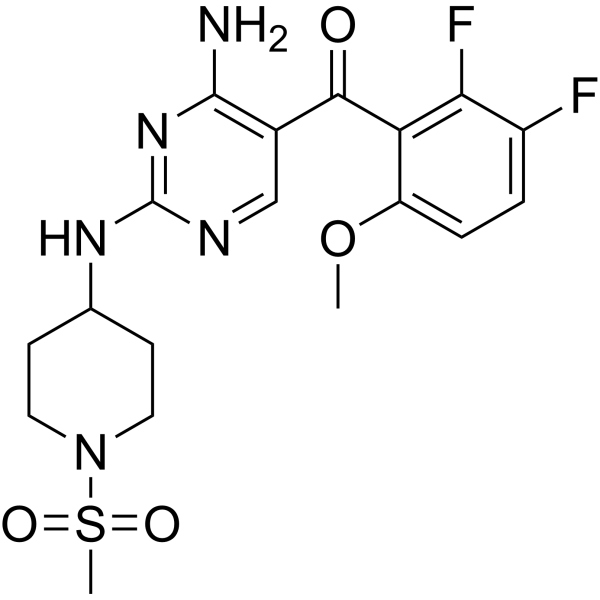
- HY-16214
-
|
LDHA inhibitor FX11
|
Lactate Dehydrogenase
Apoptosis
Reactive Oxygen Species (ROS)
|
Cancer
|
|
FX-11 is a potent, selective, reversible and competitive lactate dehydrogenase A (LDHA) inhibitor, with a Ki of 8 μM. FX-11 reduces ATP levels and induces oxidative stress, ROS production and cell death. FX-11 shows antitumor activity in lymphoma and pancreatic cancer xenografts .
|
-
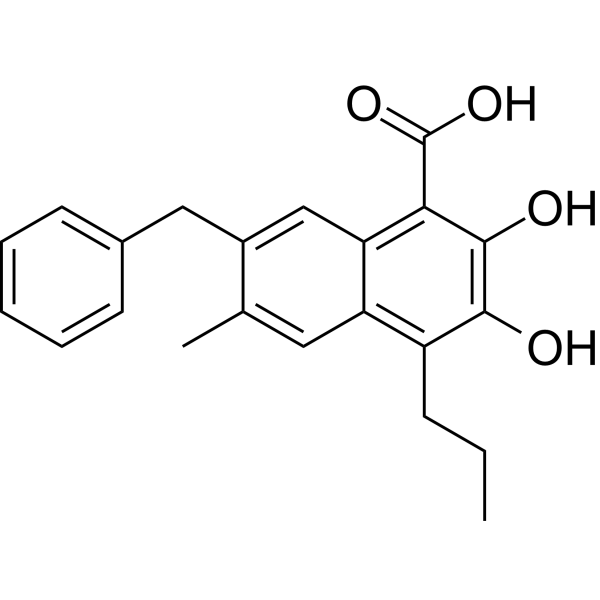
- HY-118236
-
|
(S)-KIN-193
|
PI3K
|
Cardiovascular Disease
|
|
(S)-AZD6482 ((S)-KIN-193) is a highly effective and selective ATP-competitive inhibitor of PI3Kβ, exhibiting an IC(50) of 0.01 μM, and it can reduce insulin-induced glucose uptake by human adipocytes in vitro with an IC(50) of 4.4 μM.
|
-

- HY-100429
-
CAN508
1 Publications Verification
|
CDK
|
Cancer
|
|
CAN508 is a potent, ATP-competitive CDK9/cyclin T1 inhibitor with an IC50 of 0.35 μM. CAN508 exhibits a 38-fold selectivity for CDK9/cyclin T over other CDK/cyclin complexes. Antitumor activity .
|
-
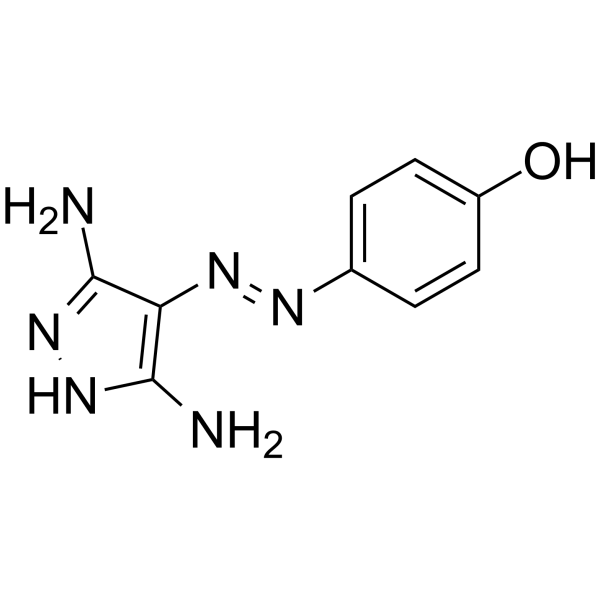
- HY-100888R
-
|
TAK-931 (Standard)
|
CDK
Reference Standards
|
Cancer
|
|
Simurosertib (Standard) is the analytical standard of Simurosertib (HY-100888). This product is intended for research and analytical applications. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM. Simurosertib has anti-cancer activity .
|
-

- HY-10119A
-
|
SCH 530348 sulfate
|
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Vorapaxar sulfate (SCH 530348 sulfate), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar sulfate inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-
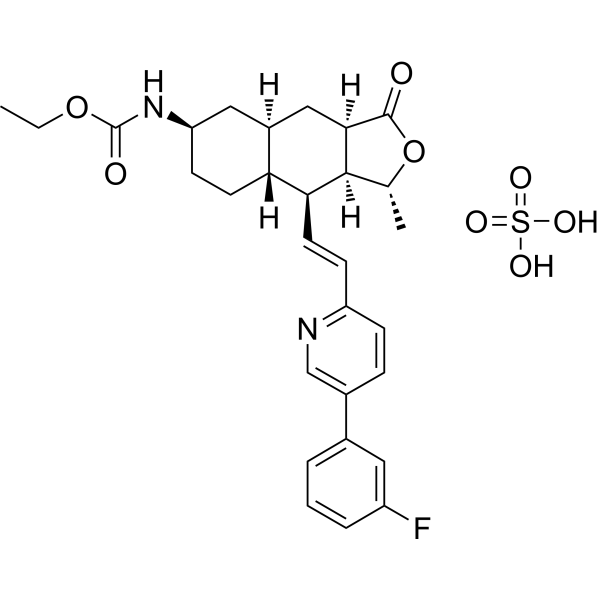
- HY-111260
-
|
|
CDK
GSK-3
Apoptosis
|
Cancer
|
|
R547 mesylate is a potent, selective and orally active ATP-competitive CDK inhibitor, with Kis of 2 nM, 3 nM and 1 nM for CDK1/cyclin B, CDK2/cyclin E and CDK4/cyclin D1, respectively .
|
-

- HY-19674
-
|
SSR182289A free base
|
Thrombin
|
Cardiovascular Disease
|
|
SSR182289 (SSR182289A free base) is a selective and potent orally active thrombin inhibitor. SSR182289 competitively and selectivity inhibits human thrombin (Ki=0.031 μM). SSR182289 demonstrates anticoagulant activity in vitro (thrombin time EC100=96 nM) and inhibits tissue factor-induced thrombin generation (IC50=0.15 μM) in human plasma. SSR182289 inhibits thrombin-induced aggregation of human platelets (IC50=32 nM), but has no effect on aggregation induced by other platelet agonists .
|
-

- HY-11107
-
|
|
c-Met/HGFR
Autophagy
Apoptosis
|
Cancer
|
|
PHA-665752 is a selective, ATP-competitive, and active-site inhibitor of the catalytic activity of c-Met kinase (Ki=4 nM; IC50=9 nM). PHA-665752 exhibits >50-fold selectivity for c-Met compared with a panel of diverse tyrosine and serine-threonine kinases. PHA-665752 induces apoptosis and cell cycle arrest, and exhibits cytoreductive antitumor activity .
|
-
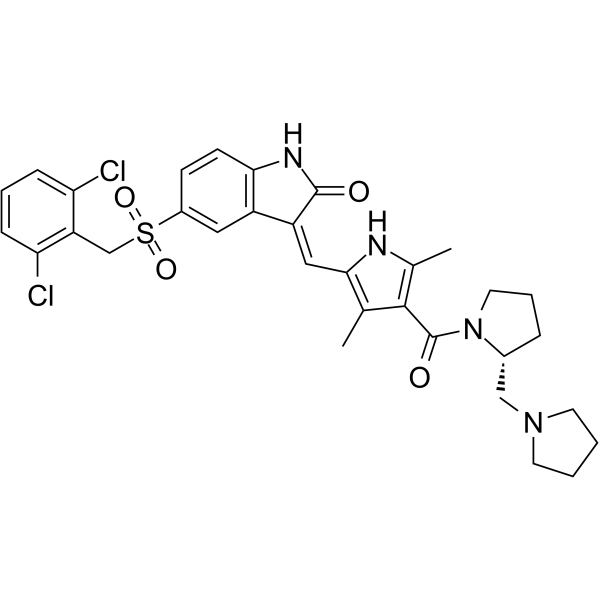
- HY-10032
-
|
PF 00477736
|
Checkpoint Kinase (Chk)
VEGFR
Src
c-Fms
Aurora Kinase
FGFR
FLT3
RET
CDK
|
Cancer
|
|
PF 477736 (PF 00477736) is a potent, selective and ATP-competitive inhibitor of Chk1, with a Ki of 0.49 nM, it is also a Chk2 inhibitor, with a Ki of 47 nM. PF 477736 shows <100-fold selectivity for Chk1 over VEGFR2, Fms, Yes, Aurora-A, FGFR3, Flt3, and Ret (IC50=8 (Ki), 10, 14, 23, 23, 25, and 39 nM, respectively). PF 477736 can enhance Gemcitabine antitumor activity in vitro and in vivo .
|
-
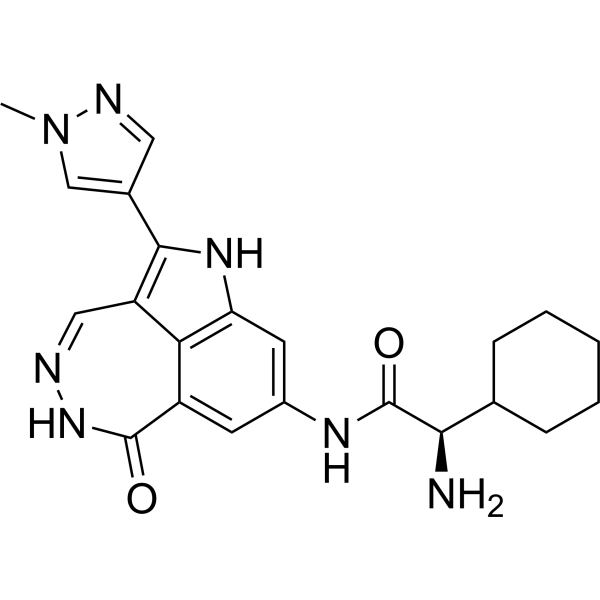
- HY-149426
-
|
|
Sirtuin
|
Inflammation/Immunology
|
|
SIRT5 inhibitor 7 (compound 58) is a substrate-competitive and selective SIRT5 inhibitor with anti-inflammatory activity. SIRT5 inhibitor 7 has renal protective effects and regulates protein succinylation and the release of pro-inflammatory cytokines. SIRT5 inhibitor 7 has in vivo activity in AKI mouse models of lipopolysaccharide (LPS) and cecal ligation/perforation (CLP)-induced sepsis-related acute kidney injury .
|
-
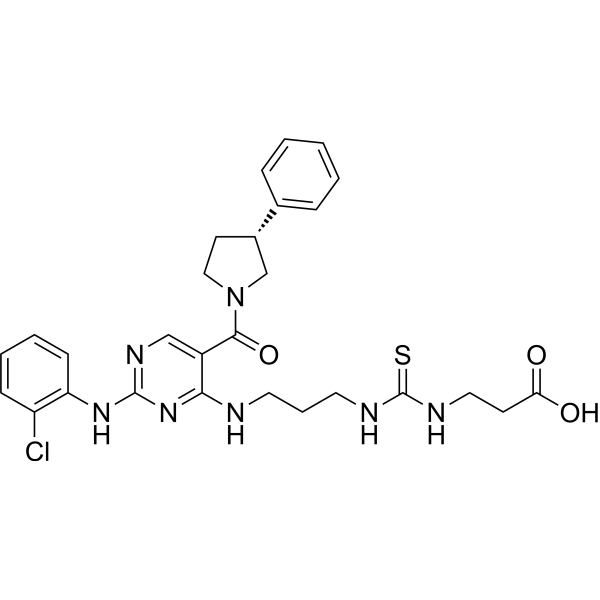
- HY-15222A
-
|
|
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
Menin-MLL inhibitor MI-2 dihydrochloride is a competitive and selective Menin-MLL interaction inhibitor with an IC50 value of 446 nM and a Ki value of 158 nM. Menin-MLL inhibitor MI-2 dihydrochloride downregulates the expression of target genes such as HOXA9 and MEIS1, inhibits proliferation of leukemia cells and induces apoptosis and differentiation. Menin-MLL inhibitor MI-2 dihydrochloride is proming for rasearch of MLL-rearranged acute leukemias (e.g., AML, ALL) .
|
-

- HY-15222
-
|
|
Epigenetic Reader Domain
Apoptosis
|
Cancer
|
|
Menin-MLL inhibitor MI-2 is a competitive and selective Menin-MLL interaction inhibitor with an IC50 value of 446 nM and a Ki value of 158 nM. Menin-MLL inhibitor MI-2 downregulates the expression of target genes such as HOXA9 and MEIS1, inhibits proliferation of leukemia cells and induces apoptosis and differentiation. Menin-MLL inhibitor MI-2 is proming for rasearch of MLL-rearranged acute leukemias (e.g., AML, ALL) .
|
-
- HY-18732B
-
|
Methylarginine citrate; Tilarginine citrate
|
NO Synthase
Endogenous Metabolite
|
Cardiovascular Disease
Cancer
|
|
L-NMMA (Tilarginine) citrate is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA citrate inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA citrate alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA citrate is applicable to research related to nociception, bone cancer pain, and myopia .
|
-

- HY-P10392B
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R TFA, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R TFA inhibits competitively the binding of β-catenin to TCF4. aStAx-35R TFA selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-

- HY-16231
-
|
|
Apoptosis
|
Cancer
|
|
GGTI-2418 is a highly potent, competitive, and selective geranylgeranyltransferase I (GGTase I) inhibitor. GGTI-2418 inhibits GGTase I and FTase activities with IC50s of 9.5 nM and 53 μM, respectively. GGTI-2418 also increases p27(Kip1) and induces significant regression of breast tumors .
|
-
- HY-W794573
-
|
|
Ser/Thr Protease
|
Cancer
|
|
APC-6860 hydrochloride is a competitive, selective arylamidine Serine protease inhibitor, with a Ki of 0.44 μM for trypsin, 0.10 μM for h-uPA, and 0.082 μM for mouse uPA. APC-6860 hydrochloride inhibits urokinase-activated plasminogen-mediated degradation of Fibronectin in cancer cells. APC-6860 hydrochloride is applicable to research related to breast cancer and prostate cancer .
|
-

- HY-16964
-
|
|
Lactate Dehydrogenase
|
Cancer
|
|
LDHA-IN-1 (Compound 1j) is a selective, competitive LDH-A inhibitor, with Ki values of 8.9 μM (NADH assay) and 4.7 μM (Pyruvate assay) against hLDH-A. LDHA-IN-1 inhibits cancer cell proliferation under hypoxic conditions. LDHA-IN-1 can be used in the research of ovarian cancer, colorectal cancer, mesothelioma and pancreatic cancer .
|
-

- HY-107794
-
|
Disodium clodronate tetrahydrate
|
Apoptosis
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
Clodronate disodium tetrahydrate (Disodium clodronate tetrahydrate) is first-generation bisphosphonate, with anti-osteoporotic, anti-inflammatory and analgesic effects. Clodronate disodium tetrahydrate is a selective, potent, reversible and Cl - competitive vesicular nucleotide transporter (VNUT) inhibitor, with an IC50 of 15.6 nM. Clodronate disodium tetrahydrate inhibits vesicular ATP release from neurons and reduces chronic neuropathic and inflammatory pain .
|
-
- HY-W179181
-
MSNBA
1 Publications Verification
|
GLUT
|
Metabolic Disease
Cancer
|
|
MSNBA is a potent and selective GLUT5 fructose transport inhibitor with an IC50 of 0.10 mM. MSNBA does not affect the transport activity of human GLUT1, GLUT2, GLUT3, GLUT4 or bacterial GlcPSe. MSNBA competitively inhibits GLUT5 fructose uptake with a Ki of 3.2 μM in MCF7 cells. MSNBA can be used for the study of cancer or diabetes .
|
-
- HY-13418R
-
|
Compound C dihydrochloride (Standard); BML-275 dihydrochloride (Standard)
|
Organoid
AMPK
TGF-β Receptor
Autophagy
Reference Standards
|
Cancer
|
|
Dorsomorphin (dihydrochloride) (Standard) is the analytical standard of Dorsomorphin (dihydrochloride). This product is intended for research and analytical applications. Dorsomorphin (Compound C) dihydrochloride is a potent, selective and ATP-competitive AMPK inhibitor, with a Ki of 109 nM. Dorsomorphin dihydrochloride inhibits BMP pathway by targeting the type I receptors ALK2, ALK3, and ALK6. Dorsomorphin dihydrochloride can reverse autophagy activation and anti-inflammatory effect of Urolithin A (HY-100599).
|
-

- HY-15999A
-
|
PRT062070 hydrochloride; PRT2070 hydrochloride
|
Syk
JAK
|
Cancer
|
|
Cerdulatinib hydrochloride (PRT062070) is a selective, oral active and reversible ATP-competitive inhibitor of dual SYK and JAK, with IC50s of 32 nM, 0.5 nM, 12 nM, 6 nM and 8 nM for SYK and Tyk2, JAK1, 2, 3, respectively. Cerdulatinib hydrochloride could be used to research autoimmune disease and B-cell malignancies .
|
-
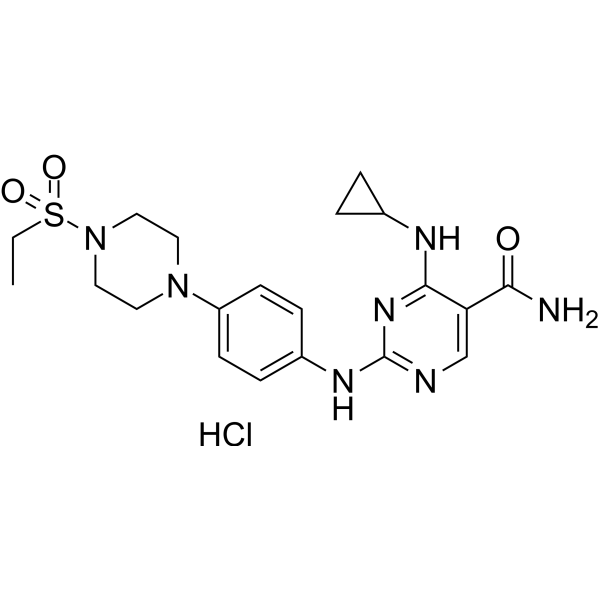
- HY-178292
-
|
|
Phosphatase
|
Cancer
|
|
VHR-IN-3 (Compound 1) is a selective vaccinia H1-related (VHR) phosphatase inhibitor with a Ki of 0.81 μM. VHR-IN-3 mimics the phosphate group through the sulfonic acid group and competitively binds to the catalytic active center of VHR. VHR-IN-3 can be used for the research of cervical cancer .
|
-

- HY-112346
-
|
|
CDK
Apoptosis
|
Cancer
|
|
RGB-286147 is a selective and ATP-competitive CDK and CDK-related kinases (CRK) inhibitor with IC50 values ranging from 9-839 nM. RGB-286147 shows less active against other non-CDK/CRK kinases. RGB-286147 induces cell apoptosis, and exhibits anti-tumor activity .
|
-
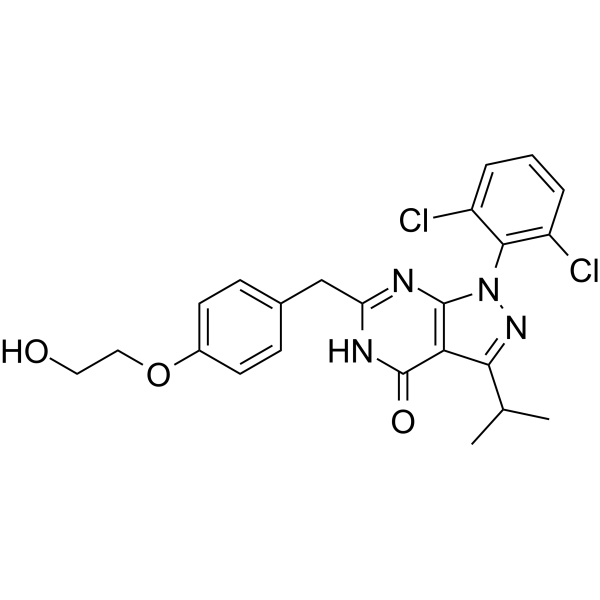
- HY-107526
-
|
(Rac)-NFPS
|
GlyT
|
Neurological Disease
|
|
NFPS is a selective, non-competitive glycine transporter-1 (GlyT1) inhibitor with IC50s of 2.8 nM and 9.8 nM for hGlyT1 and rGlyT1, respectively . NFPS exerts neuroprotection via glyR alpha1 subunit in the rat model of transient focal cerebral ischaemia and reperfusion .
|
-
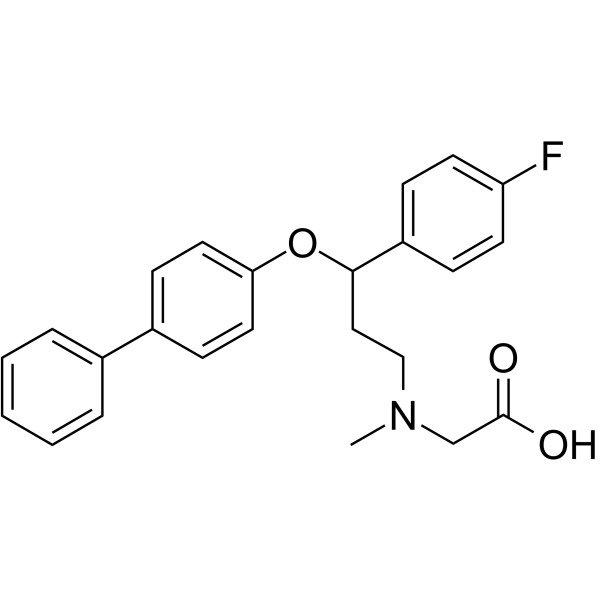
- HY-135893
-
-
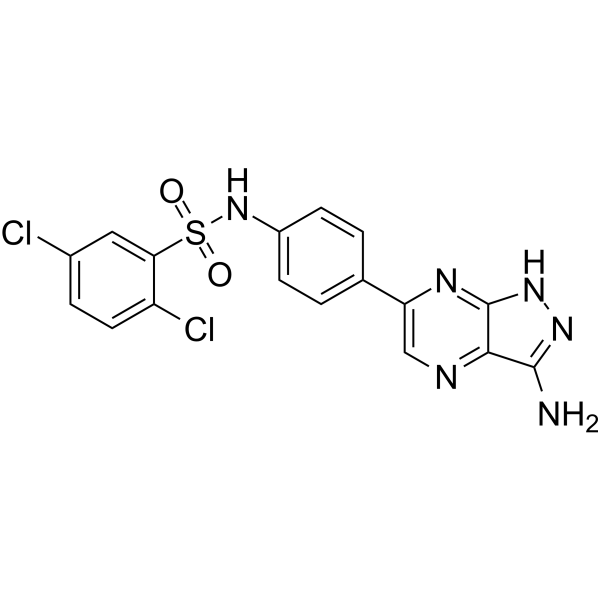
- HY-163174
-
|
|
Amine N-methyltransferase
|
Cardiovascular Disease
Neurological Disease
Metabolic Disease
Cancer
|
|
II399 is a potent, selective NNMT bisubstrate inhibitor containing an unconventional SAM mimic, with a Ki of 5.9 nM. II399 exhibits an explicit pattern of competitive inhibition for NAM. II399 occupies both the substrate and cofactor binding pockets. II399 has the potential for the research of cancers, metabolic, cardiovascular, and neurodegenerative diseases .
|
-
- HY-105101
-
|
|
Histamine Receptor
|
Inflammation/Immunology
|
|
Osutidine is a selective histamine H2 receptor antagonist, can effectively inhibit histamine-stimulated gastric acid secretion. Osutidine does not affect [ 14C]aminopyrine accumulation stimulated by carbachol or dibutyryl-cAMP. Osutidine is insurmountable and includes non-competitive inhibition. Osutidine can be used for the study of gastric mucosal injury .
|
-

- HY-149528
-
|
|
Monoamine Oxidase
Cholinesterase (ChE)
|
Cancer
|
|
MAO-B-IN-24 (compound 11h) is a selective, reversible, competitive inhibitor of MAO-B (IC50: 1.60 μM). MAO-B-IN-24 also inhibited MAO-A (22.42 μM); at 10 μM concentration, it also reduced AChE and BChE activities to 54.58% and 88.43% .
|
-
- HY-100815
-
|
|
iGluR
|
Neurological Disease
|
|
(R)-AMPA is an inactive AMPA receptor ligand that inhibits the release of excitatory amino acids from neurons. (R)-AMPA is inactive in experiments that enhance the release of [3H]D-aspartate induced by electrical stimulation. (R)-AMPA is inhibited by competitive and noncompetitive AMPA receptor selective antagonists in response to AMPA and glutamate .
|
-

- HY-110154
-
|
|
Histone Demethylase
|
Cancer
|
|
NSC636819 is a competitive and selective inhibitor of KDM4A/KDM4B. KDM4A/KDM4B are potential progression factors for prostate cancer. NSC636819 has the potential for the research of cancer diseases, especially prostate cancer .
|
-
- HY-168108
-
|
|
Fungal
|
Infection
|
|
Antifungal agent 115 (compound 8n) is a non-competitive chitin synthase (CHS) inhibitor with an IC50 of 93 μM. Antifungal agent 115 demonstrates good selectivity and broad-spectrum antifungal activity, exhibiting significant efficacy against drug-resistant fungi. Antifungal agent 115 can be utilized in fungi infection research .
|
-

- HY-105692
-
|
|
PARP
|
Neurological Disease
|
|
DR2313 is a potent, selective, competitive and brain-penetrant inhibitor of poly(ADP-ribose) polymerase (PARP), with IC50s of 0.20 μM and 0.24 μM for PARP-1 and PARP-2, respectively. DR2313 exhibits neuroprotective effects on ischemic injuries in vitro and in vivo .
|
-
- HY-182536
-
|
|
Phosphatase
|
Metabolic Disease
|
|
LMPTP IN-2 is a selective protein tyrosine phosphatase (LMPTP) inhibitor with an IC50 of 0.020 μM. LMPTP IN-2 exerts inhibitory effects through a non-competitive mechanism. LMPTP IN-2 can be used for research on obesity-related insulin resistance and type 2 diabetes .
|
-

- HY-15086
-
|
CGS 19755
|
iGluR
|
Neurological Disease
|
|
Selfotel (CGS 19755) is a selective and competitive antagonist at N-methyl-D-aspartate (NMDA)-preferring receptor. CGS 19755 inhibits the binding of [3H]-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid to NMDA-type receptors with an IC50 of 50 nM .
|
-

- HY-10229
-
|
AMG 706 Diphosphate
|
c-Kit
VEGFR
|
Cancer
|
|
Motesanib Diphosphate (AMG 706 Diphosphate) is a potent ATP-competitive inhibitor of VEGFR1/2/3 with IC50s of 2 nM/3 nM/6 nM, respectively, and has similar activity against Kit, and is approximately 10-fold more selective for VEGFR than PDGFR and Ret.
|
-
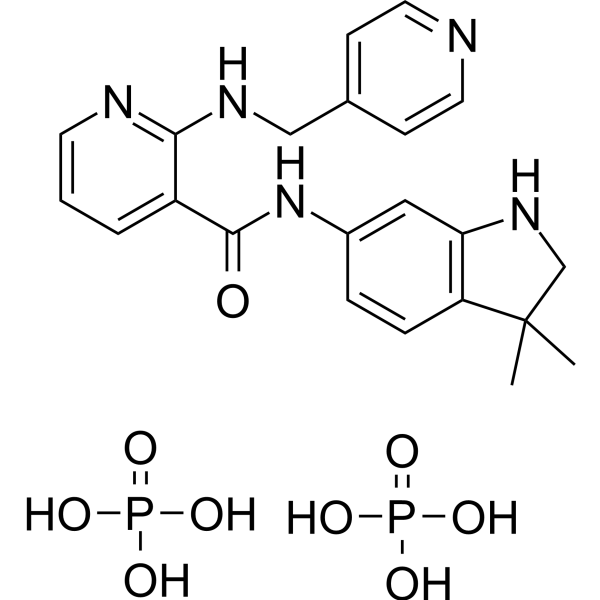
- HY-107427
-
|
|
MAPKAPK2 (MK2)
p38 MAPK
|
Inflammation/Immunology
|
|
PF-3644022 is a potent, selective, orally active and ATP-competitive MAPKAPK2 (MK2) inhibitor with an IC50 of 5.2 nM and a Ki of 3 nM. PF-3644022 also inhibits MK3 and p38 regulated/activated kinase (PRAK) with IC50s of 53 nM and 5.0 nM, respectively. PF-3644022 potently inhibits TNFα production and has anti-inflammatory effect .
|
-
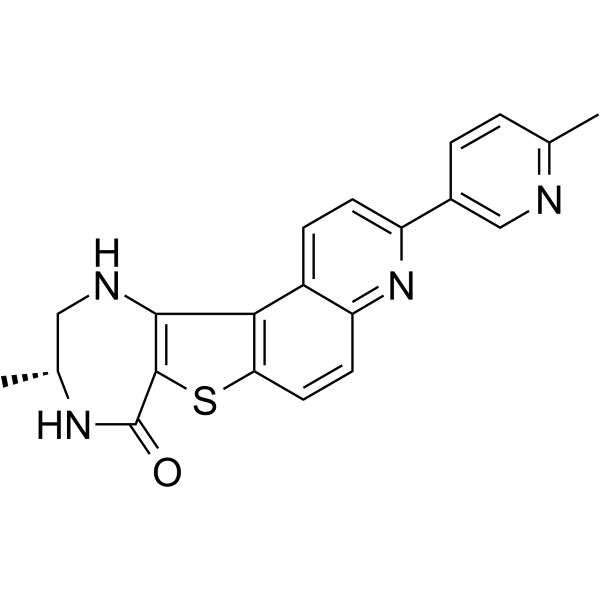
- HY-162100
-
|
|
ULK
Autophagy
|
Cancer
|
|
MR-2088 is a reversible, ATP-competitive, and selective ULK1/2 inhibitor with pEC50 values of 8.3 and 8.7 respectively. MR-2088 effectively inhibits autophagic flux and demonstrates a synergistic antiproliferative effect with Trametinib (HY-10999) (MEK inhibitor) in vitro. MR-2088 can be used for non-small lung cell cancer (NSCLC) research .
|
-
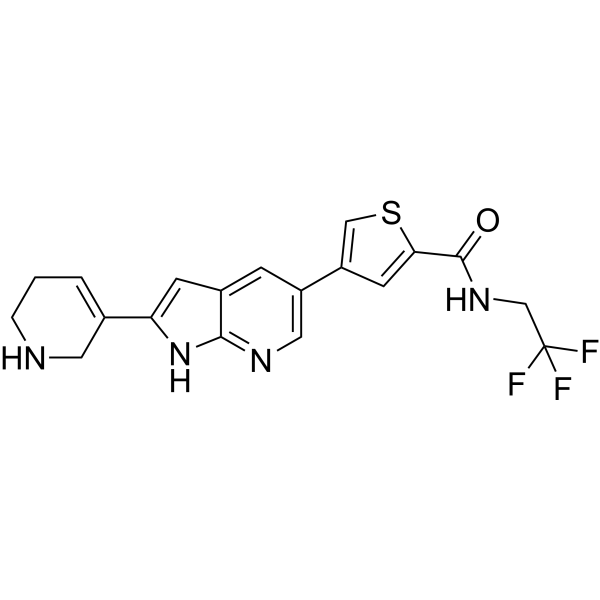
- HY-107407
-
|
|
Checkpoint Kinase (Chk)
CDK
PKC
Apoptosis
|
Cancer
|
|
SB-218078 is a potent, selective, ATP-competitive and cell-permeable checkpoint kinase 1 (Chk1) inhibitor that inhibits Chk1 phosphorylation of cdc25C with an IC50 of 15 nM. SB-218078 is less potently inhibits Cdc2 (IC50 of 250 nM) and PKC (IC50 of 1000 nM). SB-218078 causes apoptosis by DNA damage and cell cycle arrest .
|
-
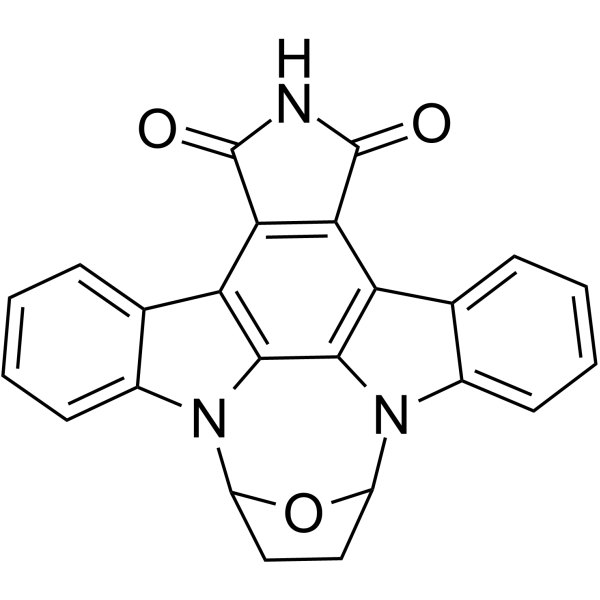
- HY-101920R
-
|
|
Reference Standards
Autophagy
PI3K
|
Neurological Disease
Cancer
|
|
Autophinib (Standard) is the analytical standard of Autophinib (HY-101920). This product is intended for research and analytical applications. Autophinib is a potent, selective autophagy inhibitor with IC50s of 90 nM and 40 nM for starvation- and Rapamycin-induced autophagy, respectively. Autophinib is also an ATP competitive Vacuolar Protein Sorting 34 (VPS34) inhibitor with an IC50 of 19 nM. Autophinib inhibits autophagy induced by starvation or Rapamycin by targeting VPS34 .
|
-

- HY-117822
-
|
|
GSK-3
|
Neurological Disease
Metabolic Disease
Cancer
|
|
BRD0209 is a potent, selective and dual inhibitor of GSK3α/β inhibitor (GSK3α IC50 = 19 nM; GSK3β IC50 = 5 nM). BRD0209 is also a reversible ATP-competitive inhibitor with fast-off kinetics (Ki = 4.2 nM, respectively). BRD0209 is a tricyclic pyrazolotetrahydroquinolinone compound. BRD0209 has the potential for the research of mood disorder diseases .
|
-
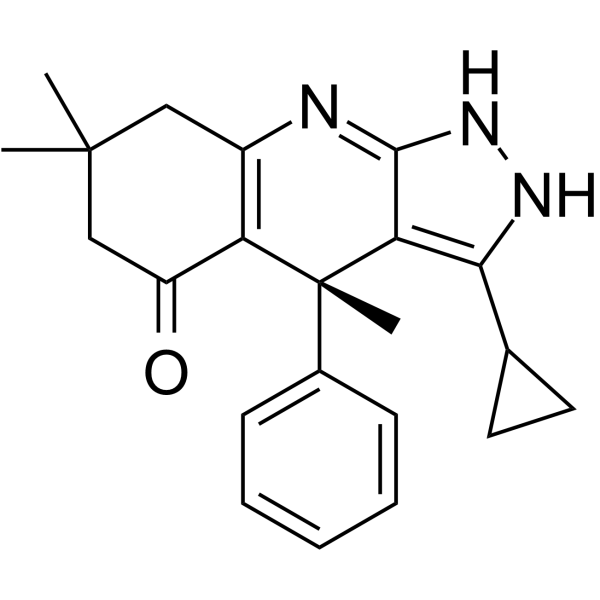
- HY-180419
-
|
|
ERK
Apoptosis
c-Myc
|
Inflammation/Immunology
Cancer
|
|
SF-3-030 is a potent, selective and non-ATP competitive ERK1/2 inhibitor. SF-3-030 selectively induces apoptosis in melanoma cells containing mutated BRaf and constitutively active ERK1/2 signalling. SF-3-030 mitigates multiple features of asthma in a murine model of asthma. SF-3-030 can be used for the research of asthma and melanomasup .
|
-

- HY-106404
-
|
|
Factor Xa
|
Cardiovascular Disease
|
|
RPR 130737 is a selective, potent and competitive inhibitor for factor Xa with a Ki of 2.4 nM. RPR 130737 shows selectivity of more than 1000-fold over thrombin, activated protein C, plasmin, tissue-plasminogen activator and trypsin. RPR 130737 can prolong plasma activated partial thromboplastin time and prothrombin time. RPR 130737 shows no effect on platelet aggregation. RPR 130737 can be used for the research of cardiovascular disease, such as thrombosis .
|
-

- HY-114209
-
MRK-740
2 Publications Verification
|
Histone Methyltransferase
|
Cancer
|
|
MRK-740, a chemical probe, is a potent, selective and substrate-competitive PRDM9 histone methyltransferase inhibitor with an IC50 of 80?nM. MRK-740 is more selective for PRDM9 than other histone methyltransferases and other non-epigenetic targets. MRK-740 reduces PRDM9-dependent trimethylation of H3K4 (IC50?=?0.8?μM) .
|
-
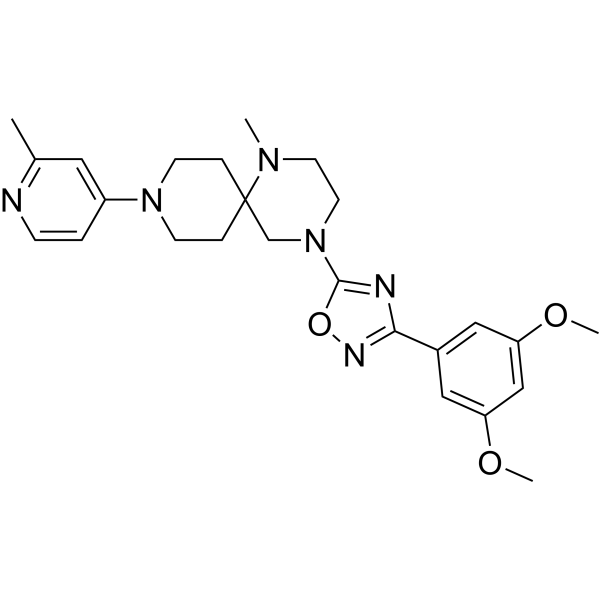
- HY-16749A
-
|
PLX-3397 hydrochloride
|
c-Fms
c-Kit
Apoptosis
|
Cancer
|
|
Pexidartinib hydrochloride (PLX-3397 hydrochloride) is a potent, orally active, selective, and ATP-competitive colony stimulating factor 1 receptor (CSF1R or M-CSFR) and c-Kit inhibitor, with IC50s of 20 and 10 nM, respectively. Pexidartinib hydrochloride exhibits 10- to 100-fold selectivity for c-Kit and CSF1R over other related kinases. Pexidartinib hydrochloride induces cell apoptosis and has anti-cancer activity .
|
-
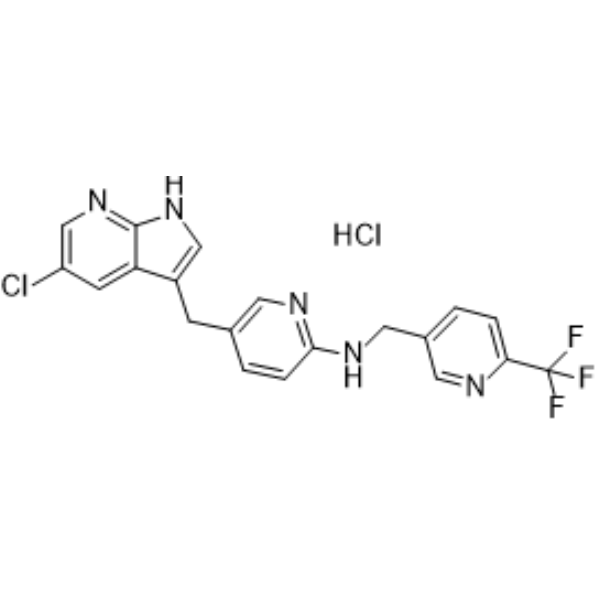
- HY-50867
-
|
CEP-701; KT-5555
|
JAK
FLT3
Trk Receptor
Apoptosis
STAT
|
Cancer
|
|
Lestaurtinib (CEP-701) is an orally active and selective RPTKs (receptor protein tyrosine kinase) inhibitor, competitively inhibits ATP binding to the TrkA/B/C domain. Lestaurtinib inhibits RPTKs phosphorylation, with IC50s of 2, 25 and 0.9 nM for FLT3, TrkA and JAK2, respectively. Lestaurtinib induces apoptosis and cycle arrest, also can inhibit growth of tumor .
|
-
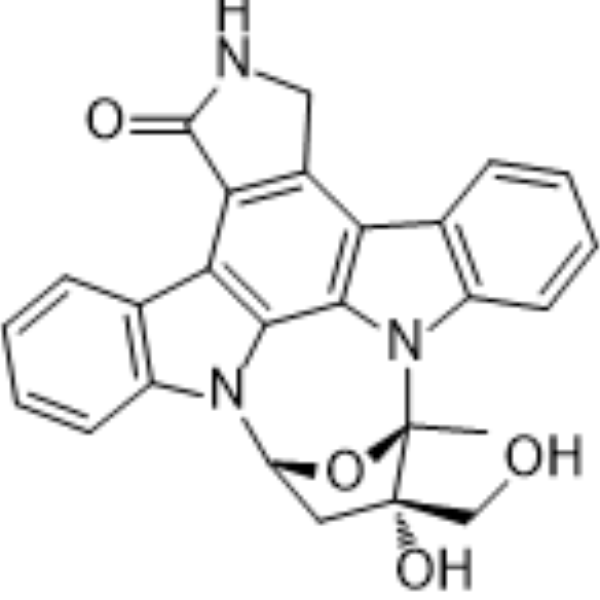
- HY-50683
-
|
|
c-Met/HGFR
|
Metabolic Disease
Cancer
|
|
JNJ-38877605 is an orally active ATP-competitive inhibitor of c-Met with an IC50 of 4 nM, 600-fold selective for c-Met than 200 other tyrosine and serine-threonine kinases . JNJ-38877605 inhibits c-Met phosphorylation and regulates lipid accumulation. JNJ-38877605 can be used for tumor and metabolic disease reseach .
|
-
- HY-13011A
-
|
CH5424802 Hydrochloride; RO5424802 Hydrochloride; AF-802 Hydrochloride
|
Anaplastic lymphoma kinase (ALK)
|
Neurological Disease
Cancer
|
|
Alectinib (CH5424802; RO5424802; RG7853) Hydrochloride is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
- HY-136270S
-
|
VX-803-d8; M4344-d8; ATR inhibitor 2-d8
|
Isotope-Labeled Compounds
ATM/ATR
|
Cancer
|
|
Gartisertib-d8 (VX-803-d8) is the deuterium labeled Gartisertib (HY-136270). Gartisertib (VX-803) is an ATP-competitive, orally active, and selective ATR inhibitor, with a Ki of <150 pM. Gartisertib potently inhibits ATR-driven phosphorylated checkpoint kinase-1 (Chk1) phosphorylation with an IC50 of 8 nM. Antitumor activity .
|
-

- HY-50892
-
|
(Rac)-Seliciclib; (Rac)-CYC202
|
CDK
Apoptosis
|
Infection
Cardiovascular Disease
Neurological Disease
Inflammation/Immunology
Cancer
|
|
(Rac)-Roscovitine ((Rac)-Seliciclib) is a selective cyclin-dependent kinases (CDKs) inhibitor. (Rac)-Roscovitine binds to the active sites of CDKs competitively with ATP, inhibiting the phosphorylation activity of CDKs. (Rac)-Roscovitine induces apoptosis in cancer cells. (Rac)-Roscovitine is promising for research of cancers or other diseases associated with CDK dysregulation, such as neurodegenerative diseases, cardiac disorders, viral and protozoan infections, glomerulonephritis, and chronic inflammation .
|
-

- HY-15816
-
|
BVD-523; VRT752271
|
ERK
|
Cancer
|
|
Ulixertinib (BVD-523; VRT752271) is a potent, orally active, highly selective, ATP-competitive and reversible covalent inhibitor of ERK1/2 kinases, with an IC50 of <0.3 nM against ERK2. Ulixertinib (BVD-523; VRT752271) inhibits the phosphorylated ERK2 (pERK) and downstream kinase RSK (pRSK) in an A375 melanoma cell line .
|
-
- HY-103021R
-
|
|
Reference Standards
TGF-β Receptor
|
Inflammation/Immunology
Cancer
|
|
LY3200882 (Standard) is the analytical standard of LY3200882 (HY-103021). This product is intended for research and analytical applications. LY3200882 is a potent, highly selective, ATP-competitive and orally active TGF-β receptor type 1 (ALK5) inhibitor with an IC50 of 38.2 nM. LY3200882 inhibits various pro-tumorigenic activities and is also used as an immune modulatory agent .
|
-

- HY-133120A
-
|
|
PROTACs
Akt
|
Cancer
|
|
INY-03-041 trihydrochloride is a potent, highly selective and PROTAC-based pan-AKT degrader consisting of the ATP-competitive AKT inhibitor Ipatasertib (HY-15186) conjugated to Lenalidomide (HY-A0003). INY-03-041 trihydrochloride inhibits AKT1, AKT2 and AKT3 with IC50s of 2.0, 6.8 and 3.5 nM, respectively .
|
-
- HY-101368A
-
|
|
Bradykinin Receptor
mAChR
|
Neurological Disease
|
|
WIN 64338 hydrochloride is a potent, selective, nonpeptide competitive antagonist of bradykinin B2 receptor. WIN 64338 hydrochloride inhibits [ 3H]-Bradykinin binding to the bradykinin B2 receptor on human IMR-90 cells with a Ki of 64 nM. WIN 64338 hydrochloride also can inhibits [ 3H]Quinuclidinyl benzilate binding to the rat brain muscarinic receptor (Ki=350 nM) .
|
-
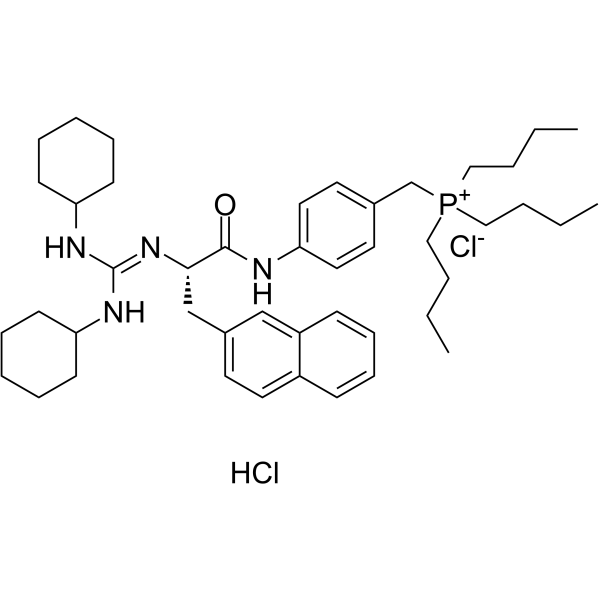
- HY-10195BR
-
|
LY333531 hydrochloride (Standard)
|
Reference Standards
PKC
|
Metabolic Disease
|
|
Ruboxistaurin hydrochloride (Standard) is the analytical standard of Ruboxistaurin hydrochloride (HY-10195B). This product is intended for research and analytical applications. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-

- HY-182721
-
|
|
PI4K
Hedgehog
|
Cancer
|
|
Pipinib is an ATP-competitive and selective phosphatidylinositol 4-kinase IIIb (PI4KB) inhibitor with an IC50 of 2.2 μM. Pipinib reduces intracellular PI4P levels. Pipinib inhibits GLI-mediated transcription, the expression of Hedgehog target genes, and blocks the trafficking of Smoothened to cilia. Pipinib can be used in the research of basal cell carcinoma and medulloblastoma .
|
-

- HY-120619
-
|
|
Neuropeptide Y Receptor
|
Neurological Disease
Endocrinology
|
|
BMS-193885 is a selective neuropeptide Y1 receptor antagonist (Ki=3.3 nM) that competitively blocks the receptor to inhibit NPY-mediated appetite regulation signaling pathways, reduce food intake and inhibit weight gain. BMS-193885 has good blood-brain barrier penetration and is mainly used in the study of obesity and related metabolic diseases .
|
-
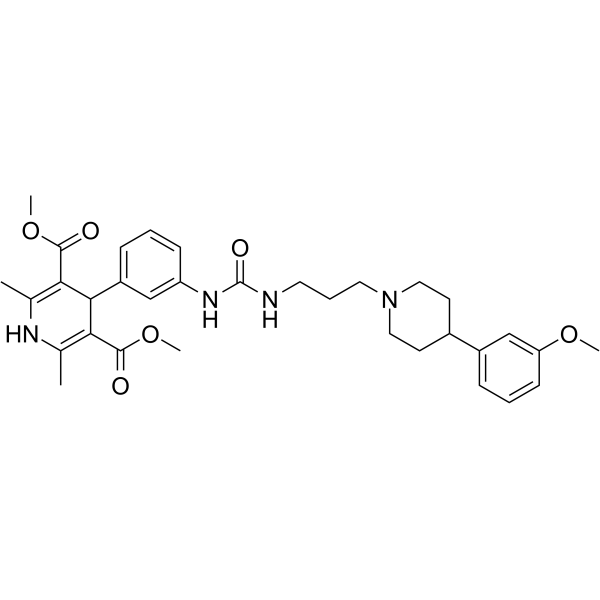
- HY-13011
-
|
CH5424802; RO5424802; RG7853
|
Anaplastic lymphoma kinase (ALK)
|
Neurological Disease
Cancer
|
|
Alectinib (CH5424802; RO5424802; RG7853) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
- HY-12830
-
|
|
Pim
|
Cancer
|
|
M-110 is a highly selective, ATP-competitive inhibitor of PIM kinases with a preference for PIM-3 (IC50=47 nM). M-110 inhibits PIM-1 and PIM-2 with similar IC50s of 2.5 μM. M-110 inhibits the proliferation of prostate cancer cell lines with IC50s of 0.6 to 0.9 μM .
|
-
- HY-10285AR
-
|
BMS-477118 hydrate (Standard)
|
Reference Standards
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin (hydrate) (Standard) is the analytical standard of Saxagliptin (hydrate). This product is intended for research and analytical applications. Saxagliptin hydrate (BMS-477118 hydrate) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin hydrate has the peotential for type 2 diabetes mellitus research .
|
-

- HY-113015R
-
|
|
Endogenous Metabolite
Reference Standards
|
Neurological Disease
|
|
Saxagliptin (hydrochloride) (Standard) is the analytical standard of Saxagliptin (hydrochloride). This product is intended for research and analytical applications. Saxagliptin hydrochloride (BMS-477118 hydrochloride) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin hydrochloride has the peotential for type 2 diabetes mellitus research .
|
-

- HY-155137
-
|
|
Monoamine Oxidase
Reactive Oxygen Species (ROS)
|
Neurological Disease
|
|
CHBO4 is a potent, reversible, competitive, and selective hMAO-B inhibitor with an IC50 value of 0.031 μM in CHBO subseries and an Ki value of 0.010 ± 0.005 μM. CHBO4 reduce cell damage by scavenging intracellular reactive oxygen species (ROS). CHBO4 can be used for Parkinson's Disease (PD) research .
|
-
- HY-16448R
-
|
BMS-477118 hydrochloride (Standard)
|
Reference Standards
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin (hydrochloride) (Standard) is the analytical standard of Saxagliptin (hydrochloride). This product is intended for research and analytical applications. Saxagliptin hydrochloride (BMS-477118 hydrochloride) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin hydrochloride has the peotential for type 2 diabetes mellitus research .
|
-

- HY-112179
-
GSK180
2 Publications Verification
|
KMO
|
Inflammation/Immunology
|
|
GSK180 is a selective, competitive, and potent inhibitor of kynurenine-3-monooxygenase (KMO), a key enzyme of tryptophan metabolism (IC50, ~6 nM), but shows negligible activity against other enzymes on the tryptophan pathway. GSK180 rapidly changes levels of kynurenine pathway metabolites, and acts as a useful tool to probe the therapeutic potential of KMO inhibition .
|
-
- HY-13302
-
|
|
VEGFR
FGFR
|
Cancer
|
|
CP-547632 is an orally active, ATP-competitive and potent VEGFR-2 and FGF kinases inhibitor with IC50s of 11 nM and 9 nM, respectively. CP-547632 is selective for VEGFR2 and bFGF over EGFR, PDGFRβ, and related tyrosine kinases (TKs). CP-547632 has antitumor efficacy .
|
-
- HY-13302B
-
|
|
VEGFR
FGFR
|
Cancer
|
|
CP-547632 hydrochloride is an orally active, ATP-competitive and potent VEGFR-2 and FGF kinases inhibitor with IC50s of 11 nM and 9 nM, respectively. CP-547632 hydrochloride is selective for VEGFR2 and bFGF over EGFR, PDGFRβ, and related tyrosine kinases (TKs). CP-547632 hydrochloride has antitumor efficacy .
|
-
- HY-100501
-
M2698
1 Publications Verification
MSC2363318A
|
Ribosomal S6 Kinase (RSK)
Akt
|
Cancer
|
|
M2698 (MSC2363318A) is an orally active, ATP competitive, selective p70S6K and Akt dual-inhibitor with IC50s of 1 nM for p70S6K, Akt1 and Akt3. M2698 can cross the blood-brain barrier and has anti-cancer activity .
|
-
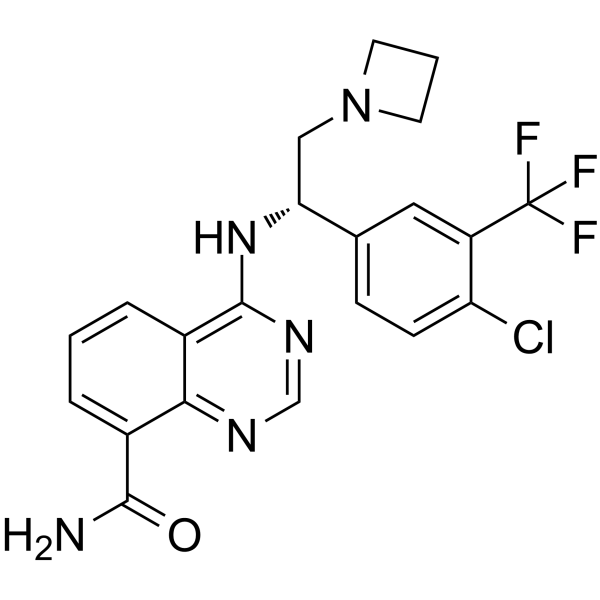
- HY-147298
-
|
CYC140
|
Polo-like Kinase (PLK)
|
Cancer
|
|
Plogosertib (CYC140) is a selective, potent, and orally active ATP-competitive PLK1 inhibitor (IC50: 3 nM). Plogosertib is an anti-cancer agent with anti-proliferative activity. Plogosertib can be used in the research of several tumors, including esophageal, gastric, leukemia, non–small cell lung cancer, ovarian, and squamous cell cancers .
|
-
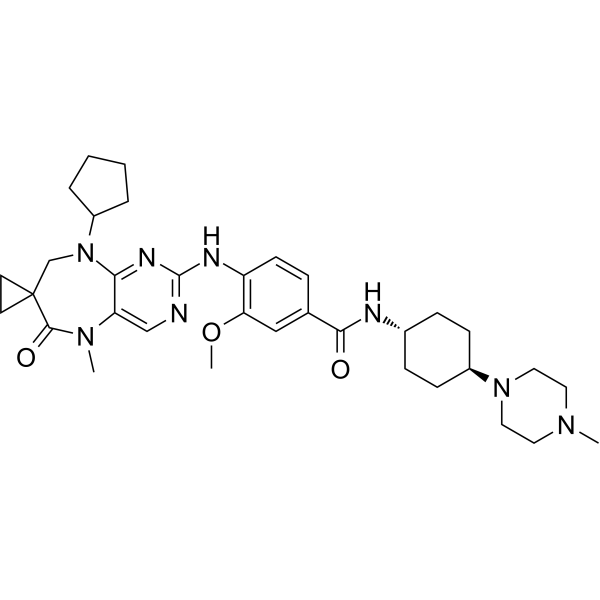
- HY-10962R
-
|
CYT387 sulfate salt (Standard)
|
JAK
Autophagy
Apoptosis
Reference Standards
|
Cancer
|
|
Momelotinib (sulfate) (Standard) is the analytical standard of Momelotinib (sulfate). This product is intended for research and analytical applications. Momelotinib sulfate (CYT387 sulfate salt) is an ATP-competitive inhibitor of JAK1/JAK2 with IC50 of 11 nM/18 nM, 10-fold selectivity versus JAK3 (IC50=155 nM).
|
-

- HY-B0290A
-
|
ONO-1078 hemihydrate
|
Leukotriene Receptor
Endogenous Metabolite
|
Inflammation/Immunology
|
|
Pranlukast hemihydrate is a highly potent, selective and competitive antagonist of peptide leukotrienes. Pranlukast inhibits [ 3H]LTE4, [ 3H]LTD4, and [ 3H]LTC4 bindings to lung membranes with Kis of 0.63±0.11, 0.99±0.19, and 5640±680 nM, respectively.
|
-
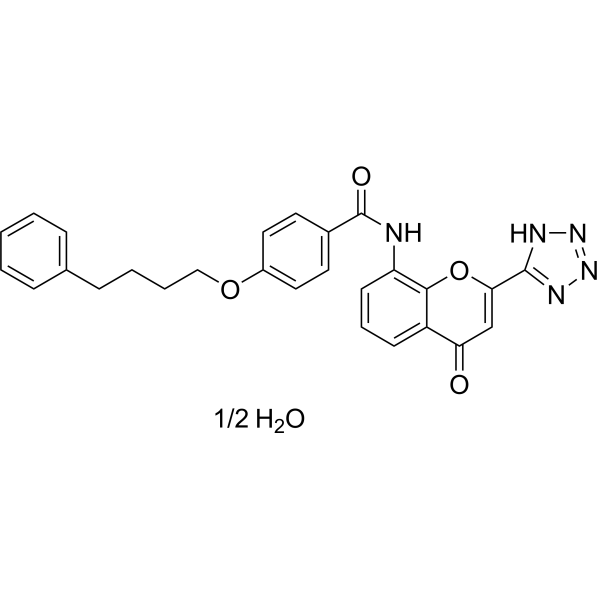
- HY-138568
-
|
|
MAP4K
|
Inflammation/Immunology
|
|
HPK1-IN-3 is a potent and selective ATP-competitive hematopoietic progenitor kinase 1 (HPK1; MAP4K1) inhibitor with an IC50 of 0.25 nM. HPK1-IN-3 has IL-2 cellular potency with an EC50 of 108 nM in human peripheral blood mononuclear cells (PBMCs) .
|
-
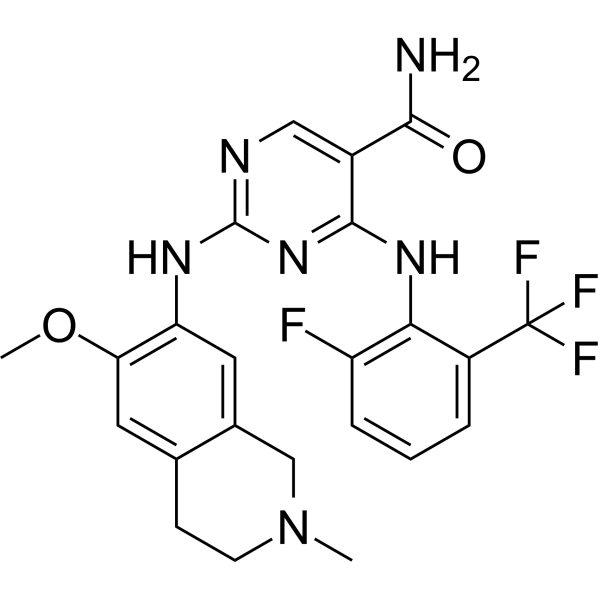
- HY-131350A
-
|
|
Proteasome
Parasite
|
Infection
|
|
LXE408 fumarate is an orally active, non-competitive and kinetoplastid-selective proteasome inhibitor. LXE408 fumarate has an IC50 of 0.04 μM for L. donovani proteasome and an EC50 of 0.04 μM for L. donovani. LXE408 fumarate has a low propensity to cross the blood brain barrier. LXE408 fumarate has the potential for visceral leishmaniasis (VL) research .
|
-

- HY-133146
-
|
|
Phosphatase
Apoptosis
|
Inflammation/Immunology
|
|
DJ001 is a highly specific, selective and non-competitive protein tyrosine phosphatase-σ (PTPσ) inhibitor with an IC50 of 1.43 μM. DJ001 displays no inhibitory activity against other phosphatases, with only modest inhibitory activity against Protein Phosphatase 5. DJ001 promotes promote hematopoietic stem cell regeneration .
|
-
- HY-B0290
-
-
- HY-128694
-
|
SR27417
|
Platelet-activating Factor Receptor (PAFR)
|
Cardiovascular Disease
|
|
Foropafant (SR27417) highly potent, competitive, selective and orally active antagonist of platelet-activating factor (PAF) receptor, with a Ki value of 57 pM for [ 3H]PAF binding, at least 5-fold lower than that of unlabeled PAF itself. Foropafant potently inhibits PAF-induced aggregation of rabbit and human platelets .
|
-
- HY-10285R
-
|
BMS-477118 (Standard)
|
Reference Standards
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin (Standard) is the analytical standard of Saxagliptin. This product is intended for research and analytical applications. Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-

- HY-145702
-
|
|
MEK
ERK
|
Cancer
|
|
MAP855 is a highly potent, selective, ATP-competitive and orally active MEK1/2 kinase inhibitor (MEK1 ERK2 cascade IC50=3 nM, pERK EC50=5 nM). MAP855 shows equipotent inhibition of wild-type and mutant MEK1/2 .
|
-
- HY-131350
-
|
|
Proteasome
Parasite
|
Infection
|
|
LXE408 is an orally active, non-competitive and kinetoplastid-selective proteasome inhibitor. LXE408 has an IC50 of 0.04 μM for L. donovani proteasome and an EC50 of 0.04 μM for L. donovani. LXE408 has a low propensity to cross the blood brain barrier. LXE408 has the potential for visceral leishmaniasis (VL) research .
|
-
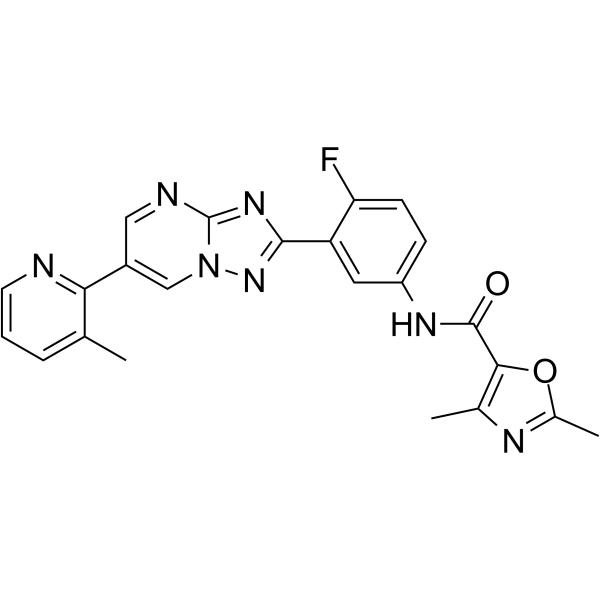
- HY-103195
-
NKY80
2 Publications Verification
|
Adenylate Cyclase
|
Metabolic Disease
|
|
NKY80 is a potent, selective and non-competitive adenylyl cyclase (AC) type V isoform inhibitor with IC50s of 8.3 µM, 132 µM and 1.7 mM for type V, III and II, respectively. NKY80 is a non-nucleoside quinazolinone and regulates the AC catalytic activity in heart and lung tissues .
|
-
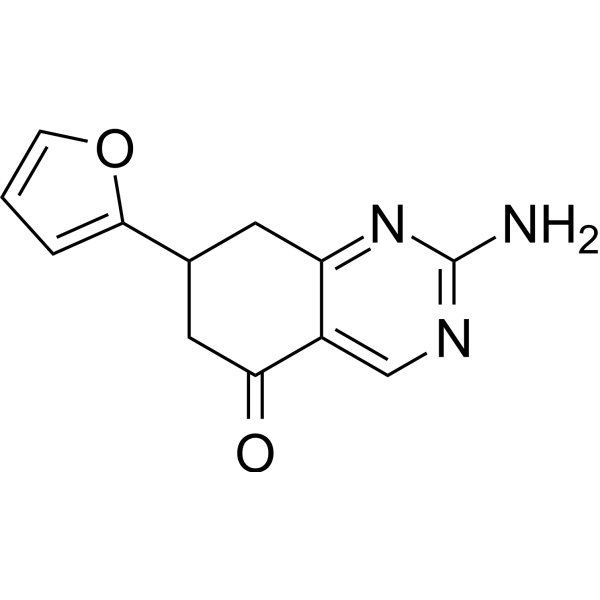
- HY-12755
-
|
CID-2950007
|
Ras
Apoptosis
|
Cancer
|
|
ML141 (CID-2950007) is a potent, allosteric, selective and reversible non-competitive inhibitor of Cdc42 GTPase. ML141 inhibits Cdc42 wild type and Cdc42 Q61L mutant with EC50s of 2.1 and 2.6 μM, respectively. ML141 shows low micromolar potency and selectivity against other members of the Rho family of GTPases (Rac1, Rab2, Rab7). ML141 do not show cytotoxicity in multiple cell lines .
|
-
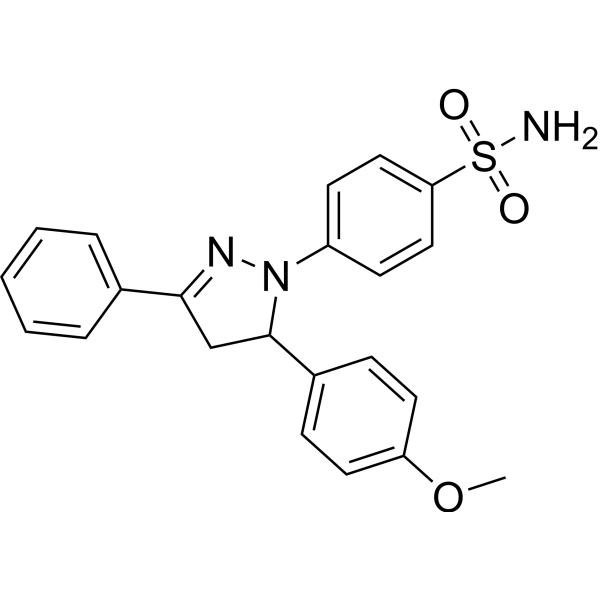
- HY-17499
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFR L858R and EGFR L861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
|
-
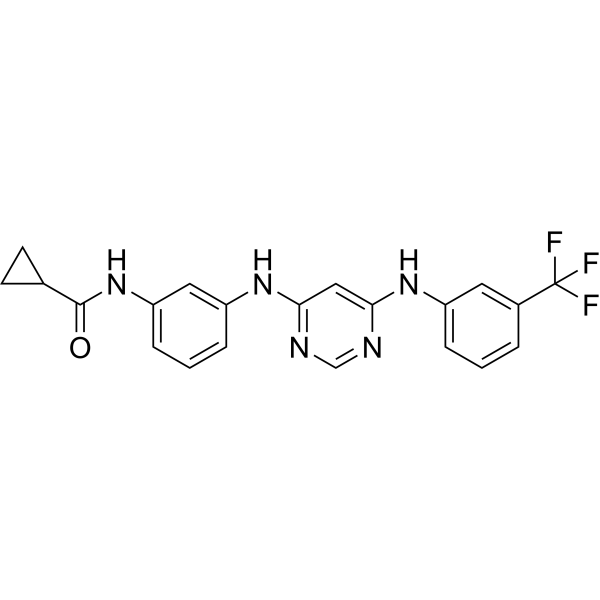
- HY-102062R
-
|
N-omega-Propyl-L-arginine (Standard)
|
Reference Standards
NO Synthase
|
Neurological Disease
|
|
Nω-Propyl-L-arginine (Standard) is the analytical standard of Nω-Propyl-L-arginine (HY-102062). This product is intended for research and analytical applications. Nω-Propyl-L-arginine (N-omega-Propyl-L-arginine) is a potent, competitive, and highly selective inhibitor of neuronal nitric oxide synthase (nNOS), with a Ki of 57 nM. Nω-Propyl-L-arginine displays a 149-fold selectivity for nNOS over endothelial NOS (eNOS) .
|
-

- HY-114263
-
NXT629
4 Publications Verification
|
PPAR
|
Cancer
|
|
NXT629 is a potent, selective, and competitive PPAR-α antagonist, with an IC50 of 77 nM for human PPARα, shows high selectivity over other nuclear hormone receptor, such as PPARδ, PPARγ, ERβ, GR and TRβ, IC50s are 6.0, 15, 15.2, 32.5 and >100 μM, respectively . NXT629 has potent anti-tumor activity and inhibits experimental metastasis of cancer cell in animal models .
|
-
- HY-119366
-
|
|
ROR
|
Inflammation/Immunology
|
|
S18-000003 is a potent, selective and orally active inhibitor of retinoic acid receptor-related orphan receptor-gamma-t (RORγt), with an IC50 of <30 nM towards human RORγt in competitive binding assays. S18-000003 shows selectivity for RORγt over other ROR family members (IC50>10 μM). S18-000003 can be used for the research of psoriasis with low risk of thymic aberrations .
|
-
- HY-124625
-
|
|
FAK
Target Protein Ligand-Linker Conjugates
|
Infection
Cancer
|
|
BI-4464 is a highly selective, ATP competitive PTK2/FAK protein kinase inhibitor with an IC50 value of 17 nM. BI-4464 is a FAK (HY-43760) ligand and linker conjugate. BI-4464 can be used to construct proteolysis targeting chimeras (PROTACs), such as PROTAC FAK degrader 4 (HY-178467). PROTAC FAK degrader 4 is a highly potent and selective FAK PROTAC degrader .
|
-
- HY-N7536
-
|
|
TRP Channel
|
Others
|
|
Voacangine is an antagonist for TRPV1 and TRPM8 but as an agonist for TRPA1 (EC50=8 μM). Voacangine competitively blockes capsaicin binding to TRPV1 (IC50=50 μM). Voacangine competitively inhibits the binding of menthol to TRPM8 (IC50=9 μM) and it shows noncompetitive inhibition against icilin (IC50=7 μM). Voacangine selectively abrogates chemical agonist-induced TRPM8 activation and did not affect cold-induced activation. Voacangine is an alkaloid isolated from the root bark of Voacanga africana .
|
-

- HY-104000
-
|
|
Ser/Thr Protease
|
Cancer
|
|
BAY-320 is a selective and ATP-competitive Bub1 inhibitor that inhibits the kinase activity of Bub1 with an IC50 of 680 nM. BAY-320 inhibits endogenous Bub1-mediated Sgo1 localization. BAY-320 affects cellular mitotic chromosome arrangement and spindle assembly. BAY-320 inhibits cell proliferation. BAY-320 can be used in the study of cancer such as ovarian cancer and cervical cancer .
|
-
- HY-W371165
-
|
|
Complement System
|
Inflammation/Immunology
|
|
C1s-IN-1 trihydrochloride (Compound A1) is the selective inhibitor for C1s protease with the Ki of 5.8 μM. C1s-IN-1 trihydrochloride inhibits cleavage of C2 by C1s with an IC50 of 85 μM, inhibits the activation of classic pathway with an IC50 of 22 μM. C1s-IN-1 trihydrochloride is the competitive inhibitor for thrombin with the Ki of 51.2 μM .
|
-

- HY-16576A
-
SMI-4a
1 Publications Verification
TCS-PIM-1-4a
|
Pim
|
Cancer
|
|
SMI-4a (TCS-PIM-1-4a) is a poten, selective, cell-permeable and ATP-competitive Pim-1 inhibitor with an IC50 of 24 μM and a Ki of 0.6 μM. SMI-4a also inhibits Pim-2 (IC50 of 100 μM), and does not significantly inhibit the other serine/threonine- or tyrosine-kinases. SMI-4a has anticancer activity .
|
-
- HY-18174A
-
|
LY2606368 dihydrochloride
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
Prexasertib dihydrochloride (LY2606368 dihydrochloride) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib dihydrochloride inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib dihydrochloride causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib dihydrochloride shows potent anti-tumor activity .
|
-
- HY-105416
-
|
UCN-1028C
|
Antibiotic
PKC
Apoptosis
|
Metabolic Disease
Cancer
|
|
Calphostin C is a highly selective PKC inhibitor (IC50=0.05 μM) and tumor apoptosis inducer. Calphostin C competitively binds to PKC and inhibits PKC-mediated phosphorylation signal transduction. Calphostin C restores Na+/K+ ATPase activity in the sciatic nerve of diabetic mice and improves neuropathy. Calphostin C can be used in the study of anti-tumor and diabetic complications .
|
-
- HY-13011S1
-
|
CH5424802-d6; RO5424802-d6; AF802-d6
|
Isotope-Labeled Compounds
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Alectinib-d6 is deuterium labeled Alectinib. Alectinib (CH5424802) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
- HY-15656R
-
-

- HY-15141R
-
|
Antibiotic AM-2282 (Standard); STS (Standard); AM-2282 (Standard)
|
Reference Standards
PKC
PKA
Apoptosis
Bacterial
Fungal
Antibiotic
|
Infection
Cancer
|
|
Staurosporine (Standard) is the analytical standard of Staurosporine. This product is intended for research and analytical applications. Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases with IC50s of 6 nM, 15 nM, 2 nM, and 3 nM for PKC, PKA, c-Fgr, and Phosphorylase kinase respectively. Staurosporine also inhibits TAOK2 with an IC50 of 3 μM. Staurosporine is an apoptosis inducer[1][2][3][4][5].
|
-

- HY-100201
-
|
|
Histone Methyltransferase
|
Cancer
|
|
A-196 is a potent and selective inhibitor of SUV420H1 and SUV420H2 with IC50 values of 25 nM and 144 nM, respectively. A-196 inhibits SUV4-20 biochemically in a substrate-competitive manner. A-196 represents a first-in-class chemical probe of SUV4-20 to investigate the role of histone methyltransferases in genomic integrity .
|
-
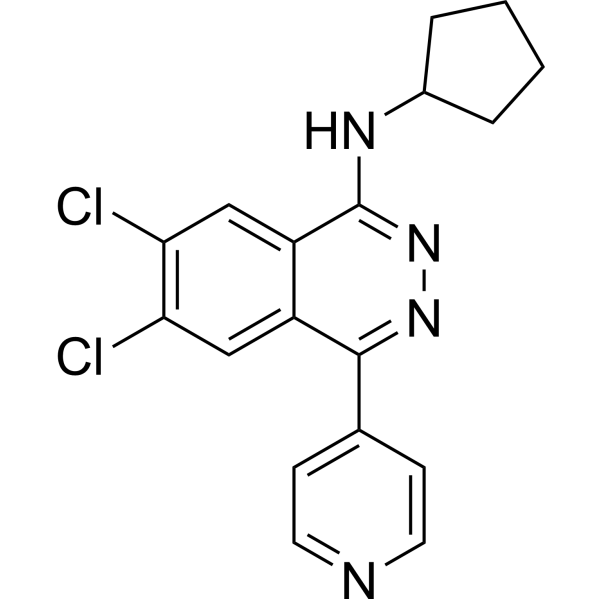
- HY-130250
-
|
|
CDK
Apoptosis
|
Cancer
|
|
SR-4835 is a potent, highly selective and ATP competitive dual inhibitor of CDK12/CDK13 (CDK12: IC50=99 nM, Kd=98 nM; CDK13: Kd=4.9 nM). SR-4835 acts in synergy with DNA-damaging chemotherapy and PARP inhibitors and provokes triple-negative breast cancer (TNBC) cell death .
|
-
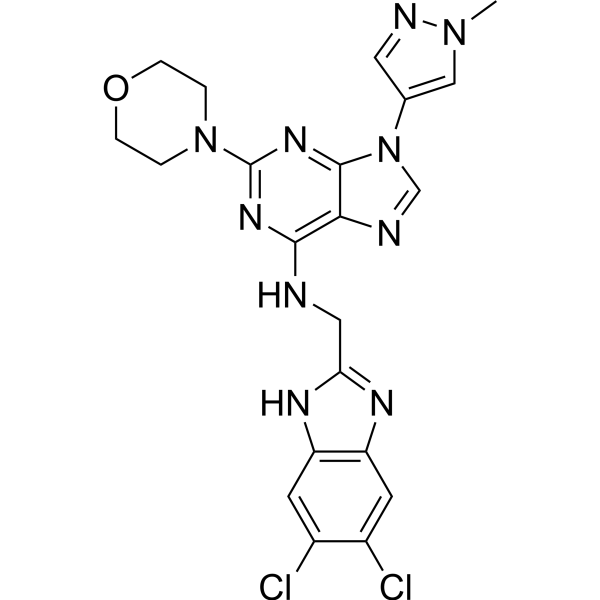
- HY-18174B
-
|
LY2606368 Mesylate Hydrate; LY2940930
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
Prexasertib Mesylate Hydrate (LY2606368 Mesylate Hydrate) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib Mesylate Hydrate inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib Mesylate Hydrate causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib Mesylate Hydrate shows potent anti-tumor activity .
|
-
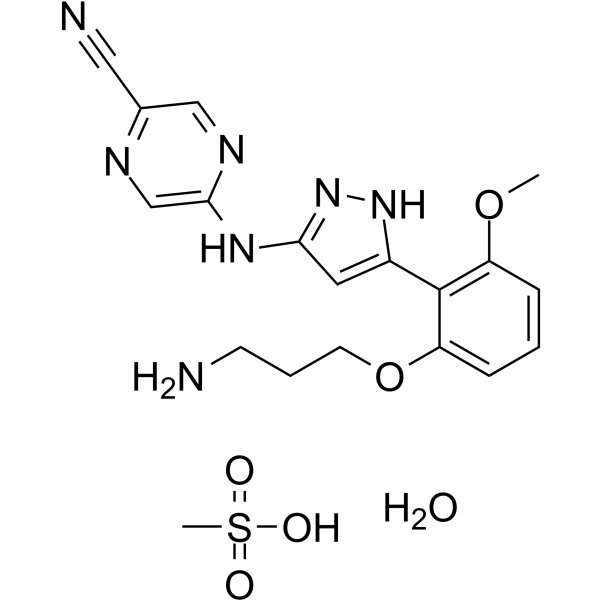
- HY-18174C
-
|
LY2606368 mesylate
|
Checkpoint Kinase (Chk)
DNA/RNA Synthesis
Apoptosis
|
Cancer
|
|
Prexasertib mesylate (LY2606368 mesylate) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib mesylate inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib mesylate causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib mesylate shows potent anti-tumor activity .
|
-
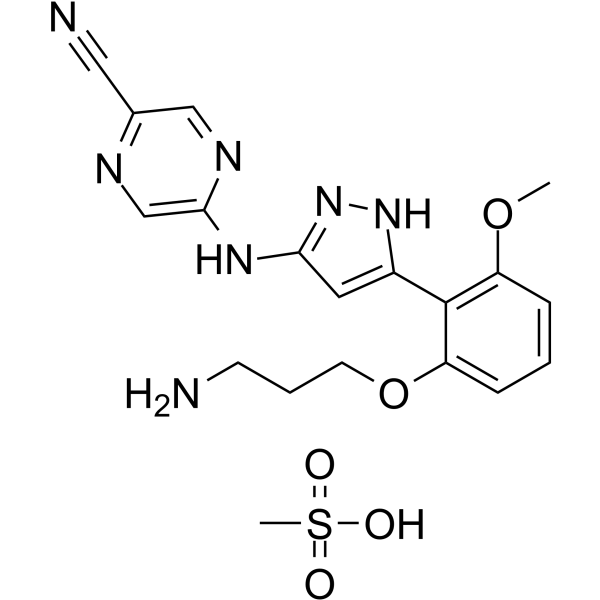
- HY-18174
-
|
LY2606368
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
Prexasertib (LY2606368) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib shows potent anti-tumor activity .
|
-
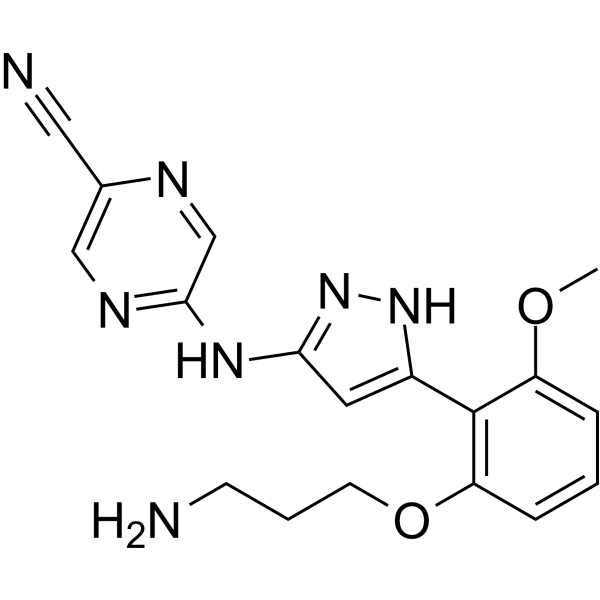
- HY-156659
-
|
|
Phosphatase
ERK
|
Inflammation/Immunology
|
|
NC1 is a selective non-competitive and allosteric lymphoid-specific tyrosine phosphatase (LYP) inhibitor, with a Ki value 4.3 μM. NC1 allosterically regulates LYP/PTPN22 activity by restricting WPD loop movement. NC1 inhibits LYP activity in lymphoid T cells and enhances T-cell receptor signaling. NC1 can be used for the research of autoimmune diseases .
|
-
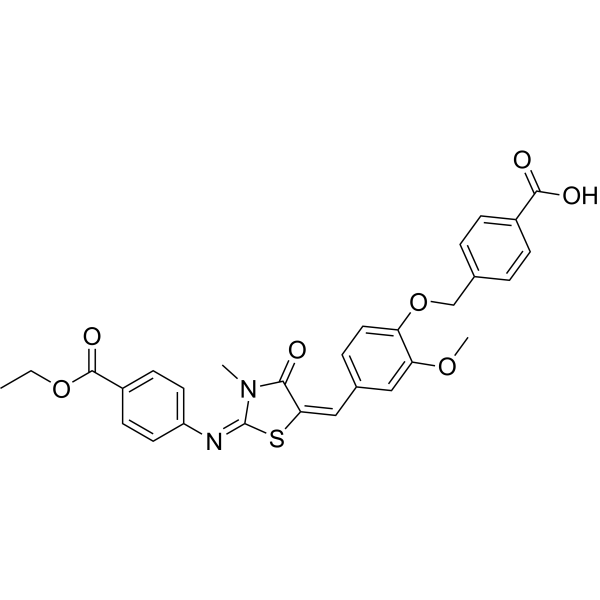
- HY-133120
-
|
|
PROTACs
Akt
|
Cancer
|
|
INY-03-041 is a potent, highly selective and PROTAC-based pan-AKT degrader consisting of the ATP-competitive AKT inhibitor Ipatasertib (HY-15186) conjugated to Lenalidomide (HY-A0003, Cereblon ligand). INY-03-041 inhibits AKT1, AKT2 and AKT3 with IC50s of 2.0, 6.8 and 3.5 nM, respectively .
|
-
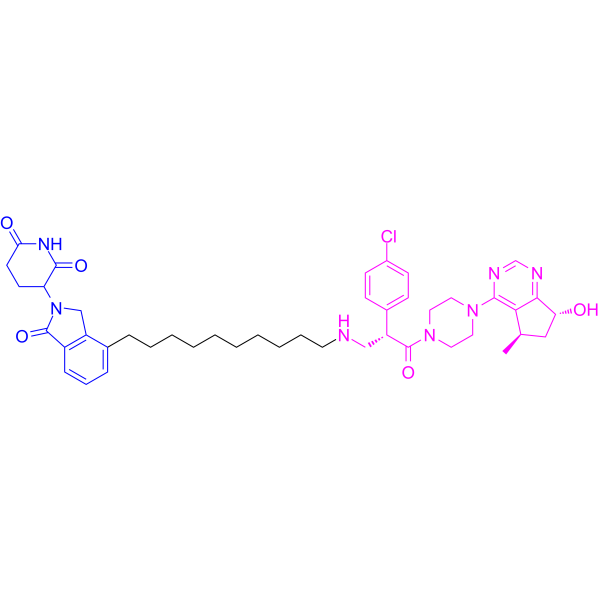
- HY-103265
-
|
FPL 67156 trisodium
|
NTPDase
|
Cardiovascular Disease
Inflammation/Immunology
|
|
ARL67156 (FPL 67156) trisodium is a selective small molecular inhibitor, targeting to ecto-ATPase, CD39, and CD73. ARL67156 trisodium is also a competitive inhibitor of NTPDase1 (CD39), NTPDase3 and NPP1, with Kis of 11, 18 and 12?μM, respectively. ARL67156 trisodium can be used in the research of calcific aortic valve disease, asthma .
|
-
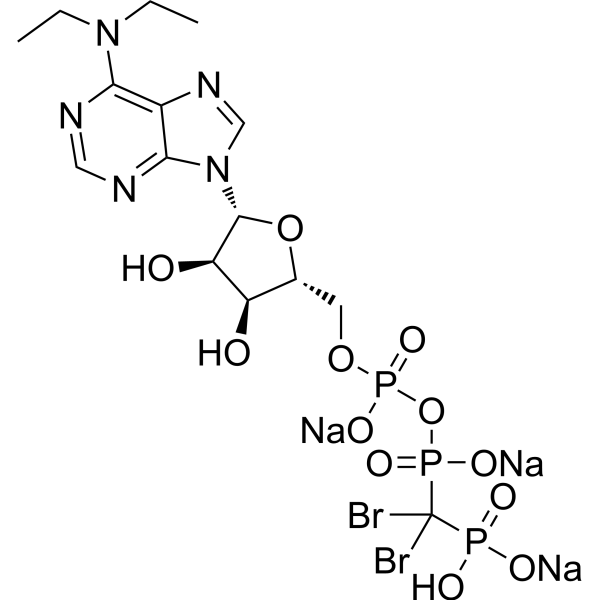
- HY-13011S
-
|
CH5424802-d8; RO5424802-d8; AF802-d8
|
Isotope-Labeled Compounds
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Alectinib-d8 is the deuterium labeled Alectinib. Alectinib (CH5424802) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
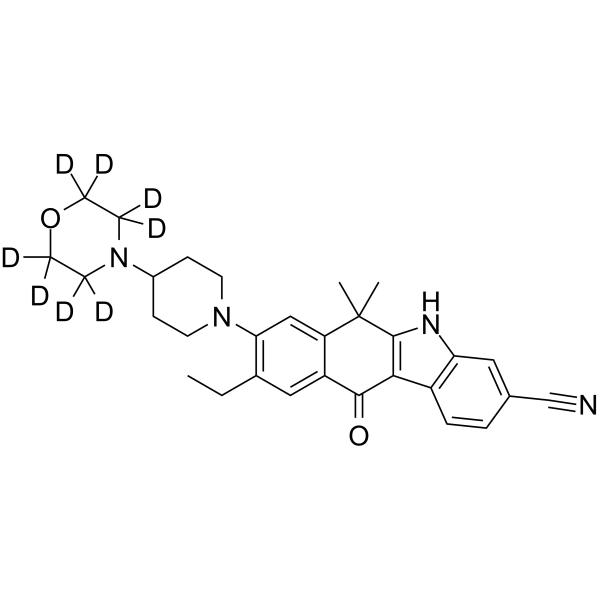
- HY-11010
-
|
|
JNK
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
AS601245 is an orally active, selective, ATP competitive JNK (c-Jun NH2-terminal protein kinase) inhibitor with IC50s of 150, 220, and 70 nM for three JNK human isoforms (hJNK1, hJNK2, and hJNK3), respectively. AS601245 exhibits 10- to 20-fold selectivity over c-src, CDK2, and c-Raf and more than 50- to 100-fold selectivity over a range of Ser/Thr- and Tyr-protein kinases. Neuroprotective properties .
|
-
- HY-11010A
-
|
|
JNK
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
AS601245 TFA is an orally active, selective, ATP competitive JNK (c-Jun NH2-terminal protein kinase) inhibitor with IC50s of 150, 220, and 70 nM for three JNK human isoforms (hJNK1, hJNK2, and hJNK3), respectively. AS601245 TFA exhibits 10- to 20-fold selectivity over c-src, CDK2, and c-Raf and more than 50- to 100-fold selectivity over a range of Ser/Thr- and Tyr-protein kinases. Neuroprotective properties .
|
-

- HY-165571
-
|
|
Carboxypeptidase
|
Neurological Disease
|
|
E2072 is a selective, orally active competitive inhibitor of glutamate carboxypeptidase II (GCPII) with a Ki of 10 nM. E2072 alleviates established thermal hyperalgesia in a rat model of chronic constriction injury. E2072 prevents oxaliplatin-induced reductions in nerve conduction velocity and amplitude in mice. E2072 is applicable to research related to neuropathic pain and neuropathy .
|
-

- HY-148318
-
|
|
Casein Kinase
|
Cancer
|
|
CK2α-IN-1 (compound 2) is a selective CK2α inhibitor (IC50=7.0 µM; Ki=1.6 µM) that exhibits a non-ATP-competitive mode of action. CK2α-IN-1 exhibits good potential for anticancer studies .
|
-
- HY-10517A
-
|
(Z)-SU6668; (Z)-TSU-68
|
VEGFR
PDGFR
FGFR
|
Cancer
|
|
(Z)-Orantinib ((Z)-SU6668) is a potent, selective, orally active and ATP competitive inhibitor of Flk‐1/KDR, PDGFRβ, and FGFR1, with IC50s of 2.1, 0.008, and 1.2 µM, respectively. (Z)-Orantinib is a potent antiangiogenic and antitumor agent that induces regression of established tumors .
|
-
- HY-112081
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
BAY-707, a chemical probe, is a substrate-competitive, highly potent and selective inhibitor of MTH1(NUDT1) with an IC50 of 2.3 nM. BAY-707 has a good pharmacokinetic (PK) profile to other MTH1 compounds and is well-tolerated in mice, but shows a clear lack of in vitro or in vivo anticancer efficacy .
|
-
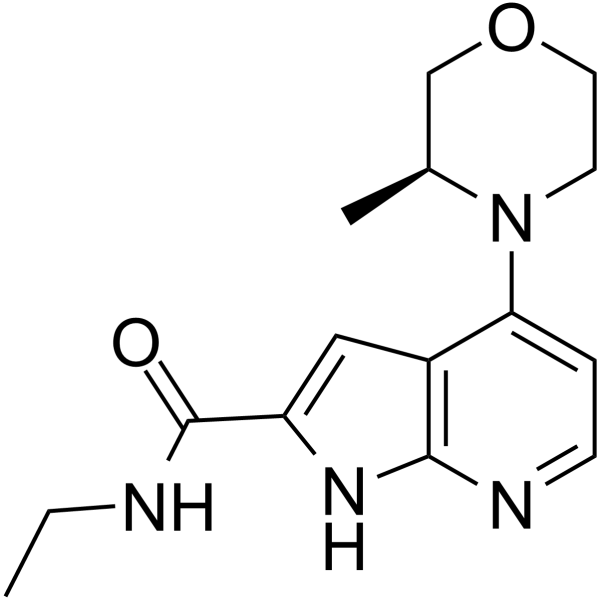
- HY-136174
-
|
|
PARP
|
Cancer
|
|
RBN-2397 is a potent, accross species and orally active NAD + competitive inhibitor of PARP7 (IC50<3 nM). RBN-2397 selectively binds to PARP7 (Kd=0.001 μM) and restores IFN signaling. RBN-2397 has the potential for the study of advanced or metastatic solid tumors .
|
-
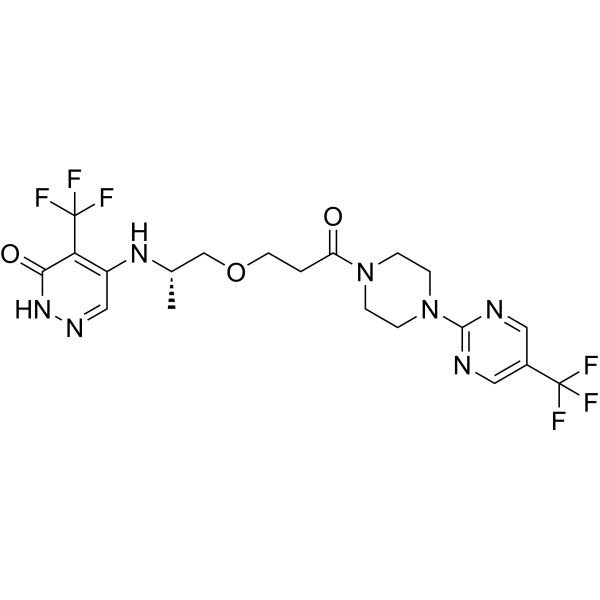
- HY-14892
-
|
LC15-0444
|
Dipeptidyl Peptidase
|
Metabolic Disease
Cancer
|
|
Gemigliptin (LC15-0444 ) is a highly selective, reversible and competitive dipeptidyl peptidase-4 (DPP-4) inhibitor, with an IC50 of 10.3 nM for human recombinant DPP-4. Gemigliptin exhibits potent anti-glycation properties. Gemigliptin can be used for the research of advanced glycation end products (AGE)-related diabetic complications .
|
-
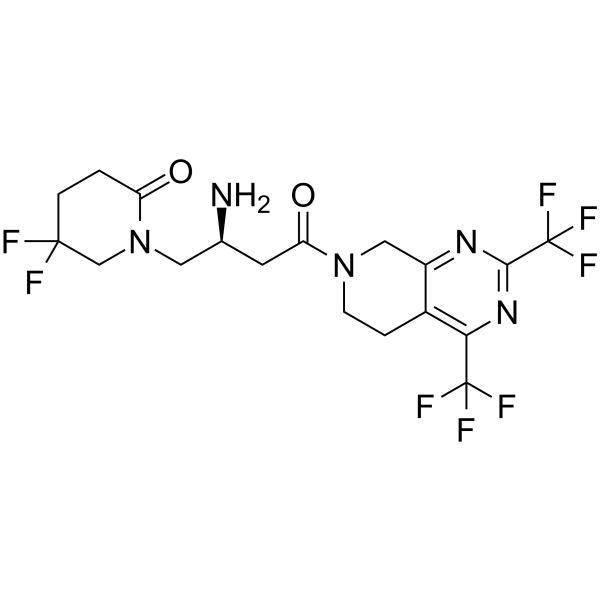
- HY-13204R
-
|
KL 373 hydrochloride (Standard)
|
Reference Standards
mAChR
|
Neurological Disease
|
|
Biperiden (hydrochloride) (Standard) is the analytical standard of Biperiden (hydrochloride). This product is intended for research and analytical applications. Biperiden (KL 373) hydrochloride is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden hydrochloride has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-
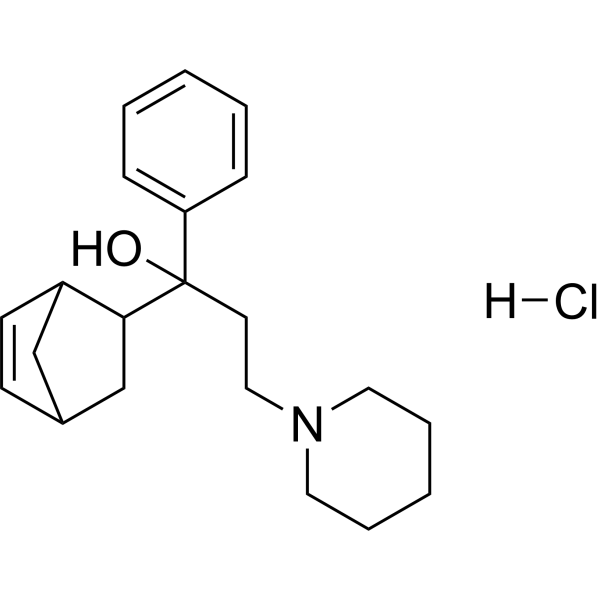
- HY-13204B
-
|
KL 373 lactate
|
mAChR
|
Neurological Disease
|
|
Biperiden (KL 373) lactate is an orally active non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors. Biperiden (KL 373) lactate inhibits acetylcholine and enhances dopamine signaling in the central nervous system. Biperiden (KL 373) lactate has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-
- HY-147040
-
|
|
c-Met/HGFR
Apoptosis
|
Cancer
|
|
ABN401 is an orally active and selective ATP-competitive c-MET inhibitor with an IC50 of 10 nM. ABN401 is cytotoxic to MET-addicted cancer cells with the IC50 of 2-43 nM. ABN401 has bioavailability in rats and dogs of 42.1-56.2% and 27.4-37.7%, respectively. ABN401 has antitumor activity .
|
-
- HY-112683
-
|
|
ASCT
|
Cancer
|
|
V-9302 is a competitive antagonist of transmembrane glutamine flux. V-9302 selectively and potently targets the amino acid transporter ASCT2 (SLC1A5) not ASCT1. V-9302 inhibits ASCT2-mediated glutamine uptake (IC50=9.6 μM) in HEK-293 cells .
|
-
- HY-13302C
-
|
|
VEGFR
FGFR
|
Cancer
|
|
CP-547632 TFA is an orally active, ATP-competitive and potent VEGFR-2 and FGF kinases inhibitor with IC50s of 11 nM and 9 nM, respectively. CP-547632 TFA is selective for VEGFR2 and bFGF over EGFR, PDGFRβ, and related tyrosine kinases (TKs). CP-547632 TFA has antitumor efficacy .
|
-
- HY-19432
-
|
|
iGluR
|
Neurological Disease
|
|
UBP-282 is a potent, selective and competitive AMPA and kainate receptor antagonist. UBP-282 inhibits the fast component of the dorsal root-evoked ventral root potential (fDR-VRP) with an IC50 value of 10.3 μM. UBP-282 antagonizes kainate-induced depolarisations of dorsal roots with a pA2 value of 4.96 .
|
-
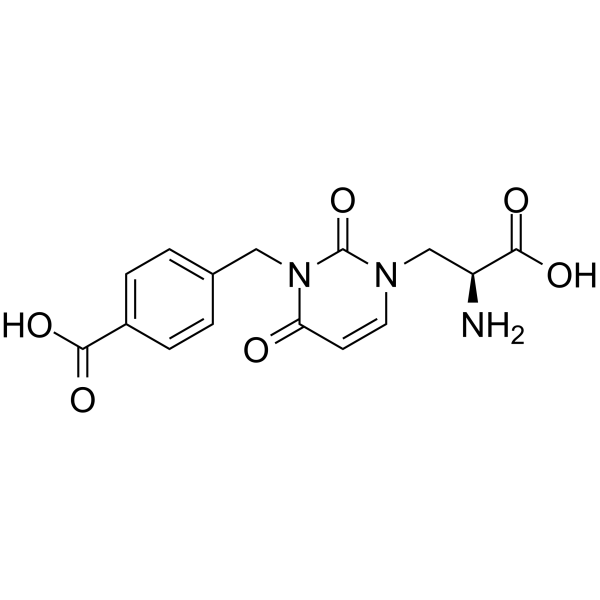
- HY-117583A
-
|
|
HDAC
Histone Methyltransferase
|
Neurological Disease
|
|
BG47 is a prototypical histone deacetylases HDAC1 and HDAC2 selective, optoepigenetic probe. BG47 can bind to and competitively inhibits the deacetylase activity of HDAC targets upon a light-induced trans-to-cis isomerization, and increases Histone Methyltransferase H3K9 acetylation. BG47 can be used for neurological disease research .
|
-

- HY-112683A
-
|
|
ASCT
|
Cancer
|
|
V-9302 hydrochloride is a competitive antagonist of transmembrane glutamine flux. V-9302 hydrochloride selectively and potently targets the amino acid transporter ASCT2 (SLC1A5) not ASCT1. V-9302 hydrochloride inhibits ASCT2-mediated glutamine uptake (IC50=9.6 µM) in HEK-293 cells .
|
-
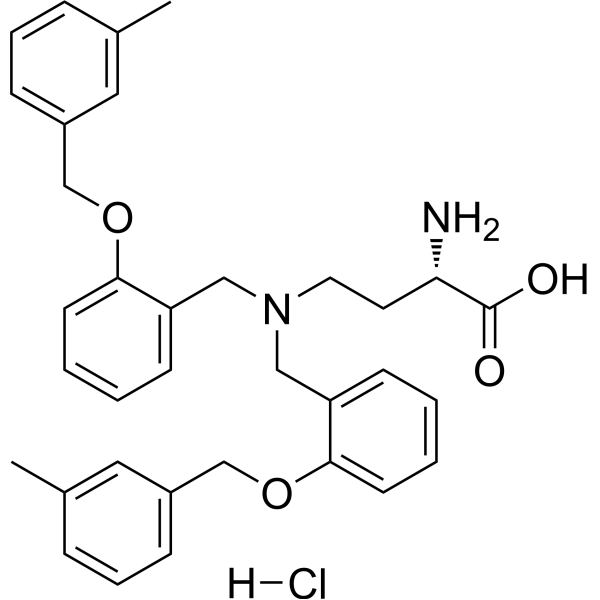
- HY-13418G
-
|
Compound C dihydrochloride; BML-275 dihydrochloride
|
AMPK
|
Neurological Disease
Cancer
|
|
Dorsomorphin dihydrochloride (GMP) is the GMP level of Dorsomorphin dihydrochloride (HY-13418). GMP guidelines are used to produce Dorsomorphin dihydrochloride (GMP). GMP small molecules works appropriately as an auxiliary reagent for cell research manufacture. Dorsomorphin dihydrochloride (GMP) is a potent, selective and ATP-competitive AMPK inhibitor. Dorsomorphin dihydrochloride (GMP) can be used for the research of induced differentiation of pluripotent stem cells (PSCs) .
|
-
- HY-12447
-
|
|
MEK
Apoptosis
|
Cancer
|
|
SMK-17 is a selective, non-ATP-competitive MEK1/MEK2 inhibitor with IC50s of 62 nM and 56 nM, respectively. SMK-17 binds to the allosteric pocket of MEK1/2. SMK-17 induces apoptosis in tumor cell lines harboring β-catenin mutations .
|
-

- HY-12866AR
-
|
LOXO-101 sulfate (Standard); ARRY-470 sulfate (Standard)
|
Trk Receptor
Apoptosis
Reference Standards
|
Cancer
|
|
Larotrectinib sulfate (Standard) is the analytical standard of Larotrectinib sulfate. This product is intended for research and analytical applications. Larotrectinib sulfate (LOXO-101 sulfate; ARRY-470 sulfate) is an ATP-competitive oral, selective inhibitor of the tropomyosin-related kinase (TRK) family receptors, with low nanomolar 50% inhibitory concentrations against all three isoforms (TRKA, B, and C).
|
-

- HY-10716AR
-
|
|
Reference Standards
GlyT
|
Neurological Disease
|
|
PF-03463275 (Standard) is the analytical standard of PF-03463275 (HY-10716A). This product is intended for research and analytical applications. PF-03463275 is a centrally penetrant, orally available, selective, and competitive GlyT1 (glycine transporter-1) reversible inhibitor, with a Ki of 11.6 nM. PF-03463275 has the potential for Schizophrenia research .
|
-

- HY-100888AR
-
|
|
CDK
Reference Standards
|
Others
|
|
(R)-Simurosertib (Standard) is the analytical standard of (R)-Simurosertib (HY-100888A). This product is intended for research and analytical applications. (R)-Simurosertib ((R)-TAK-931) is the (R)-enantiomer of Simurosertib. Simurosertib (TAK-931) is an orally active, selective and ATP-competitive cell division cycle 7 (CDC7) kinase inhibitor, with an IC50 of <0.3 nM .
|
-

- HY-13204AR
-
|
KL 373 (Standard)
|
Reference Standards
mAChR
|
Neurological Disease
|
|
Biperiden (Standard) is the analytical standard of Biperiden. This product is intended for research and analytical applications. Biperiden (KL 373) is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-

- HY-I0096
-
|
|
iGluR
HIV
HIV Integrase
|
Neurological Disease
|
|
Indole-2-carboxylic acid (I2CA) is a competitive antagonist of the glycine site of the NMDA receptor (Ki=15 μM, 5-fluoro-I2CA) and an inhibitor of HIV-1 integrase. Indole-2-carboxylic acid is selective for the glycine site of the NMDA receptor and blocks the enhancement of NMDA receptor by competitively inhibiting the binding of glycine to the NMDA receptor. Indole-2-carboxylic acid can also inhibit the strand transfer activity of HIV-1 integrase by chelating Mg 2+ at the active site of integrase and interacting with the hydrophobic cavity. Indole-2-carboxylic acid can be used in the study of neurological diseases (such as stroke, epilepsy) and HIV-1 infection .
|
-
- HY-103712AR
-
|
CT7001 hydrochloride (Standard); ICEC0942 hydrochloride (Standard)
|
Reference Standards
CDK
Apoptosis
|
Cancer
|
|
Samuraciclib hydrochloride (Standard) is the analytical standard of Samuraciclib hydrochloride (HY-103712A). This product is intended for research and analytical applications. Samuraciclib hydrochloride (CT7001 hydrochloride) is a potent, selective, ATP-competitive and orally active CDK7 inhibitor, with an IC50 of 41 nM. Samuraciclib hydrochloride displays 45-, 15-, 230- and 30-fold selectivity over CDK1, CDK2 (IC50 of 578 nM), CDK5 and CDK9, respectively. Samuraciclib hydrochloride inhibits the growth of breast cancer cell lines with GI50 values between 0.2-0.3 µM. Samuraciclib hydrochloride has anti-tumor effects .
|
-

- HY-108601A
-
|
|
PKC
|
Neurological Disease
Inflammation/Immunology
|
|
(S)-Ro 32-0432 is a potent, selective, ATP-competitive and orally active PKC inhibitor. The IC50 values of (S)-Ro 32-0432 for PKCα, PKCβI, PKCβII, PKCγ and PKCε are 9.3 nM, 28 nM, 30 nM, 36.5 nM and 108.3 nM, respectively. (S)-Ro 32-0432 is also a selective G protein-coupled receptor kinase 5 (GRK5) inhibitor. (S)-Ro 32-0432 prevents T-cell activation and has the potential for chronic inflammatory and autoimmune diseases research .
|
-
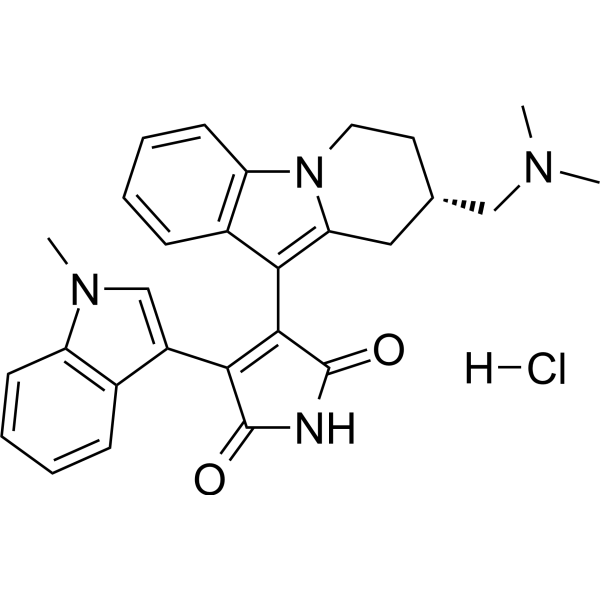
- HY-18299
-
|
NG 95
|
CDK
Parasite
|
Infection
Cancer
|
|
Purvalanol B (NG 95) is a potent, selective, reversible and ATP-competitive inhibitor CDK, with IC50s of 6 nM, 6 nM, 9 nM, 6 nM for cdc2-cyclin B, CDK2-cyclin A, CDK2-cyclin E and CDK5-p35, respectively. Purvalanol B shows selectivity for CDK over a range of other protein kinases (IC50>1000 nM). Purvalanol B inhibits the growth a chloroquine-resistant strain of P. falciparum .
|
-
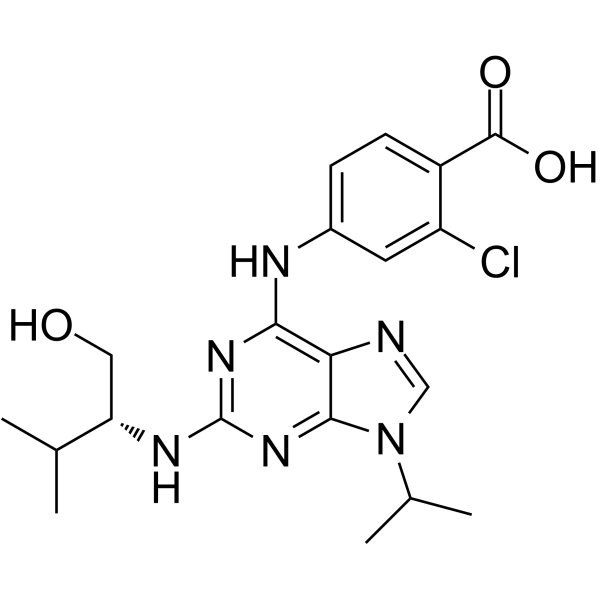
- HY-100707
-
|
|
DNA-PK
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
IC 86621 is a potent DNA-dependent protein kinase (DNA-PK) inhibitor, with an IC50 of 120 nM. IC 86621 also acts as a selective and reversible ATP-competitive inhibitor.IC 86621 inhibits DNA-PK mediated cellular DNA double-strand break (DSB) repair (EC50=68 µM). IC 86621 increases DSB-induced antitumor activity without cytotoxic effects. IC 86621 can protects rheumatoid arthritis (RA) T cells from apoptosis .
|
-
- HY-103566
-
|
|
mGluR
EGFR
p38 MAPK
Apoptosis
|
Infection
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
LY456236 is a selective, non-competitive and orally active antagonist of glutamate receptor 1 (mGlu1), which can inhibit phosphatidylinositol hydrolysis with an IC50 of 0.145 μM. LY456236 can also inhibit EGFR, with an IC50 of 0.918 μM. LY456236 blocks cell proliferation by inhibiting the MAPK pathway, reversing the anti-apoptotic effect of DHPG (HY-12598A). LY456236 can be used in epilepsy research .
|
-
- HY-18768
-
NCT-501
5 Publications Verification
|
Aldehyde Dehydrogenase (ALDH)
Akt
β-catenin
Necroptosis
|
Cancer
|
|
NCT-501 is a reversible, non-competitive, selective, blood-brain barrier-permeable ALDH1A1 inhibitor with an IC50 of 40 nM. NCT-501 inhibits the AKT-β-catenin signaling pathway, induces necroptosis in nasopharyngeal carcinoma cells, suppresses their proliferation and inhibits stem cell spheroid formation. NCT-501 can be used in research related to nasopharyngeal carcinoma, esophageal squamous cell carcinoma and malignant tumors .
|
-
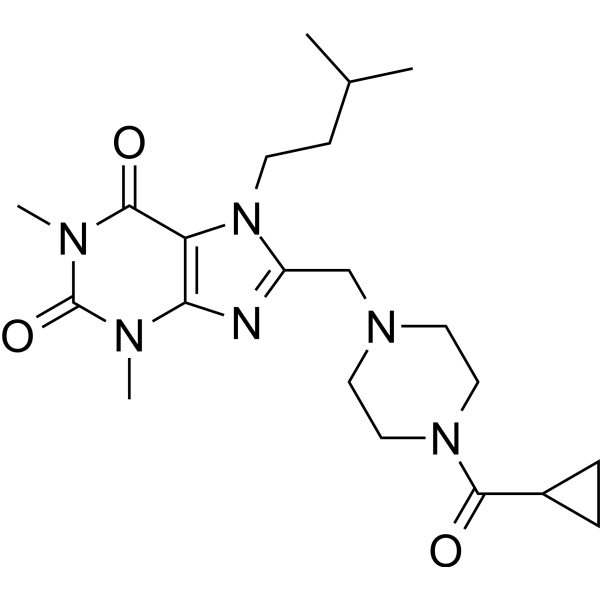
- HY-172747
-
|
|
PARP
|
Cancer
|
|
TNKS-2-IN-3 (Compound 5) is a selective competitive tankyrase 2 (TNKS2) inhibitor with an IC50 value of 0.3 nM, showing over 20-fold selectivity over TNKS1 and more than 100-fold selectivity over PARP1/2. TNKS-2-IN-3 stabilizes axin and suppresses the Wnt/β-catenin pathway by inhibiting TNKS2-mediated ADP-ribosylation, exhibiting antiproliferative activity in colorectal cancer cells. TNKS-2-IN-3 is proming for rasearch of solid tumors with aberrant Wnt pathway activation, such as colorectal cancer .
|
-

- HY-116815
-
|
|
Beta-lactamase
Bacterial
|
Infection
Neurological Disease
|
|
Lalistat 1 is a potent, selective, and competitive inhibitor of lysosomal acid lipase (LAL) and against purified human LAL (phLAL) with an IC50 of 68 nM. Lalistat 1 is a inhibitor of immunoglobulin A1 protease (IgA1P) proteases for H. influenzae, has less effects on other serine hydrolases (trypsin or β-lactamase, etc.). Lalistat 1 can be used for the research of niemann-pick type C (NPC) disease .
|
-
- HY-175270
-
|
|
MAGL
Keap1-Nrf2
NF-κB
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
MAGL-IN-22 (Compound 40) is a reversible, competitive, selective and BBB-penetrable MAGL inhibitor with an IC50 of 0.34 μM for hMAGL. MAGL-IN-22 has significant antioxidant and anti-inflammatory activity, and activates the Nrf2 pathway and significantly inhibits NFκB-mediated inflammation, without inducing cytotoxic effects. MAGL-IN-22 can be used for neurodegenerative diseases, chronic pain and cancers research .
|
-

- HY-182519
-
|
|
NO Synthase
|
Neurological Disease
|
|
HMN-1180 is a selective, competitive neuronal nitric oxide synthase (nNOS) inhibitor with a Ki value of 5.4 μM against rat nNOS. HMN-1180 exerts no significant effect on endothelial nitric oxide synthase (eNOS) or inducible nitric oxide synthase (iNOS). HMN-1180 inhibits nitric oxide production. HMN-1180 can be used for the study of nNOS-related neuronal functional physiology .
|
-

- HY-183978
-
|
|
Renin
|
Cardiovascular Disease
|
|
KRI-1314 is an orally active human renin inhibitor with selectivity for primate renin over non-primate renin. KRI-1314 competitively inhibits the binding of recombinant human renin to its substrate, reduces plasma renin activity, lowers blood pressure, and exhibits high stability in tissue homogenates. KRI-1314 is applicable to research on renin-dependent hypertension and hypertension-related studies .
|
-

- HY-W354203
-
|
1,2-Diisonicotinoylhydrazine
|
MOFs
Reactive Oxygen Species (ROS)
Bacterial
|
Infection
|
|
N'-Isonicotinoylisonicotinohydrazide (Compound 7, 1,2-Diisonicotinoylhydrazine) is a competitive inhibitor (IC50=5-30 μM) of bacterial heme oxygenase (HO). N'-Isonicotinoylisonicotinohydrazide inhibits iron release and bacterial iron acquisition. N'-Isonicotinoylisonicotinohydrazide exhibits selective activity against HO enzymes from Pseudomonas aeruginosa and Neisseria meningitidis. N'-Isonicotinoylisonicotinohydrazide is promising for research of multidrug-resistant Gram-negative bacterial infections .
|
-

- HY-15656AR
-
|
LDK378 dihydrochloride (Standard)
|
Anaplastic lymphoma kinase (ALK)
Insulin Receptor
IGF-1R
Reference Standards
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Ceritinib (LDK378) dihydrochloride (Standard) is the analytical standard of Ceritinib dihydrochloride (HY-15656A). This product is intended for research and analytical applications. Ceritinib dihydrochloride is a selective, orally bioavailable, and ATP-competitive ALK tyrosine kinase inhibitor with an IC50 of 200 pM. Ceritinib dihydrochloride also inhibits IGF-1R, InsR, and STK22D with IC50 values of 8, 7, and 23 nM, respectively. Ceritinib shows great antitumor potency .
|
-

- HY-107597R
-
|
SU3327 (Standard)
|
Reference Standards
JNK
|
Metabolic Disease
Inflammation/Immunology
|
|
Halicin (Standard) is the analytical standard of Halicin (HY-107597). This product is intended for research and analytical applications. Halicin (SU3327) is a potent, selective and substrate-competitive JNK inhibitor with an IC50 of 0.7 μM. Halicin also inhibits protein-protein interactions between JNK and JNK Interacting Protein (JIP) with an IC50 of 239 nM. Halicin shows less active against p38α and Akt Kinase .
|
-

- HY-170322
-
|
|
Indoleamine 2,3-Dioxygenase (IDO)
|
Metabolic Disease
Cancer
|
|
TDO2-IN-1 is a selective TDO2 inhibitor. TDO2-IN-1 binds to the active pocket, heme-binding pocket and substrate-binding pocket of apo-TDO2, interacts with key residues, competitively inhibits heme insertion, and suppresses intracellular TDO2 activity. TDO2-IN-1 can be used in research related to cancer, metabolism and other fields .
|
-

- HY-10253R
-
|
Tyrphostin AG 1024 (Standard)
|
Reference Standards
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
AG1024 (Standard) is the analytical standard of AG1024 (HY-10253). This product is intended for research and analytical applications. AG1024 (Tyrphostin AG 1024) is a reversible, competitive and selective IGF-1R inhibitor with an IC50 of 7 μM. AG1024 inhibits phosphorylation of IR (IC50=57 μM). AG1024 induces apoptosis and has anti-cancer activity .
|
-

- HY-101440A
-
|
CHF-3381
|
iGluR
Monoamine Oxidase
|
Neurological Disease
|
|
Indantadol hydrochloride (CHF-3381) is an orally active, non-selective NMDA antagonist and MAO inhibitor. Indantadol hydrochloride blocks the binding of [³H]-MK-801 to NMDA receptors in a non-competitive manner, with an IC50 of 8.1 μM. Indantadol hydrochloride completely inhibits dopamine release caused by NMDA. Indantadol hydrochloride protects neurons, with an ED₅₀ of 35 μM. Indantadol hydrochloride has anticonvulsant and anti-high pain hypersensitivity activities .
|
-

- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-10409AR
-
|
TG-101348 hydrochloride hydrate (Standard); SAR 302503 hydrochloride hydrate (Standard)
|
Reference Standards
JAK
Apoptosis
|
Cancer
|
|
Fedratinib (hydrochloride hydrate) (Standard) is the analytical standard of Fedratinib (hydrochloride hydrate). This product is intended for research and analytical applications. Fedratinib hydrochloride hydrate (TG-101348 hydrochloride hydrate) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib hydrochloride hydrate shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib hydrochloride hydrate induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-

- HY-128129
-
|
|
Urea Transporter
|
Metabolic Disease
|
|
UT-B-IN-1 (UTBINH-14) is a reversible, competitive and selective urea transporter-B (UT-B) inhibitor with IC50 values of 10 and 25 nM for human and mouse UT-B, respectively. UT-B-IN-1 shows low toxicity and high selectivity for UT-B over UT-A isoforms. UT-B-IN-1 increases urine output and reduces urine osmolality of mice. UT-B-IN-1 can be used for diuretic mechanism research .
|
-
- HY-N13917
-
|
|
Proteasome
Bacterial
|
Infection
Cancer
|
|
Argyrin B, a natural product cyclic peptide, is a reversible, non-competitive immunoproteasome inhibitor. Argyrin B shows selective inhibition of the β5i and β1i sites of the immunoproteasome over the β5c and β1c sites of the constitutive proteasome with nearly 20-fold selective inhibition of β1i over the homologous β1c. Argyrin B has antibacterial effects .
|
-

- HY-104066
-
|
Xiliertinib; HMPL-309
|
EGFR
|
Cancer
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-100195
-
|
|
Checkpoint Kinase (Chk)
|
Cancer
|
|
SAR-020106 is an ATP-competitive, potent, and selective CHK1 inhibitor with an IC50 of 13.3 nM for human CHK1. SAR-020106 shows excellent selectivity over CHK2. SAR-020106 significantly enhances the cell killing of Gemcitabine and SN38 by 3- to 29-fold in several colon tumor lines and in a p53-dependent fashion. SAR-020106 can enhance antitumor activity with selected anticancer agents .
|
-
- HY-100414R
-
|
BYK61359 (Standard)
|
Proton Pump
Reference Standards
|
Metabolic Disease
|
|
Soraprazan (Standard) is the analytical standard of Soraprazan (HY-100414). This product is intended for research and analytical applications. Soraprazan (BYK61359) is a selective, reversible K-competitive inhibitor of the H,K-ATPase (Ki=6.4 nM), with an IC50 of 0.19 μM in gastric glands. Soraprazan binds to the H,K-ATPase with a Kd of 28.27 nM. Soraprazan shows immediate inhibition of acid secretion and is more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases .
|
-

- HY-10409R
-
|
TG-101348 (Standard); SAR 302503 (Standard)
|
Reference Standards
JAK
Apoptosis
|
Cancer
|
|
Fedratinib (Standard) is the analytical standard of Fedratinib. This product is intended for research and analytical applications. Fedratinib (TG-101348) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-

- HY-18944
-
|
|
CDK
HSV
CMV
DNA/RNA Synthesis
|
Infection
|
|
FIT-039 is a selective, ATP-competitive and orally active CDK9 inhibitor with an IC50 of 5.8 μM for CKD9/cyclin T1. FIT-039 does not inhibit other CDKs and other kinases. FIT-039 inhibits replication of HSV-1 (IC50 of 0.69 μM), HSV-2, human adenovirus, and human CMV. FIT-039 is a promising antiviral agent for inhibiting drug-resistant HSVs and other DNA viruses.
|
-
- HY-18263
-
|
SB-656933 hydrochloride
|
CXCR
Interleukin Related
|
Inflammation/Immunology
|
|
Elubrixin (SB-656933) hydrochloride is a potent, selective, competitive, reversible and orally active CXCR2 antagonist and an IL-8 receptor antagonist. Elubrixin hydrochloride inhibits neutrophil CD11b upregulation (IC50 of 260.7 nM) and shape change (IC50 of 310.5 nM). Elubrixin hydrochloride has the potential for inflammatory diseases research, such as inflammatory bowel disease and airway inflammation .
|
-
- HY-10254
-
|
PD0325901; PD325901
|
MEK
Autophagy
Apoptosis
|
Cancer
|
|
Mirdametinib (PD0325901) is an orally active, selective and non-ATP-competitive MEK inhibitor with an IC50 of 0.33 nM. Mirdametinib exhibits a Ki app of 1 nM against activated MEK1 and MEK2. Mirdametinib suppresses the expression of p-ERK1/2 and induces apoptosis. Mirdametinib has anti-cancer activity for a broad spectrum of human tumor xenografts .
|
-
- HY-17623D
-
|
CJ-12420 Benzoate; RQ-00000004 Benzoate
|
Proton Pump
|
Metabolic Disease
|
|
Tegoprazan Benzoate is the benzoate form of Tegoprazan (HY-17623). Tegoprazan (CJ-12420), a potassium-competitive acid blocker, is a potent, oral active and highly selective inhibitor of gastric H +/K +-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H +/K +-ATPases in vitro .
|
-
- HY-155102
-
|
|
PROTACs
Glutaminase
|
Cancer
|
|
PROTAC TG2 degrader-2 (compound 7) is a selective, competitive degrader targeting Transglutaminase 2 (TG2), with Kd > 100 μM. PROTAC TG2 degrader-2 inhibits the cell migration and decreases the level of TG2 in ovarian cancer cells. PROTAC TG2 degrader-2 can be used for ovarian cancer study .
|
-
- HY-18263C
-
|
SB-656933 tosylate
|
CXCR
Interleukin Related
|
Inflammation/Immunology
|
|
Elubrixin tosylate (SB-656933 tosylate) is a potent, selective, competitive, reversible and orally active CXCR2 antagonist and an IL-8 receptor antagonist. Elubrixin tosylate inhibits neutrophil CD11b upregulation (IC50 of 260.7 nM) and shape change (IC50 of 310.5 nM). Elubrixin tosylate has the potential for inflammatory diseases research, such as inflammatory bowel disease and airway inflammation .
|
-
- HY-N18289
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
8-Hydroxydihydrochelerythrine is a benzophenanthridine alkaloid-derived, selective and competitive AChE inhibitor (IC50=0.61 μM) isolated from the roots of Zanthoxylum nitidum (Roxb.) DC. 8-Hydroxydihydrochelerythrine increases the content of acetylcholine in the synapses of cholinergic neurons and enhances cholinergic neurotransmission. 8-Hydroxydihydrochelerythrine is applicable to the research of neurodegenerative diseases with dementia such as Alzheimer's disease .
|
-

- HY-10285S2
-
|
BMS-477118-13C2
|
Isotope-Labeled Compounds
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin- 13C2 (BMS-477118- 13C2) is 13C labeled Saxagliptin. Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-

- HY-128306
-
|
|
HCV Protease
DNA/RNA Synthesis
|
Infection
|
|
HCV-IN-50 (Compound 2) is a competitive and selective HCV NS5B RNA-dependent RNA polymerase inhibitor with an IC50 of 0.3 μM for NS5B △C21 enzyme over △C55 enzyme. HCV-IN-50 has an antiviral activity and efficiently blocks replication of HCV subgenomic replicons especially mutant replicons .
|
-

- HY-14892C
-
|
LC15-0444 hydrochloride
|
Dipeptidyl Peptidase
|
Metabolic Disease
Cancer
|
|
Gemigliptin hydrochloride is the hydrochloride salt of Gemigliptin (HY-14892). Gemigliptin hydrochloride is a highly selective, reversible and competitive dipeptidyl peptidase-4 (DPP-4) inhibitor, with an IC50 of 10.3 nM for human recombinant DPP-4. Gemigliptin hydrochloride exhibits potent anti-glycation properties. Gemigliptin hydrochloride can be used for the research of advanced glycation end products (AGE)-related diabetic complications .
|
-

- HY-14892B
-
|
LC15-0444 tartrate hydrate
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Gemigliptin (LC15-0444 ) tartrate hydrate is a selective, reversible and competitive dipeptidyl peptidase-4 (DPP-4) inhibitor, with an IC50 of 10.3 nM for human recombinant DPP-4. Gemigliptin tartrate hydrate exhibits potent anti-glycation properties. Gemigliptin tartrate hydrate can be used for the research of advanced glycation end products (AGE)-related diabetic complications .
|
-

- HY-P1173
-
|
Myristoylated L 803; GSK-3β inhibitor XIII
|
GSK-3
Amyloid-β
|
Neurological Disease
|
|
L803-mts (Myristoylated L 803) is a selective and substrate-competitive GSK-3 peptide inhibitor (IC50: 40 μM). L803-mts also reduces Aβ deposits and ameliorates cognitive deficits in 5XFAD mice. L803-mts shows antidepressive effect in the forced swimming test .
|
-
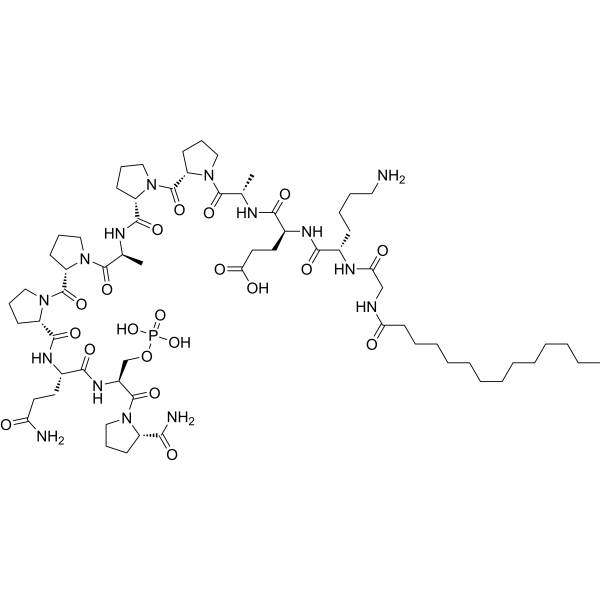
- HY-100714AR
-
|
D-APV (Standard); D-2-Amino-5-phosphonovaleric acid (Standard)
|
iGluR
Reference Standards
|
Neurological Disease
|
|
D-AP5 (Standard) is the analytical standard of D-AP5 (HY-100714A). This product is intended for research and analytical applications. D-AP5 (D-APV) is a selective and competitive NMDA receptor antagonist with a Kd of 1.4 μM. D-AP5 (D-APV) inhibits the glutamate binding site of NMDA receptors .
|
-

- HY-18263A
-
|
SB-656933
|
CXCR
Interleukin Related
|
Inflammation/Immunology
|
|
Elubrixin (SB-656933) is a potent, selective, competitive, reversible and orally active CXCR2 antagonist and an IL-8 receptor antagonist. Elubrixin inhibits neutrophil CD11b upregulation (IC50 of 260.7 nM) and shape change (IC50 of 310.5 nM). Elubrixin has the potential for inflammatory diseases research, such as inflammatory bowel disease and airway inflammation .
|
-
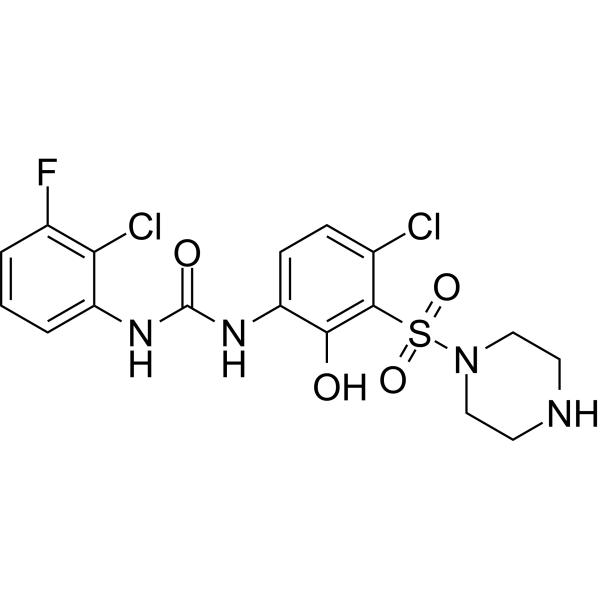
- HY-12344A
-
|
|
FLT3
|
Cancer
|
|
UNC2025 hydrochloride is a potent, ATP-competitive, highly orally active and BBB-permeable Mer/Flt3 inhibitor with IC50 values of 0.74 nM and 0.8 nM, respectively. UNC2025 hydrochloride is >45-fold selectivity for MERTK relative to Axl (IC50= 122 nM; Ki = 13.3 nM). UNC2025 hydrochloride exhibits an excellent PK properties, and can be used for the investigation of acute leukemia .
|
-
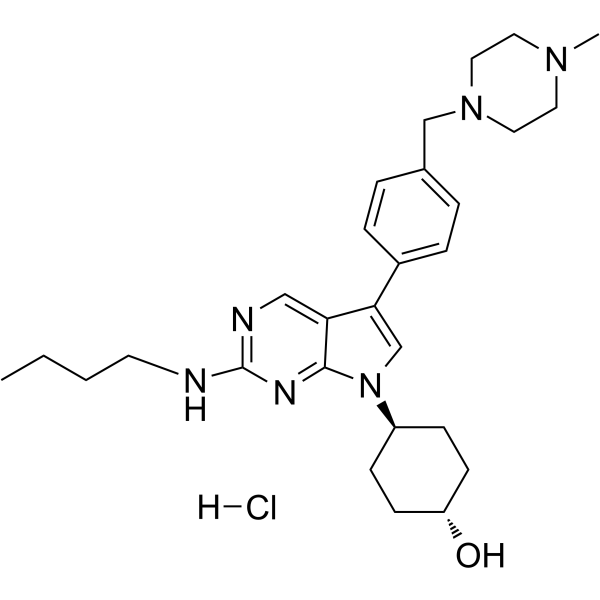
- HY-12854
-
|
GRN163L
|
Telomerase
Apoptosis
|
Cardiovascular Disease
Cancer
|
|
Imetelstat (GRN163L) is a 13-mer oligonucleotide and competitive Telomerase inhibitor. Imetelstat binds with high affinity to the template region of the RNA component of human telomerase. Imetelstat induces Apoptosis. Imetelstat is capable of selectively eliminating myelofibrosis hematopoietic stem cells. Imetelstat leads to the loss of a cancer cell's ability to maintain telomere length, resulting in the inhibition of cell proliferation .
|
-

- HY-178367
-
|
|
Phosphatase
Apoptosis
Caspase
PARP
|
Cancer
|
|
PFKFB4-IN-1 is a potent and selective ATP-competitive PFKFB4 inhibitor (IC50 = 4.50 μM) that reduces intracellular PFKFB4 protein levels. PFKFB4-IN-1 exhibits >12-fold selectivity over PFKFB1/4 and PFKFB3/4. PFKFB4-IN-1 inhibits cancer cell proliferation, induces apoptosis, and inhibits cell migration. PFKFB4-IN-1 inhibits tumor growth in the MDA-MB-231 xenograft mouse model. PFKFB4-IN-1 can be used for breast, lung and liver cancer research .
|
-

- HY-119601
-
|
|
Phosphodiesterase (PDE)
|
Cancer
|
|
GRI918013 (compound 1) is a selective and competitive autotaxin (ATX/NPP2) inhibitor with anti-invasive and anti-metastatic activity. GRI918013 competitively binds to ATX, blocking lipid substrates such as lysophosphatidylcholine (LPC) from entering the ATX active site, thereby inhibiting ATX-mediated hydrolysis of LPC to lysophosphatidic acid (LPA), and consequently inhibiting ATX-LPA axis-related tumor cell invasion and metastasis. GRI918013 inhibits ATX-mediated hydrolysis of the LPL substrate FS-3 (IC50=31.42 nM, Ki=12.98 nM). GRI918013 can be used in research on cancer invasion and metastasis, such as melanoma, and can also serve as a tool compound for ATX-LPA axis-related diseases such as fibrotic diseases, neuropathic pain, and cholestatic pruritus .
|
-

- HY-143288
-
|
|
NTPDase
|
Infection
Inflammation/Immunology
Cancer
|
|
NTPDase-IN-2 (compound 5g) is a selective NTPDase inhibitor with IC50s of 0.04 and 2.27 µM for h-NTPDase-2/-8, respectively. NTPDase-IN-2 non-competitively inhibits h-NTPDase-1/-2 with a Km of 74 µM for h-NTPDase-2. NTPDase-IN-2 can be used in studies of cancer, immunologic disorders as well as bacterial infections .
|
-
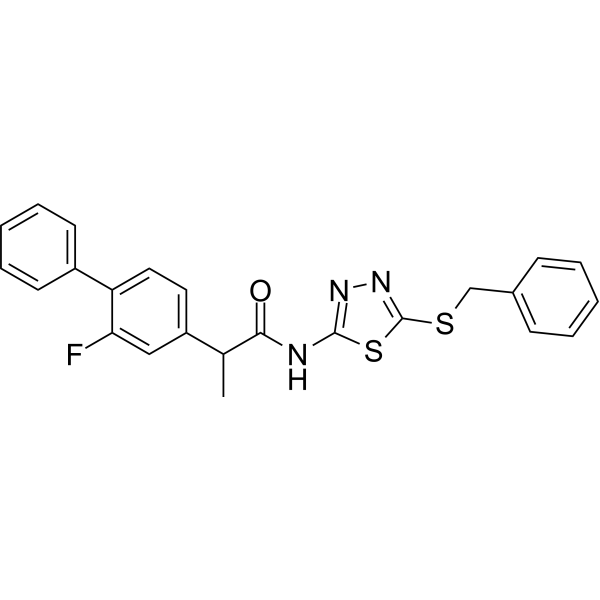
- HY-10403R
-
|
|
Reference Standards
p38 MAPK
Autophagy
|
Inflammation/Immunology
Cancer
|
|
PH-797804 (Standard) is the analytical standard of PH-797804 (HY-10403). This product is intended for research and analytical applications. PH-797804 is a ATP-competitive, selective p38α/p38β inhibitor (IC50=26 nM and Ki=5.8 nM for p38α; Ki=40 nM for p38β) and does not inhibit JNK2.
|
-

- HY-10780
-
|
|
Endogenous Metabolite
|
Cardiovascular Disease
|
|
JTV-803 mesylate is a human factor Xa inhibitor with oral anticoagulant activity. JTV-803 exhibits competitive inhibition of human factor Xa, with a Ki value of 0.019μM and IC50Value is 0.081μM. JTV-803 is 100 times more selective at inhibiting human factor Xa than its comparator. JTV-803 is an effective oral anticoagulant for the prevention of thrombosis .
|
-

- HY-10280S
-
|
|
Isotope-Labeled Compounds
Factor Xa
|
Others
|
|
YM-60828-d3 is the deuterium labeled YM-60828 (HY-10280). YM-60828 is an orally active, selective and competitive factor Xa inhibitor with a Ki of 1.3 nM and an IC50 of 2.3 nM. YM-60828 inhibits thrombus formation and platelet aggregation. YM-60828 can be used for the research of venous thrombosis, arterial thrombosis, and thromboembolic disorders .
|
-
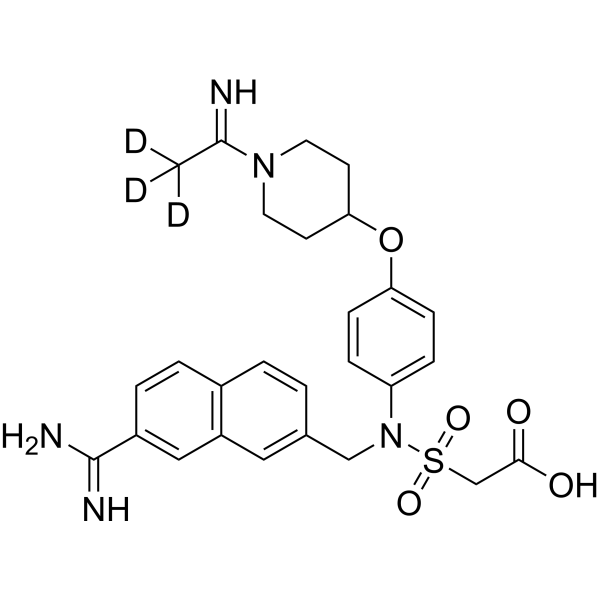
- HY-101440
-
|
CHF-3381 free base
|
iGluR
Monoamine Oxidase
|
Neurological Disease
|
|
Indantadol (the free base of CHF-3381) is an orally active, non-selective NMDA antagonist and MAO inhibitor. Indantadol blocks the binding of [³H]-MK-801 to NMDA receptors in a non-competitive manner, with an IC50 of 8.1 μM. Indantadol completely inhibits dopamine release caused by NMDA. Indantadol protects neurons, with an ED₅₀ of 35 μM. Indantadol has anticonvulsant and anti-high pain hypersensitivity activities .
|
-

- HY-103441
-
|
|
EGFR
|
Cancer
|
|
JNJ28871063 hydrochloride is an orally active, highly selective and ATP competitive pan-ErbB kinase inhibitor with IC50 values of 22 nM, 38 nM, and 21 nM for ErbB1, ErbB2, and ErbB4, respectively. JNJ28871063 hydrochloride inhibits phosphorylation of functionally important tyrosine residues in both EGFR and ErbB2. JNJ28871063 hydrochloride crosses the blood-brain barrier and has antitumor activity in human tumor xenograft models that overexpress EGFR and ErbB2 .
|
-
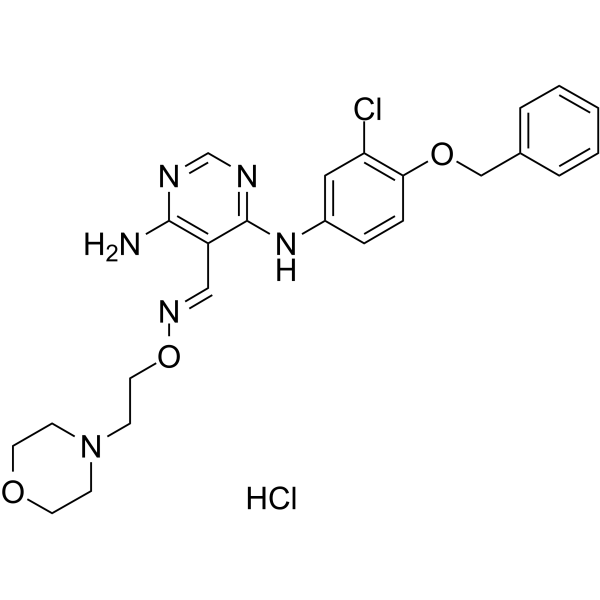
- HY-18174E
-
|
LY2606368 dimesylate
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
Prexasertib dimesylate (LY2606368 dimesylate) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib dimesylate inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib dimesylate causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib dimesylate shows potent anti-tumor activity .
|
-
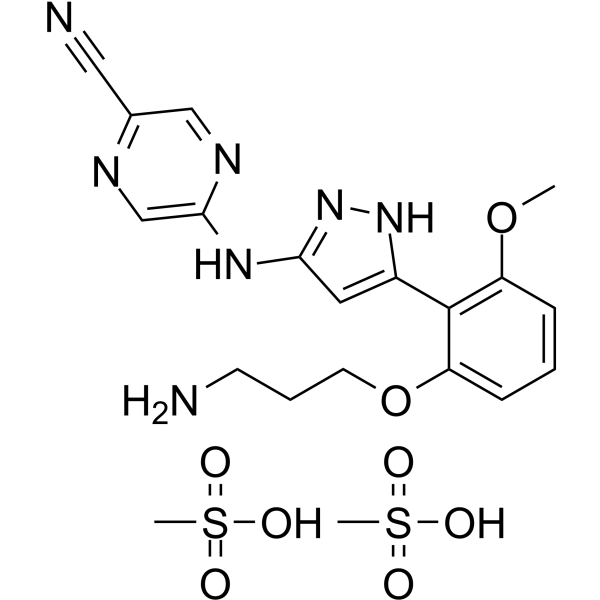
- HY-148105
-
|
|
MNK
Eukaryotic Initiation Factor (eIF)
Apoptosis
FLT3
DYRK
|
Cancer
|
|
DS12881479 is a selective non-ATP-competitive MNK1 inhibitor with an IC50 value of 387 nM. DS12881479 stabilizes MNK1 in its autoinhibited DFD-out conformation, blocks eIF4E phosphorylation, suppresses tumor cell proliferation and induces weak apoptosis. DS12881479 also inhibits FLT3 and DYRK1a kinase activity at high concentrations. DS12881479 can be used for the research of cancer, such as leukemia .
|
-
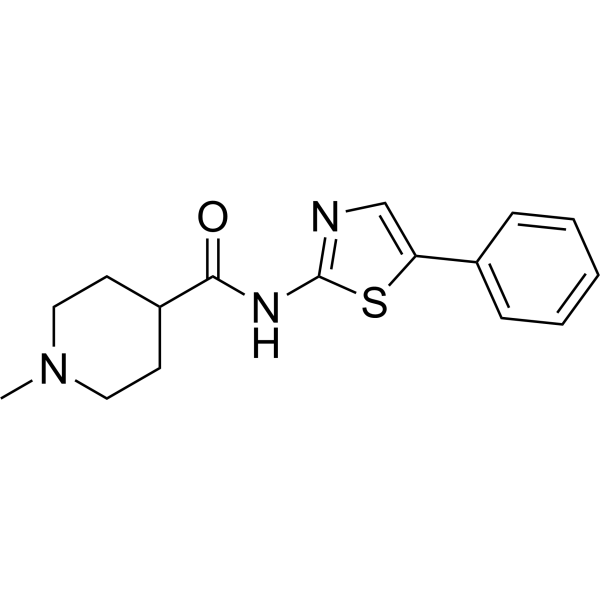
- HY-124267
-
|
SOMG-833
|
c-Met/HGFR
|
Cancer
|
|
Zgwatinib (SOMG-833) is a potent, selective, and ATP-competitive c-MET inhibitor, with an IC50 of 0.93 nM against c-MET, over 10,000-fold more potent compared with 19 tyrosine kinases (including c-MET family members and highly homologous kinases). Zgwatinib potently inhibits c-MET-driven cell proliferation. Zgwatinib as a potential candidate agent for c-MET-driven human cancers research .
|
-
- HY-10580
-
|
6-Bromoindirubin-3'-oxime; BIO; MLS 2052
|
GSK-3
CDK
Apoptosis
|
Cancer
|
|
GSK 3 Inhibitor IX (6-Bromoindirubin-3'-oxime; BIO) is a potent, selective, reversible and ATP-competitive inhibitor of GSK-3α/β and CDK1-cyclinB complex with IC50s of 5 nM/320 nM/80 nM for (GSK-3α/β)/CDK1/CDK5, respectively.
|
-
- HY-120812
-
|
|
HIV Protease
|
Infection
|
|
HIV-IN-11 is part of the hydroxylaminoglutaramide (HAPA) transition state isomeric series of HIV protease inhibitors and is a potent and selective inhibitor of HIV-1 protease. HIV-IN-11 competitively inhibits HIV-1 PR (Ki: 0.049 nM) and potently inhibits replication of HIV(IIIb)-infected MT4 lymphocytes at concentrations of 25.0-50.0 nM. HIV-IN-11 displays a longer half-life than indinavir sulfate in animal models and serves as a promising second-generation HIV protease inhibitor .
|
-

- HY-110137A
-
|
DB75; NSC 305831
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine (DB75) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine is also an antiparasite agent .
|
-
- HY-10032R
-
|
PF 00477736 (Standard)
|
Checkpoint Kinase (Chk)
Reference Standards
VEGFR
Src
c-Fms
Aurora Kinase
FGFR
FLT3
RET
CDK
|
Cancer
|
|
PF 477736 (Standard) is the analytical standard of PF 477736 (HY-10032). This product is intended for research and analytical applications. PF 477736 (PF 00477736) is a potent, selective and ATP-competitive inhibitor of Chk1, with a Ki of 0.49 nM, it is also a Chk2 inhibitor, with a Ki of 47 nM. PF 477736 shows <100-fold selectivity for Chk1 over VEGFR2, Fms, Yes, Aurora-A, FGFR3, Flt3, and Ret (IC50=8 (Ki), 10, 14, 23, 23, 25, and 39 nM, respectively). PF 477736 can enhance Gemcitabine antitumor activity in vitro and in vivo .
|
-

- HY-179599
-
|
|
DYRK
EGFR
|
Neurological Disease
Cancer
|
|
Dyrk1A-IN-15 is a selective, brain-penetrant and ATP-competitive Dyrk1A inhibitor with a IC50 of 19 nM. Dyrk1A-IN-15 displays high selectivity across a broad kinase panel (specific for DYRK kinases) with nanomolar potency. Dyrk1A-IN-15 impairs neurosphere self-renewal, cell invasion, and EGFR stability in vitro. Dyrk1A-IN-15 inhibits tumor growth and prolongs survival in vivo. Dyrk1A-IN-15 has potential for glioblastoma (GBM) research .
|
-

- HY-179600
-
|
|
DYRK
EGFR
|
Neurological Disease
Cancer
|
|
Dyrk1A-IN-16 is a selective, brain-penetrant and ATP-competitive Dyrk1A inhibitor with a IC50 of 53 nM. Dyrk1A-IN-16 displays high selectivity across a broad kinase panel (specific for DYRK kinases) with nanomolar potency. Dyrk1A-IN-16 impairs neurosphere self-renewal, cell invasion, and EGFR stability in vitro. Dyrk1A-IN-16 inhibits tumor growth and prolongs survival in vivo. Dyrk1A-IN-16 has potential for glioblastoma (GBM) research .
|
-

- HY-110137
-
|
DB75 dihydrochloride; NSC 305831 dihydrochloride
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine dihydrochloride (DB75 dihydrochloride) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine dihydrochloride is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine dihydrochloride is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine dihydrochloride is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine dihydrochloride is also an antiparasite agent .
|
-
- HY-12354
-
|
|
MMP
|
Cancer
|
|
SB-3CT is a potent and competitive matrix metalloproteinase MMP-2 and MMP-9 inhibitor with Ki values of 13.9 and 600 nM, respectively. SB-3CT has high selectivity for gelatinases. SB-3CT shows blood-brain barrier permeability and has neuroprotective effects and anticancer activity .
|
-
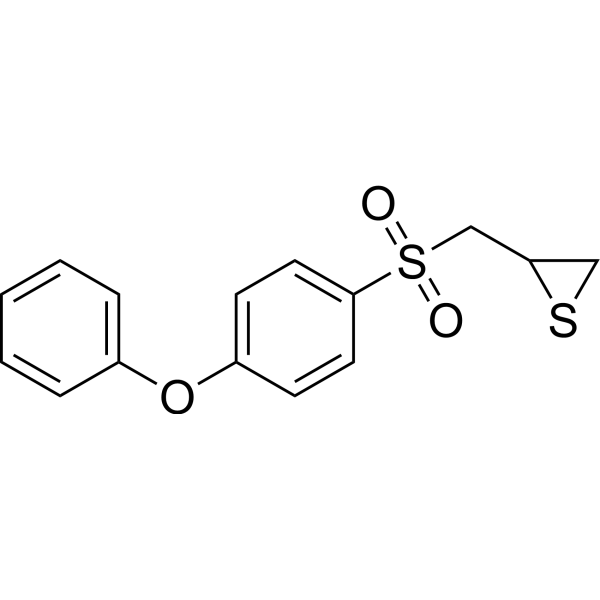
- HY-12344
-
|
|
FLT3
|
Neurological Disease
Cancer
|
|
UNC2025 is a potent, ATP-competitive, highly orally active and BBB-permeable Mer/Flt3 inhibitor with IC50 values of 0.74 nM and 0.8 nM, respectively. UNC2025 is >45-fold selectivity for MERTK relative to Axl (IC50= 122 nM; Ki = 13.3 nM). UNC2025 exhibits an excellent PK properties, and can be used for the investigation of acute leukemia .
|
-
- HY-130608
-
|
|
EGFR
|
Cancer
|
|
Mutated EGFR-IN-3 (compound 3) is a potent, ATP-competitive and highly selective allosteric dibenzodiazepinone inhibitor of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with IC50 values of 12 nM and 13 nM, respectively .
|
-
- HY-10119R
-
|
SCH 530348 (Standard)
|
Reference Standards
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Vorapaxar (Standard) is the analytical standard of Vorapaxar. This product is intended for research and analytical applications. Vorapaxar (SCH 530348), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar (SCH 530348) inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-

- HY-10119S
-
|
SCH 530348-d5
|
Protease Activated Receptor (PAR)
Isotope-Labeled Compounds
|
Cardiovascular Disease
|
|
Vorapaxar-d5 is deuterated labeled Vorapaxar (HY-10119). Vorapaxar (SCH 530348), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar (SCH 530348) inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-
- HY-177784
-
|
|
Molecular Glues
Indoleamine 2,3-Dioxygenase (IDO)
|
Infection
Neurological Disease
Cancer
|
|
iDeg-3 is a selective molecular glue degrader that targets IDO1. iDeg-3 can competitively bind to the heme binding site of apo-IDO1, preventing heme binding and inhibiting the enzymatic reaction that converts tryptophan into kynurenine by IDO1 (IC50 = 46 nM). iDeg-3 can be used for the researches of cancer, infection and neurological disease, such as melanoma .
|
-

- HY-155374
-
|
|
Phosphatase
Apoptosis
Caspase
|
Cancer
|
|
PP5-IN-1 is a competitive and selective serine/threonine protein phosphatase 5 (PP5) inhibitor with a Ki value of 244 nM. PP5-IN-1 induces extrinsic apoptosis (Apoptosis), disrupts the integrity of complex II, and activates Caspase 8. PP5-IN-1 can be used in research related to clear cell renal cell carcinoma .
|
-
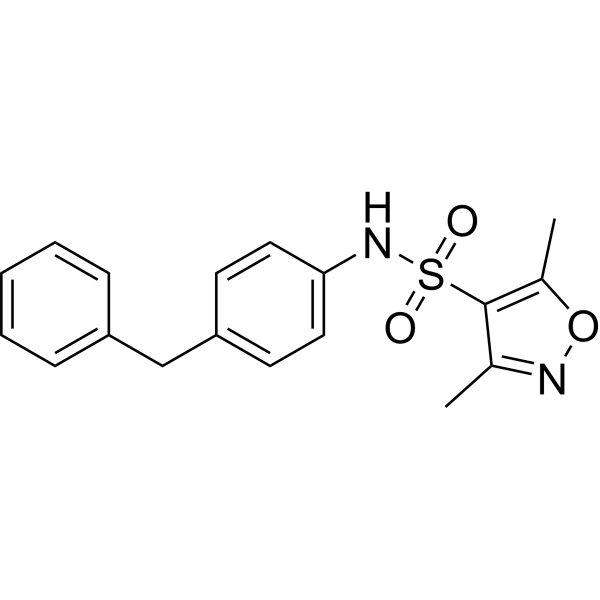
- HY-10287A
-
|
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Gosogliptin hydrochloride is the hydrochloride of Gosogliptin (HY-10287). Gosogliptin (PF-00734200) is a potent, orally active, selective, and competitive inhibitor of DPP-IV, the enzyme mainly responsible for the degradation of the incretin peptides GLP-1 and glucose-dependent insulinotropic polypeptide. Gosogliptin demonstrates rapid and reversible inhibition of plasma DPP-4 activity. Gosogliptin stimulates insulin secretion and improves glucose tolerance .
|
-

- HY-13204AS
-
|
KL 373-d5
|
Isotope-Labeled Compounds
mAChR
|
Neurological Disease
|
|
Biperiden-d5 (KL 373-d5) is deuterium labeled Biperiden. Biperiden (KL 373) is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-

- HY-17623R
-
|
CJ-12420 (Standard); RQ-00000004 (Standard)
|
Reference Standards
Proton Pump
|
Metabolic Disease
|
|
Tegoprazan (Standard) is the analytical standard of Tegoprazan. This product is intended for research and analytical applications. Tegoprazan (CJ-12420), a potassium-competitive acid blocker, is a potent, oral active and highly selective inhibitor of gastric H +/K +-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H +/K +-ATPases in vitro .
|
-

- HY-14892A
-
|
LC15-0444 tartrate
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Gemigliptin tartrate (LC15-0444 tartrate) is a highly selective, reversible and competitive dipeptidyl peptidase-4 (DPP-4) inhibitor, with an IC50 of 10.3 nM for human recombinant DPP-4. Gemigliptin tartrate exhibits potent anti-glycation properties. Gemigliptin tartrate can be used for the research of advanced glycation end products (AGE)-related diabetic complications .
|
-
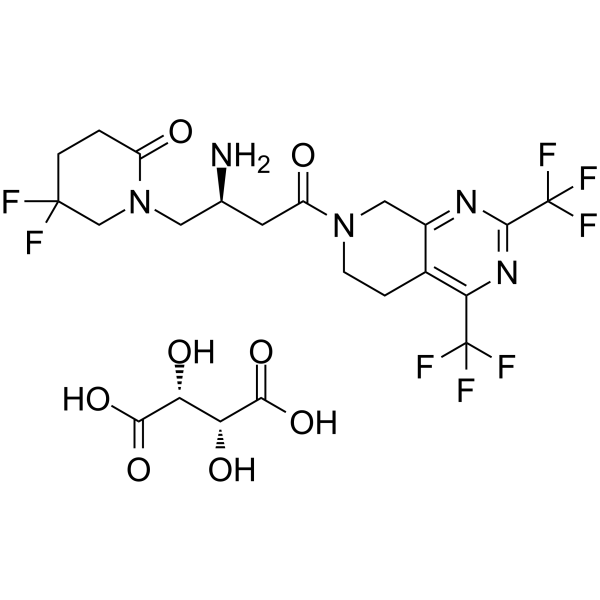
- HY-175856
-
|
|
Cholinesterase (ChE)
|
Neurological Disease
|
|
AChE-IN-95 (Compound 7) is a highly selective competitive acetylcholinesterase (AChE) inhibitor (IC50=17.87 μM, Ki=19.48 μM). AChE-IN-95 exhibits strong cytotoxicity against SH-SY5Y neuroblastoma cells. AChE-IN-95 is promising for research of Alzheimer’s disease and neurodegenerative disorders .
|
-

- HY-101925
-
CM-272
3 Publications Verification
|
Histone Methyltransferase
DNA Methyltransferase
Apoptosis
|
Cancer
|
|
CM-272 is a first-in-class, potent, selective, substrate-competitive and reversible dual G9a/DNA methyltransferases (DNMTs) inhibitor with antitumor activities. CM-272 inhibits G9a, DNMT1, DNMT3A, DNMT3B and GLP with IC50s of 8 nM, 382 nM, 85 nM, 1200 nM and 2 nM, respectively. CM-272 inhibits cell proliferation and promotes apoptosis, inducing IFN-stimulated genes and immunogenic cell death .
|
-
- HY-50878A
-
|
PF-02341066 hydrochloride
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
- HY-181022
-
|
|
Casein Kinase
|
Cancer
|
|
BMS-135 is a potenr, selective and ATP-competitive casein kinase 2 (CK2) inhibitor with IC50 of 0.8 and 0.3 nM for CK2αandCK2α′ isoforms. BMS-135 can simulate the structure of ATP and bind to the active pocket of CK2, thereby inhibiting its serine/threonine phosphorylation function. BMS-135 can inhibit cells proliferation and shows anti-tumor effect. BMS-135 can be used for research of colon cancer .
|
-

- HY-123981
-
5MPN
3 Publications Verification
|
Phosphatase
|
Cancer
|
|
5MPN is a first-in-class, potent, orally active and selective 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) inhibitor. 5MPN appears to be a competitive inhibitor of the F6P binding site (Ki=8.6 μM). 5MPN does not inhibit PFK-1 or PFKFB3. 5MPN targets the sugar metabolism of tumors and suppresses proliferation of multiple human cancer cell lines .
|
-
- HY-15196
-
TAK-285
3 Publications Verification
|
EGFR
|
Cancer
|
|
TAK-285 is a potent, selective, ATP-competitive and orally active HER2 and EGFR(HER1) inhibitor with IC50 of 17 nM and 23 nM, respectively. TAK-285 is >10-fold selectivity for HER1/2 than HER4, and less potent to MEK1/5, c-Met, Aurora B, Lck, CSK etc. TAK-285 has effective antitumor activity . TAK-285 can cross the blood-brain barrier (BBB) .
|
-
- HY-16749AR
-
|
PLX-3397 hydrochloride (Standard)
|
c-Fms
c-Kit
Apoptosis
Reference Standards
|
Cancer
|
|
Pexidartinib (hydrochloride) (Standard) is the analytical standard of Pexidartinib (hydrochloride). This product is intended for research and analytical applications. Pexidartinib hydrochloride (PLX-3397 hydrochloride) is a potent, orally active, selective, and ATP-competitive colony stimulating factor 1 receptor (CSF1R or M-CSFR) and c-Kit inhibitor, with IC50s of 20 and 10 nM, respectively. Pexidartinib hydrochloride exhibits 10- to 100-fold selectivity for c-Kit and CSF1R over other related kinases. Pexidartinib hydrochloride induces cell apoptosis and has anti-cancer activity .
|
-

- HY-10409S
-
|
TG-101348-d9; SAR 302503-d9
|
Isotope-Labeled Compounds
Apoptosis
JAK
|
Cancer
|
|
Fedratinib-d9 (TG-101348-d9) is deuterium labeled Fedratinib. Fedratinib (TG-101348) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-

- HY-13011AR
-
|
CH5424802 Hydrochloride (Standard); RO5424802 Hydrochloride (Standard); AF-802 Hydrochloride (Standard)
|
Anaplastic lymphoma kinase (ALK)
Reference Standards
|
Cancer
|
|
Alectinib (Hydrochloride) (Standard) is the analytical standard of Alectinib (Hydrochloride). This product is intended for research and analytical applications. Alectinib Hydrochloride (CH5424802 Hydrochloride; RO5424802 Hydrochloride; AF-802 Hydrochloride) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-

- HY-B0442C
-
|
|
Endogenous Metabolite
Phosphodiesterase (PDE)
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
Cancer
|
|
Vardenafil dihydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil dihydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM respectively, while IC50s are >1000 nM for PDE3 and PDE4. Vardenafil dihydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil dihydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-
- HY-B0442
-
|
|
Phosphodiesterase (PDE)
Endogenous Metabolite
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
Cancer
|
|
Vardenafil is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-
- HY-103442R
-
|
DAPH (Standard)
|
Reference Standards
EGFR
Amyloid-β
|
Neurological Disease
Cancer
|
|
CGP52411 (Standard) is the analytical standard of CGP52411 (HY-103442). This product is intended for research and analytical applications. CGP52411 (DAPH) is a high selective, potent, orally active and ATP-competitive EGFR inhibitor with an IC50 of 0.3 μM. CGP52411 blocks the toxic influx of Ca2+ ions into neuronal cells, and dramatic inhibits and reverses the formation of β-amyloid (Aβ42) fibril aggregates associated with Alzheimer's disease .
|
-

- HY-160215A
-
|
|
TGF-β Receptor
p38 MAPK
TGF-beta/Smad
Interleukin Related
|
Cancer
|
|
GFH018 is an orally active, selective and ATP-competitive TGF-βR1 inhibitor with an IC50 of 40 nM. GFH018 reactivates the immune system by blocking the immunosuppression mediated by regulatory T cells and M2 macrophages. GFH018 inhibits tumor angiogenesis. GFH018 suppresses tumor growth in mouse tumor models. GFH018 can be used for the research of solid tumors, hepatocellular carcinoma, colorectal cancer, breast cancer, and relapsed/metastatic nasopharyngeal carcinoma .
|
-
- HY-P0097A
-
|
Melanostatine-5 acetate salt
|
Melanocortin Receptor
|
Metabolic Disease
Endocrinology
Cancer
|
|
Nonapeptide-1 (Melanostatine-5) acetate salt, a peptide hormone, is a selective antagonist of MC1R (Ki: 40 nM). Nonapeptide-1 acetate salt is a competitive α-MSH antagonist that potently inhibits intracellular cAMP and melanosome dispersion induced by α-MSH in melanocytes (IC50: 2.5 nM and 11 nM, respectively). Nonapeptide-1 acetate salt inhibits melanin synthesis, and can be used in the research of skin pigmentation and regulation of steroid production in the adrenal gland, skin cancer .
|
-
- HY-146332
-
|
|
P-selectin
|
Cardiovascular Disease
|
|
Collagen-IN-1 (compound 3), an ortho-carbonyl hydroquinone derivative, is a selective inhibitor on collagen. Collagen-IN-1 inhibits agonist-induced platelet aggregation in a non-competitive manner with an IC50 value of 1.77 μM. Collagen-IN-1 reduces the expression of P-selectin, activation of glycoprotein IIb/IIIa, and release of adenosine triphosphate and CD63 from platelet. Collagen-IN-1 has the potential for platelet-related thrombosis diseases research .
|
-
- HY-15186
-
|
GDC-0068; RG7440
|
Organoid
Akt
Apoptosis
|
Cancer
|
|
Ipatasertib (GDC-0068) is an orally active, highly selective and ATP-competitive pan-Akt inhibitor with IC50 values of 5, 18, 8 nM for Akt1/2/3, respectively. Ipatasertib synchronously activates FoxO3a and NF-κB through inhibition of Akt leading to p53-independent activation of PUMA. Ipatasertib also induces apoptosis in cancer cells and inhibits tumor growth in xenograft mouse models .
|
-
- HY-19989
-
|
L-660711
|
P-glycoprotein
LPL Receptor
Leukotriene Receptor
|
Inflammation/Immunology
|
|
MK-571 (L-660711) is an orally active, potent and selective competitive leukotriene D4 (LTD4) receptor antagonist, with Ki values of 0.22 and 2.1 nM in guinea pig and human lung membranes, respectively. MK-571 is also a MRP4 and ABCC1 (MRP1) inhibitor. MK-571 inhibits constitutive and antigen-stimulated S1P (sphingosine-1-phosphate) release .
|
-
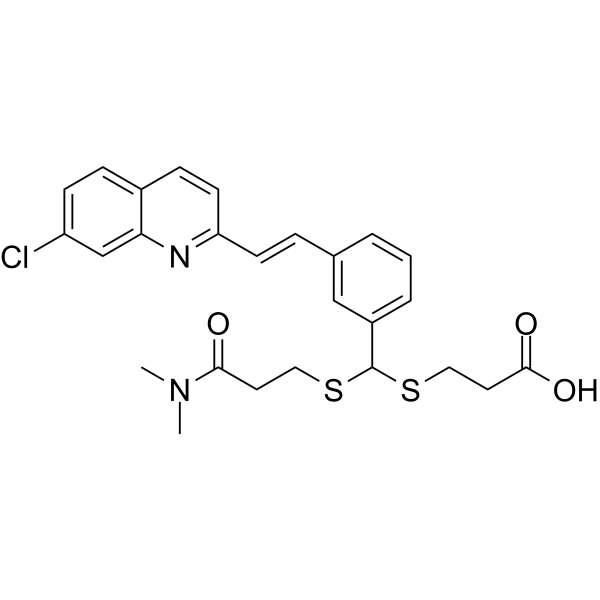
- HY-B0442A
-
|
|
Phosphodiesterase (PDE)
Endogenous Metabolite
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
Cancer
|
|
Vardenafil hydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-
- HY-15186C
-
|
GDC-0068 tosylate; RG7440 tosylate
|
Organoid
Akt
Apoptosis
|
Cancer
|
|
Ipatasertib (GDC-0068) tosylate is an orally active, highly selective and ATP-competitive pan-Akt inhibitor with IC50 values of 5, 18, 8 nM for Akt1/2/3, respectively. Ipatasertib tosylate synchronously activates FoxO3a and NF-κB through inhibition of Akt leading to p53-independent activation of PUMA. Ipatasertib tosylate also induces apoptosis in cancer cells and inhibits tumor growth in xenograft mouse models .
|
-

- HY-160215
-
|
|
TGF-β Receptor
p38 MAPK
TGF-beta/Smad
Interleukin Related
|
Cancer
|
|
GFH018 is an orally active, selective and ATP-competitive TGF-βR1 inhibitor with an IC50 of 40 nM. GFH018 reactivates the immune system by blocking the immunosuppression mediated by regulatory T cells and M2 macrophages. GFH018 inhibits tumor angiogenesis. GFH018 suppresses tumor growth in mouse tumor models. GFH018 can be used for the research of solid tumors, hepatocellular carcinoma, colorectal cancer, breast cancer, and relapsed/metastatic nasopharyngeal carcinoma .
|
-
- HY-18732AR
-
|
Tilarginine acetate (Standard); Methylarginine acetate (Standard)
|
Reference Standards
NO Synthase
|
Cardiovascular Disease
Cancer
|
|
L-NMMA (Tilarginine) acetate (Standard) is the analytical standard of L-NMMA acetate (HY-18732A). This product is intended for research and analytical applications. L-NMMA (Tilarginine) acetate is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA acetate inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA acetate alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA acetate is applicable to research related to nociception, bone cancer pain, and myopia .
|
-

- HY-10044R
-
|
WYE-125132 (Standard)
|
mTOR
Reference Standards
Apoptosis
|
Cancer
|
|
WYE-132 (Standard) is the analytical standard of WYE-132 (HY-10044). This product is intended for research and analytical applications. WYE-132 (WYE-125132) is a highly potent, ATP-competitive, and specific mTOR kinase inhibitor (IC50: 0.19±0.07 nM; >5,000-fold selective versus PI3Ks). WYE-132 (WYE-125132) inhibits mTORC1 and mTORC2.
|
-

- HY-P0097
-
|
Melanostatine-5
|
Melanocortin Receptor
|
Metabolic Disease
Endocrinology
Cancer
|
|
Nonapeptide-1 (Melanostatine-5), a peptide hormone, is a selective antagonist of MC1R (Ki: 40 nM). Nonapeptide-1 is a competitive α-MSH antagonist that potently inhibits intracellular cAMP and melanosome dispersion induced by α-MSH in melanocytes (IC50: 2.5 nM and 11 nM, respectively). Nonapeptide-1 inhibits melanin synthesis, and can be used in the research of skin pigmentation and regulation of steroid production in the adrenal gland, skin cancer .
|
-
- HY-143287
-
|
|
NTPDase
|
Infection
Inflammation/Immunology
Cancer
|
|
NTPDase-IN-1 (compound 5a) is a selective NTPDase inhibitor with IC50s of 0.05, 0.23 and 0.54 µM for h-NTPDase-1/-2/-8, respectively. NTPDase-IN-1 non-competitively inhibits h-NTPDase-1/-2 with a Km of 21 µM for h-NTPDase-1. NTPDase-IN-1 can be used in studies of cancer, immunologic disorders as well as bacterial infections .
|
-
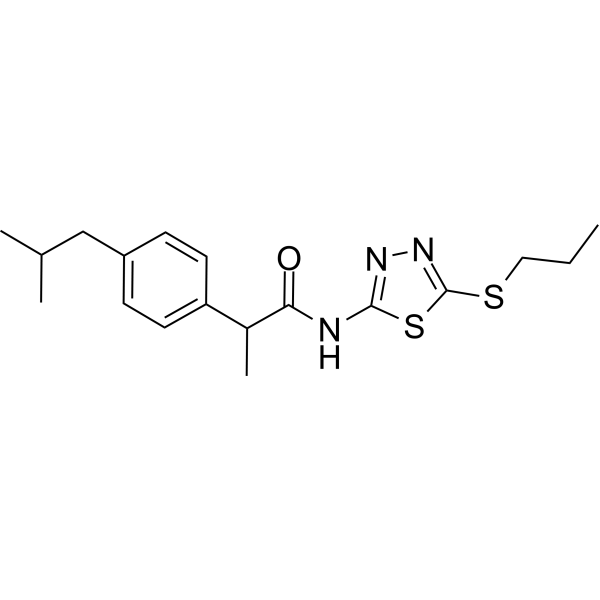
- HY-15816R
-
|
BVD-523 (Standard); VRT752271 (Standard)
|
ERK
Reference Standards
|
Cancer
|
|
Ulixertinib (Standard) is the analytical standard of Ulixertinib. This product is intended for research and analytical applications. Ulixertinib (BVD-523; VRT752271) is a potent, orally active, highly selective, ATP-competitive and reversible covalent inhibitor of ERK1/2 kinases, with an IC50 of <0.3 nM against ERK2. Ulixertinib (BVD-523; VRT752271) inhibits the phosphorylated ERK2 (pERK) and downstream kinase RSK (pRSK) in an A375 melanoma cell line .
|
-

- HY-112291A
-
|
|
p38 MAPK
Src
PKC
|
Inflammation/Immunology
|
|
SB 220025 trihydrochloride is a reversible, orally active, cell-permeable, ATP-competitive and selective human p38 MAPK inhibitor (IC50 = 60 nM). SB 220025 trihydrochloride also inhibits p56 Lck and PKC with IC50 values of 3.5 and 2.89 μM, respectively. SB 220025 trihydrochloride inhibits the expression of IL-8 gene in response to globular adiponectin (gAd), reduces inflammatory cytokine production and inhibits angiogenesis. SB 220025 trihydrochloride effectively prevents the progression of arthritis in a chronic inflammatory disease model and can be used in the study of inflammation .
|
-

- HY-112291
-
|
|
p38 MAPK
Src
PKC
|
Inflammation/Immunology
|
|
SB 220025 is a reversible, orally active, cell-permeable, ATP-competitive and selective human p38 MAPK inhibitor (IC50 = 60 nM). SB 220025 also inhibits p56 Lck and PKC with IC50 values of 3.5 and 2.89 µM, respectively. SB 220025 inhibits the expression of IL-8 gene in response to globular adiponectin (gAd), reduces inflammatory cytokine production and inhibits angiogenesis. SB 220025 effectively prevents the progression of arthritis in a chronic inflammatory disease model and can be used in the study of inflammation .
|
-
- HY-12583
-
|
|
Histone Methyltransferase
Epigenetic Reader Domain
|
Cancer
|
|
A-366, a chemical probe, is a potent, highly selective, peptide-competitive histone methyltransferase G9a inhibitor with IC50s of 3.3 and 38 nM for G9a and GLP (EHMT1), respectively. A-366 shows >1000-fold selectivity over 21 other methyltransferases. A-366 is also a potent, nanomolar inhibitor of the Spindlin1-H3K4me3-interaction (IC50=182.6 nM). A-366 displays high affinity at human histamine H3 receptor (Ki=17 nM) and shows subtype selectivity among subsets of the histaminergic and dopaminergic receptor families .
|
-
- HY-133123
-
|
|
Prostaglandin Receptor
|
Inflammation/Immunology
Cancer
|
|
EP4 receptor antagonist 1 is a highly potent and selective competitive prostanoid EP4 receptor antagonist for cancer immunotherapy. EP4 receptor antagonist 1 inhibits human and mouse EP4 receptor with IC50s of 6.1 nM and 16.2 nM, respectively. IC50s >10 μM for human EP1, EP2,and EP3 receptors .
|
-
- HY-178980
-
|
|
Casein Kinase
|
Cancer
|
|
APL-5125 (Compound 61f) is a potent, selective and orally active ATP-competitive CK2α inhibitor with an IC50 of 0.348 nM and a Ki of 0.095 nM. APL-5125 binds to CK2α in a bivalent manner, simultaneously interacting with the ATP-binding site and the αD pocket. APL-5125 exhibits antitumor activity and can be used for the research of cancer, such as colon cancer .
|
-

- HY-103721
-
|
|
Sirtuin
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
SIRT6-IN-2 (Compound 5) is a selective and competitive SIRT6 inhibitor (IC50: 34 μM). SIRT6-IN-2 increases acetylation of H3K9 and increases glucose uptake in cultured cells. SIRT6-IN-2 also reduces T cell proliferation. SIRT6-IN-2 has immunosuppressive and chemosensitizing effects .
|
-
- HY-A0009
-
|
Galantamine hydrobromide
|
Cholinesterase (ChE)
nAChR
|
Neurological Disease
|
|
Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 µM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
- HY-125159
-
|
PF-00520904
|
Parasite
nAChR
|
Infection
|
|
Derquantel, a spirocyclic anthelmintic, is a competitive, orally active nicotinic acetylcholine receptor (nAChR) antagonist. Derquantel inhibits ACh-induced depolarization with an IC50 of 0.22 μM. By selectively antagonizing nAChRs on the somatic muscles of nematodes, Derquantel causes flaccid paralysis of muscles, thereby dislodging parasites from the host's gastrointestinal tract. Derquantel is applicable to research related to Haemonchus contortus infection and Ascaris suum infection .
|
-
- HY-15883
-
|
|
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
GNE-900 is a an ATP-competitive, selective, and orally active ChK1 inhibitor with IC50s of 0.0011, 1.5 μM for ChKl, ChK2, respectively. GNE-900 abrogates the G2-M checkpoint, enhances DNA damage, and induces Apoptosis. Gemcitabine (HY-17026) and GNE-900 administration shows anti-tumor activity .
|
-
- HY-10423R
-
|
ASP7486 (Standard)
|
Reference Standards
mTOR
Autophagy
|
Cancer
|
|
OSI-027 (Standard) is the analytical standard of OSI-027 (HY-10423). This product is intended for research and analytical applications. OSI-027 (ASP7486) is a potent, selective, orally active and ATP-competitive mTOR kinase activity inhibitor with an IC50 of 4 nM. OSI-027 targets both mTORC1 and mTORC2 with IC50s of 22 nM and 65 nM, respectively .
|
-

- HY-183657A
-
|
|
|
Neurological Disease
|
|
GPR3 inverse agonist-1 TFA is a selective and competitive GPR3 inverse agonist with an EC50 of 5 μM. GPR3 inverse agonist-1 TFA inhibits GPR3-dependent Gs activity, stabilizes a specific receptor conformation, and reduces cAMP levels. GPR3 inverse agonist-1 TFA can be used for the research of Alzheimer's disease .
|
-

- HY-174231
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-163 (Compound 13) is a competitive epidermal growth factor receptor (EGFR) inhibitor (IC50=0.079 μM, selective for HER-2 inhibition). EGFR-IN-163 induces tumor cell apoptosis and cell cycle arrest at G₂/M phase. EGFR-IN-163 is promising for research of estrogen receptor-positive (ER+) breast cancer .
|
-

- HY-10119AR
-
|
SCH 530348 sulfate (Standard)
|
Reference Standards
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Vorapaxar (sulfate) (Standard) is the analytical standard of Vorapaxar (sulfate). This product is intended for research and analytical applications. Vorapaxar sulfate (SCH 530348 sulfate), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar sulfate inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-

- HY-182921
-
|
|
GPR84
Enterovirus
|
Inflammation/Immunology
|
|
GPR84 antagonist 11 is a highly selective GPR84 antagonist, with a human pA2 of 8.41 and a pKi of 8.16. GPR84 antagonist 11 competitively inhibits the binding of agonists to the orthosteric site of GPR84 and has improved druglike properties, though its metabolic stability still requires optimization. GPR84 antagonist 11 can be used in the research of autoimmune diseases and fibrotic diseases .
|
-

- HY-130553
-
|
β-NAAG; β-N-Acetylaspartylglutamic acid
|
Aminopeptidase
mGluR
|
Neurological Disease
|
|
β-Spaglumic acid (β-NAAG) is a competitive NAAG peptidase inhibitor (Ki=1 µM) that protects spinal cord neurons from excitotoxicity and hypoxic damage. β-Spaglumic acid is also a selective mGluR3 antagonist (mGluR3 receptor functions to regulate activity-dependent synaptic potentiation in the hippocampus). β-Spaglumic acid can be used in neuroprotection-related studies .
|
-
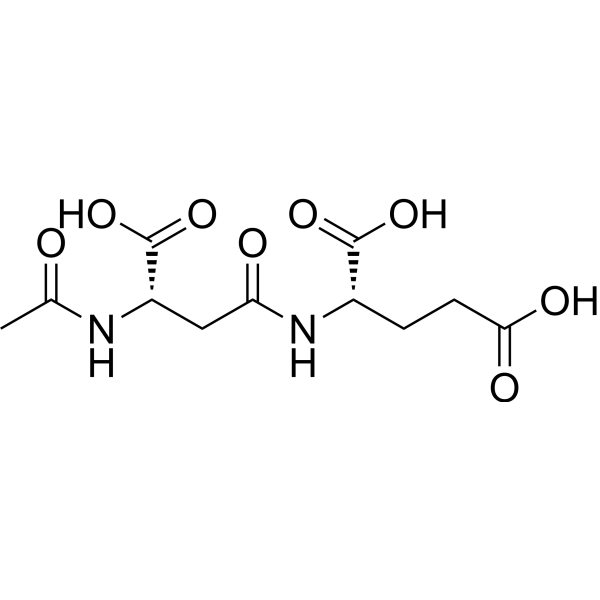
- HY-152208
-
|
|
SHP1
SHP2
|
Cancer
|
|
BPDA2 is a highly selective and competitive active site SHP2 inhibitor with IC50s of 92.0 nM, 33.39 μM, 40.71 μM for SHP2, SHP1, SHP1B, respectively. DBDA2 downregulates mitogenic and cell survival signaling and RTK expression. BPDA2 suppresses SHP2 mediated signaling and breast cancer cell phenotypes .
|
-
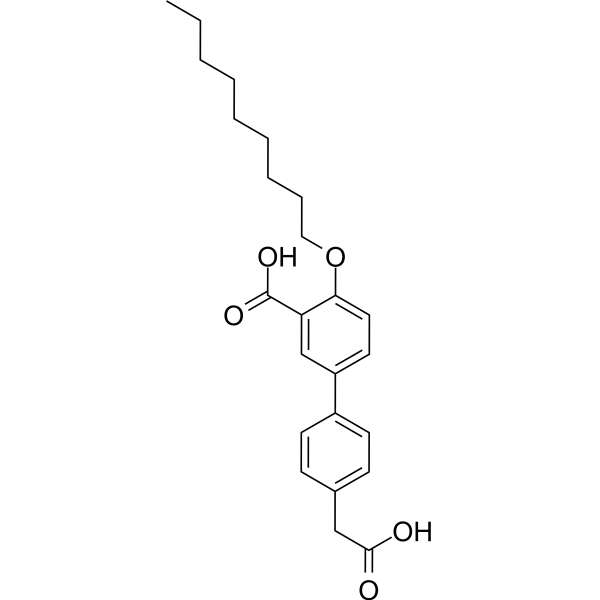
- HY-B0290S1
-
-
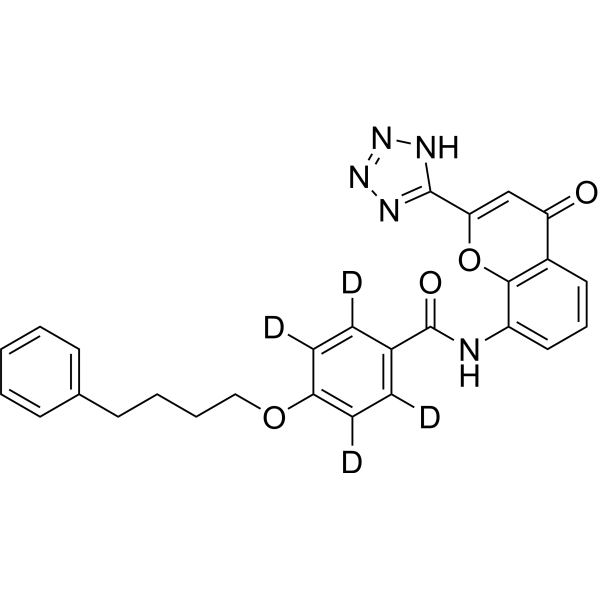
- HY-108601
-
|
|
PKC
|
Inflammation/Immunology
|
|
(S)-Ro 32-0432 free base is a potent, selective, ATP-competitive and orally active PKC inhibitor. The IC50 values of (S)-Ro 32-0432 free base for PKCα, PKCβI, PKCβII, PKCγ and PKCε are 9.3 nM, 28 nM, 30 nM, 36.5 nM and 108.3 nM, respectively. (S)-Ro 32-0432 free base is also a selective G protein-coupled receptor kinase 5 (GRK5) inhibitor. (S)-Ro 32-0432 free base prevents T-cell activation and has the potential for chronic inflammatory and autoimmune diseases research .
|
-
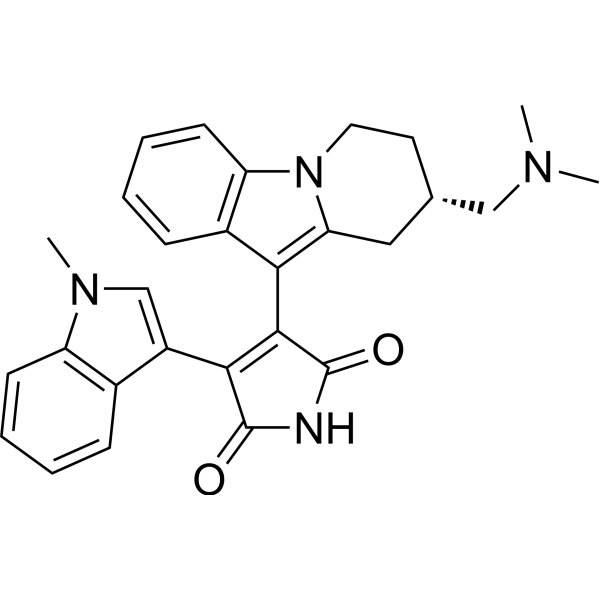
- HY-P5158
-
|
|
Adrenergic Receptor
|
Others
|
|
Conopeptide rho-TIA is a peptide derived from the venom contained in the predatory sea snail Conus tulipa, has highly selective and noncompetitive inhibitor at human α1B-Adrenergic Receptor. Conopeptide rho-TIA acts a competitive inhibitor at human α1A-Adrenergic Receptor and α1D-Adrenergic Receptor. Conopeptide rho-TIA binds to each subtype and may provide useful information for the development of novel α1-Adrenergic Receptor subtype-selective drugs .
|
-
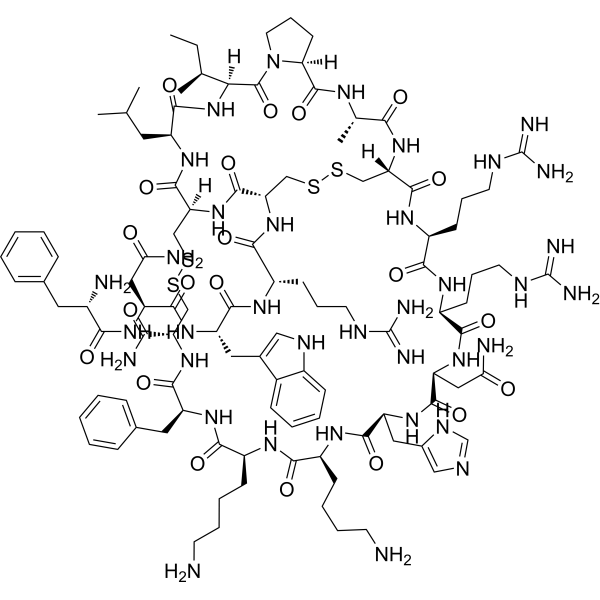
- HY-173162
-
|
|
P-glycoprotein
Potassium Channel
|
Cardiovascular Disease
Cancer
|
|
GPV0057 (Compound 5d) is a selective and potent P-glycoprotein (P-gp) inhibitor. GPV0057 is also a selective potassium channel Kir2.1 activator. GPV0057 competitively binds to the substrate-binding site of P-gp, inhibiting ATP-dependent drug efflux to reverse multidrug resistance in tumor cells. GPV0057 can also stabilizes the open state of Kir2.1 and promotes potassium ion influx. GPV0057 is promising for research of tumors with high P-gp expression, Kir2.1-deficient diseases such as heart failure and Andersen-Tawil Syndrome .
|
-

- HY-17499R
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-12 (Standard) is the analytical standard of EGFR-IN-12. This product is intended for research and analytical applications. EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFRL858R and EGFRL861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
|
-

- HY-117626
-
|
|
AAK1
Cyclin G-associated Kinase (GAK)
SARS-CoV
|
Infection
Neurological Disease
Inflammation/Immunology
|
|
LP-935509 is an orally active, potent, selective, ATP-competitive and brain-penetrant inhibitor of adaptor protein-2 associated kinase 1 (AAK1) with an IC50 of 3.3 nM and a Ki of 0.9 nM, respectively. LP-935509 is also a potent inhibitor of BIKE (IC50=14 nM) and a modest inhibitor of GAK (IC50=320 nM). LP-935509 shows antinociceptive activity. LP-935509 can be used for neuropathic pain and SARS-CoV-2 research .
|
-
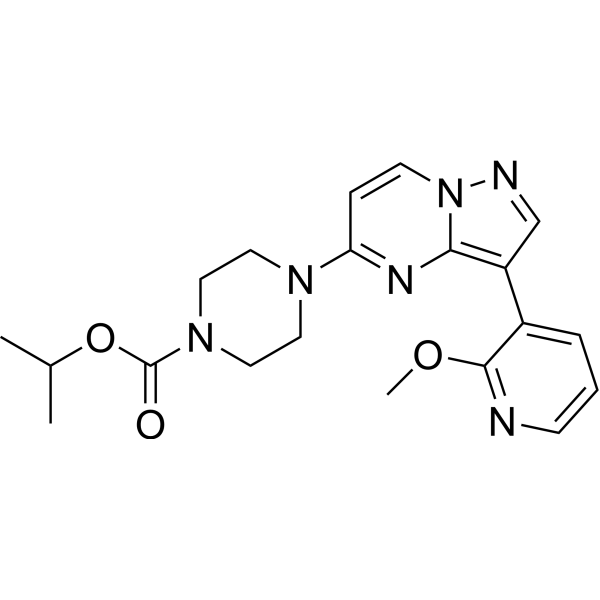
- HY-103566A
-
|
|
mGluR
EGFR
p38 MAPK
Apoptosis
|
Neurological Disease
|
|
LY456236 free base is a selective, non-competitive and orally active antagonist of glutamate receptor 1 (mGlu1), which can inhibit phosphatidylinositol hydrolysis with an IC50 of 0.145 μM. LY456236 free base can also inhibit EGFR, with an IC50 of 0.918 μM. LY456236 free base blocks cell proliferation by inhibiting the MAPK pathway, reversing the anti-apoptotic effect of DHPG (HY-12598A). LY456236 free base can be used in epilepsy research .
|
-

- HY-164036
-
|
|
Antibiotic
Bacterial
|
Infection
|
|
Lolamicin is an orally effective inhibitor that specifically targets the Gram-negative bacteria lipoprotein transport system LolCDE complex. It selectively inhibits the transmembrane transport of outer membrane lipoproteins by competitively binding to lipoprotein binding sites. Lolamicin destroys the integrity of the bacterial outer membrane, leading to cell death, and has both bactericidal and antibacterial activity. It has significant effects on multidrug-resistant Enterobacteriaceae pathogens (such as Escherichia coli and Klebsiella pneumoniae). Lolamicin can be used to inhibit the study of acute pneumonia, sepsis and other infections caused by Gram-negative bacteria .
|
-

- HY-110294
-
CM037
1 Publications Verification
A37
|
Aldehyde Dehydrogenase (ALDH)
|
Cancer
|
|
CM037 is a highly selective and competitive ALDH1A1 inhibitor (IC50=4.6 μM). CM037 blocks the catalytic activity of ALDH1A1, thereby inhibiting the activation of the downstream HIF-1α/VEGF pathway. CM037 is mainly used to study the ALDH1A1-mediated regulation of cancer stem cells (CSCs) and angiogenesis, especially in breast cancer, showing the potential to inhibit tumor angiogenesis and stem cell characteristics .
|
-
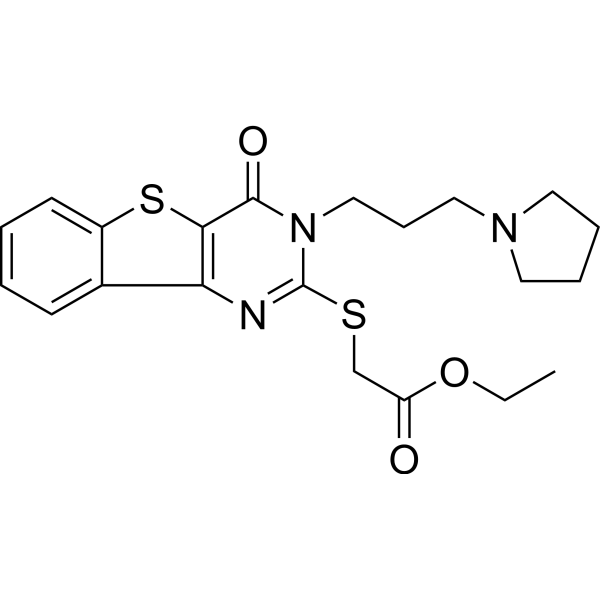
- HY-105349
-
|
|
Phosphodiesterase (PDE)
|
Neurological Disease
|
|
T-0156 is a potent and selective phosphodiesterase type 5 (PDE5) inhibitor. T-0156 specifically inhibits the hydrolysis of cyclic guanosine monophosphate (cGMP) by PDE5 in a competitive manner (IC50=0.23 nM). T-0156 inhibits PDE6 (IC50=56 nM) and has low potencies against PDE1, PDE2, PDE3, and PDE4 (IC50>10 μM). T-0156 enhances the nitric oxide (NO)/cGMP pathway .
|
-
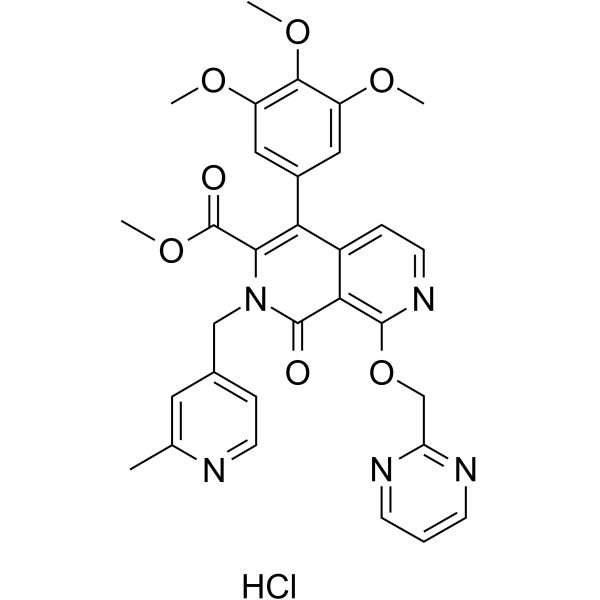
- HY-16749R
-
|
PLX-3397 (Standard)
|
c-Fms
c-Kit
Apoptosis
Reference Standards
|
Cancer
|
|
Pexidartinib (Standard) is the analytical standard of Pexidartinib. This product is intended for research and analytical applications. Pexidartinib (PLX-3397) is a potent, orally active, selective, and ATP-competitive colony stimulating factor 1 receptor (CSF1R or M-CSFR) and c-Kit inhibitor, with IC50s of 20 and 10 nM, respectively. Pexidartinib (PLX-3397) exhibits 10- to 100-fold selectivity for c-Kit and CSF1R over other related kinases. Pexidartinib (PLX-3397) induces cell apoptosis and has anti-tumor activity .
|
-

- HY-15346
-
|
BAY 80-6946
|
PI3K
Apoptosis
|
Cancer
|
|
Copanlisib (BAY 80-6946) is a potent, selective and ATP-competitive pan-class I PI3K inhibitor, with IC50s of 0.5 nM, 0.7 nM, 3.7 nM and 6.4 nM for PI3Kα, PI3Kδ, PI3Kβ and PI3Kγ, respectively. Copanlisib has more than 2,000-fold selectivity against other lipid and protein kinases, except for mTOR. Copanlisib has superior antitumor activity .
|
-
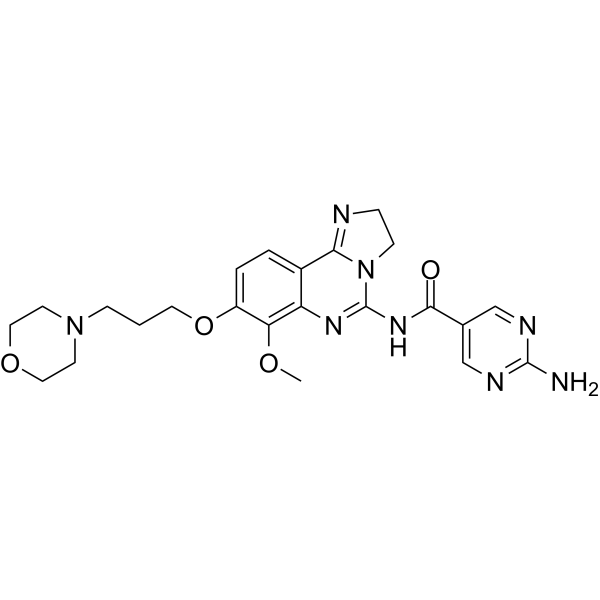
- HY-15346A
-
|
BAY 80-6946 dihydrochloride
|
PI3K
Apoptosis
|
Cancer
|
|
Copanlisib dihydrochloride (BAY 80-6946 dihydrochloride) is a potent, selective and ATP-competitive pan-class I PI3K inhibitor, with IC50s of 0.5 nM, 0.7 nM, 3.7 nM and 6.4 nM for PI3Kα, PI3Kδ, PI3Kβ and PI3Kγ, respectively. Copanlisib dihydrochloride has more than 2,000-fold selectivity against other lipid and protein kinases, except for mTOR. Copanlisib dihydrochloride has superior antitumor activity .
|
-
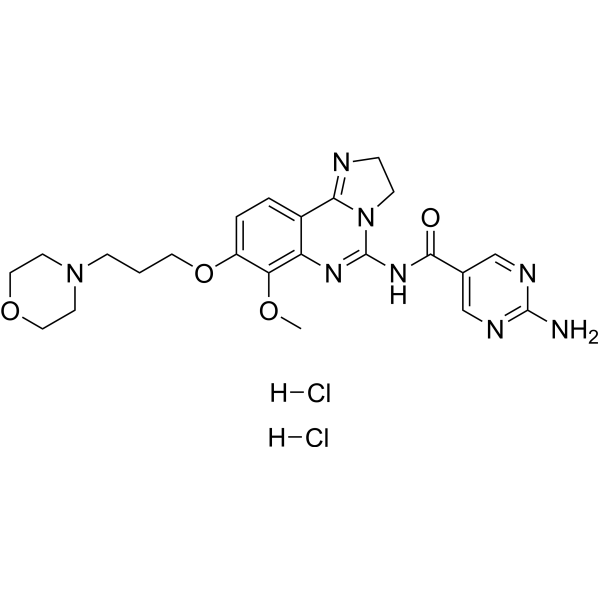
- HY-12983B
-
|
|
Drug Metabolite
DNA/RNA Synthesis
|
Infection
|
|
ALS-8112-TP is the 5'-triphosphate metabolite of ALS-8112 (HY-12983). ALS-8112-TP is a potent, selective and competitive respiratory syncytial virus (RSV) RNA polymerase inhibitor, with selectivity against polymerases from host or viruses unrelated to RSV such as hepatitis C virus (HCV). ALS-8112-TP can be efficiently recognized by the recombinant RSV polymerase complex, causing chain termination of RNA synthesis. ALS-8112-TP can be used for RSV-infection research .
|
-

- HY-19487
-
|
|
Bacterial
|
Infection
|
|
Ribocil is a selective inhibitor targeting the bacterial FMN riboswitch, regulating the bacterial riboflavin riboswitch. Ribocil competitively binds to the FMN binding site, mimicking the natural ligand FMN to induce conformational changes in the riboswitch, inhibiting ribB gene expression, reducing riboflavin synthesis, and thus inhibiting bacterial growth. Ribocil strongly inhibits GFP expression (EC50=0.3 μM). Ribocil exhibits in vivo antibacterial activity in a mouse model and can be used to study antibacterial drugs related to drug-resistant bacterial infections and bacterial riboflavin metabolic pathways[1][2].
|
-
- HY-124131
-
DS-437
1 Publications Verification
|
Histone Methyltransferase
|
Inflammation/Immunology
Cancer
|
|
DS-437 is a dual PRMT5/7 inhibitor (IC50s of PRMT5/7=6 μM). DS-437 is selective for PRMT5 and PRMT7 over 29 other human protein-, DNA-, and RNA-methyltransferases. DS-437 is a S-adenosylmethionine (SAM)-competitive inhibitor of PRMT5. DS-437 also inhibits DNMT3A and DNMT3B, with IC50s of 52 and 62 μM, respectively. DS-437 inhibits the methylation of FOXP3 .
|
-
- HY-10683
-
|
|
PI3K
mTOR
|
Cancer
|
|
PKI-402 is a selective, reversible, ATP-competitive inhibitor of PI3K, including PI3K-α mutants, and mTOR (IC50=2, 3, 7,14 and 16 nM for PI3Kα, mTOR, PI3Kβ, PI3Kδ and PI3Kγ).
|
-
- HY-181959
-
|
|
Eukaryotic Initiation Factor (eIF)
|
Cancer
|
|
APL-4098 is an orally active, selective, ATP-competitive GCN2 inhibitor with a Ki of 4.39 nM and a Kd of 2.9 nM. APL-4098 reduces the phosphorylation level of eIF2α and the expression level of ATF4. APL-4098 impairs mitochondrial function and exerts cytotoxic effects on primary acute myeloid leukemia cells. APL-4098 is applicable to research related to acute myeloid leukemia .
|
-

- HY-B0290R
-
|
ONO-1078 (Standard)
|
Reference Standards
Leukotriene Receptor
Endogenous Metabolite
|
Inflammation/Immunology
|
|
Pranlukast (Standard) is the analytical standard of Pranlukast. This product is intended for research and analytical applications. Pranlukast is a highly potent, selective and competitive antagonist of peptide leukotrienes. Pranlukast inhibits [ 3H]LTE4, [ 3H]LTD4, and [ 3H]LTC4 bindings to lung membranes with Kis of 0.63±0.11, 0.99±0.19, and 5640±680 nM, respectively.
|
-

- HY-10229R
-
|
AMG 706 Diphosphate (Standard)
|
Reference Standards
c-Kit
VEGFR
|
Cancer
|
|
Motesanib (Diphosphate) (Standard) is the analytical standard of Motesanib (Diphosphate). This product is intended for research and analytical applications. Motesanib Diphosphate (AMG 706 Diphosphate) is a potent ATP-competitive inhibitor of VEGFR1/2/3 with IC50s of 2 nM/3 nM/6 nM, respectively, and has similar activity against Kit, and is approximately 10-fold more selective for VEGFR than PDGFR and Ret.
|
-

- HY-139467
-
|
|
SHP2
PKC
|
Metabolic Disease
|
|
PF-04577806 is a potent, selective and ATP competitive PKC inhibitor. PF-04577806 shows potent inhibitory activity towards PKCα, PKCβI, PKCβII, PKCγ, and PKCθ with IC50s of 2.4 nM, 8.1 nM, 6.9 nM, 45.9 nM, and 29.5 nM, respectively. PF-04577806 can reverse retinal vascular leakage in diabetic rats .
|
-
- HY-178969
-
|
|
FAK
Pyk2
Apoptosis
|
Neurological Disease
Cancer
|
|
GZD-257 is a brain-penetrant, ATP-competitive FAK inhibitor (IC50 = 14.3 nM), performing 4.77-fold selectivity with FAK to Pyk2 (IC50 = 68.2 nM). GZD-257 can significantly induce apoptosis of U118MG cells and arrest the cell cycle at the G2/M phase. GZD-257 can be used for the study of Glioblastoma (GBM) .
|
-

- HY-B0290AR
-
|
ONO-1078 hemihydrate (Standard)
|
Leukotriene Receptor
Endogenous Metabolite
Reference Standards
|
Inflammation/Immunology
|
|
Pranlukast (hemihydrate) (Standard) is the analytical standard of Pranlukast (hemihydrate). This product is intended for research and analytical applications. Pranlukast hemihydrate is a highly potent, selective and competitive antagonist of peptide leukotrienes. Pranlukast inhibits [3H]LTE4, [3H]LTD4, and [3H]LTC4 bindings to lung membranes with Kis of 0.63±0.11, 0.99±0.19, and 5640±680 nM, respectively.
|
-

- HY-10228R
-
|
AMG 706 (Standard)
|
Reference Standards
c-Kit
VEGFR
|
Cancer
|
|
Motesanib (Standard) is the analytical standard of Motesanib. This product is intended for research and analytical applications. Motesanib (AMG 706) is a potent ATP-competitive inhibitor of?VEGFR1/2/3?with?IC50s?of 2 nM/3 nM/6 nM, respectively, and has similar activity against Kit, and is appr 10-fold more selective for VEGFR than PDGFR and Ret.
|
-

- HY-19340
-
|
(E)-2,3',4,5'-tetramethoxystilbene
|
Cytochrome P450
|
Cancer
|
|
TMS ((E)-2,3',4,5'-tetramethoxystilbene) is a selective and competitive CYP1B1 inhibitor with an IC50 of 6 nM and a Ki value of 3 nM. TMS shows a lesser extent inhibitory effect on CYP1A1 (IC50=300 nM) and CYP1A2 (IC50=3.1 μM). TMS is a methylated derivative of resveratrol and has anti-cancer activity .
|
-
- HY-163457
-
|
|
Aminoacyl-tRNA Synthetase
Parasite
|
Infection
|
|
Antileishmanial agent-27 (compound 7j) is a benzothiazolo-coumarin derivative. Antileishmanial agent-27 is a competitive inhibitor of arginyl-tRNA synthetases (ArgRSs). Antileishmanial agent-27 shows selectivity toward ArgRS of Leishmania donovani (LdArgRS) than its human counterpart (HsArgRS), with IC50 values of 1.2 and 19 μM, respectively. Antileishmanial agent-27 possesses high pharmacokinetic properties .
|
-
- HY-181094
-
|
|
Glycosidase
|
Metabolic Disease
|
|
α-Glucosidase-IN-110 is a selective and orally active competitive inhibitor of α-Glucosidase with an IC50 of 7.09 μM and a Ki of 6.9 μM. α-Glucosidase-IN-110 can reduce fasting blood glucose levels, improve glucose tolerance, and restore the histomorphology of liver and pancreatic tissues in diabetic rat models. α-Glucosidase-IN-110 can be used for the research of diabetes .
|
-

- HY-103374B
-
|
(±)-Eseroline phenylcarbamate
|
Cholinesterase (ChE)
Amyloid-β
|
Neurological Disease
|
|
(±)Phenserine ((±)-Eseroline phenylcarbamate) is the racemic form of Phenserine (HY-103374). Phenserine is a derivative of Physostigmine (HY-N6608) and is an effective, non-competitive, long-acting and selective AChE inhibitor. Phenserine can reduce the formation of β-amyloid precursor protein (APP) and β-amyloid peptide (Aβ). Phenserine can improve cognitive ability and slow down the progression of Alzheimer's disease .
|
-

- HY-125102
-
|
|
IGF-1R
|
Cancer
|
|
AZ12253801 is an ATP-competitive IGF-1R tyrosine kinase inhibitor that shows ∼10-fold selectivity over the insulin receptor. AZ12253801 inhibits IGF-1R–driven proliferation in 3T3 mouse fibroblasts (transfected with human IGF-1R) with an IC50 of 17 nmol/L. The IC50 for epidermal growth factor receptor (EGFR)–driven proliferation is 440 nmol/L. Anti-tumor activity.
|
-
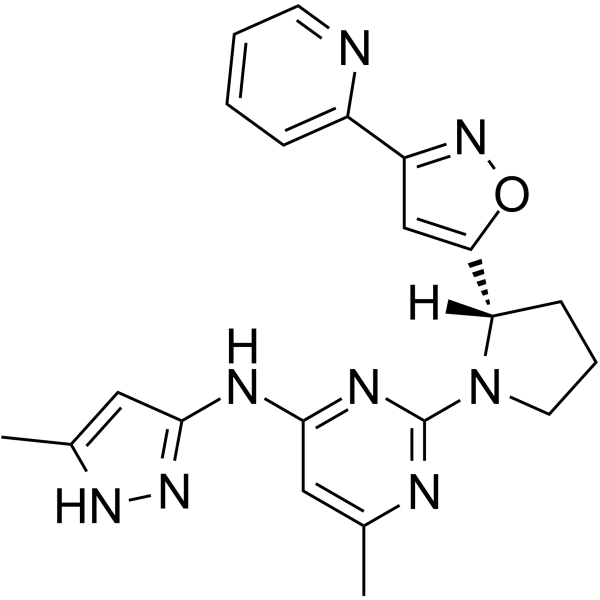
- HY-160735
-
|
|
Herbicide
Glutaminase
|
Others
|
|
L-Phosphinothricin is a glutamine synthetase inhibitor and a non-selective herbicide. L-Phosphinothricin acts as a competitive inhibitor, induces toxic ammonium ion accumulation in plants and bacteria, and indirectly blocks the photosynthesis process. L-Phosphinothricin exerts herbicidal activity against both monocotyledonous and dicotyledonous plant species, is primarily absorbed through plant leaves, exhibits limited translocation in plants, and undergoes rapid degradation by soil microorganisms with no root uptake. L-Phosphinothricin can be used for research on weed control in agricultural and non-crop scenarios .
|
-

- HY-101903A
-
|
|
FABP
|
Cardiovascular Disease
Metabolic Disease
|
|
BMS-309403 sodium is a potent, orally active, and selective adipocyte fatty acid binding protein (also known as FABP4, aP2) inhibitor, with Kis of <2, 250, and 350 nM for FABP4, FABP3, and FABP5, respectively. BMS-309403 sodium interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. BMS-309403 sodium improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells .
|
-
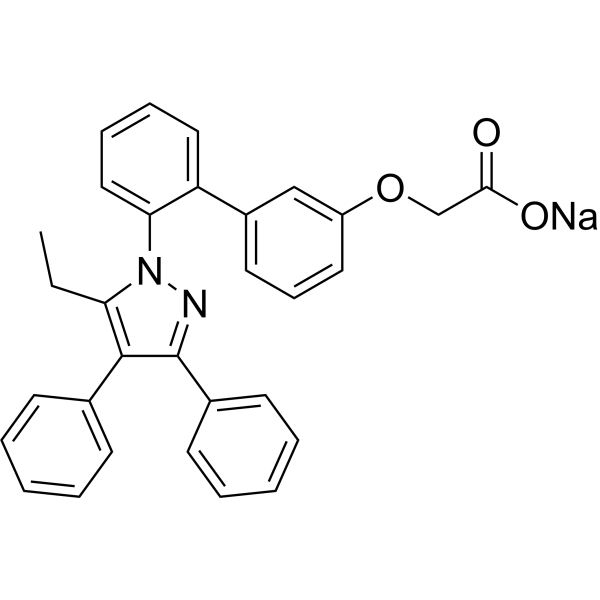
- HY-17623
-
|
CJ-12420; RQ-00000004
|
Proton Pump
Potassium Channel
Na+/K+ ATPase
|
Inflammation/Immunology
|
Tegoprazan (CJ-12420), a potassium-competitive acid blocker, is a reversible, orally active and highly selective inhibitor of gastric H +/K +-ATPase. Tegoprazan inhibits gastric acid secretion and motility against porcine, canine and human H +/K +-ATPase with IC50 values ranging from 0.29-0.52 μM in vitro. Tegoprazan significantly improves colitis and enhances the intestinal epithelial barrier function in mice. Tegoprazan is promising for research of Inflammatory bowel, gastric acid-related, motilityimpaired diseases .
|
-
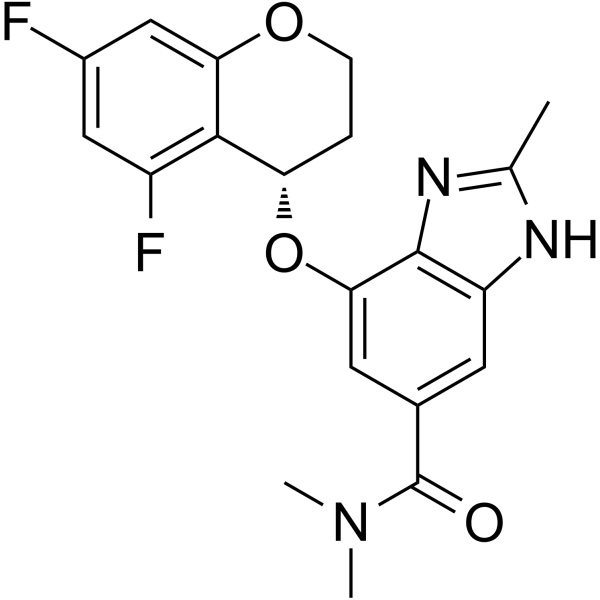
- HY-101903
-
|
|
FABP
|
Cardiovascular Disease
Metabolic Disease
|
|
BMS-309403 is a potent, orally active and selective adipocyte fatty acid binding protein (also known as FABP4, aP2) inhibitor with Kis of <2, 250, and 350 nM for FABP4, FABP3, and FABP5, respectively. BMS-309403 interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. BMS-309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells .
|
-
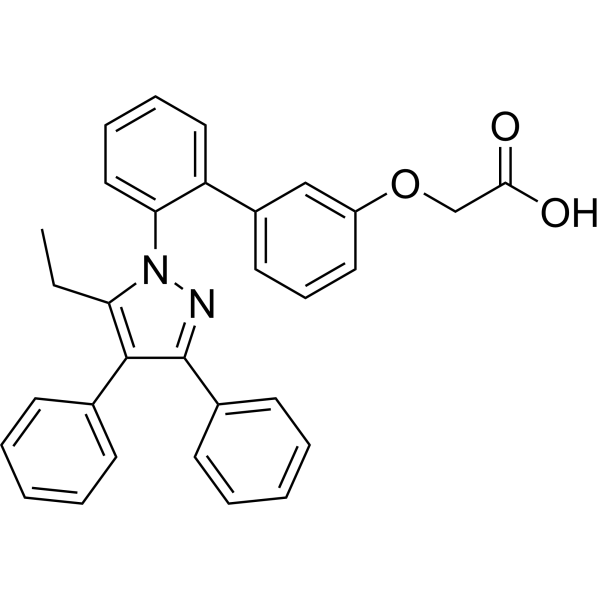
- HY-18174H
-
|
LY2606368 lactate
|
Checkpoint Kinase (Chk)
Apoptosis
DNA/RNA Synthesis
|
Cancer
|
|
Prexasertib lactate (LY2606368 lactate) is the lactate form of Prexasertib (HY-18174). Prexasertib lactate is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib lactate inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib lactate causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib lactate shows potent anti-tumor activity .
|
-

- HY-108907
-
|
|
Casein Kinase
|
Cancer
|
|
SR-1277 is a potent, selective and ATP competitive CK1δ/ε inhibitor, with IC50s of 49 nM and 260 nM, respectively. SR-1277 also inhibits FLT3, CDK4/cyclin D1, CDK6/cyclin D3 and CDK9/cyclin K, with IC50s of 305 nM, 1340 nM, 311 nM and 109 nM, respectively. SR-1277 can be used for the research of cancer .
|
-
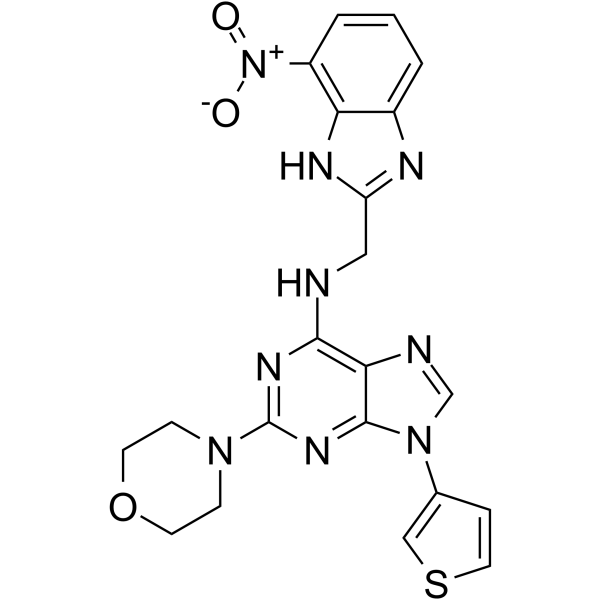
- HY-174310
-
|
|
Glycosidase
|
Metabolic Disease
|
|
α-Glucosidase-IN-91 (Compound 15j) is a competitive and potent inhibitor for α-Glucosidase (IC50 = 6.6 μM). α-Glucosidase-IN-91 has a potent binding affinity towards α-Glucosidase. α-Glucosidase-IN-91 exhibits high stability, interacting and inhibiting α-Glucosidase selectively. α-Glucosidase-IN-91 can be studied in research for type 2 diabetes and blood glucose control .
|
-

- HY-12964
-
|
|
TAM Receptor
|
Cancer
|
|
SGI-7079 is a selective, ATP-competitive, orally active inhibitor of the receptor tyrosine kinase Axl. SGI-7079 blocks Axl-mediated signaling pathways such as NF-κB activation and MMP-9 expression, thereby inhibiting tumor cell proliferation, migration and invasion. SGI-7079 is mainly used in the research of malignant tumors such as inflammatory breast cancer and bladder cancer, as well as in combination with immunization (used in combination with PD-1 therapy)[1][2][3].
|
-
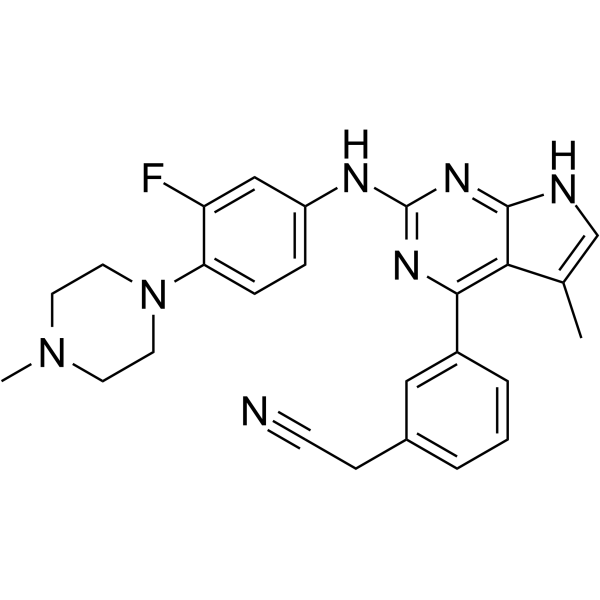
- HY-B0442B
-
|
|
Endogenous Metabolite
Phosphodiesterase (PDE)
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
|
|
Vardenafil hydrochloride trihydrate is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride trihydrate shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride trihydrate competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride trihydrate can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-
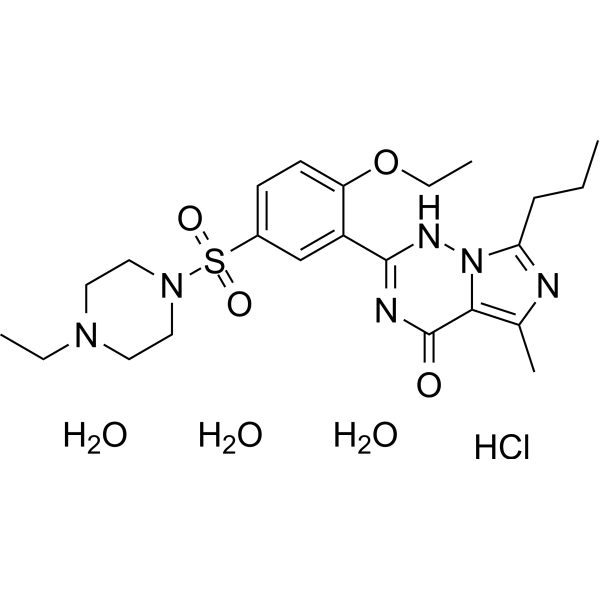
- HY-13011R
-
|
CH5424802 (Standard); RO5424802 (Standard); RG7853 (Standard)
|
Reference Standards
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Alectinib (CH5424802; RO5424802; RG7853) (Standard) is the analytical standard of Alectinib. This product is intended for research and analytical applications. Alectinib (CH5424802) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-

- HY-107794R
-
|
Disodium clodronate tetrahydrate (Standard)
|
Others
Reference Standards
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
Clodronate (disodium tetrahydrate) (Standard) is the analytical standard of Clodronate (disodium tetrahydrate). This product is intended for research and analytical applications. Clodronate disodium tetrahydrate (Disodium clodronate tetrahydrate) is first-generation bisphosphonate, with anti-osteoporotic, anti-inflammatory and analgesic effects. Clodronate disodium tetrahydrate is a selective, potent, reversible and Cl-competitive vesicular nucleotide transporter (VNUT) inhibitor, with an IC50 of 15.6 nM. Clodronate disodium tetrahydrate inhibits vesicular ATP release from neurons and reduces chronic neuropathic and inflammatory pain .
|
-

- HY-103430
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 hydrobromide is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for the research of parkinson’s disease and nicotine craving alleviation .
|
-
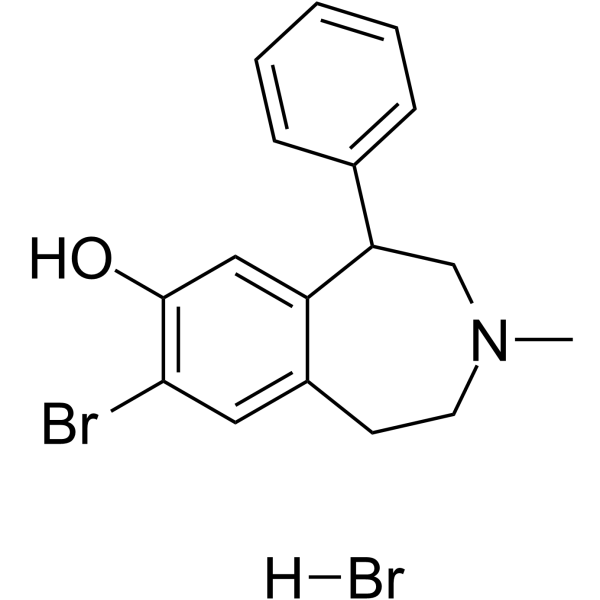
- HY-182764A
-
|
|
CDK
|
Cancer
|
|
CDK11-IN-1 hydrochloride is a potent, highly selective, and orally active CDK11 inhibitor with an IC50 of 4 nM, showing 32.5-fold and 2700-fold selectivity over CDK7 and CDK9, respectively. CDK11-IN-1 hydrochloride binds competitively to the ATP-binding pocket of CDK11 and forms a hydrogen bond with the hinge region residue Val163. It inhibits tumor cell proliferation and exhibits antitumor activity in lung cancer xenograft models. CDK11-IN-1 hydrochloride can be used for studies on the pathophysiology of CDK11-mediated tumors, as well as research on malignant tumors such as lung cancer .
|
-

- HY-110137R
-
|
DB75 dihydrochloride (Standard); NSC 305831 dihydrochloride (Standard)
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
Reference Standards
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine (dihydrochloride) (Standard) is the analytical standard of Furamidine (dihydrochloride). This product is intended for research and analytical applications. Furamidine dihydrochloride (DB75 dihydrochloride) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine dihydrochloride is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 μM, 283 μM, and >400 μM, respectively). Furamidine dihydrochloride is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine dihydrochloride is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine dihydrochloride is also an antiparasite agent .
|
-

- HY-10014R
-
|
|
CDK
Reference Standards
GSK-3
Apoptosis
|
Cancer
|
|
R547 (Standard) is the analytical standard of R547 (HY-10014). This product is intended for research and analytical applications. R547 is a potent, selective and orally active ATP-competitive CDK inhibitor, with Kis of 2 nM, 3 nM and 1 nM for CDK1/cyclin B, CDK2/cyclin E and CDK4/cyclin D1, respectively .
|
-

- HY-76474A
-
|
|
Apoptosis
Syk
|
Inflammation/Immunology
Cancer
|
|
BAY 61-3606 hydrochloride is an orally available, ATP-competitive, reversible and highly selective Syk inhibitor with a Ki of 7.5 nM and an IC50 of 10 nM . BAY 61-3606 hydrochloride reduces ERK1/2 and Akt phosphorylation in neuroblastoma cell. BAY 61-3606 hydrochloride induces a large decrease of Syk phosphorylation in K-rn cell lysates. BAY 61-3606 hydrochloride sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells.
|
-

- HY-105327R
-
|
|
Reference Standards
Cholinesterase (ChE)
|
Neurological Disease
|
|
P11149 (Standard) is the analytical standard of P11149 (HY-105327). This product is intended for research and analytical applications. P11149 is a competitive, BBB-penetarated weakly, orally active and selective inhibitor of AChE. P11149 exhibits an IC50 of 1.3 μM for rat BChE/AChE. P11149, a Galanthamine derivative, demonstrates central cholinergic activity, behavioral efficacy and safety. P11149 is used in the study for Alzheimer's disease .
|
-

- HY-15844
-
-
- HY-117583
-
|
|
HDAC
Histone Methyltransferase
|
Neurological Disease
|
|
cis-BG47 is an cis-isomer of BG47, BG47 is a prototypical histone deacetylases HDAC1 and HDAC2 selective, optoepigenetic probe. BG47 can bind to and competitively inhibits the deacetylase activity of HDAC targets upon a light-induced trans-to-cis isomerization, and increases Histone Methyltransferase H3K9 acetylation. cis-BG47 can be used for neurological disease research .
|
-
- HY-10524
-
|
|
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
GSK1904529A is a potent, selective, orally active, and ATP-competitive inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR), with IC50s of 27 and 25 nM, respectively. GSK1904529A shows poor activity (IC50>1 μM) in 45 other serine/threonine and tyrosine kinases. GSK1904529A exhibits anti-tumor activity .
|
-
- HY-125957
-
|
|
PKA
Casein Kinase
CaMK
PKC
|
Others
|
|
A-3 hydrochloride is a potent, cell-permeable, reversible, ATP-competitive non-selective antagonist of various kinases. It against PKA (Ki=4.3 µM), casein kinase II (Ki=5.1 µM) and myosin light chain kinase (MLCK) (Ki=7.4 µM). A-3 hydrochloride also inhibits PKC and casein kinase I with Ki values of 47 µM and 80 µM, respectively .
|
-
- HY-177785
-
|
|
Molecular Glues
Indoleamine 2,3-Dioxygenase (IDO)
|
Infection
Neurological Disease
Cancer
|
|
iDeg-6 is a selective molecular glue degrader that targets IDO1 with a DC50 of 6.5 nM. iDeg-6 can competitively bind to the heme binding site of apo-IDO1, preventing heme binding and inhibiting the enzymatic reaction that converts tryptophan into kynurenine by IDO1 (IC50 = 1.6 μM). iDeg-6 can be used for the researches of cancer, infection and neurological disease, such as melanoma .
|
-

- HY-120214
-
|
|
Syk
RET
|
Inflammation/Immunology
|
|
TAS05567 is a potent, highly selective, ATP-competitive and orally active Syk inhibitor with an IC50 of 0.37 nM. In a panel of 192 kinases, TAS05567 only shows >70% inhibition of Syk and 4 other kinases (FLT3, JAK2, KDR and RET with IC50s of 10 nM, 4.8 nM, 600 nM and 29 nM, respectively). TAS05567 can be used for humoral immune-mediated inflammatory conditions such as autoimmune and allergic diseases .
|
-
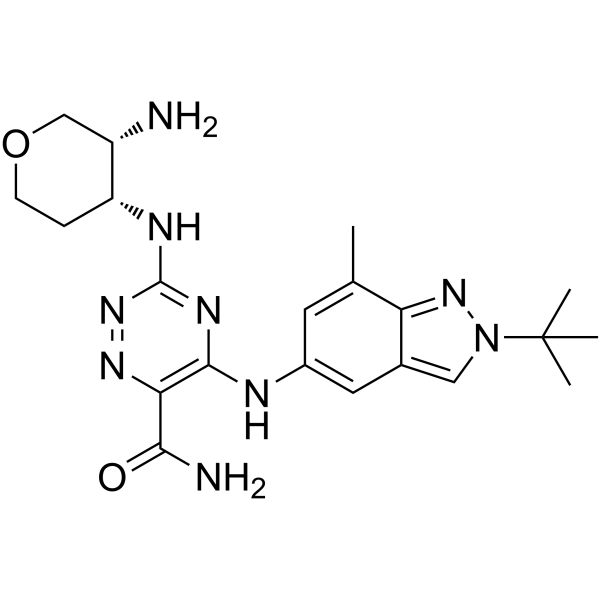
- HY-10262R
-
|
|
Reference Standards
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
BMS-536924 (Standard) is the analytical standard of BMS-536924 (HY-10262). This product is intended for research and analytical applications. BMS-536924 is an orally active, competitive and selective insulin-like growth factor receptor (IGF-1R) kinase and insulin receptor (IR) inhibitor with IC50s of 100 nM and 73 nM, respectively. BMS-536924 has anti-cancer activity .
|
-

- HY-W040220
-
|
|
Bacterial
|
Infection
|
|
N-(3-Hydroxyoctanoyl)-DL-homoserine lactone (Compound 40) is a competitive inhibitor of the quorum sensing receptor LuxR with an IC50 value of 4 μM. N-(3-Hydroxyoctanoyl)-DL-homoserine lactone shows selective inhibition toward quorum sensing systems in Gram-negative bacteria like Pseudomonas aeruginosa. N-(3-Hydroxyoctanoyl)-DL-homoserine lactone is promising for research of bacterial infections .
|
-

- HY-W011266
-
|
|
PDGFR
|
Cancer
|
|
JNJ-10198409 is a relatively selective, orally active, and ATP competitive PDGF-RTK (platelet-derived growth factor receptor tyrosine kinase) inhibitor (IC50=2 nM). It is a dual-mechanism, antiangiogenic, and tumor cell antiproliferative agent. JNJ-10198409 has good activity against PDGFR-β kinase (IC50=4.2 nM) and PDGFR-α kinase (IC50=45 nM) .
|
-
- HY-W011109
-
CKI-7
1 Publications Verification
|
Casein Kinase
CDK
SGK
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
CKI-7 is a potent and ATP-competitive casein kinase 1 (CK1) inhibitor with an IC50 of 6 μM and a Ki of 8.5 μM. CKI-7 is a selective Cdc7 kinase inhibitor. CKI-7 also inhibits SGK, ribosomal S6 kinase-1 (S6K1) and mitogen- and stress-activated protein kinase-1 (MSK1). CKI-7 has a much weaker effect on casein kinase II and other protein kinases .
|
-
- HY-13404A
-
|
INC280 dihydrochloride; INCB28060 dihydrochloride
|
c-Met/HGFR
Apoptosis
|
Cancer
|
|
Capmatinib (INC280; INCB28060) dihydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-
- HY-111033
-
|
|
MEK
ERK
Apoptosis
p38 MAPK
CDK
PARP
|
Inflammation/Immunology
Cancer
|
|
RO5068760 is a potent, orally active and selective non-ATP-competitive MEK1/2 inhibitor with an IC50 of 0.025 μM for MEK1. RO5068760 significantly inhibits MAPK pathway activity, thereby inducing G1 cell cycle arrest and apoptosis to inhibit cancer cell growth. RO5068760 exhibits significant efficacy in a broad spectrum of tumors with aberrant MAPK pathway activation. RO5068760 can be used for melanoma, colorectal cancer, non-small cell lung cancer (NSCLC), and pancreatic cancer research .
|
-

- HY-13404C
-
|
INC280 dihydrochloride hydrate; INCB-28060 dihydrochloride hydrate
|
c-Met/HGFR
Apoptosis
|
Cancer
|
|
Capmatinib (INC280; INCB28060) dihydrochloride hydrate is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride hydrate can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride hydrate potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride hydrate is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-
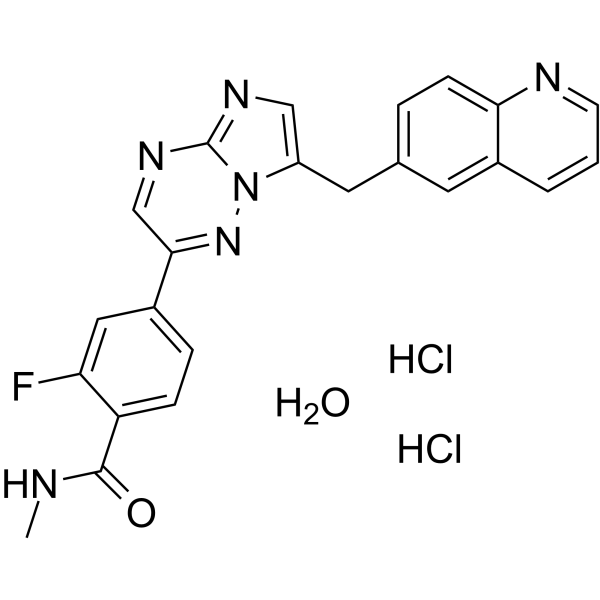
- HY-B1036
-
|
|
Parasite
|
Infection
|
|
Decoquinate is an orally active, selective inhibitor of the mitochondrial bc1 complex, targeting Eimeria spp. sporozoites and first generation schizonts, and Plasmodium spp. Decoquinate inhibits electron transfer by competitively binding to the mitochondrial cytochrome b system, blocking the parasite's energy metabolism, thereby inhibiting its development and reproduction. Decoquinate has significant anticoccidial activity, preventing intestinal damage and improving host growth performance, and also has inhibitory effects on the liver and blood stages of Plasmodium. Decoquinate is mainly used in veterinary research to prevent and treat coccidiosis in ruminants and poultry .
|
-
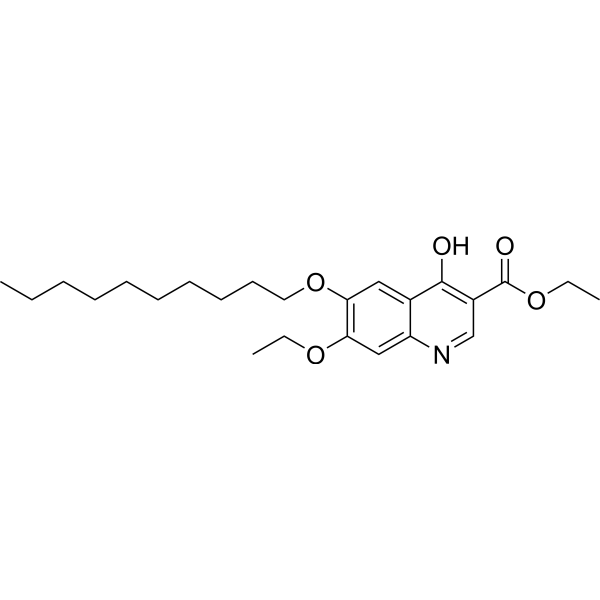
- HY-13404
-
|
INC280; INCB28060
|
c-Met/HGFR
Apoptosis
|
Cancer
|
|
Capmatinib (INC280; INCB28060) is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-
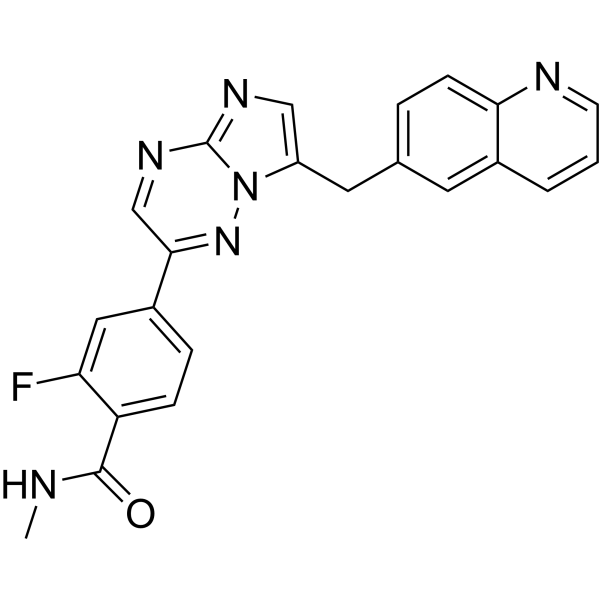
- HY-107427R
-
|
|
Reference Standards
MAPKAPK2 (MK2)
p38 MAPK
|
Inflammation/Immunology
|
|
PF-3644022 (Standard) is the analytical standard of PF-3644022 (HY-107427). This product is intended for research and analytical applications. PF-3644022 is a potent, selective, orally active and ATP-competitive MAPKAPK2 (MK2) inhibitor with an IC50 of 5.2 nM and a Ki of 3 nM. PF-3644022 also inhibits MK3 and p38 regulated/activated Kinase (PRAK) with IC50s of 53 nM and 5.0 nM, respectively. PF-3644022 potently inhibits TNFα production and has anti-inflammatory effect .
|
-

- HY-13404B
-
|
INC280 hydrochloride; INCB-28060 hydrochloride
|
c-Met/HGFR
Apoptosis
|
Cancer
|
|
Capmatinib (INC280; INCB28060) hydrochloride is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib hydrochloride can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib hydrochloride potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib hydrochloride is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-
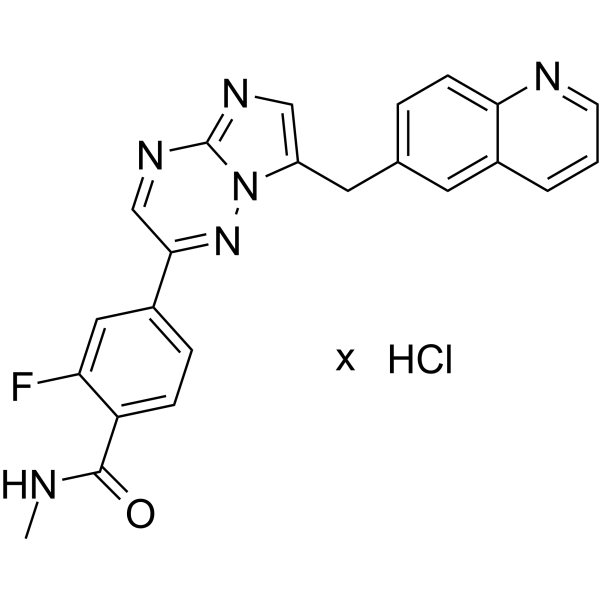
- HY-P1137
-
10Panx
1 Publications Verification
|
Gap Junction Protein
|
Others
|
|
10Panx is a competitive inhibitor of selective Pannexin 1 (PANX1) channels. 10Panx blocks the opening of PANX1 channels, inhibits ATP release and downstream P2X7 receptor-mediated signaling pathways, thereby reducing cell death and inflammatory responses. 10Panx can be used in the study of diseases such as neuropathic pain, inflammatory bowel disease, and Clostridioides difficile infection. 10Panx can effectively reduce mechanical hyperalgesia and enhanced C-reflexes, and inhibit the expression of pro-inflammatory factors such as IL-6[1][2][3].
|
-
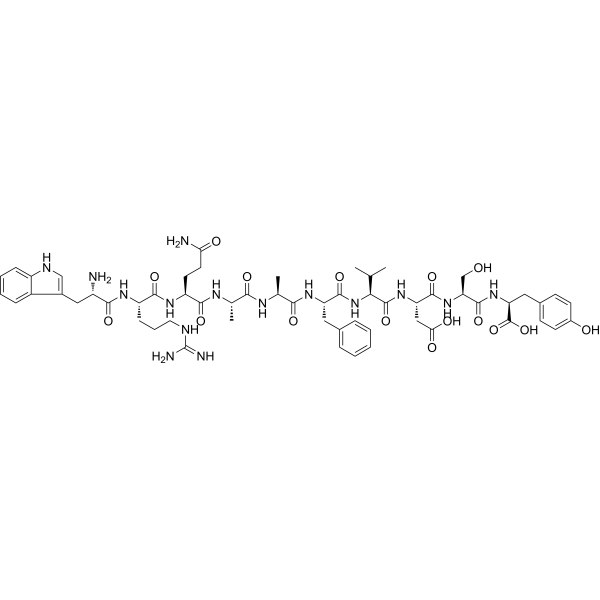
- HY-129980A
-
|
|
|
Neurological Disease
Cancer
|
|
NPC-15437 is a selective PKC inhibitor with an IC50 of 19 µM. NPC-15437 competitively inhibits phorbol ester- (Ki of 5 µM) and phosphatidylserine-induced (Ki of 12 µM) PKC activity. NPC-15437 does not inhibits cAMP-dependent or calcium/calmodulin-dependent protein kinases. NPC-15437 augments TRAIL-induced cell death in non-small cell lung cancer and medulloblastoma cells. NPC-15437 can be used for the research of non-small cell lung cancer, medulloblastoma, and neurological disease .
|
-

- HY-104066R
-
|
Xiliertinib (Standard); HMPL-309 (Standard)
|
Reference Standards
EGFR
|
Cancer
|
|
Theliatinib (Standard) is the analytical standard of Theliatinib (HY-104066). This product is intended for research and analytical applications. Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-

- HY-10580R
-
|
6-Bromoindirubin-3'-oxime (Standard); BIO (Standard); MLS 2052 (Standard)
|
Reference Standards
GSK-3
CDK
Apoptosis
|
Cancer
|
|
GSK 3 Inhibitor IX (Standard) is the analytical standard of GSK 3 Inhibitor IX (HY-10580). This product is intended for research and analytical applications. GSK 3 Inhibitor IX (6-Bromoindirubin-3'-oxime; BIO) is a potent, selective, reversible and ATP-competitive inhibitor of GSK-3α/β and CDK1-cyclinB complex with IC50s of 5 nM/320 nM/80 nM for (GSK-3α/β)/CDK1/CDK5, respectively.
|
-

- HY-118835
-
|
|
Serotonin Transporter
|
Neurological Disease
|
|
Zimelidine is an orally active selective serotonin reuptake inhibitor. Zimelidine competitively inhibits central 5-HT uptake and desensitizes 5-HT autoreceptors in dorsal raphe nucleus. Zimelidine time-dependently modulates 5-HT neuronal firing and hippocampal CA3 responses. Zimelidine strengthens central serotonergic neurotransmission and produces related behavioral changes. Zimelidine exerts anxiolytic, analgesic, feeding-suppressive and tolerance-attenuating effects. Zimelidine is used for the study of depressive disorders and analgesic tolerance .
|
-
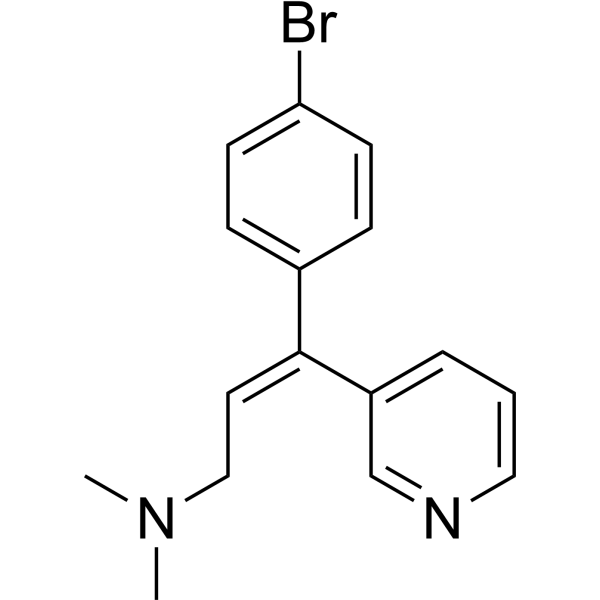
- HY-180407
-
|
|
Bacterial
|
Infection
|
|
UGM-IN-1 (compound 107) is a selective competitive inhibitor of Mycobacterium tuberculosis UDP-Galp mutase (UGM), encoded by Rv3809c. UGM-IN-1 inhibits the conversion of UDP-galactopyranose (UDP-Galp) to UDP-galactofuranose (UDP-Galf), thereby blocking the synthesis of key components of the mycobacterial cell wall, including mycolic acid-arabinogalactan (mAG) and liparabinomannan (LAM), leading to anti-mycobacterial activity against Mycobacterium tuberculosis. UGM-IN-1 is useful for research on tuberculosis, including drug-resistant tuberculosis .
|
-

- HY-101523R
-
|
|
CDK
Reference Standards
|
Cancer
|
|
Cdc7-IN-1 (Standard) is the analytical standard of Cdc7-IN-1 (HY-101523). This product is intended for research and analytical applications. Cdc7-IN-1 (Compound 13) is a highly potent, selective and ATP competitive inhibitor of Cdc7 kinase, with an IC50 value of 0.6 nM at 1 mM ATP and with slow off-rate characteristics. Cdc7-IN-1 potently inhibits Cdc7 activity in cancer cells, and effectively induces cell death .
|
-

- HY-19989A
-
|
L-660711 sodium
|
P-glycoprotein
LPL Receptor
Leukotriene Receptor
|
Inflammation/Immunology
|
|
MK-571 (L-660711) sodium is an orally active, potent and selective competitive leukotriene D4 (LTD4) receptor antagonist, with Ki values of 0.22 and 2.1 nM in guinea pig and human lung membranes, respectively. MK-571 sodium is also a inhibitor of multidrug resistance-associated protein MRP4 (ABCC4) and ABCC1 (MRP1). MK-571 sodium inhibits constitutive and antigen-stimulated S1P (sphingosine-1-phosphate) release .
|
-
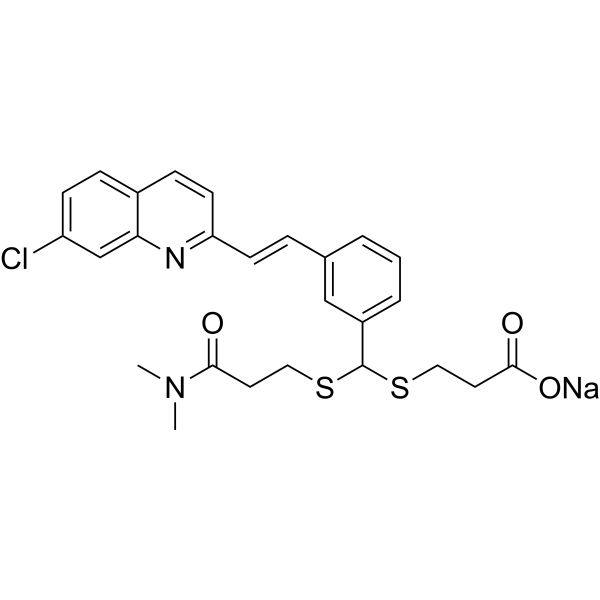
- HY-112654
-
|
|
Eukaryotic Initiation Factor (eIF)
Glutathione Peroxidase
|
Cardiovascular Disease
Metabolic Disease
Cancer
|
|
GCN2iB is an ATP-competitive, selective GCN2 inhibitor with an IC50 of 2.4 nM. GCN2iB inhibits the activation of the GCN2 pathway and upregulates GPX4. GCN2iB enhances the anticancer effect of ASNase against acute lymphoblastic leukemia. GCN2iB increases left ventricular ejection fraction, while reducing fasting blood glucose and myocardial fibrosis. GCN2iB can be used in research related to acute lymphoblastic leukemia, acute myeloid leukemia and diabetic cardiomyopathy .
|
-
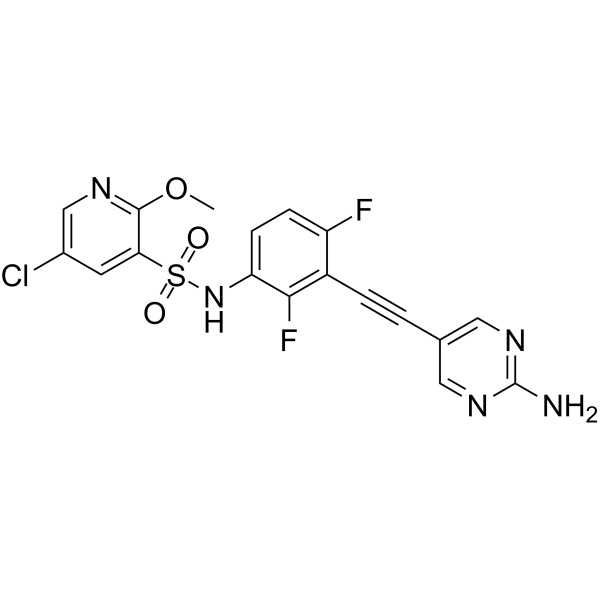
- HY-162642
-
|
|
Bcl-2 Family
|
Cancer
|
|
Bfl-1-IN-3 (Compound 56) is a selective, competitive inhibitor for Bfl-1 on BID binding site with Ki of 105 nM. Bfl-1-IN-3 inhibits the proliferation of cell pfeiffer and MV4-11, with IC50 of 6.92 μM and 12.6 μM. Bfl-1-IN-3 induces apoptosis in pfeiffer cells. Bfl-1-IN-3 overcomes Venetoclax (HY-15531) resistance at the cellular level, and shows synergistically enhanced anti-tumor activity with Venetoclax .
|
-

- HY-110023
-
|
|
Serotonin Transporter
|
Neurological Disease
|
|
Zimelidine dihydrochloride is an orally active selective serotonin reuptake inhibitor. Zimelidine dihydrochloride competitively inhibits central 5-HT uptake and desensitizes 5-HT autoreceptors in dorsal raphe nucleus. Zimelidine dihydrochloride time-dependently modulates 5-HT neuronal firing and hippocampal CA3 responses. Zimelidine dihydrochloride strengthens central serotonergic neurotransmission and produces related behavioral changes. Zimelidine dihydrochloride exerts anxiolytic, analgesic, feeding-suppressive and tolerance-attenuating effects. Zimelidine dihydrochloride is used for the study of depressive disorders and analgesic tolerance .
|
-
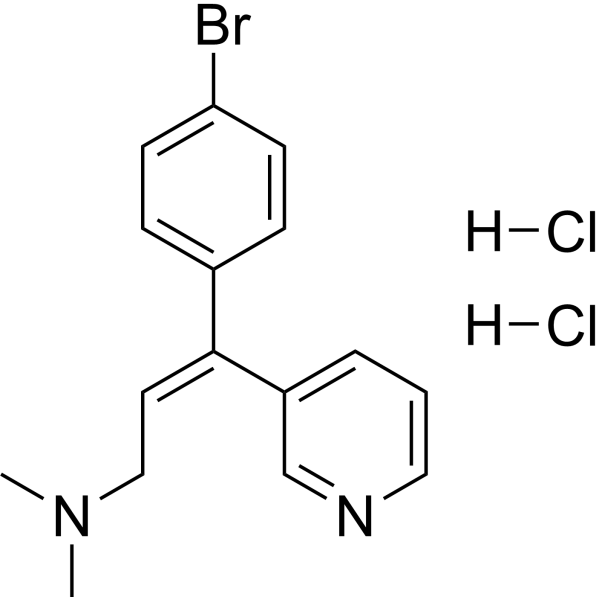
- HY-112654A
-
|
|
Eukaryotic Initiation Factor (eIF)
Glutathione Peroxidase
|
Cardiovascular Disease
Metabolic Disease
Cancer
|
|
GCN2iB acetate is an ATP-competitive, selective GCN2 inhibitor with an IC50 of 2.4 nM. GCN2iB acetate inhibits the activation of the GCN2 pathway and upregulates GPX4. GCN2iB acetate enhances the anticancer effect of ASNase against acute lymphoblastic leukemia. GCN2iB acetate increases left ventricular ejection fraction, while reducing fasting blood glucose and myocardial fibrosis. GCN2iB acetate can be used in research related to acute lymphoblastic leukemia, acute myeloid leukemia and diabetic cardiomyopathy .
|
-

- HY-100201R
-
|
|
Histone Methyltransferase
Reference Standards
|
Cancer
|
|
A-196 (Standard) is the analytical standard of A-196 (HY-100201). This product is intended for research and analytical applications. A-196 is a potent and selective inhibitor of SUV420H1 and SUV420H2 with IC50 values of 25 nM and 144 nM, respectively. A-196 inhibits SUV4-20 biochemically in a substrate-competitive manner. A-196 represents a first-in-class chemical probe of SUV4-20 to investigate the role of histone methyltransferases in genomic integrity .
|
-

- HY-181937
-
|
|
Monoamine Oxidase
Reactive Oxygen Species (ROS)
NO Synthase
Interleukin Related
|
Neurological Disease
|
|
Multi-target kinase-IN-10 (Compound 6l) is an orally active, blood-brain barrier-permeable, selective, reversible, and competitive MAO-B inhibitor with an IC50 of 0.0053 μM. Multi-target kinase-IN-10 competes with substrates for binding to the active site of MAO-B, chelates Cu 2+ ions, inhibits Cu 2+-induced ROS production, and reduces the release of NO, TNF-α, and IL-1β. Multi-target kinase-IN-10 ameliorates Parkinson's disease .
|
-

- HY-18174S
-
|
LY2606368-d4
|
Isotope-Labeled Compounds
Checkpoint Kinase (Chk)
Apoptosis
|
Cancer
|
|
Prexasertib-d4 (LY2606368-d4) is the deuterium labeled Prexasertib (HY-18174). Prexasertib (LY2606368) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib shows potent anti-tumor activity .
|
-

- HY-121916
-
|
|
Isocitrate Dehydrogenase (IDH)
|
Cancer
|
|
ML309 is a highly selective and potent inhibitor of the R132H mutant isocitrate dehydrogenase 1 (IDH1 R132H), with an IC50 of 96 nM. ML309 is a competitive inhibitor of α-KG, with a Ki value of 156 nM, and its inhibitory activity against the wild-type IDH1 is greater than 35 μM. ML309 effectively reduces the production of the tumor metabolite 2-HG in U87MG cells. ML309 can be used as a chemical probe to study the role of the mutant IDH1 in cancer .
|
-

- HY-B0273B
-
|
|
Parasite
Bacterial
Environmental Pollutants
|
Infection
|
|
Sulfadiazine 100 µg/mL in methanol is an orally active sulfonamide antibiotic. Sulfadiazine 100 µg/mL in methanol competitively inhibits p-aminobenzoic acid in the folic-acid-metabolism cycle, inhibiting multiplication of most Gram-positive and many Gram-negative bacteria. Sulfadiazine 100 µg/mL in methanol persists in soil long-term, and exerts selective pressure for sulfonamide-resistant microbial populations. Sulfadiazine 100 µg/mL in methanol targets Toxoplasma gondii DHPS enzyme. Sulfadiazine 100 µg/mL in methanol can be used for the research of congenital toxoplasmosis and bacterial infection .
|
-

- HY-103430A
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for research of parkinson’s disease and nicotine craving alleviation .
|
-
- HY-N14094
-
|
|
JAK
Apoptosis
Autophagy
|
Cancer
|
|
Tubulosine is an alkaloid. Tubulosine can be isolated from Pogonopus tubulosus (DC.) Schumann. Tubulosine is an ATP-competitive, selective JAK3 inhibitor with an IC50 value of 9.9 nM. Tubulosine also inhibits the kinase activities of other JAK family members, the extent of inhibition is less than that of JAK3, with IC50 values of 69.5, 84.9 and 76.3 nM for JAK1, JAK2 and TYK2, respectively. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. Tubulosine induces apoptotic and necrotic/autophagic cell death. Tubulosine inhibits the process of peptide chain elongation by eukaryotic polysomes by, specifically preventing the elongation-factor-2-dependent step of translocation. Tubulosine exhibits anticancer activity in breast cancer cells .
|
-

- HY-A0009S
-
|
Galantamine-d3 hydrobromide
|
Isotope-Labeled Compounds
Cholinesterase (ChE)
nAChR
|
Neurological Disease
|
|
Galanthamine-d3 (hydrobromide) is deuterium labeled Galanthamine (hydrobromide). Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 μM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
- HY-180420
-
|
|
Cytochrome P450
|
Cancer
|
|
Tan-931 is a non-competitive and selective inhibitor of aromatase , with an IC50 is 17.2 μM and a Ki of 40 μM for human aromatase, and an IC50 of 162 μM for rat aromatase. Tan-931 reduces plasma estradiol-17β level and weight of ovaries and uterus in PMSG (HY-N12634)-treated female rats. Tan-931 can be used for the research of estrogen-dependent metastatic breast cancer .
|
-

- HY-100501R
-
|
MSC2363318A (Standard)
|
Ribosomal S6 Kinase (RSK)
Reference Standards
Akt
|
Cancer
|
|
M2698 (Standard) is the analytical standard of M2698 (HY-100501). This product is intended for research and analytical applications. M2698 (MSC2363318A) is an orally active, ATP competitive, selective p70S6K and Akt dual-inhibitor with IC50s of 1 nM for p70S6K, Akt1 and Akt3. M2698 can cross the blood-brain barrier and has anti-cancer activity .
|
-

- HY-76474
-
|
|
Syk
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
BAY 61-3606 is an orally available, ATP-competitive, reversible and highly selective Syk inhibitor with a Ki of 7.5 nM and an IC50 of 10 nM . BAY 61-3606 reduces ERK1/2 and Akt phosphorylation in neuroblastoma cell . BAY 61-3606 induces a large decrease of Syk phosphorylation in K-rn cell lysates . Bay 61-3606 sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells .
|
-
- HY-19347
-
|
WD-Repeat Protein 5-0103
|
WDR5
|
Neurological Disease
Cancer
|
|
WDR5-0103 (WD-Repeat Protein 5-0103) is a potent and selective WD repeat-containing protein 5 (WDR5) antagonist with a Kd of 450 nM. WDR5-0103 competitively binds to the peptide-binding pocket of WDR5, blocking the interaction between WDR5 and mixed-lineage leukemia (MLL) protein and inhibiting the methyltransferase activity of MLL. WDR5-0103 is mainly used in the research of cancer and neurodegenerative diseases .
|
-
- HY-10285S
-
|
BMS-477118-15N,d2 Hydrochloride
|
Isotope-Labeled Compounds
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Saxagliptin-15N,d2 Hydrochloride (BMS-477118-15N,d2 Hydrochloride) is the 15N and deuterium labeled isotope of Saxagliptin (HY-10285). Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-

- HY-117026
-
LKY-047
1 Publications Verification
|
Cytochrome P450
|
Metabolic Disease
Inflammation/Immunology
|
|
LKY-047, a Decursin derivative, is a potent and selective reversible competitive cytochrome P45022J2 (CYP2J2) inhibitor with an IC50 of 1.7 μM. LKY-047 is inactive against other human P450s, such as CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A .
|
-
- HY-145578
-
|
X842
|
Drug Intermediate
Proton Pump
|
Metabolic Disease
|
|
Linaprazan glurate (X842) is an orally atcive prodrug of Linaprazan (HY-100412) with a potent and prolonged inhibitory effect on gastric acid secretion. Linaprazan glurate is rapidly transformed by enzymatic cleavage into its active metabolite, linaprazan. Linaprazan glurate is a potassium-competitive acid blocker. Linaprazan glurate selectively inhibites acid formation from gastric H⁺/K⁺-ATPase in a potassium-dependent manner (IC50 = 436.2 nM). Linaprazan glurate can be used for the studies of erosive esophagitis (EE) .
|
-
- HY-14985
-
|
|
Syk
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
BAY 61-3606 dihydrochloride is an orally available, ATP-competitive, reversible and highly selective Syk inhibitor with a Ki of 7.5 nM an IC50 of 10 nM . BAY 61-3606 dihydrochloride reduces ERK1/2 and Akt phosphorylation in neuroblastoma cell . BAY 61-3606 dihydrochloride induces a large decrease of Syk phosphorylation in K-rn cell lysates . Bay 61-3606 dihydrochloride sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells .
|
-
- HY-18263CR
-
|
SB-656933 tosylate (Standard)
|
CXCR
Interleukin Related
Reference Standards
|
Inflammation/Immunology
|
|
Elubrixin (tosylate) (Standard) is the analytical standard of Elubrixin (tosylate). This product is intended for research and analytical applications. Elubrixin tosylate (SB-656933 tosylate) is a potent, selective, competitive, reversible and orally active CXCR2 antagonist and an IL-8 receptor antagonist. Elubrixin tosylate inhibits neutrophil CD11b upregulation (IC50 of 260.7 nM) and shape change (IC50 of 310.5 nM). Elubrixin tosylate has the potential for inflammatory diseases research, such as inflammatory bowel disease and airway inflammation .
|
-

- HY-181700
-
|
|
Phosphatase
GLUT
|
Metabolic Disease
|
|
PTP1B-IN-34 is an orally active, selective, non-competitive PTP1B inhibitor, with an IC50 value of 0.64 μM and a Ki value of 1.15 μM against human PTP1B. PTP1B-IN-34 reduces blood glucose levels in diabetic mice. PTP1B-IN-34 can be used in research related to type 2 diabetes .
|
-

- HY-117053
-
|
|
Neurokinin Receptor
|
Neurological Disease
|
|
ZM 253270 is a species-selective non-peptide NK-2 receptor (NK-2R) antagonist. ZM 253270 competitively inhibits the binding of [ 3H]NKA to native or cloned NK-2R from hamster bladder (Ki=2 nM), but has a weaker inhibitory effect (48-fold) on the binding of [ 3H]NKA to cloned human NK-2R .
|
-

- HY-122262
-
|
|
Beta-lactamase
Bacterial
|
Infection
|
|
NDM-1-IN-6 (Compound 1) is a potent, selective and competitive New Delhi Metallo-β-lactamase-1 (NDM-1) inhibitor with a Ki of 0.72 μM. NDM-1-IN-6 has a synergistic antibacterial effect with the carbapenem antibiotic Meropenem (HY-13678). NDM-1-IN-6 is mainly used for research on NDM-1-mediated multidrug-resistant bacterial infections .
|
-

- HY-161227
-
|
|
17β-HSD
|
Metabolic Disease
Inflammation/Immunology
|
|
HSD17B13-IN-43 is a selective inhibitor of HSD17B13 that competitively blocks the activity of this enzyme. HSD17B13-IN-43 exhibits an IC50 ≤ 0.1 µM in in vitro assays. HSD17B13-IN-43 can be used in studies of non-alcoholic steatohepatitis, fatty liver disease and hepatic fibrosis .
|
-
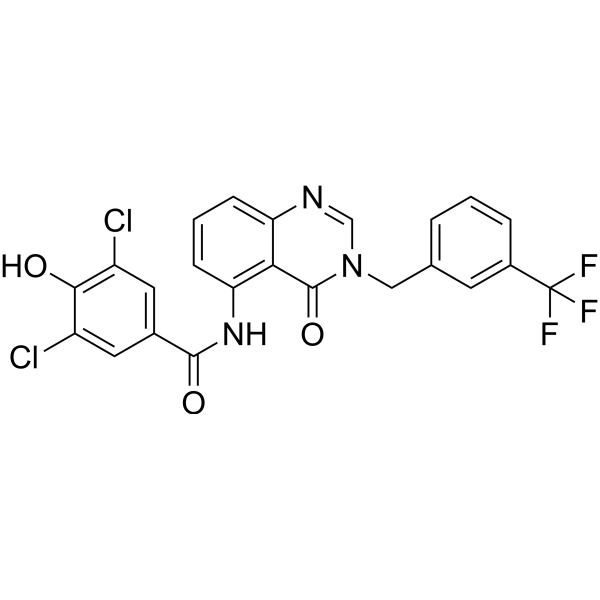
- HY-13285
-
|
Debio 0719
|
LPL Receptor
YAP
|
Neurological Disease
Cancer
|
|
Ki16425 (Debio 0719) is a subtype-selective, competitive antagonist of the EDG-family receptors, LPA1 and LPA3 with Kis of 0.34 μM and 0.93 μM, respectively. Ki16425 (Debio 0719) reduces the LPA-induced activation of p42/p44 MAPK . Ki16425 can also inhibit LPA-induced dephosphorylation of Yes-associated protein (YAP)/TAZ in HEK293A cells .
|
-
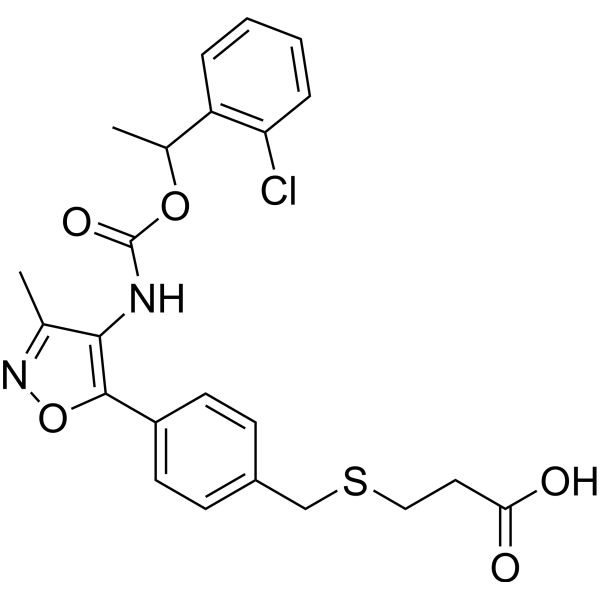
- HY-103195R
-
|
|
Reference Standards
Adenylate Cyclase
|
Metabolic Disease
|
|
NKY80 (Standard) is the analytical standard of NKY80 (HY-103195). This product is intended for research and analytical applications. NKY80 is a potent, selective and non-competitive adenylyl cyclase (AC) type V isoform inhibitor with IC50s of 8.3 µM, 132 µM and 1.7 mM for type V, III and II, respectively. NKY80 is a non-nucleoside quinazolinone and regulates the AC catalytic activity in heart and lung tissues .
|
-

- HY-112457
-
|
|
MAPKAPK2 (MK2)
|
Inflammation/Immunology
|
|
MK2-IN-3 hydrate (compound 16) is an orally active, selective, and ATP-competitive MAPKAP-K2 (MK-2) inhibitor with an IC50 of 8.5 nM.MK2-IN-3 hydrate is exceptional selectivity against MK-3 (IC50=0.21 μM), MK-5 (IC50=0.081 μM), ERK2 (IC50=3.44 μM), MNK1(IC50=5.7 μM) as well as CDK2, JNK2, IKK2, MSK1, and MSK2 .
|
-
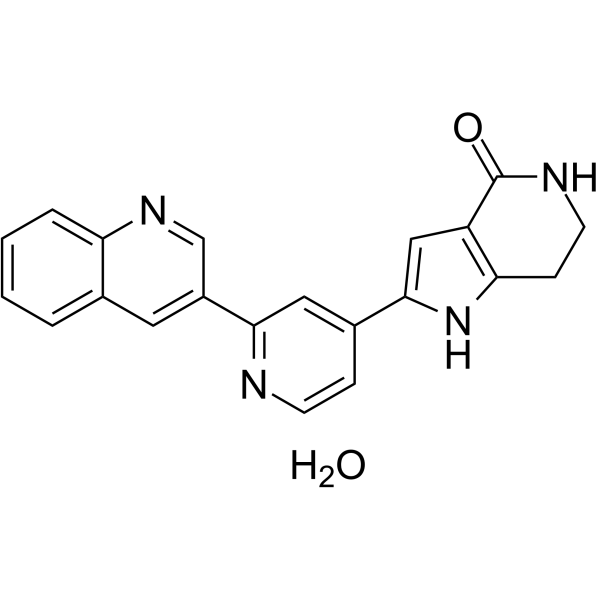
- HY-16749
-
|
PLX-3397
|
c-Fms
c-Kit
Apoptosis
|
Cancer
|
|
Pexidartinib (PLX-3397) is a potent, orally active, selective, and ATP-competitive colony stimulating factor 1 receptor (CSF1R or M-CSFR) and c-Kit inhibitor, with IC50s of 20 and 10 nM, respectively. Pexidartinib (PLX-3397) exhibits 10- to 100-fold selectivity for c-Kit and CSF1R over other related kinases. Pexidartinib (PLX-3397) induces cell apoptosis and has anti-tumor activity. Pexidartinib has limited permeability to the blood-brain barrier, primarily through ABCB1 .
|
-
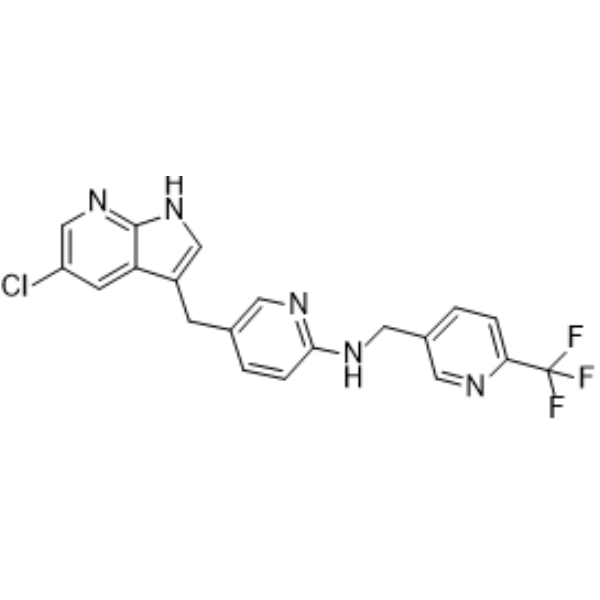
- HY-50878AS
-
|
PF-02341066-d9 hydrochloride
|
c-Met/HGFR
Autophagy
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Isotope-Labeled Compounds
|
Cancer
|
|
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
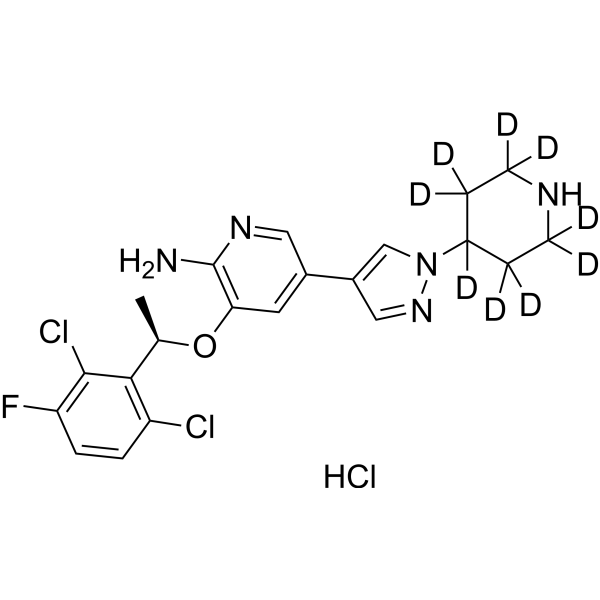
- HY-103566R
-
|
|
Reference Standards
mGluR
EGFR
p38 MAPK
Apoptosis
|
Infection
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
LY456236 (Standard) is the analytical standard of LY456236 (HY-103566). This product is intended for research and analytical applications. LY456236 is a selective, non-competitive and orally active antagonist of glutamate receptor 1 (mGlu1), which can inhibit phosphatidylinositol hydrolysis with an IC50 of 0.145 μM. LY456236 can also inhibit EGFR, with an IC50 of 0.918 μM. LY456236 has anticonvulsant effects and blocks cell proliferation by inhibiting the MAPK pathway, reversing the anti-apoptotic effect of DHPG (HY-12598A). LY456236 can be used in epilepsy research .
|
-

- HY-164685
-
|
|
Phosphodiesterase (PDE)
|
Neurological Disease
|
|
T-0156 free base is a potent and selective phosphodiesterase type 5 (PDE5) inhibitor. T-0156 free base specifically inhibits the hydrolysis of cyclic guanosine monophosphate (cGMP) by PDE5 in a competitive manner (IC50=0.23 nM). T-0156 free base inhibits PDE6 (IC50=56 nM) and has low potencies against PDE1, PDE2, PDE3, and PDE4 (IC50>10 μM). T-0156 free base enhances the nitric oxide (NO)/cGMP pathway .
|
-

- HY-16461
-
|
(-)-Solenopsin A
|
Akt
Ribosomal S6 Kinase (RSK)
PI3K
PDK-1
FOXO
Mitophagy
Reactive Oxygen Species (ROS)
Mitochondrial Metabolism
|
Cardiovascular Disease
|
|
Solenopsin ((-)-Solenopsin A) is an ATP-competitive and selective Akt-1 inhibitor with an IC50 of 5-10 μM, and also acts as an RSK1 inhibitor. Solenopsin inhibits the activities of PDK1 in lipid rafts, downregulates PI3K, blocks PI3K-dependent generation of 3-phosphoinositides, and suppresses the phosphorylation of FOXO1a. Solenopsin induces Mitophagy and ROS production, reduces mitochondrial oxygen consumption, and exhibits antiproliferative and antiangiogenic activities. Solenopsin can be used in research related to hyperproliferative skin diseases and malignant diseases .
|
-
- HY-W013375
-
|
|
Neprilysin
|
Neurological Disease
|
|
Thiorphan is a selective neprilysin (NEP) inhibitor with an IC50 of 6.9 nM. Thiorphan competitively binds to NEP and blocks its activity, preventing the degradation of neuropeptides such as substance P (SP) and neurokinin NKA. In the field of neonatal brain injury research, Thiorphan can increase the levels of SP and NKA, activate NK1 and NK2 receptors and downstream transduction pathways, and inhibit excessive activation of NMDA receptors. Thus, Thiorphan can protect neocortical neurons from excitotoxic cell death. Thiorphan may also inhibit NEP from enhancing bronchoconstriction and can be used in the study of respiratory diseases .
|
-
- HY-N2554
-
|
Ostenol
|
Monoamine Oxidase
PI3K
Akt
Apoptosis
|
Infection
Neurological Disease
Cancer
|
|
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cell cycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
|
-
- HY-18944R
-
|
|
CDK
HSV
CMV
DNA/RNA Synthesis
|
Infection
|
|
FIT-039 (Standard) is the analytical standard of FIT-039. This product is intended for research and analytical applications. FIT-039 is a selective, ATP-competitive and orally active CDK9 inhibitor with an IC50 of 5.8 μM for CKD9/cyclin T1. FIT-039 does not inhibit other CDKs and other kinases. FIT-039 inhibits replication of HSV-1 (IC50 of 0.69 μM), HSV-2, human adenovirus, and human CMV. FIT-039 is a promising antiviral agent for inhibiting drug-resistant HSVs and other DNA viruses.
|
-

- HY-171835
-
|
|
HIV
HIV Protease
|
Infection
|
|
DPC 684 is a potent and selective HIV-1 protease inhibitor (IC90 = 5.7-40 nM, Ki = 0.021 nM). DPC 684 competitively inhibits HIV-1 protease and blocks viral polyprotein cleavage. DPC 684 has low protein binding and broad-spectrum inhibition against a variety of wild-type and mutant HIV-1 proteases. DPC 684 has low protein binding and broad-spectrum inhibition (IC90 = 1.9-6.3 nM). DPC 684 has research significance for HIV .
|
-

- HY-173334
-
|
|
Monoamine Oxidase
Interleukin Related
TNF Receptor
|
Neurological Disease
|
|
hMAO-B-IN-11 (Compound 12) is a selective and reversible inhibitor of human monoamine oxidase B (hMAO-B) with an IC50 of 0.11 µM. hMAO-B-IN-11 acts through competitive binding to the hMAO-B active site, preventing oxidative deamination of monoamines and reducing hydrogen peroxide production. hMAO-B-IN-11 also inhibits pro-inflammatory mediators (NO, TNF-α, IL-1β) in activated microglia, hMAO-B-IN-11 is promising for research of neurodegenerative diseases like Parkinson’s and Alzheimer’s .
|
-

- HY-B0442CR
-
|
|
Reference Standards
Endogenous Metabolite
Phosphodiesterase (PDE)
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
Cancer
|
|
Vardenafil (dihydrochloride) (Standard) is the analytical standard of Vardenafil (dihydrochloride). This product is intended for research and analytical applications. Vardenafil dihydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil dihydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM respectively, while IC50s are >1000 nM for PDE3 and PDE4. Vardenafil dihydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil dihydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-

- HY-B0442AR
-
|
|
Phosphodiesterase (PDE)
Endogenous Metabolite
Reference Standards
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
Cancer
|
|
Vardenafil (hydrochloride) (Standard) is the analytical standard of Vardenafil (hydrochloride). This product is intended for research and analytical applications. Vardenafil hydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-

- HY-110206
-
|
|
Cannabinoid Receptor
|
Metabolic Disease
|
|
AM6545 is a highly selective, brain-free (peripherally active) CB1 receptor antagonist (Ki=1.7 nM). AM6545 inhibits endocannabinoid signaling by competitively antagonizing CB1 receptors, inhibiting CB1-mediated appetite stimulation and inflammatory responses without affecting cAMP levels. AM6545 significantly reduces food intake and body weight in mice, while improving metabolic syndrome-related renal impairment (such as proteinuria, fibrosis) and insulin resistance. AM6545 can be used in the study of obesity and its complications .
|
-
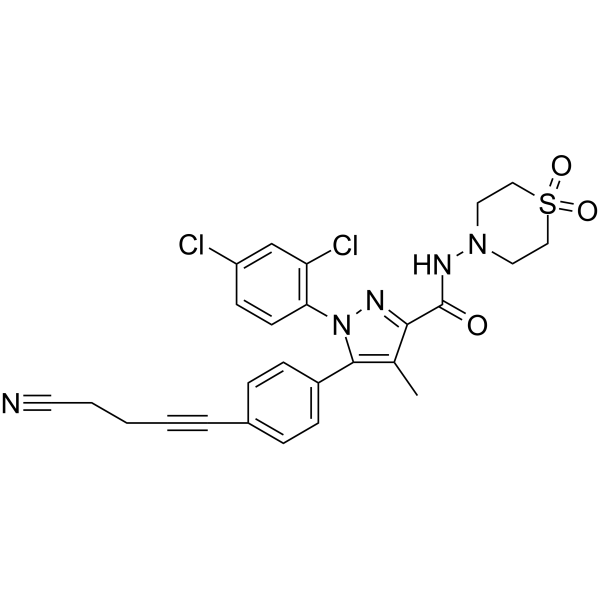
- HY-174332
-
|
|
Cytochrome P450
|
Neurological Disease
|
|
Cholesterol 24-hydroxylase-IN-3 is an orally active, selective, and blood-brain barrier-penetrant CH24H inhibitor (IC50 = 23 nM) belonging to 1,3-oxazole derivatives. Cholesterol 24-hydroxylase-IN-3 competitively inhibits CH24H enzyme activity by using the 1,3-oxazole nitrogen atom to coordinate the heme iron and the cyclopropyl group occupying the hydrophobic pocket. Cholesterol 24-hydroxylase-IN-3 can be used for research on epilepsy and other neurological diseases.
|
-

- HY-10256A
-
|
SB 203580 hydrochloride; RWJ 64809 hydrochloride
|
Organoid
p38 MAPK
Autophagy
Mitophagy
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (SB 203580; RWJ 64809) hydrochloride is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod hydrochloride inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod hydrochloride does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-
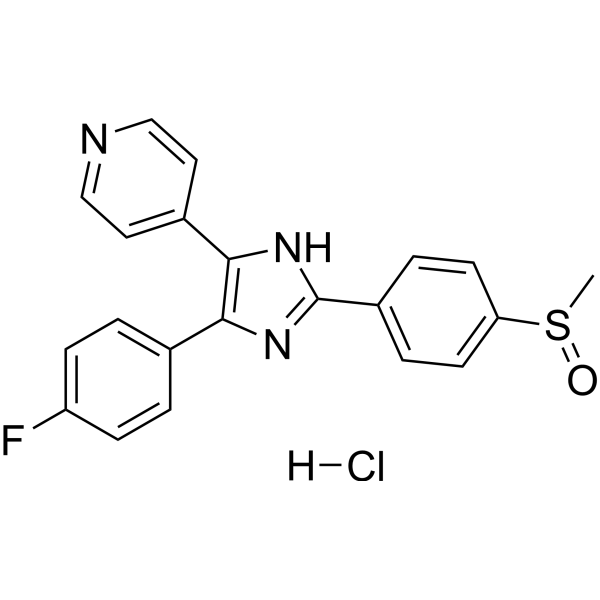
- HY-B0442R
-
|
|
Reference Standards
Phosphodiesterase (PDE)
Endogenous Metabolite
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
|
|
Vardenafil (Standard) is the analytical standard of Vardenafil. This product is intended for research and analytical applications. Vardenafil is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-

- HY-11009
-
|
|
CDK
PKC
|
Cancer
|
|
CGP60474, a highly potent anti-endotoxemic agent, is a potent cyclin-dependent kinase (CDK) inhibitor (IC50 values are 26, 3, 4, 216, 10, 200 and 13 nM for CDK1/B, CDK2/E, CDK2/A, CDK4/D, CDK5/p25, CDK7/H and CDK9/T, respectively). CGP60474 is a selective and ATP-competitive PKC inhibitor .
|
-
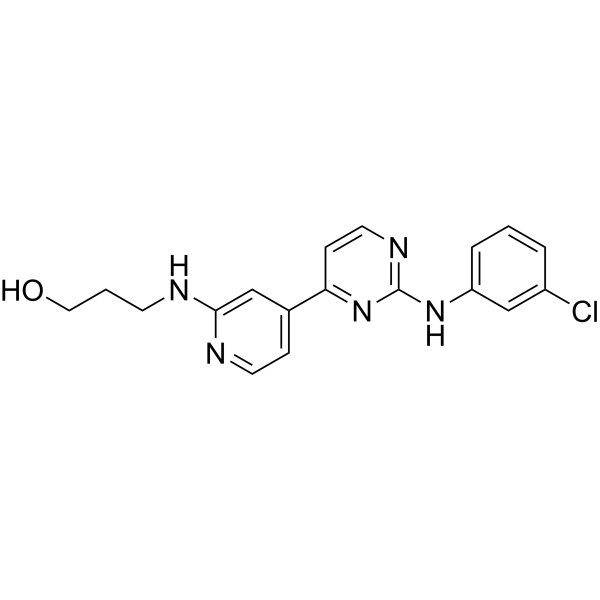
- HY-W049735R
-
|
|
Drug Metabolite
Reference Standards
Endogenous Metabolite
|
Metabolic Disease
|
|
Vardenafil (dihydrochloride) (Standard) is the analytical standard of Vardenafil (dihydrochloride). This product is intended for research and analytical applications. Vardenafil dihydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil dihydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM respectively, while IC50s are >1000 nM for PDE3 and PDE4. Vardenafil dihydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil dihydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-

- HY-B0442AS
-
|
|
Endogenous Metabolite
Phosphodiesterase (PDE)
Isotope-Labeled Compounds
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
|
|
Vardenafil-d5 hydrochloride is deuterated labeled Vardenafil hydrochloride (HY-B0442A). Vardenafil hydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-
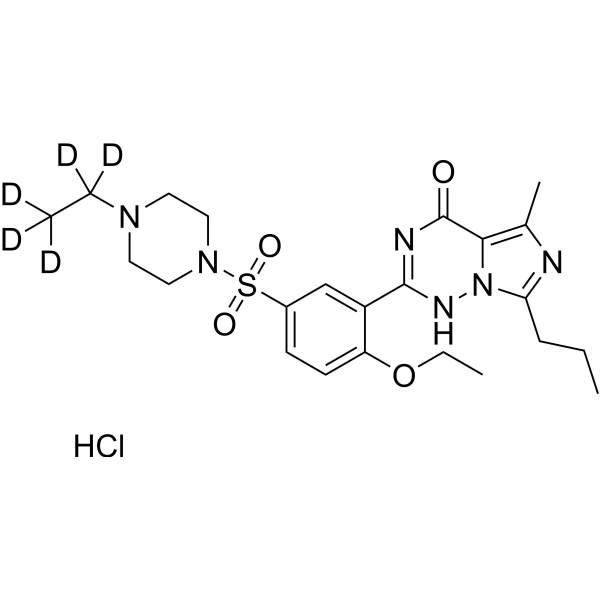
- HY-107691
-
|
|
Neurokinin Receptor
|
Neurological Disease
Endocrinology
Cancer
|
|
GR 159897 is a highly potent, selective, competitive, brain-penetrated non-peptide neurokinin 2 (NK2) receptor antagonist. GR 159897 has little or no affinity for NK1 and NK3 receptors. GR 159897 inhibits binding of [ 3H]GR100679 to human NK2 (hNK2)-CHO cells and rat colon membranes with pKis of 9.51 and 10, respectively. Antagonizes bronchoconstriction. Anxiolytic-like and anti-tumor effects .
|
-
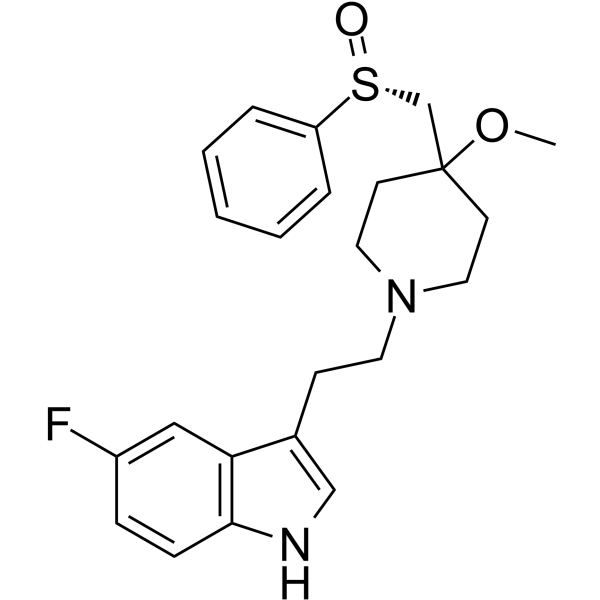
- HY-10517AR
-
|
(Z)-SU6668 (Standard); (Z)-TSU-68 (Standard)
|
Reference Standards
VEGFR
PDGFR
FGFR
|
Cancer
|
|
(Z)-Orantinib (Standard) is the analytical standard of (Z)-Orantinib (HY-10517A). This product is intended for research and analytical applications. (Z)-Orantinib ((Z)-SU6668) is a potent, selective, orally active and ATP competitive inhibitor of Flk‐1/KDR, PDGFRβ, and FGFR1, with IC50s of 2.1, 0.008, and 1.2 μM, respectively. (Z)-Orantinib is a potent antiangiogenic and antitumor agent that induces regression of established tumors .
|
-

- HY-P991869
-
|
AQmabAM; rAb-53
|
Aquaporin
|
Neurological Disease
|
|
Aquaporumab (AQmabAM) is an engineered human monoclonal IgG antibody. Aquaporumab is a selective inhibitor of aquaporin 4 (AQP4), binding tightly to AQP4 and competitively displacing AQP4-IgG from serum [1][2]. Aquaporumab significantly reduces neuromyelitis optica lesions in spinal cord slice cultures and in mice receiving intracerebral injection of AQP4-IgG and complement [1][2]. Aquaporumab can be used for research on neuromyelitis optica spectrum disorders [1][2]."}
|
-

- HY-181135
-
|
|
GABA Receptor
|
Neurological Disease
|
|
Bicyclo-GABA is a selective betaine/GABA transporter 1 (BGT1) competitive, non-transported inhibitor with an IC50 of 590 nM. Bicyclo-GABA exhibits low micromolar agonistic activity at α1β2γ2 GABAA receptors with an EC50 of 5.1 μM. Bicyclo-GABA displays 129 times higher activity for BGT1 than GAT3. Bicyclo-GABA serves as a valuable tool compound for deciphering its elusive pharmacological role in the brain and periphery .
|
-

- HY-182701
-
-

- HY-113914
-
|
Elraglusib
|
GSK-3
Apoptosis
Autophagy
|
Cancer
|
|
9-ING-41 (Elraglusib) is a maleimide-based ATP-competitive and selective glycogen synthase kinase-3β (GSK-3β) inhibitor with an IC50 of 0.71 μM. 9-ING-41 significantly leads to cell cycle arrest, autophagy and apoptosis in cancer cells. 9-ING-41 has anticancer activity and has the potential for enhancing the antitumor effects of chemotherapeutic agents .
|
-
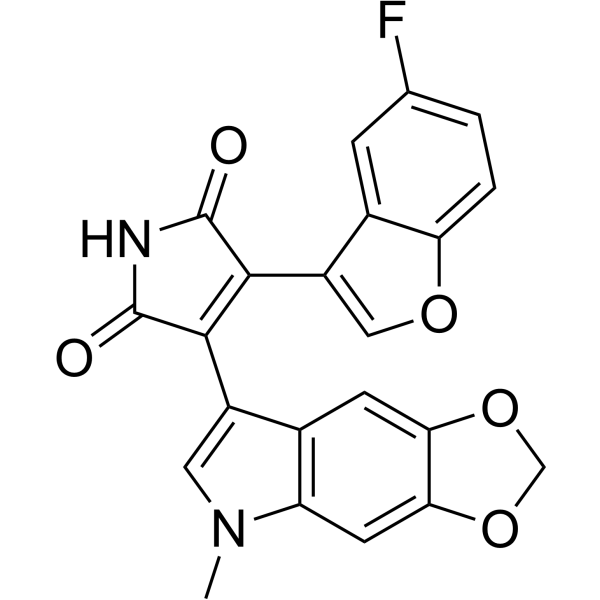
- HY-10254R
-
|
PD0325901 (Standard); PD325901 (Standard)
|
Reference Standards
MEK
Autophagy
Apoptosis
|
Cancer
|
|
Mirdametinib (Standard) is the analytical standard of Mirdametinib (HY-10254). This product is intended for research and analytical applications. Mirdametinib (PD0325901) is an orally active, selective and non-ATP-competitive MEK inhibitor with an IC50 of 0.33 nM. Mirdametinib exhibits a Kiapp of 1 nM against activated MEK1 and MEK2. Mirdametinib suppresses the expression of p-ERK1/2 and induces apoptosis. Mirdametinib has anti-cancer activity for a broad spectrum of human tumor xenografts .
|
-

- HY-W013268
-
|
(+)-N-3-Benzylnirvanol
|
Cytochrome P450
|
Metabolic Disease
|
|
(S)-(+)-N-3-Benzylnirvanol ((+)-N-3-Benzylnirvanol) is a selective and competitive cytochrome P450 (CYP) isoform CYP2C19 inhibitor with a Ki of 250 nM. (S)-(+)-N-3-Benzylnirvanol has low activity against CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2D6, CYP2E1, and CYP3A4 .
|
-
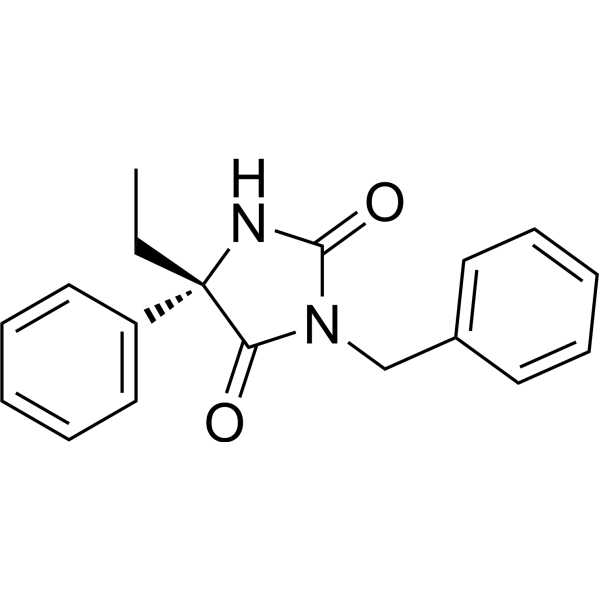
- HY-B1164A
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
Bromopride hydrochloride is a selective, irreversible, competitive, and orally active dopamine D2 receptor antagonist. Bromopride hydrochloride can pass through the blood-brain barrier, inhibit the vomiting center, and enhance gastrointestinal motility, exerting antiemetic and gastrointestinal motility effects. Bromopride hydrochloride antagonizes dopamine-mediated vomiting reflexes and promotes gastrointestinal smooth muscle contraction, and has no adverse effects on abdominal wall healing in rats with postoperative abdominal infection. Bromopride hydrochloride can be used for the study of digestive system diseases (such as gastric hypomotility, nausea and vomiting) .
|
-

- HY-P5756
-
|
|
Opioid Receptor
|
Neurological Disease
|
|
CSD-CH2(1,8)-NH2 is a selective and competitive KOR antagonist (Ki: 6.8 nM). CSD-CH2(1,8)-NH2 inhibits calcium mobilization in DRG neurons. CH2(1,8)-NH2 antagonizes the antinociceptive effect of U50,488. CSD-CH2(1,8)-NH2 can be used for research of neuropsychiatric disorders .
|
-
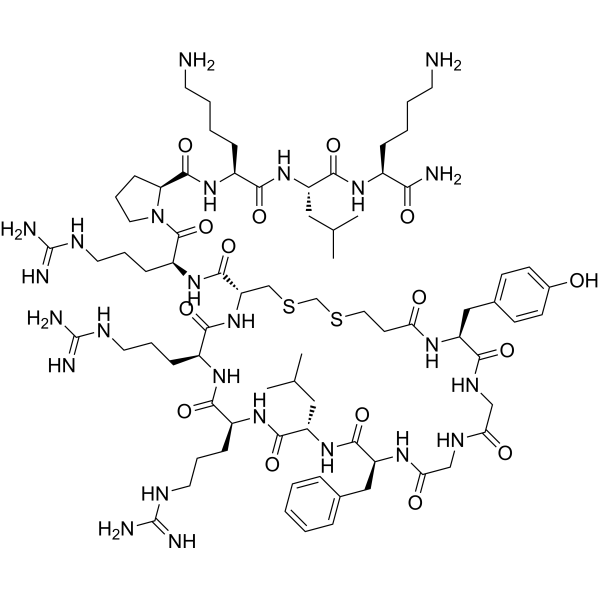
- HY-206735
-
|
|
Phosphatase
|
Cancer
|
|
CDC14A/B-IN-2, phosphotyrosine mimetic, is a competitive human CDC14A/B phosphatase inhibitor with IC50 values of 10.4 and 11.2 μM, and Ki values of 5.8 and 7.3 μM. CDC14A/B-IN-2 shows at least 20-fold selectivity over other protein tyrosine phosphatases. CDC14A/B-IN-2 can be used for the research of cancer .
|
-

- HY-B1164
-
|
|
Dopamine Receptor
|
Neurological Disease
|
|
Bromopride is a selective, irreversible, competitive, and orally effective dopamine D2 receptor antagonist. Bromopride can pass through the blood-brain barrier, inhibit the vomiting center, and enhance gastrointestinal motility, exerting antiemetic and gastrointestinal motility effects. Bromopride antagonizes dopamine-mediated vomiting reflexes and promotes gastrointestinal smooth muscle contraction, and has no adverse effects on abdominal wall healing in rats with postoperative abdominal infection. Bromopride can be used for the study of digestive system diseases (such as gastric hypomotility, nausea and vomiting) .
|
-
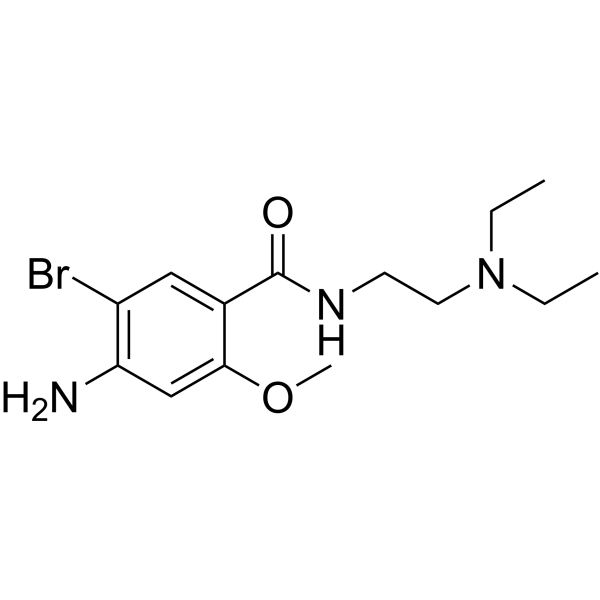
- HY-116143
-
|
|
MAGL
|
Metabolic Disease
|
|
SAR127303 is an orally active, selective, competitive monoacylglycerol lipase (MAGL) covalent inhibitor with IC50s of 3.8 nM and 29 nM for mouse and human MAGL, respectively. SAR127303 potently elevates hippocampal levels of 2-AG in mice. SAR127303 decreased long term potentiation (LTP) of CA1 synaptic transmission and acetylcholine release in the hippocampus. SAR127303 produces antinociceptive effects in assays of inflammatory and visceral pain. SAR127303 slows down epileptogenesis .
|
-
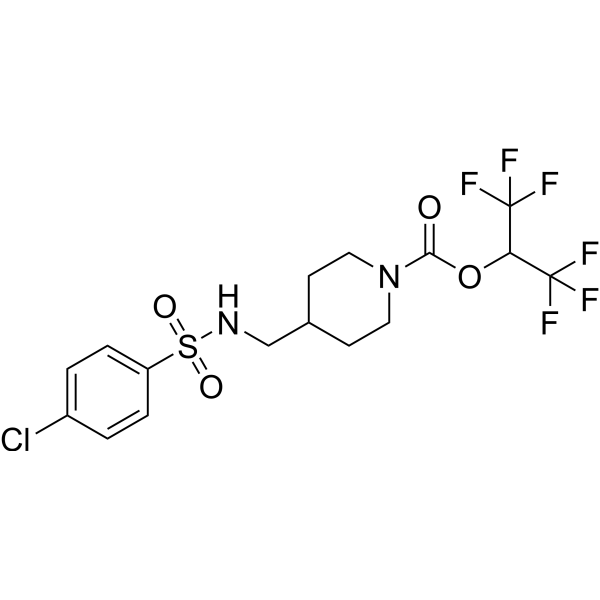
- HY-183932
-
|
|
mAChR
|
Neurological Disease
Inflammation/Immunology
|
|
NPC-14695 is a competitive and selective M3 muscarinic receptor antagonist, with a Kd value of 15 nM for guinea pig M3, 60 nM for guinea pig M2, and 25 nM for rabbit M1. NPC-14695 exhibits higher activity towards M3 receptors in bronchial smooth muscle than towards those regulating salivary secretion. NPC-14695 inhibits Carbachol (HY-B1208)-induced contraction of isolated rabbit iris smooth muscle .
|
-

- HY-A0009R
-
|
Galantamine hydrobromide (Standard)
|
nAChR
Cholinesterase (ChE)
Reference Standards
|
Neurological Disease
|
|
Galanthamine (hydrobromide) (Standard) is the analytical standard of Galanthamine (hydrobromide). This product is intended for research and analytical applications. Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 µM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
- HY-15346R
-
|
BAY 80-6946 (Standard)
|
Reference Standards
PI3K
Apoptosis
|
Cancer
|
|
Copanlisib (Standard) is the analytical standard of Copanlisib. This product is intended for research and analytical applications. Copanlisib (BAY 80-6946) is a potent, selective and ATP-competitive pan-class I PI3K inhibitor, with IC50s of 0.5 nM, 0.7 nM, 3.7 nM and 6.4 nM for PI3Kα, PI3Kδ, PI3Kβ and PI3Kγ, respectively. Copanlisib has more than 2,000-fold selectivity against other lipid and protein kinases, except for mTOR. Copanlisib has superior antitumor activity .
|
-

- HY-103430AR
-
|
|
Reference Standards
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 (Standard) is the analytical standard of SKF-83566 (HY-103430A). This product is intended for research and analytical applications. SKF-83566 is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for research of parkinson’s disease and nicotine craving alleviation .
|
-

- HY-133028
-
|
|
Casein Kinase
CDK
SGK
Ribosomal S6 Kinase (RSK)
|
Cancer
|
|
CKI-7 free base is a potent and ATP-competitive casein kinase 1 (CK1) inhibitor with an IC50 of 6 μM and a Ki of 8.5 μM. CKI-7 free base is a selective Cdc7 kinase inhibitor. CKI-7 free base also inhibits SGK, ribosomal S6 kinase-1 (S6K1) and mitogen- and stress-activated protein kinase-1 (MSK1). CKI-7 free base has a much weaker effect on casein kinase II and other protein kinases .
|
-
- HY-B0927
-
|
(-)-β-Hydrastine; (1R,9S)-β-Hydrastine
|
Tyrosine Hydroxylase
Dopamine Receptor
OAT
|
Neurological Disease
|
|
Hydrastine ((-)-β-Hydrastine; (1R,9S)-β-Hydrastine) is a selective competitive inhibitor of tyrosine hydroxylase (TH), inhibiting dopamine biosynthesis (IC50=20.7 μM, PC12 cells). Hydrastine also inhibits the organic cation transporter OCT1 (IC50=6.6 μM). Hydrastine may cause neuronal toxicity through mitochondrial dysfunction rather than oxidative stress damage, and can aggravate cell apoptosis when combined with L-DOPA. Hydrastine can be used to study Parkinson's disease-related dopaminergic neuronal damage .
|
-
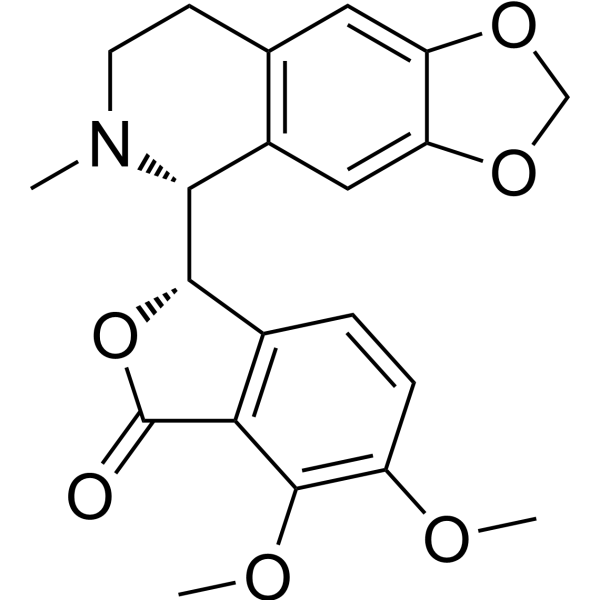
- HY-101925R
-
|
|
Reference Standards
Histone Methyltransferase
DNA Methyltransferase
Apoptosis
|
Cancer
|
|
CM-272 (Standard) is the analytical standard of CM-272 (HY-101925). This product is intended for research and analytical applications. CM-272 is a first-in-class, potent, selective, substrate-competitive and reversible dual G9a/DNA methyltransferases (DNMTs) inhibitor with antitumor activities. CM-272 inhibits G9a, DNMT1, DNMT3A, DNMT3B and GLP with IC50s of 8 nM, 382 nM, 85 nM, 1200 nM and 2 nM, respectively. CM-272 inhibits cell proliferation and promotes apoptosis, inducing IFN-stimulated genes and immunogenic cell death .
|
-

- HY-N8540
-
|
|
Phosphoglycerate Kinase (PGK)
Fungal
Apoptosis
|
Infection
Cancer
|
|
Ilicicolin H is a selective and non-ATP-competitive phosphoglycerate kinase 1 (PGK1) (IC50 = 9.02 μM) and mitochondrial cytochrome bc1 reductase (IC50 = 2-3 ng/mL) inhibitor. Ilicicolin H directly binds to PGK1 with KD of 60 μM .Ilicicolin H can inhibit cell proliferation and induce apoptosis. Ilicicolin H can inhibit the lactate production and glucose uptake of hepatocellular carcinoma (HCC) cells. Ilicicolin H has a broad antifungal spectrum including C. albicans, Cryptococcus and A. fumigatus. Ilicicolin H can be used for the researches of cancer and infection, such as hepatocellular carcinoma (HCC) and C. albicans infection .
|
-

- HY-W251428
-
|
Egg PG
|
Biochemical Assay Reagents
|
Others
|
|
Phosphatidylglycerols (PG) is a selective inhibitor targeting the TLR4 accessory protein CD14/MD-2 complex, inhibiting LPS or virus (such as RSV)-mediated inflammatory signaling pathways through competitive binding. Phosphatidylglycerols directly bind to viral particles to block infection, inhibit COX-2 expression to reduce the release of inflammatory factors (IL-6, IL-8), and improve oxidative stress by regulating mitochondrial membrane phospholipid remodeling. Phosphatidylglycerols can be taken orally or by inhalation and can be used in the study of chronic inflammatory diseases (such as atherosclerosis) and respiratory viral infections (such as RSV) .
|
-

- HY-103490
-
|
EDHS-206
|
MAP3K
Apoptosis
|
Infection
Inflammation/Immunology
Cancer
|
|
Takinib (EDHS-206) is an orally active and selective TAK1 inhibitor (IC50=9.5 nM), more than 1.5 log more potent than the second and third ranked targets, IRAK4 (120 nM) and IRAK1 (390 nM), respectively. Takinib is an inhibitor of autophosphorylated TAK1 that non-competitively binds within the ATP binding pocket. Takinib induces apoptosis following TNFα stimulation in cell models of rheumatoid arthritis and metastatic breast cancer. Takinib is also a P. falciparum protein kinase 9 (PfPK9) inhibitor (KD(app) of 0.46 μM) .
|
-
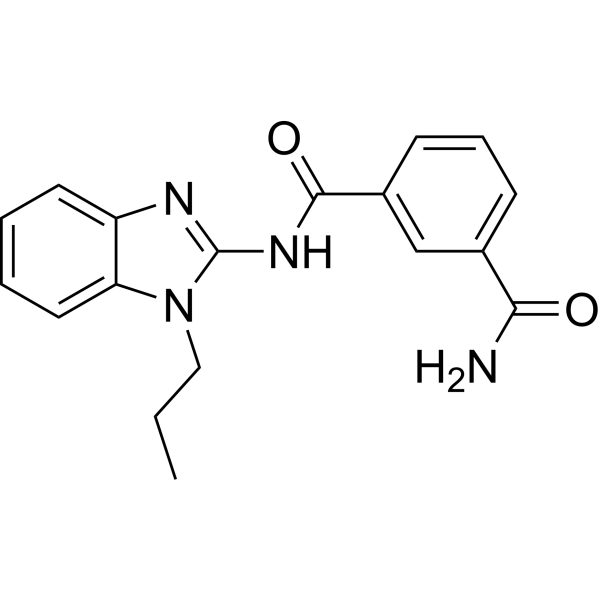
- HY-112631
-
|
|
Phosphodiesterase (PDE)
|
Cardiovascular Disease
|
|
OPC-33540 is a highly selective and competitive PDE3 inhibitor with IC50 values of 0.32 nM (PDE3A) and 1.5 nM (PDE3B). OPC-33540 exhibits IC50s against PDE1, PDE2, PDE4, PDE5, and PDE7 of 42.9, 52.3, 100.8, 2.5, and 51.3 μM, respectively. OPC-33540 significantly enhances cAMP accumulation in platelets and effectively inhibits thrombin-induced platelet aggregation. OPC-33540 can be used in antithrombotic studies .
|
-

- HY-B0442BR
-
|
|
Reference Standards
Endogenous Metabolite
Phosphodiesterase (PDE)
|
Metabolic Disease
Inflammation/Immunology
Endocrinology
|
|
Vardenafil (hydrochloride trihydrate) (Standard) is the analytical standard of Vardenafil (hydrochloride trihydrate). This product is intended for research and analytical applications. Vardenafil hydrochloride trihydrate is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride trihydrate shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride trihydrate competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride trihydrate can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-

- HY-179723
-
|
|
GSK-3
Tau Protein
|
Neurological Disease
|
|
GSK3β-IN-4 is a selective, potent, orally active and brain-penetrant ATP-competitive GSK3β inhibitor with an IC50 of 0.37 nM. GSK3β-IN-4 shows an IC50 of 2.75 nM and SI of 7.4 for GSK3α. GSK3β-IN-4 reduces tau phosphorylation at Ser396 by inhibiting GSK3β and imoroves cognitive deficits in Alzheimer's disease models. GSK3β-IN-4 can be used for the research of Alzheimer's disease .
|
-

- HY-182474
-
|
Indolopyridone-1
|
Reverse Transcriptase
DNA/RNA Synthesis
HIV
|
Infection
|
|
INDOPY-1 (Indolopyridone-1) is a selective, reversible, and competitive HIV-1 reverse transcriptase inhibitor. INDOPY-1 reversibly binds to the active site of reverse transcriptase. INDOPY-1 inhibits DNA synthesis. INDOPY-1 exhibits antiviral activity against various retroviruses, including HIV-1 IIIB, HIV-1 HXB2 K103N Y181C, HIV-2 ROD, and SIV Mac251. INDOPY-1 can be used in the research of immunodeficiency virus infection .
|
-

- HY-111388A
-
|
SEL120-34A monohydrochloride
|
CDK
|
Cancer
|
|
SEL120-34A monohydrochloride is an ATP-competitive and selective CDK8 inhibitor, inhibits kinase activities of CDK8/CycC and CDK19/CycC complexes with IC50s of 4.4 nM and 10.4 nM, respectively, with a Kd of 3 nM for CDK8. SEL120-34A monohydrochloride weakly inhibits CDK9 (calculated IC50=1070 nM), but shows no obvious activity against CDK1, 2, 4, 6, 5, 7. SEL120-34A monohydrochloride inhibits phosphorylation of STAT1 S727 and STAT5 S726 . Has anti-tumor activity .
|
-
- HY-106969A
-
|
|
Glycine Receptor (GlyR)
iGluR
|
Cardiovascular Disease
Neurological Disease
|
|
ZD 9379 sodium is a competitive glycine/NMDA receptor antagonist, with an IC50 value of 75 nM (glutamate site). ZD 9379 sodium selectively antagonizes the glycine binding site (GlyB site) on the NMDA receptor, inhibiting the binding of glycine to the NMDA receptor and alleviating excitotoxicity. ZD 9379 sodium reduces the frequency of cortical spreading depression (SDs), alleviates energy depletion in the ischemic penumbra, and delays the expansion of infarction. ZD 9379 sodium reduces the infarct volume and improves neurological function in mouse models. ZD 9379 sodium can be used in studies of acute ischemic stroke, etc .
|
-

- HY-12354R
-
|
|
MMP
|
Cancer
|
|
SB-3CT (Standard) is the analytical standard of SB-3CT. This product is intended for research and analytical applications. SB-3CT is a potent and competitive matrix metalloproteinase MMP-2 and MMP-9 inhibitor with Ki values of 13.9 and 600 nM, respectively. SB-3CT has high selectivity for gelatinases. SB-3CT shows blood-brain barrier permeability and has neuroprotective effects and anticancer activity .
|
-

- HY-122595
-
|
|
Histone Methyltransferase
|
Cancer
|
|
EZH2-IN-1 (compound 3) is a selective and SAM-competitive EZH2 and EZH1 inhibitor with an IC50s of 32 nM, 197 nM and 213 nM for EZH2wt, EZH2 Y641N mutant and EZH1, respectively. EZH2-IN-1 reduces bulk H3K27me3 and H3K27me2 levels. EZH2-IN-1 has the potential for diffuse large B cell lymphoma research .
|
-
- HY-W074912
-
|
(S)-5,5,5-Trifluoronorvaline; H-Nva(5,5,5-triF)-OH
|
mTOR
|
Neurological Disease
Metabolic Disease
Cancer
|
|
(S)-2-Amino-5,5,5-trifluoropentanoic acid ((S)-5,5,5-Trifluoronorvaline; H-Nva (5,5,5-triF)-OH) is a selective Sestrin-GATOR2 modulator that indirectly inhibits mTORC1 activity via a competitive binding mechanism. (S)-2-Amino-5,5,5-trifluoropentanoic acid can be used in research on cancer, metabolic diseases, neurodegenerative diseases, and muscle atrophy .\n
|
-

- HY-101190A
-
|
(3R,4S,5S)-SHP626; (3R,4S,5S)-LUM002
|
Drug Isomer
|
Inflammation/Immunology
|
|
(3R,4S,5S)-Volixibat ((3R,4S,5S)-SHP626) is an isomer of Volixibat (HY-101190). Volixibat is a highly selective, minimally absorbed, and competitive apical sodium-dependent bile acid transporter (ASBT) inhibitor. (3R,4S,5S)-Volixibat may be used in research on non-alcoholic steatohepatitis (NASH) .
|
-

- HY-12214A
-
|
|
CDK
Apoptosis
|
Cancer
|
|
NVP-2 is a potent and selective ATP-competitive cyclin dependent kinase 9 (CDK9) probe, inhibits CDK9/CycT activity with an IC50 of 0.514 nM. NVP-2 displays inhibitory effcts on CDK1/CycB, CDK2/CycA and CDK16/CycY kinases with IC50 values of 0.584 μM, 0.706 μM, and 0.605 μM, respectively. NVP-2 induces cell apoptosis.
|
-
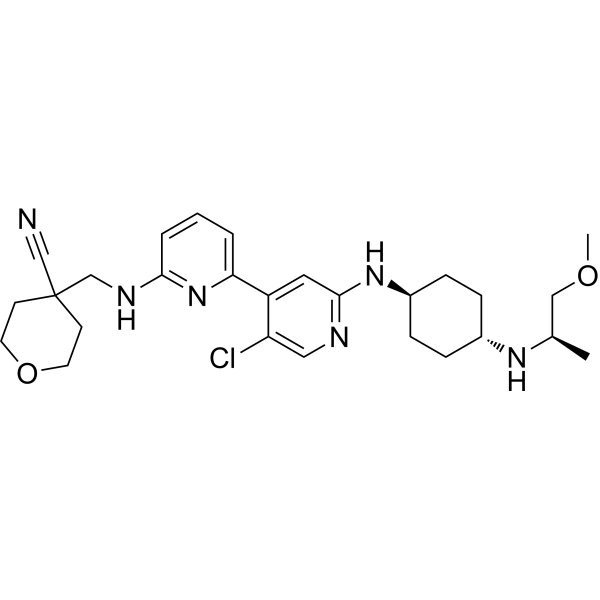
- HY-119413
-
|
|
Adenosine Receptor
|
Neurological Disease
|
|
9-Ethyladenine is a precursor of competitive antagonists of adenosine receptors (A1, A2, A3), with no significant inhibitory effect on adenine phosphoribosyltransferase (APRT). 9-Ethyladenine derivatives have high affinity and selectivity for A1 (Ki=27 nM), A2A (Ki=46 nM), and A3 (Ki=86 nM) receptors. 9-Ethyladenine does not inhibit brain APRT activity, can be used in the study of adenosine receptor-related diseases (such as nervous system diseases) models .
|
-
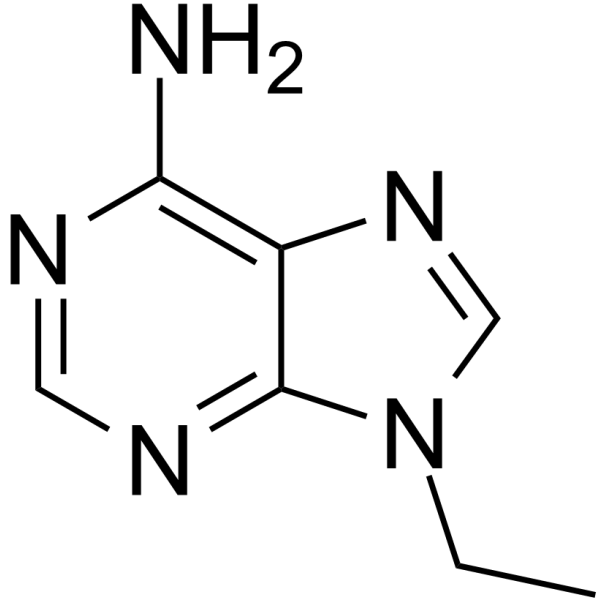
- HY-17623S
-
|
CJ-12420-d6; RQ-00000004-d6
|
Proton Pump
Na+/K+ ATPase
|
Metabolic Disease
|
|
Tegoprazan (CJ-12420; RQ-00000004), a potassium-competitive acid blocker, is a reversible, oral active and highly selective inhibitor of gastric H+/K+-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H +/K +-ATPases in vitro. Tegoprazan significantly improves colitis in mice and enhances the intestinal epithelial barrier function. Tegoprazan is promising for research of Inflammatory bowel, gastric acid-related, motilityimpaired diseases .
|
-
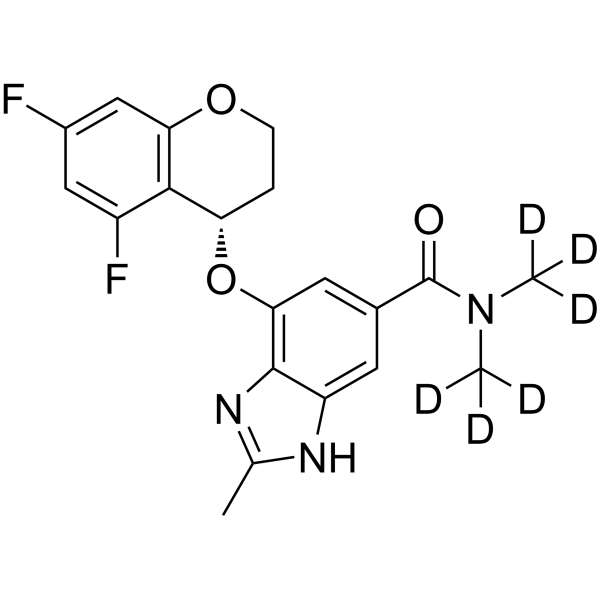
- HY-10256
-
|
SB 203580; RWJ 64809
|
Organoid
p38 MAPK
Autophagy
Mitophagy
HSP
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (SB 203580) is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod can inhibit p38 MAPK and lead to the inhibition of downstream HSP27 phosphorylation. Adezmapimod does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-
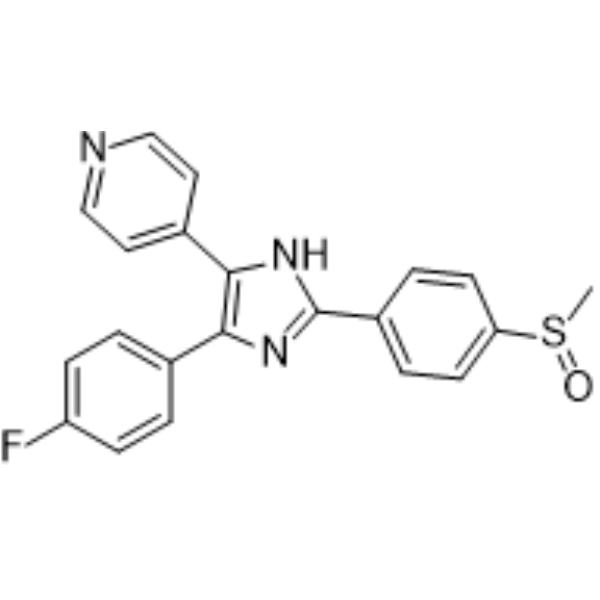
- HY-13404CR
-
|
INC280 dihydrochloride hydrate (Standard); INCB-28060 dihydrochloride hydrate (Standard)
|
c-Met/HGFR
Apoptosis
Reference Standards
|
Cancer
|
|
Capmatinib (dihydrochloride hydrate) (Standard) is the analytical standard of Capmatinib (dihydrochloride hydrate). This product is intended for research and analytical applications. Capmatinib (INC280; INCB28060) dihydrochloride hydrate is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib dihydrochloride hydrate can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib dihydrochloride hydrate potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib dihydrochloride hydrate is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-

- HY-13404R
-
|
INC280 (Standard); INCB28060 (Standard)
|
Reference Standards
c-Met/HGFR
Apoptosis
|
Cancer
|
|
Capmatinib (Standard) is the analytical standard of Capmatinib. This product is intended for research and analytical applications. Capmatinib (INC280; INCB28060) is a potent, orally active, selective, and ATP competitive c-Met kinase inhibitor (IC50=0.13 nM). Capmatinib can inhibit phosphorylation of c-MET as well as c-MET pathway downstream effectors such as ERK1/2, AKT, FAK, GAB1, and STAT3/5. Capmatinib potently inhibits c-MET-dependent tumor cell proliferation and migration and effectively induces apoptosis. Antitumor activity. Capmatinib is largely metabolized by CYP3A4 and aldehyde oxidase .
|
-

- HY-181861
-
|
|
Cholinesterase (ChE)
Monoamine Oxidase
Amyloid-β
Tau Protein
|
Neurological Disease
|
|
AChE/MAO-B-IN-9 (Compound E12) is an orally active, selective, reversible, non-competitive AChE and MAO-B inhibitor, with an IC50 of 0.156 μM against electric eel AChE. AChE/MAO-B-IN-9 inhibits Aβ40/42 fibril formation, promotes Aβ fibril depolymerization, and inhibits Tau protein fibril formation. AChE/MAO-B-IN-9 exerts antioxidant and neuroprotective effects, and improves scopolamine (HY-N0296)-induced memory impairment in mice. AChE/MAO-B-IN-9 can be used for the research of Alzheimer's disease .
|
-

- HY-W661499
-
|
|
Phosphatase
Apoptosis
Reactive Oxygen Species (ROS)
Caspase
|
Cancer
|
|
Orellanine, a nephrotoxic alkaloid found in Cortinarius orellanus, is an orally active and selective non-competitive inhibitor of alkaline phosphatase. Orellanine chelates iron, generates reactive oxygen species (ROS), induces DNA scission, forms ortho-semiquinone radicals, downregulates antioxidant defenses, and inhibits mitochondrial function. Orellanine induces caspase 8/9-mediated apoptosis. Orellanine inhibits synthesis of proteins, RNA, DNA, and mitochondrial protein synthesis, with metabolic activation required for cell-free protein synthesis inhibition. Orellanine can be used for the research of metastatic clear cell renal cell carcinoma, acute renal failure, chronic renal insufficiency, and kidney damage .
|
-
- HY-119080
-
|
|
Progesterone Receptor
|
Endocrinology
Cancer
|
|
CP8754 is an orally active, selective human progesterone receptor (hPR) antagonist that blocks progesterone-mediated signaling pathways. CP8754 competitively inhibits the binding of [ 3H]-progesterone to hPR. And in vitro, CP8754 inhibits progesterone-dependent exogenous luciferase and endogenous alkaline phosphatase expression; while in vivo, CP8754 inhibits rabbit endometrial transformation. CP8754 does not significantly bind to human glucocorticoid receptors (hGR), estrogen receptors (hER), or rat androgen receptors (rAR). CP8754 can be used in the study of progesterone-related diseases such as breast cancer, endometriosis, uterine fibroids, and meningioma, as well as hormone-dependent tumors .
|
-

- HY-W657887
-
|
|
Histone Methyltransferase
GSK-3
Tau Protein
Amyloid-β
Glucocorticoid Receptor
Androgen Receptor
Adrenergic Receptor
|
Neurological Disease
|
|
GSK-3β/G9a-IN-1 (Compound T2) is an orally active, selective, blood-brain-barrier permeable, competitive G9a (substrate-competitive, IC50: 1.1 μM) and GSK-3β (ATP competitive, IC50: 0.8 μM) inhibitor. GSK-3β/G9a-IN-1 is a potent H3K9me2 inhibitor that reshapes chromatin landscape. GSK-3β/G9a-IN-1 lowers tau phosphorylation, reduces Aβ aggregation. GSK-3β/G9a-IN-1 displays inhibition toward glucocorticoid receptor, androgen receptor, and alpha-2A adrenergic receptor. GSK-3β/G9a-IN-1 also upregulates SAGA complex members such as Eny2 and Sgf29. GSK-3β/G9a-IN-1 markedly improves memory, restores social behaviors, and increases synaptic complexity in late-onset Alzheimer’s disease .
|
-

- HY-101903R
-
|
|
Reference Standards
FABP
|
Cardiovascular Disease
Metabolic Disease
|
|
BMS-309403 (Standard) is the analytical standard of BMS-309403. This product is intended for research and analytical applications. BMS-309403 is a potent, orally active and selective adipocyte fatty acid binding protein (also known as FABP4, aP2) inhibitor with Kis of <2, 250, and 350 nM for FABP4, FABP3, and FABP5, respectively. BMS-309403 interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. BMS-309403 improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells .
|
-

- HY-110023R
-
|
|
Reference Standards
Serotonin Transporter
|
Neurological Disease
|
|
Zimelidine dihydrochloride (Standard) is the analytical standard of Zimelidine dihydrochloride (HY-110023). This product is intended for research and analytical applications. Zimelidine dihydrochloride is an orally active selective serotonin reuptake inhibitor. Zimelidine dihydrochloride competitively inhibits central 5-HT uptake and desensitizes 5-HT autoreceptors in dorsal raphe nucleus. Zimelidine dihydrochloride time-dependently modulates 5-HT neuronal firing and hippocampal CA3 responses. Zimelidine dihydrochloride strengthens central serotonergic neurotransmission and produces related behavioral changes. Zimelidine dihydrochloride exerts anxiolytic, analgesic, feeding-suppressive and tolerance-attenuating effects. Zimelidine dihydrochloride is used for the study of depressive disorders and analgesic tolerance .
|
-

- HY-P1291
-
|
|
PKA
Epigenetic Reader Domain
Flavivirus
|
Infection
Neurological Disease
|
|
PKI 14-22 amide, myristoylated is a selective, cAMP-dependent, competitive PKA inhibitor with Ki=~36 nM. The myristoylation modification of PKI 14-22 amide, myristoylated makes it more permeable to cell membranes and blood-brain barriers than the precursor molecule. PKI 14-22 amide, myristoylated can block the phosphorylation of cAMP-dependent downstream targets (such as CREB). PKI 14-22 amide, myristoylated can prevent the development of morphine analgesic tolerance in mice, and also inhibits protein translation and negative-strand RNA synthesis of Zika virus. PKI 14-22 amide, myristoylated can be used in research fields such as opioid tolerance mechanisms and antiviral drugs .
|
-
- HY-P2055
-
|
|
Endogenous Metabolite
|
Endocrinology
|
|
A-57696 is a cholecystokinin antagonist with selective activity at cortical CCK-B receptors (IC50 = 25 nM). A-57696 behaves as a competitive antagonist in reversing CCK8-stimulated pancreatic alpha-amylase secretion and phosphatidylinositol degradation. A-57696 fails to induce gallbladder contraction and inhibits CCK8-induced contraction. A-57696 behaves as a partial agonist at CCK-B/gastrin receptors on NCI-H345 cells, achieving 80% of the maximal CCK8 response. A-57696 and CCK8 inhibit each other in a calcium mobilization assay .
|
-

- HY-12215
-
|
PF-06463922
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Apoptosis
|
Cancer
|
|
Lorlatinib (PF-06463922) is a selective, orally active, brain-penetrant and ATP-competitive ROS1/ALK inhibitor with anticancer activity. Lorlatinib has Kis of <0.025 nM, <0.07 nM, and 0.7 nM for ROS1, wild type ALK, and ALK L1196M, respectively. Lorlatinib targets to EML4-ALK, and inhibits ALK phosphorylation with IC50s of 15-43 nM (ALK L1196), 14-80 nM (ALK G1269A), 38-50 nM (ALK 1151Tins), 77-113 nM (ALK G1202R), respectively .
|
-
- HY-101903AR
-
|
|
Reference Standards
FABP
|
Cardiovascular Disease
Metabolic Disease
|
|
BMS-309403 (sodium) (Standard) is the analytical standard of BMS-309403 (sodium). This product is intended for research and analytical applications. BMS-309403 sodium is a potent, orally active, and selective adipocyte fatty acid binding protein (also known as FABP4, aP2) inhibitor, with Kis of <2, 250, and 350 nM for FABP4, FABP3, and FABP5, respectively. BMS-309403 sodium interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. BMS-309403 sodium improves endothelial function in apolipoprotein E-deficient mice and in cultured human endothelial cells .
|
-

- HY-135509
-
|
PF-06463922 acetate
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Apoptosis
|
Cancer
|
|
Lorlatinib (PF-06463922) acetate is a selective, orally active, brain-penetrant and ATP-competitive ROS1/ALK inhibitor with anticancer activity. Lorlatinib acetate has Kis of <0.025 nM, <0.07 nM, and 0.7 nM for ROS1, wild type ALK, and ALK L1196M, respectively. Lorlatinib acetate targets to EML4-ALK, and inhibits ALK phosphorylation with IC50s of 15-43 nM (ALK L1196), 14-80 nM (ALK G1269A), 38-50 nM (ALK 1151Tins), 77-113 nM (ALK G1202R), respectively .
|
-

- HY-118835S
-
|
|
Isotope-Labeled Compounds
Serotonin Transporter
|
Neurological Disease
|
|
Zimeldine-d6 is the deuterium labeled Zimeldine (HY-118835) . Zimelidine is an orally active selective serotonin reuptake inhibitor. Zimelidine competitively inhibits central 5-HT uptake and desensitizes 5-HT autoreceptors in dorsal raphe nucleus. Zimelidine time-dependently modulates 5-HT neuronal firing and hippocampal CA3 responses. Zimelidine strengthens central serotonergic neurotransmission and produces related behavioral changes. Zimelidine exerts anxiolytic, analgesic, feeding-suppressive and tolerance-attenuating effects. Zimelidine is used for the study of depressive disorders and analgesic tolerance .
|
-

- HY-403733C
-
|
|
Androgen Receptor
|
Cancer
|
|
JJ-450 is a non-competitive antagonist androgen receptor (AR) that inhibits the transcriptional activity of wild-type AR and mutant AR F876L. JJ-450 has an IC50 of approximately 1-10 μM in inhibiting AR transcriptional activity in PC3 cells. It is selective for AR binding and does not compete with androgens for binding to the ligand binding domain (LBD) of AR. JJ-450 inhibits the transcriptional activity of AR and its splice variants (such as AR F876L) by inhibiting AR nuclear translocation and promoting the degradation of unliganded AR in the nucleus. JJ-450 can be used in castration-resistant prostate cancer (CRPC) studies that are resistant to Enzalutamide (MDV3100) (HY-70003) .
|
-

- HY-153718
-
|
|
Ligands for Target Protein for PROTAC
CDK
c-Myc
|
Cancer
|
|
KI-ARv-03 is a potent and selective ATP-competitive CDK9 inhibitor with an IC₅₀ of 0.15 μM (at 45 μM ATP), exhibiting over 130-fold selectivity against other CDKs (including CDK1-7). KI-ARv-03 reduces androgen receptor (AR)-driven transcription and proliferation in prostate cancer cells. KI-ARv-03 can be used for leukemia, pancreatic cancer, alveolar rhabdomyosarcoma (ARMS) and castration-resistant prostate cancer (CRPC) research. KI-ARv-03 is a ligand for target protein for PROTAC. KI-ARv-03 can be used to synthesize PROTAC KI-CDK9d-32 (HY-173523) [1][2].
|
-

- HY-180197
-
|
|
PKC
iGluR
Reactive Oxygen Species (ROS)
|
Cardiovascular Disease
Neurological Disease
|
|
PICK1 PDZ-IN-1 (Compound 6b) is a selective and brain-penetrant protein interacting with C kinase 1 (PICK1) PDZ domain inhibitor with a Ki of 27.73 μM. PICK1 PDZ-IN-1 can competitively inhibit the interaction between PICK1 and the GluA2 subunit of AMPA receptors. PICK1 PDZ-IN-1 can increase the survival rate of HT22 cells and primary cortical neuron cells induced by glutamate and inhibit ROS production. PICK1 PDZ-IN-1 exhibits neuroprotective effect and reduces the area of cerebral infarction. PICK1 PDZ-IN-1 can be used for the research of ischemic stroke .
|
-

- HY-P99463
-
|
AVB-500; AVB-S6-500
|
TAM Receptor
PI3K
Akt
p38 MAPK
|
Cancer
|
|
Batiraxcept (AVB-500; AVB-S6-500) is a selective, soluble AXL receptor and GAS6 inhibitor that targets the GAS6-AXL signaling axis. Batiraxcept is orally inactive and does not cross the blood-brain barrier. Batiraxcept competitively binds to GAS6 ((KD <1 nM), preventing its interaction with the AXL receptor tyrosine kinase, thereby inhibiting downstream PI3K/AKT and MAPK signaling pathways, reducing tumor cell glycolysis, angiogenesis, and metastatic potential. Batiraxcept has demonstrated antitumor activity in preclinical models of endometrial, cholangiocarcinoma, and ovarian cancer by inhibiting tumor growth, invasion, and metastasis .
|
-
- HY-126328
-
|
|
PKC
Interleukin Related
|
Inflammation/Immunology
|
|
PKC-theta inhibitor 1 is an orally active and selective ATP-competitive inhibitor of Protein Kinase Cθ (PKCθ), with a Ki value of 6 nM. PKC-theta inhibitor 1 inhibits T-cell-mediated inflammatory responses by suppressing the release of proinflammatory cytokines (IL-2 IC50 = 0.21 μM in anti-CD3/CD28-stimulated PBMCs; IL-17 IC50 = 1 μM in CD3/CD28-stimulated Th17 cells) PKC-theta inhibitor 1 significantly reduces symptoms in mice with ongoing experimental autoimmune encephalomyelitis (EAE). PKC-theta inhibitor 1 can be used for the study of T-cell-mediated inflammatory diseases such as multiple sclerosis .
|
-
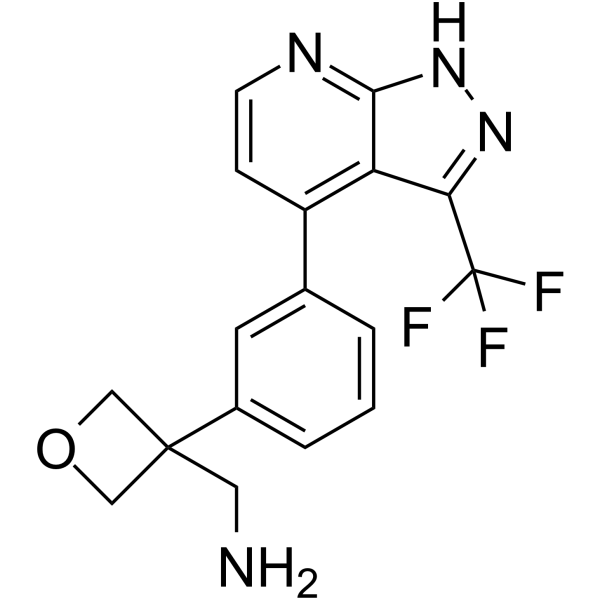
- HY-153789
-
|
|
PI5P4K
mTOR
|
Cancer
|
|
PI5P4Kγ-IN-1 is an ATP-competitive, highly selective chemical probe for PI5P4Kγ, with a Kd of 19 nM and an IC50 of 67 nM. PI5P4Kγ-IN-1 effectively inhibits PI5P4Kγ function and activates the mTORC1 signaling pathway in cells. PI5P4Kγ-IN-1 can be used in studies related to diseases such as breast cancer .
|
-
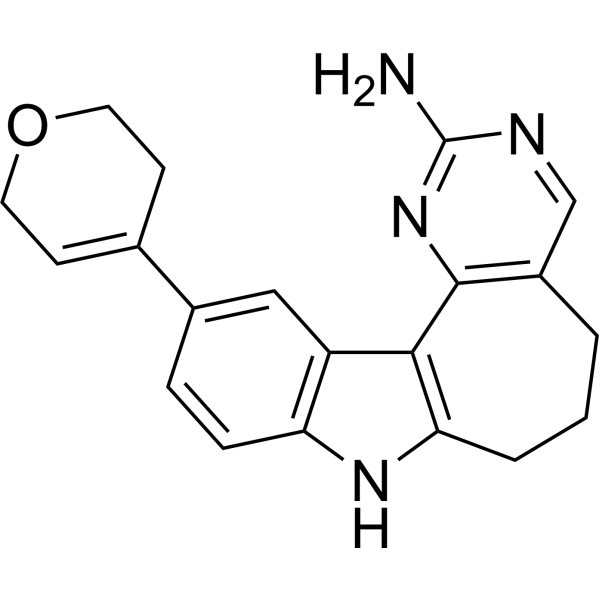
- HY-13285R
-
|
Debio 0719 (Standard)
|
LPL Receptor
YAP
Reference Standards
|
Neurological Disease
Cancer
|
|
Ki16425 (Standard) is the analytical standard of Ki16425. This product is intended for research and analytical applications. Ki16425 (Debio 0719) is a subtype-selective, competitive antagonist of the EDG-family receptors, LPA1 and LPA3 with Kis of 0.34 μM and 0.93 μM, respectively. Ki16425 (Debio 0719) reduces the LPA-induced activation of p42/p44 MAPK . Ki16425 can also inhibit LPA-induced dephosphorylation of Yes-associated protein (YAP)/TAZ in HEK293A cells .
|
-

- HY-I1070
-
|
(R)-Isoleucine
|
ASCT
|
Neurological Disease
|
|
D-Isoleucine is a selective competitive activator of the Asc-1 antiporter (Ki=0.98 mM). D-Isoleucine promotes the release of D-serine and glycine by binding to the Asc-1 protein on the neuronal cell membrane, and enhances NMDA receptor-dependent synaptic plasticity. D-Isoleucine can be used in the study of neurodegenerative diseases (such as Alzheimer's disease and schizophrenia). D-Isoleucine also acts as a non-classical D-amino acid, interferes with bacterial peptidoglycan synthesis, and inhibits the formation of Staphylococcus aureus biofilm, and has potential antibacterial application value[1][2].
|
-
- HY-146691
-
|
|
Monoamine Oxidase
|
Neurological Disease
|
|
hMAO-B-IN-2 (compound 6j) is an orally active, potent, selective and BBB penetrated and competitive reversible hMAO-B inhibitor, with an IC50 of 4 nM. hMAO-B-IN-2 shows low toxicity and good neuroprotective effects in SH-SY5Y cell. hMAO-B-IN-2 can be used for alzheimer’s disease research . hMAO-B-IN-2 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-100903
-
|
nor-Binaltorphimine dihydrochloride; nor-BNI dihydrochloride
|
Opioid Receptor
|
Neurological Disease
|
|
Norbinaltorphimine dihydrochloride (nor-Binaltorphimine dihydrochloride; nor-BNI dihydrochloride) is a selective, long-acting competitive antagonist of the κ-opioid receptor. Norbinaltorphimine dihydrochloride blocks κ-opioid receptor-mediated analgesic effects, and inhibits butorphanol-induced changes in κ-opioid receptor binding kinetics, desensitization and down-regulation. Norbinaltorphimine dihydrochloride suppresses specific opioid withdrawal symptoms, precipitates withdrawal behaviors in butorphanol-dependent rats, and serves as a molecular probe for studying κ-opioid receptor-agonist interactions. Norbinaltorphimine dihydrochloride is applicable to research related to neurological disorders such as pain .
|
-
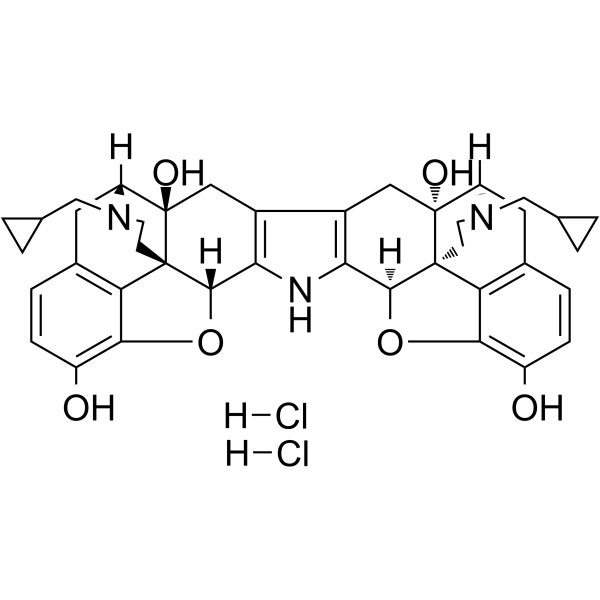
- HY-117089
-
|
|
Environmental Pollutants
Fungal
|
Infection
|
|
Tetraconazole is a selective irreversible inhibitor of 14-α-sterol demethylase (CYP51) with antifungal activity. Tetraconazole competitively binds to the enzyme to block fungal ergosterol synthesis, resulting in cell membrane damage. The EC50 of tetraconazole against wheat pathogens is 0.382-0.802 mg/L, and the EC50 against onion root tip meristem cell growth is 6.7 mg/L, and (R)-(+)-Tetraconazole is 1.49-1.98 times more active than (S)-(-)-Tetraconazole. Tetraconazole can also induce oxidative stress and chromosomal aberrations in plant cells .
|
-
- HY-10524R
-
|
|
Reference Standards
IGF-1R
Insulin Receptor
Apoptosis
|
Endocrinology
Cancer
|
|
GSK1904529A (Standard) is the analytical standard of GSK1904529A (HY-10524). This product is intended for research and analytical applications. GSK1904529A is a potent, selective, orally active, and ATP-competitive inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR), with IC50s of 27 and 25 nM, respectively. GSK1904529A shows poor activity (IC50>1 μM) in 45 other serine/threonine and tyrosine kinases. GSK1904529A exhibits anti-tumor activity .
|
-

- HY-182430
-
|
|
JAK
STAT
Apoptosis
|
Cancer
|
|
NVP-BVB808 is a selective and ATP-competitive JAK2 inhibitor with an IC50 of 0.35 nM. NVP-BVB808 binds to JAK2’s ATP-binding site, stabilizes JAK2’s active conformation, increases JAK2 activation loop phosphorylation, and blocks downstream kinase function. NVP-BVB808 exhibits antiproliferative and pro-apoptosis effects, suppresses constitutive STAT5a phosphorylation. NVP-BVB808 can be used for the research of cancer, such as leukemia .
|
-

- HY-10383
-
|
|
Endothelin Receptor
|
Cardiovascular Disease
Metabolic Disease
|
|
J-104132 (L-753037) is a potent, orally active, selective and competitive ETA/ETB receptor antagonist with Ki of 0.034 nM for ETA and 0.104 nM for ETB receptors. J-104132 inhibits Endothelin-1 (ET-1) (HY-P71446)-induced signaling and vascular contractions in vitro. J-104132 alleviates hypertension, vascular remodeling, and diabetic endothelial dysfunction in vivo by dual ETA/ETB blockade. J-104132 can be used for research on diabetic vascular complications [1][3].
|
-

- HY-10683R
-
|
|
Reference Standards
PI3K
mTOR
|
Cancer
|
|
PKi-402 (Standard) is the analytical standard of PKi-402 (HY-10683). This product is intended for research and analytical applications. PKi-402 is a selective, reversible, ATP-competitive inhibitor of PI3K, including PI3K-α mutants, and mTOR (IC50=2, 3, 7,14 and 16 nM for PI3Kα, mTOR, PI3Kβ, PI3Kδ and PI3Kγ).
|
-

- HY-133178
-
|
3,4,8,9-Tetrahydroxy urolithin
|
Ephrin Receptor
PPAR
AMPK
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Urolithin D (3,4,8,9-Tetrahydroxy urolithin) is a colonic metabolite of Ellagitannins and a competitive, reversible, and selective antagonist of the EphA receptor. Urolithin D inhibits EphA2-ephrin-A1 binding with an IC50 of 0.9 μM. Urolithin D is also a potent antioxidant that scavenges free radicals and repairs oxidized DNA damage. Additionally, Urolithin D suppresses triglyceride accumulation and promotes fatty acid oxidation by activating the AMPK signaling pathway. Urolithin D can be used for research on tumors, metabolic, and inflammatory diseases .
|
-
- HY-117040
-
|
Norbinaltorphimine; NorBNI
|
Opioid Receptor
|
Neurological Disease
|
|
nor-Binaltorphimine (Norbinaltorphimine; NorBNI) is a selective, long-acting competitive antagonist of the κ-opioid receptor. nor-Binaltorphimine blocks κ-opioid receptor-mediated analgesic effects, and inhibits butorphanol-induced changes in κ-opioid receptor binding kinetics, desensitization and down-regulation. nor-Binaltorphimine suppresses specific opioid withdrawal symptoms, precipitates withdrawal behaviors in butorphanol-dependent rats, and serves as a molecular probe for studying κ-opioid receptor-agonist interactions. nor-Binaltorphimine is applicable to research related to neurological disorders such as pain .
|
-
- HY-125959
-
Ucf-101
1 Publications Verification
|
Apoptosis
|
Cardiovascular Disease
Neurological Disease
|
|
Ucf-101 is a selective and competitive inhibitor of pro-apoptotic protease Omi/HtrA2, with an IC50 of 9.5 μM for His-Omi. Ucf-101 exhibits very little activity against various other serine proteases (IC50>200 μM). Ucf-101 has a natural red fluorescence at 543 nm that is used to monitor its ability to enter mammalian cells. Ucf-101 has a significant cardioprotective effect against MI/R injury and also has certain neuroprotective effect .
|
-
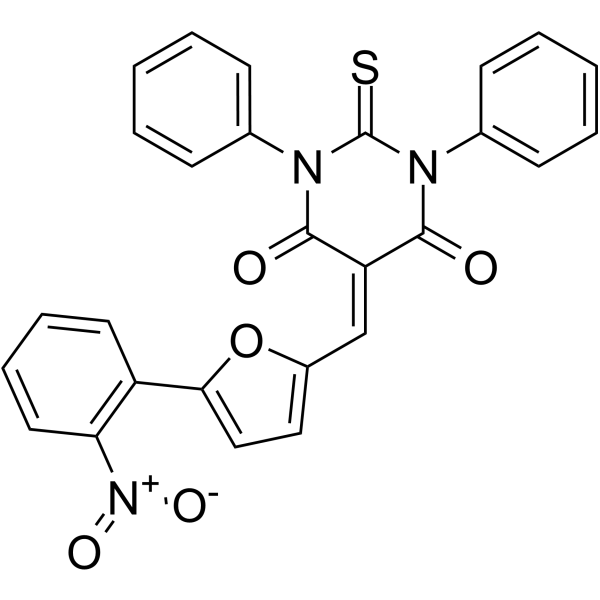
- HY-10256AR
-
|
SB 203580 hydrochloride (Standard); RWJ 64809 hydrochloride (Standard)
|
Organoid
Reference Standards
p38 MAPK
Autophagy
Mitophagy
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (hydrochloride) (Standard) is the analytical standard of Adezmapimod (hydrochloride). This product is intended for research and analytical applications. Adezmapimod (SB 203580; RWJ 64809) hydrochloride is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod hydrochloride inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod hydrochloride does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-

- HY-100607A
-
|
ONO1101 hydrochloride
|
Adrenergic Receptor
Calcium Channel
Potassium Channel
Reactive Oxygen Species (ROS)
|
Cardiovascular Disease
Inflammation/Immunology
Endocrinology
|
|
Landiolol (ONO1101) hydrochloride is a highly selective, ultra-short-acting competitive inhibitor of β1 adrenergic receptors. Landiolol hydrochloride specifically blocks cardiac β1 receptors, reducing heart rate and myocardial oxygen consumption. Landiolol hydrochloride inhibits TNF-α-induced excessive mitochondrial oxygen consumption and reactive oxygen species production in a sepsis model, alleviating renal injury. Landiolol hydrochloride has little effect on cardiac ion channels (such as L-type calcium current and inward rectifier potassium current) and has a weak negative inotropic effect. Landiolol hydrochloride can be used for perioperative tachycardia control and protection studies of sepsis-related acute kidney injury .
|
-
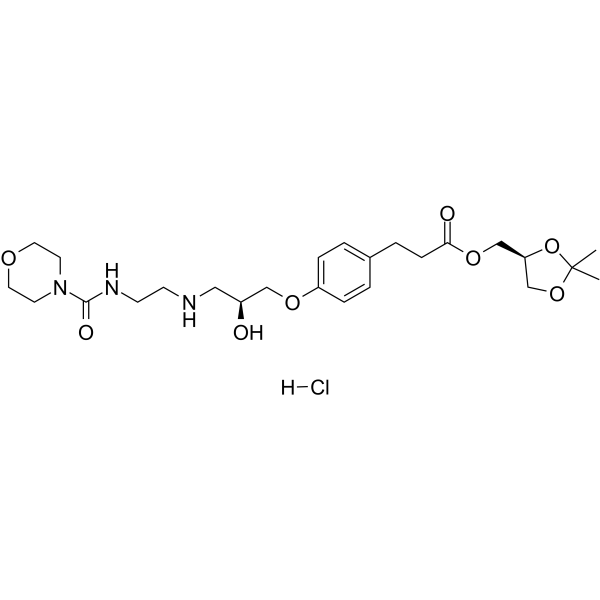
- HY-111126
-
K67
1 Publications Verification
|
p62
Keap1-Nrf2
|
Cancer
|
|
K67 is a selective the interaction between Keap1 and S349 phosphorylated p62 inhibitor, with an IC50 of 1.5 μM. K67 has a weaker inhibitory effect on the interaction between Keap1 and Nrf2 (IC50 is 6.2 μM). K67 competitively binds to the binding site of Keap1 with p-p62, blocking the abnormal activation of the p62-dependent Nrf2 pathway. K67 inhibits tumor cell proliferation and enhances the sensitivity of hepatocellular carcinoma (HCC) to chemotherapeutic drugs by restoring Keap1-mediated ubiquitination and degradation of Nrf2 .
|
-
- HY-100607
-
|
ONO1101
|
Adrenergic Receptor
Calcium Channel
Potassium Channel
Reactive Oxygen Species (ROS)
|
Cardiovascular Disease
Inflammation/Immunology
Endocrinology
|
|
Landiolol (ONO1101) is a highly selective, ultra-short-acting competitive inhibitor of β1 adrenergic receptors. Landiolol specifically blocks cardiac β1 receptors, reducing heart rate and myocardial oxygen consumption. Landiolol inhibits TNF-α-induced excessive mitochondrial oxygen consumption and reactive oxygen species production in a sepsis model, alleviating renal injury. Landiolol has little effect on cardiac ion channels (such as L-type calcium current and inward rectifier potassium current) and has a weak negative inotropic effect. Landiolol can be used for perioperative tachycardia control and protection studies of sepsis-related acute kidney injury .
|
-

- HY-183942
-
|
|
Deubiquitinase
Apoptosis
MDM-2/p53
PARP
Caspase
|
Cancer
|
|
USP7-IN-20 is a highly selective USP7 inhibitor that binds allosterically to the allosteric pocket of USP7 in a non-competitive and reversible manner. USP7-IN-20 downregulates MDM2 protein levels and stabilizes p53, thereby inducing p21 expression and enhancing the ubiquitin-dependent degradation of MDM2. USP7-IN-20 effectively binds endogenous USP7 to inhibit cancer cell proliferation, and also induces apoptosis by promoting the cleavage of PARP and caspase 3. USP7-IN-20 can be widely used in cancer-related basic and translational research.
|
-

- HY-P1291A
-
|
|
PKA
Epigenetic Reader Domain
Flavivirus
|
Infection
Neurological Disease
|
|
PKI 14-22 amide, myristoylated TFA is a selective, cAMP-dependent, competitive PKA inhibitor with Ki=~36 nM. The myristoylation modification of PKI 14-22 amide, myristoylated TFA makes it more permeable to cell membranes and blood-brain barriers than the precursor molecule. PKI 14-22 amide, myristoylated TFA can block the phosphorylation of cAMP-dependent downstream targets (such as CREB). PKI 14-22 amide, myristoylated TFA can prevent the development of analgesic tolerance in mice, and also inhibits protein translation and negative-strand RNA synthesis of Zika virus. PKI 14-22 amide, myristoylated TFA can be used in research fields such as opioid tolerance mechanisms and antiviral drugs .
|
-
- HY-161449
-
|
|
11β-HSD
|
Metabolic Disease
|
|
JTT-654 is an orally active, potent and selective11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor. The IC50 of JTT-654 for 11β-HSD1 is 4.65, 0.97, and 0.74 nM in human, rat, and mouse recombinant enzymes, respectively. JTT-654 showed competitive inhibition against human recombinant enzyme. The IC50 value for human 11β-HSD2 is > 30 μM (human 11β-HSD2 is responsible for the reverse reaction against human 11β-HSD1). JTT-654 ameliorates insulin resistance and non-obese type 2 diabetes by inhibiting adipose tissue and liver 11β-HSD1 .
|
-

- HY-19261
-
|
|
Cholecystokinin Receptor
|
Metabolic Disease
|
|
T-0632 is a CCK A receptor antagonist that exhibits significant pharmacological properties in in vitro studies. T-0632 competitively inhibits the binding of [125I]CCK-8 to rat pancreatic CCK A receptors with a K_i value of 0.24 nM, which is significantly lower than the K_i value for guinea pig CCK B receptors. T-0632 has higher selectivity in inhibiting CCK-8-stimulated pancreatic enzyme release, with an IC_50 value of 5.0 nM, which is more advantageous than L-364,718 and loxiglumide. In rabbit gallbladder smooth muscle, the antagonistic effects of T-0632 and loxiglumide are reversible, while L-364,718 shows a persistent inhibitory effect. These results indicate that T-0632 is a highly potent, reversible and more selective CCK A receptor antagonist.
|
-

- HY-10256R
-
|
SB 203580 (Standard); RWJ 64809 (Standard)
|
Organoid
Reference Standards
p38 MAPK
Autophagy
Mitophagy
HSP
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (Standard) is the analytical standard of Adezmapimod. This product is intended for research and analytical applications. Adezmapimod (SB 203580) is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod can inhibit p38 MAPK and lead to the inhibition of downstream HSP27 phosphorylation. Adezmapimod does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-

- HY-103490R
-
|
EDHS-206 (Standard)
|
Reference Standards
MAP3K
Apoptosis
|
Infection
Inflammation/Immunology
Cancer
|
|
Takinib (Standard) is the analytical standard of Takinib. This product is intended for research and analytical applications. Takinib (EDHS-206) is an orally active and selective TAK1 inhibitor (IC50=9.5 nM), more than 1.5 log more potent than the second and third ranked targets, IRAK4 (120 nM) and IRAK1 (390 nM), respectively. Takinib is an inhibitor of autophosphorylated TAK1 that non-competitively binds within the ATP binding pocket. Takinib induces apoptosis following TNFα stimulation in cell models of rheumatoid arthritis and metastatic breast cancer. Takinib is also a P. falciparum protein kinase 9 (PfPK9) inhibitor (KD(app) of 0.46 μM) .
|
-

- HY-B0927R
-
|
(-)-β-Hydrastine (Standard); (1R,9S)-β-Hydrastine (Standard)
|
Reference Standards
Tyrosine Hydroxylase
Dopamine Receptor
OAT
|
Others
|
|
Hydrastine (Standard) is the analytical standard of Hydrastine (HY-B0927). This product is intended for research and analytical applications. Hydrastine ((-)-β-Hydrastine; (1R,9S)-β-Hydrastine) is a selective competitive inhibitor of tyrosine hydroxylase (TH), inhibiting dopamine biosynthesis (IC50=20.7 μM, PC12 cells). Hydrastine also inhibits the organic cation transporter OCT1 (IC50=6.6 μM). Hydrastine may cause neuronal toxicity through mitochondrial dysfunction rather than oxidative stress damage, and can aggravate cell apoptosis when combined with L-DOPA. Hydrastine can be used to study Parkinson's disease-related dopaminergic neuronal damage .
|
-

- HY-19633A
-
|
|
Endogenous Metabolite
|
Neurological Disease
|
|
CS-003 is a triple neurokinin receptor antagonist with activity in inhibiting neurokinin-related respiratory diseases. CS-003 exhibits high affinity for human neurokinin 1, 2 and 3 receptors, withKi values of 2.3 nM, 0.54 nM and 0.74 nM respectively. The Ki values of CS-003 on the guinea pig neurokinin receptor are 5.2 nM, 0.47 nM and 0.71 nM respectively, showing superior inhibitory effect. CS-003 significantly inhibits the formation of inositol phosphate involving substance P, neurokinin A and neurokinin B through competitive antagonism. CS-003 significantly inhibits citric acid-induced cough, and its effect is better than other selective neurokinin receptor antagonists .
|
-

- HY-15574
-
|
SB-207266
|
5-HT Receptor
|
Cardiovascular Disease
|
|
Piboserod is an orally available selective antagonist of the 5-HT4 receptor, with a Ki value of approximately 0.1 nM for human 5-HT4 receptors. Piboserod can competitively bind to the 5-HT4 receptor and block the activation of the 5-HT4 receptor. Piboserod can inhibit the enhancing effect of 5-HT on the nerve-mediated contraction response of the human bladder detrusor muscle. Piboserod is mainly used in the research of urinary system diseases (such as overactive bladder) and cardiovascular diseases (such as chronic heart failure) .
|
-
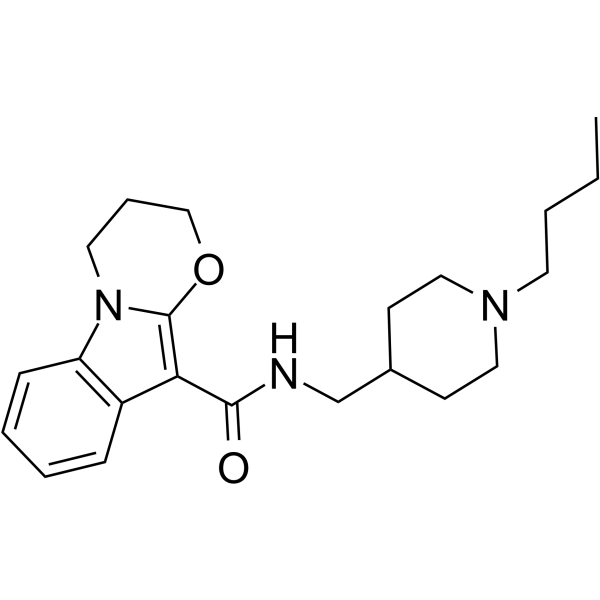
- HY-B1194
-
|
(±)-Tetramisole hydrochloride; DL-Tetramisole hydrochloride; R-829
|
Potassium Channel
Parasite
PKA
|
Infection
Cardiovascular Disease
|
|
Tetramisole hydrochloride is an orally active, selective inward rectifier potassium channel agonist with an EC50 of approximately 30 μM for the Kir2.1 subunit. Tetramisole hydrochloride is also an anti-nematode agent that blocks neuromuscular transmission by non-competitive depolarization. Tetramisole hydrochloride promotes the forward transport of Kir2.1 channels, hyperpolarizes the resting potential (RP), shortens the action potential duration (APD), inhibits intracellular calcium overload and the PKA signaling pathway, and exerts anti-arrhythmic and anti-myocardial remodeling activities. Tetramisole hydrochloride can be used in cardiac electrophysiology research and research related to myocardial ischemia and heart failure .
|
-
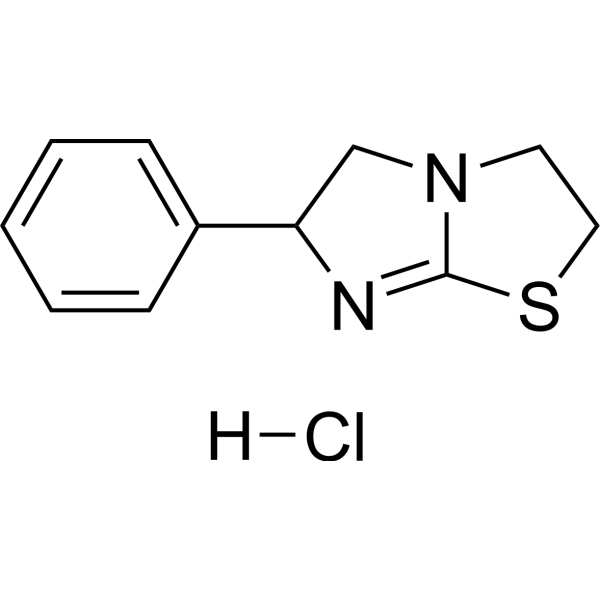
- HY-103721R
-
|
|
Sirtuin
Reference Standards
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
SIRT6-IN-2 (Standard) is the analytical standard of SIRT6-IN-2 (HY-103721). This product is intended for research and analytical applications. SIRT6-IN-2 (Compound 5) is a selective and competitive SIRT6 inhibitor (IC50: 34 μM). SIRT6-IN-2 increases acetylation of H3K9 and increases glucose uptake in cultured cells. SIRT6-IN-2 also reduces T cell proliferation. SIRT6-IN-2 has immunosuppressive and chemosensitizing effects .
|
-

- HY-180195
-
|
|
HSP
Integrin
|
Neurological Disease
Cancer
|
|
Grp94-IN-3 (Compound 47) is a selective ATP competitive inhibitor of Grp94, with a Kd value of 76 nM. Grp94-IN-3 has a much lower affinity for Hsp90α, with a Kd value of 9.17 μM. Grp94-IN-3 induces the degradation of integrin α2 (Integrin α2) in MDA-MB-231 cells and reduces the intracellular accumulation of mutant cardiac proteins in human trabecular meshwork cells. Grp94-IN-3 can be used for the study of metastatic cancer and open-angle glaucoma .
|
-

- HY-W414915
-
|
CGP 48933 methyl ester
|
Drug Derivative
Angiotensin Receptor
|
Cardiovascular Disease
Cancer
|
|
Valsartan (CGP 48933) methyl ester is the methyl ester derivative of Valsartan (HY-18204). Valsartan is a selective and orally active angiotensin II type 1 (AT1) receptor blocker (ARB) with potent antihypertensive and cardioprotective effects. Valsartan competitively binds to AT1 receptors, inhibiting the binding of angiotensin II to AT1 receptors, thereby blocking angiotensin II-mediated vasoconstriction, sodium retention, and myocardial hypertrophy signaling pathways. Valsartan reduces systolic blood pressure in L-NAME-induced hypertensive rats. Valsartan can be used for the study and treatment of arterial hypertension, hypertensive heart disease, and heart failure .
|
-

- HY-B1194A
-
|
|
Potassium Channel
Parasite
PKA
|
Infection
Cardiovascular Disease
|
|
Tetramisole is an orally active, selective inward rectifier potassium channel agonist with an EC50 of approximately 30 μM for the Kir2.1 subunit. Tetramisole is also an anti-nematode agent that blocks neuromuscular transmission by non-competitive depolarization. Tetramisole promotes the forward transport of Kir2.1 channels, hyperpolarizes the resting potential (RP), shortens the action potential duration (APD), inhibits intracellular calcium overload and the PKA signaling pathway, and exerts anti-arrhythmic and anti-myocardial remodeling activities. Tetramisole can be used in cardiac electrophysiology research and research related to myocardial ischemia and heart failure .
|
-

- HY-139481
-
TL-895
1 Publications Verification
|
Btk
BMX Kinase
Interleukin Related
TNF Receptor
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
TL-895 is a potent, orally active, ATP-competitive, and highly selective irreversible BTK inhibitor. TL-895 is active against recombinant BTK (average IC50: 1.5 nM) and inhibits only three additional kinases BLK, BMX (IC50 = 1.6 nM) and TXK with IC50 within tenfold of BTK activity. TL-895 inhibits BTK auto-phosphorylation at the Y223 phosphorylation site (IC50: 1-10 nM). The TL-895 effectively inhibits the production of inflammatory factors such as IL-8, IL-1β, MCP-1 and TNF-α by monocytes or macrophages, and reduces the chemotactic migration of MF cells towards SDF-1. TL-895 is used be for studies of chronic lymphocytic leukemia (CLL), myelofibrosis (MF), and B-cell malignancies .
|
-
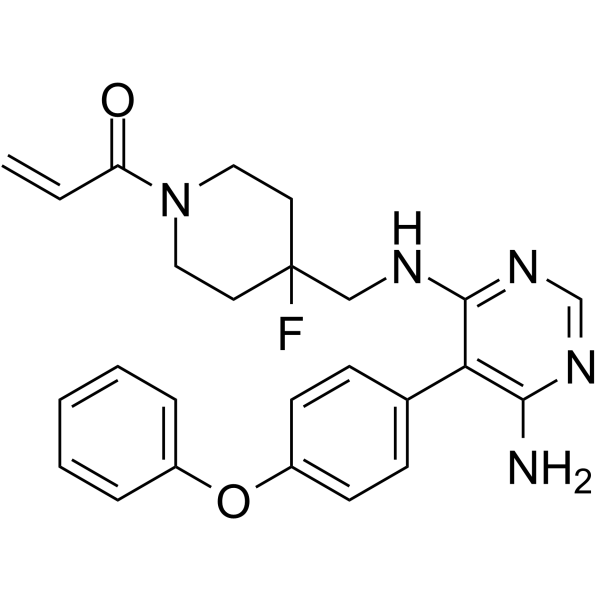
- HY-W380450
-
|
Viloxazin; Emovit
|
5-HT Receptor
|
Neurological Disease
|
|
Viloxazine is a non-brain-penetrant, selective norepinephrine transporter (NET) inhibitor (IC50= 0.26 μM) and 5-HT receptor modulator. Viloxazine antagonizes 5-HT2B receptors (Ki=4.2 μM) and agonizes 5-HT2C receptors (EC50= 32 μM), respectively, and enhances 5-HT neurotransmission by modulating 5-HT2B/C receptors. Viloxazine also competitively inhibits NET from increasing NE and DA levels in the synaptic cleft, and can be used in the study of attention deficit hyperactivity disorder (ADHD) .
|
-
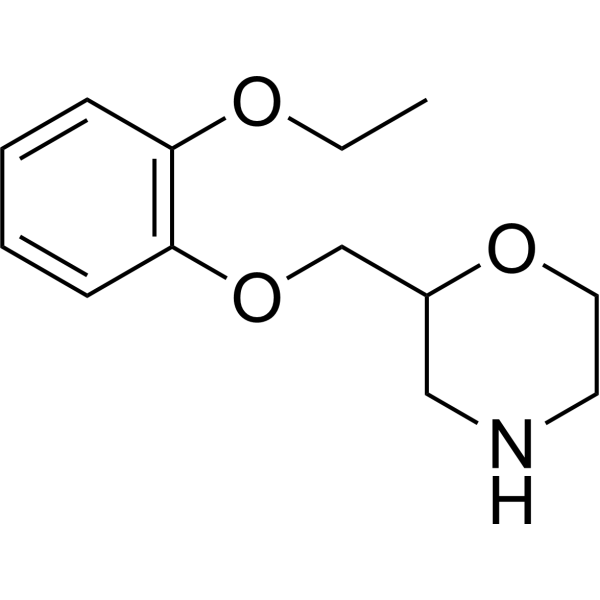
- HY-153228
-
|
PBI-0451
|
SARS-CoV
|
Infection
|
|
Pomotrelvir is a selective, competitive, orally active covalent inhibitor of the SARS-CoV-2 main protease (M pro), with an IC50 of 24 nM for wild-type SARS-CoV-2 M pro. Pomotrelvir inhibits viral polyprotein processing, thereby preventing viral replication. Pomotrelvir has shown broad antiviral activity against multiple SARS-CoV-2 variants (including Omicron) in cell-based experiments, and has an additive effect when combined with nucleoside analogs that target viral RNA synthesis. Pomotrelvir is primarily used for the research and development of COVID-19 antiviral drugs, especially for infections caused by SARS-CoV-2 and its variants .
|
-
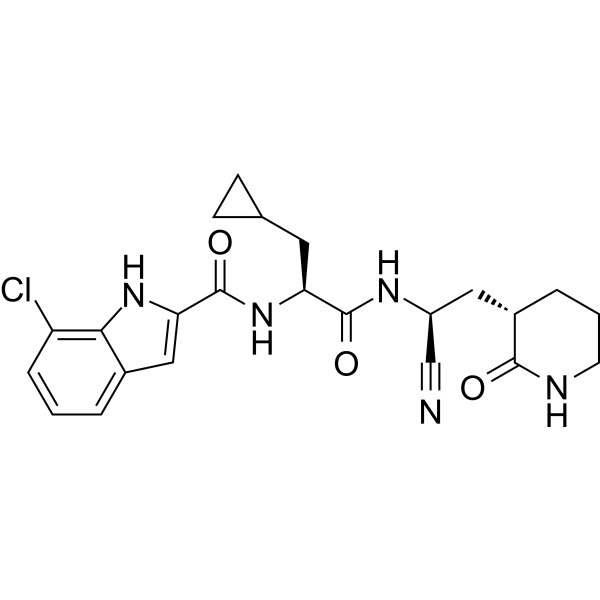
- HY-W380450R
-
|
Viloxazin (Standard); Emovit (Standard)
|
Reference Standards
5-HT Receptor
|
Neurological Disease
|
|
Viloxazine is a non-brain-penetrant, selective norepinephrine transporter (NET) inhibitor (IC50= 0.26 μM) and 5-HT receptor modulator. Viloxazine antagonizes 5-HT2B receptors (Ki=4.2 μM) and agonizes 5-HT2C receptors (EC50= 32 μM), respectively, and enhances 5-HT neurotransmission by modulating 5-HT2B/C receptors. Viloxazine also competitively inhibits NET from increasing NE and DA levels in the synaptic cleft, and can be used in the study of attention deficit hyperactivity disorder (ADHD) .
|
-

- HY-160696
-
|
|
CD73
|
Cancer
|
|
ORIC-533 is an orally active, highly selective, AMP-competitive CD73 inhibitor that potently blocks adenosine production with sub-nanomolar affinity (Ka=0.03 nM). In multiple myeloma, ORIC-533 restores and enhances the cytotoxicity of the immune system against tumor cells through multiple immunological mechanisms, including reversing the immunosuppressive microenvironment, inducing immunogenic cell death, and activating dendritic cells, T cells and NK cells, with no direct toxicity to normal cells. The combination of ORIC-533 with Daratumumab (HY-P9915) synergistically enhances anti-tumor efficacy, significantly increases intratumoral CD8 + T cell infiltration and inhibits tumor growth in vivo .
|
-
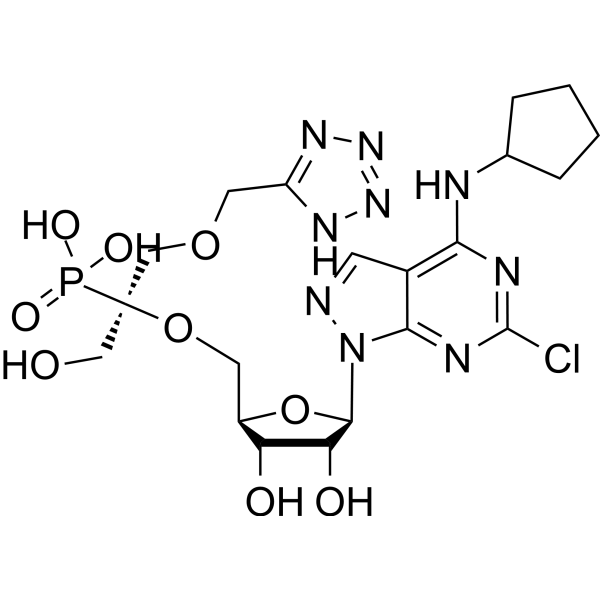
- HY-12215R
-
|
PF-06463922 (Standard)
|
Reference Standards
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Apoptosis
|
Cancer
|
|
Lorlatinib (Standard) is the analytical standard of Lorlatinib. This product is intended for research and analytical applications. Lorlatinib (PF-06463922) is a selective, orally active, brain-penetrant and ATP-competitive ROS1/ALK inhibitor with anticancer activity. Lorlatinib has Kis of <0.025 nM, <0.07 nM, and 0.7 nM for ROS1, wild type ALK, and ALK L1196M, respectively. Lorlatinib targets to EML4-ALK, and inhibits ALK phosphorylation with IC50s of 15-43 nM (ALK L1196), 14-80 nM (ALK G1269A), 38-50 nM (ALK 1151Tins), 77-113 nM (ALK G1202R), respectively .
|
-

- HY-P5542
-
|
SB-01; Peniel 2000
|
Factor Xa
TGF-beta/Smad
|
Inflammation/Immunology
|
|
Vicatertide (SB-01, Peniel 2000) is a polypeptide with both competitive inhibitory activity against TGF-β1 and selective inhibitory activity against human factor XIa (hFXIa, with a Ka of 80 nM for hFXIa). Vicatertide binds allosterically to the two binding sites of dimeric hFXI/hFXIa, while directly binding to activated TGF-β1, selectively blocking the Smad1/5/8 pathway and maintaining low-level activation of the Smad2 pathway to enhance the synthesis of type Ⅱ collagen and aggrecan. Vicatertide inhibits thrombus formation in arteriovenous thrombosis models, and also reduces thrombus weight and thrombus incidence in mouse lung cancer models. Vicatertide can be used for research on degenerative disc disease and thrombosis-related diseases .
|
-
- HY-101308
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
MRS2179 tetrasodium is a competitive P2Y1 receptor antagonist, with a Kb of 102 nM and a pA2 of 6.99 for turkey P2Y1 receptor. MRS2179 tetrasodium is selective for P2Y1 over P2X1 (IC50=1.15 µM), P2X3 (12.9 µM), P2X2, P2X4, P2Y2, P2Y4, and P2Y6 receptors . MRS2179 tetrasodium inhibits platelet aggregation .
|
-
- HY-N1151
-
|
|
Bacterial
Cholinesterase (ChE)
MMP
TNF Receptor
|
Infection
Neurological Disease
Inflammation/Immunology
|
|
Thunberginol C is an orally active, selective, and non-competitive inhibitor of AChE and BChE, with IC50 values of 41.96 and 42.36 μM, respectively. Thunberginol C exerts cytoprotective, pro-collagen type I restorative, MMP-1 inhibitory, hyaluronic acid restorative, anti-photoaging effects in skin cells. Thunberginol C exerts neuroprotective, anxiolytic, TNF-α inhibitory, neuroinflammation inhibitory, and oxidative stress inhibitory effects. Thunberginol C can be used for the research of Alzheimer’s disease, UVB-induced skin photoaging, allergic reactions, oral bacterial infections, and stress-induced anxiety .
|
-
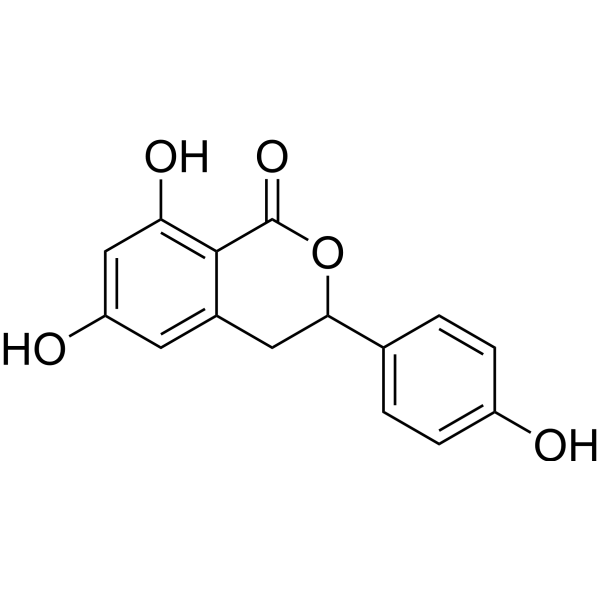
- HY-W856819
-
|
|
Opioid Receptor
Cholinesterase (ChE)
Drug Metabolite
nAChR
|
Neurological Disease
|
|
Eseroline is a potent μ-opioid receptor agonist, which is the hydrolytic metabolite of Physostigmine (HY-N6608). Eseroline is a selective and competitive acetylcholinesterase (AChE) inhibitor, with its Ki values for AChE and BuChE being 0.1 μM and 200 μM respectively. Eseroline has nicotinic acetylcholine receptor allosteric enhancing ligand (nAChR-APL) activity, meaning it does not activate the receptor but significantly enhances the signal transduction of Ach triggered by the receptor. Eseroline is neurotoxic, causing cell membrane damage (LDH leakage) and energy metabolism collapse (ATP depletion). Eseroline can be used for the study of Alzheimer's disease .
|
-

- HY-101308A
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
MRS2179 tetrasodium hydrate is a competitive P2Y1 receptor antagonist, with a Kb of 102 nM and a pA2 of 6.99 for turkey P2Y1 receptor. MRS2179 tetrasodium hydrate is selective for P2Y1 over P2X1 (IC50=1.15 µM), P2X3 (12.9 µM), P2X2, P2X4, P2Y2, P2Y4, and P2Y6 receptors . MRS2179 tetrasodium hydrate inhibits platelet aggregation .
|
-
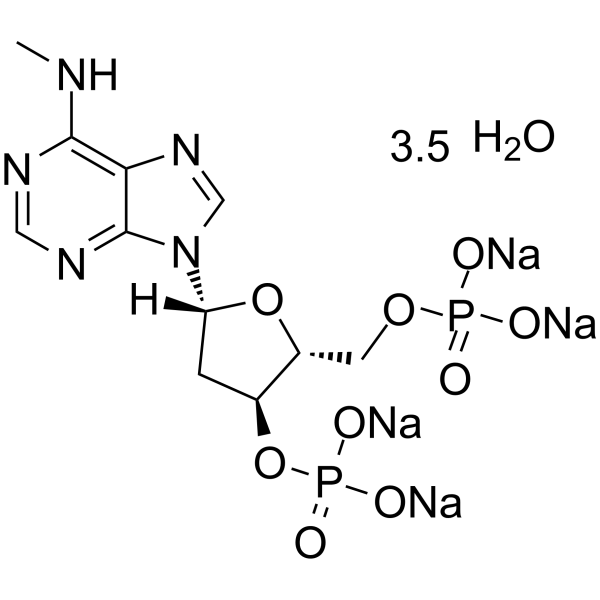
- HY-159829
-
|
NBI-1117568; HTL-0016878
|
mAChR
|
Neurological Disease
|
|
Direclidine (NBI-1117568, HTL-0016878) is a selective orthosteric agonist targeting the muscarinic acetylcholine M4 receptor, exhibiting very low affinity for M1, M2, M3, and M5 receptors. It binds to the orthosteric site of the M4 receptor in a non-covalent, competitive manner. Direclidine specifically activates the M4 receptor, inhibiting the release of acetylcholine from striatal cholinergic interneurons, thereby regulating the balance of the dopaminergic system and reducing psychiatric symptoms associated with excessive dopamine release. Direclidine can improve symptoms associated with neuropsychiatric disorders and is used in research on schizophrenia and other neuropsychiatric disorders .
|
-

- HY-125784
-
|
Viloxazin hydrochloride; Emovit hydrochloride
|
5-HT Receptor
|
Neurological Disease
|
|
Viloxazine hydrochloride is a non-brain-penetrant, selective norepinephrine transporter (NET) inhibitor (IC50=0.26 μM) and 5-HT receptor modulator. Viloxazine antagonizes 5-HT2B receptors (Ki=4.2 μM) and agonizes 5-HT2C receptors (EC50=32 μM), respectively, and enhances 5-HT neurotransmission by modulating 5-HT2B/C receptors. Viloxazine also competitively inhibits NET from increasing NE and DA levels in the synaptic cleft, and can be used in the study of attention deficit hyperactivity disorder (ADHD) .
|
-
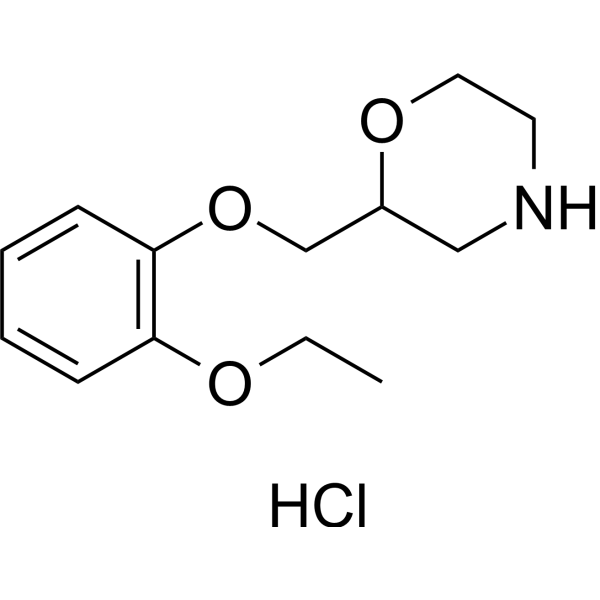
- HY-15322
-
|
P505-15; PRT-2607; BIIB-057
|
Syk
Apoptosis
Caspase
|
Inflammation/Immunology
Cancer
|
|
PRT062607 (P505-15; PRT-2607) is an orally active ATP-competitive Syk inhibitor with an IC50 value of 1 nM, and exhibits at least 80-fold selectivity over other kinases. PRT062607 blocks B cell antigen receptor-mediated activation, Fcε receptor 1-mediated basophil degranulation and microglial phagocytosis, and induces caspase-dependent apoptosis and microglial death. PRT062607 inhibits tumor growth and peripheral nerve injury-induced mechanical allodynia, and prevents neuronal loss. PRT062607 can be used in research related to rheumatoid arthritis, chronic lymphocytic leukemia, non-Hodgkin's lymphoma, neurodegenerative diseases and neuropathic pain .
|
-
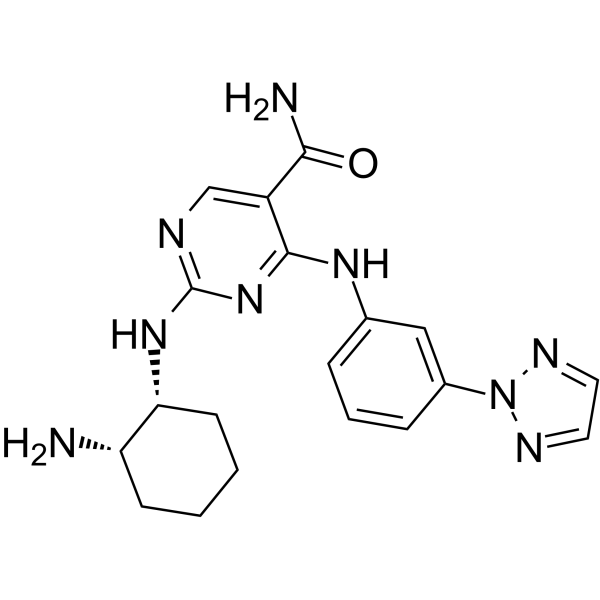
- HY-120877
-
|
|
MARK
Salt-inducible Kinase (SIK)
AMPK
Apoptosis
|
Cancer
|
|
MRT199665 is a potent and ATP-competitive, selective MARK/SIK/AMPK inhibitor with IC50s of 2/2/3/2 nM, 10/10 nM, and 110/12/43 nM for MARK1/MARK2/MARK3/MARK14, AMPKα1/AMPKα2, and SIK1/SIK2/SIK3, respectively . MRT199665 causes apoptosis in MEF2C-activated human acute myeloid leukemias (AML) cells . MRT199665 inhibits the phosphorylation of SIK substrate CRTC3 at S370 .
|
-
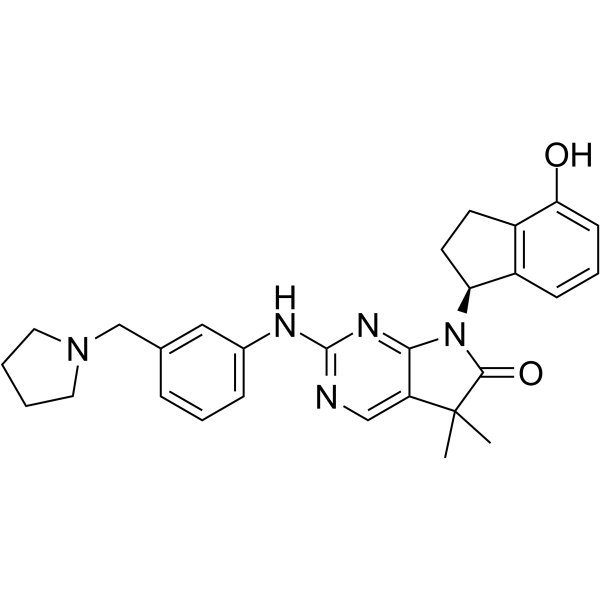
- HY-171216
-
|
|
Integrin
|
Neurological Disease
|
|
GW559090 is a selective, competitive, and high-affinity α4 integrin antagonist with a Kd of 0.19 nM for α4β1. GW559090 can effectively block the binding of α4β1 to vascular cell adhesion molecule 1 (VCAM-1) and fibronectin with IC50 values of 7.72 and 8.04 nM. GW559090 also inhibits the interaction between α4β7 and mucosal addressin cell adhesion molecule 1 (MAdCAM-1) (IC50 = 23 nM). GW559090 can inhibit inflammatory infiltration in the eyes, repair the corneal barrier and restore the function of goblet cells. GW559090 can be used for research of Sjögren's syndrome associated dry eye .
|
-

- HY-110036
-
|
L768242
|
Cannabinoid Receptor
Endogenous Metabolite
HIF/HIF Prolyl-Hydroxylase
TNF Receptor
Interleukin Related
|
Neurological Disease
Inflammation/Immunology
|
|
GW-405833 (L768242) is a potent, selective cannabinoid receptor 2 (CB2) agonist. GW405833 has EC50 and Ki values of 0.65 nM and 3.92 nM for CB2, and EC50 and Ki values of 16.1 μM and 4772 nM for CB1. GW-405833 also exhibits non-competitive CB1 antagonist, exerting its analgesic and and anti-inflammatory effect through a CB1 receptor (rather than CB2) dependent mechanism. GW-405833 can significantly inhibit the production of cAMP stimulated by Forskolin (HY-15371). GW405833 inhibits glycolysis by down-regulating HIF-1α, thereby alleviating acute liver failure (ALF) .
|
-
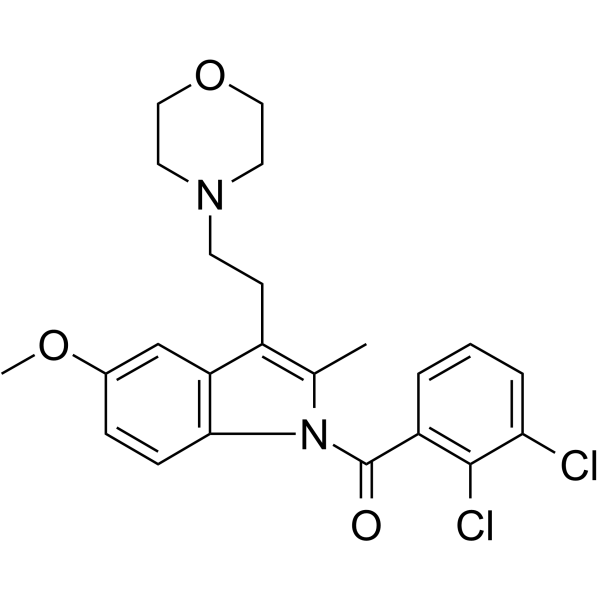
- HY-110036A
-
|
L768242 hydrochloride
|
Cannabinoid Receptor
Endogenous Metabolite
HIF/HIF Prolyl-Hydroxylase
TNF Receptor
Interleukin Related
|
Neurological Disease
Inflammation/Immunology
|
|
GW405833 (L768242) hydrochloride is a potent, selective cannabinoid receptor 2 (CB2) agonist. GW405833 has EC50 and Ki values of 0.65 nM and 3.92 nM for CB2, and EC50 and Ki values of 16.1 μM and 4772 nM for CB1. GW405833 hydrochloride also exhibits non-competitive CB1 antagonist, exerting its analgesic effect through a CB1 receptor (rather than CB2) dependent mechanism. GW405833 hydrochloride can significantly inhibit the production of cAMP stimulated by Forskolin (HY-15371). GW405833 hydrochloride inhibits glycolysis by down-regulating HIF-1α, thereby alleviating acute liver failure (ALF) .
|
-
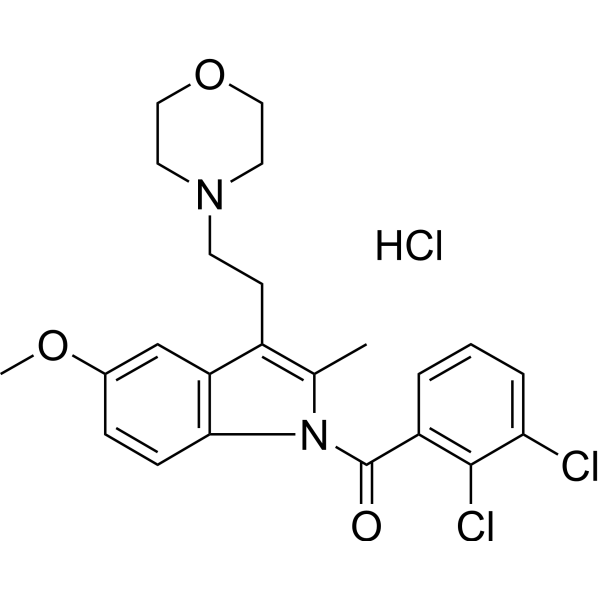
- HY-130841
-
|
|
APC
Ligands for Target Protein for PROTAC
|
Cancer
|
|
Apcin-A is a small molecule inhibitor that selectively targets the cell division cycle protein Cdc20 and is a derivative of Apcin (HY-110287). Apcin-A competitively binds to the D-box binding pocket of Cdc20 and inhibits substrate ubiquitination mediated by the anaphase promoting complex APC/C-Cdc20. Apcin-A also blocks the binding of Cdc20 to substrates (such as securin and cyclin B1), inhibiting anaphase initiation and cell cycle exit. Apcin-A can promote or prolong mitotic slippage in coordination with p31 comet under conditions of high spindle assembly checkpoint (SAC) activity. Apcin-A can be used to develop anti-mitotic drugs and overcome tumor chemotherapy resistance. Apcin-A can be used to synthesize PROTAC CP5V (HY-130257)[1][2][3].
|
-
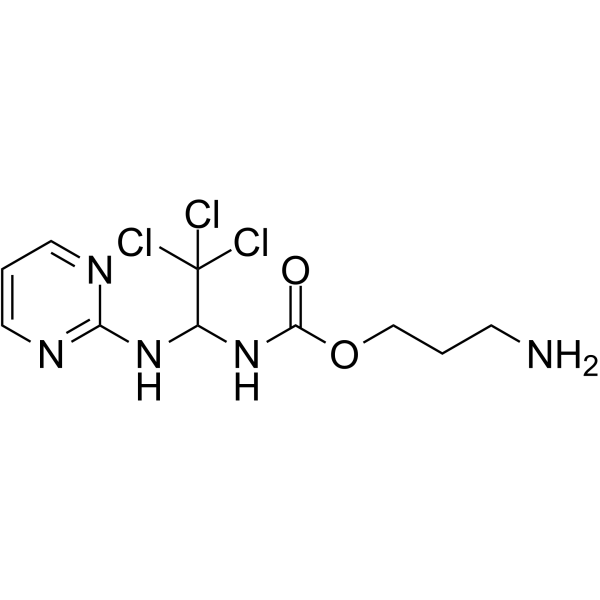
- HY-W700452
-
|
|
Isotope-Labeled Compounds
Apoptosis
ROCK
|
Cancer
|
|
Y-27632-d4 hydrochloride hydrate is the deuterium labeled Y-27632 hydrochloride hydrate (HY-10071A). Y-27632 hydrochloride hydrate is an orally active, ATP-competitive inhibitor of ROCK-I and ROCK-II, with Kis of 220 and 300 nM, respectively. Y-27632 hydrochloride hydrate attenuates Doxorubicin-induced apoptosis of human cardiac stem cells. Y-27632 hydrochloride hydrate also suppresses dissociation-induced apoptosis of murine prostate stem/progenitor cells. Y-27632 hydrochloride hydrate primes human induced pluripotent stem cells (hIPSCs) to selectively differentiate towards mesendodermal lineage via epithelial-mesenchymal transition-like modulation .
|
-

- HY-116142
-
|
|
iGluR
|
Neurological Disease
|
|
CP-283097 is an orally active and conformationally restricted and NR2B subtype-selective NMDA antagonist. CP-283097 efficiently competitively inhibits the binding of [³H]CP-101,606 to the rat meninges, with an IC50 value of 18 nM. CP-283097 exhibits nearly complete inhibition of the current mediated by the NR2B receptor (IC50 = 206 nM), while the inhibitory effect on the NR2A or NR2C receptors is very weak. CP-283097 demonstrates excellent central nervous system permeability and in vivo efficacy in animal models. CP-283097 can be used for neurological diseases related to excessive activation of NMDA receptors .
|
-

- HY-100903R
-
|
nor-Binaltorphimine dihydrochloride (Standard); nor-BNI dihydrochloride (Standard)
|
Opioid Receptor
Reference Standards
|
Neurological Disease
|
|
Norbinaltorphimine dihydrochloride (Standard) is the analytical standard of Norbinaltorphimine dihydrochloride (HY-100903). This product is intended for research and analytical applications. Norbinaltorphimine dihydrochloride (nor-Binaltorphimine dihydrochloride; nor-BNI dihydrochloride) is a selective, long-acting competitive antagonist of the κ-opioid receptor. Norbinaltorphimine dihydrochloride blocks κ-opioid receptor-mediated analgesic effects, and inhibits butorphanol-induced changes in κ-opioid receptor binding kinetics, desensitization and down-regulation. Norbinaltorphimine dihydrochloride suppresses specific opioid withdrawal symptoms, precipitates withdrawal behaviors in butorphanol-dependent rats, and serves as a molecular probe for studying κ-opioid receptor-agonist interactions. Norbinaltorphimine dihydrochloride is applicable to research related to neurological disorders such as pain .
|
-

- HY-135439
-
|
|
5-HT Receptor
|
Neurological Disease
|
|
SB-203186 is a highly selective 5-HT4 receptor antagonist. SB-203186 exhibits a potent competitive antagonistic effect, with its pKB value being 8.3 in the isolated right atrium model of piglets. SB-203186 can dose-dependently shift the 5-HT-induced tachycardia curve to the right, and does not inhibit the maximum response. SB-203186 is an efficient 5-HT₄ antagonist in pig and human atria, but has no significant inhibitory effect in rat atria. SB-203186 can be used for the study of diseases such as arrhythmias and abnormal myocardial contraction .
|
-

- HY-P99014
-
|
ARGX-110
|
Fc Receptor (FcR)
NF-κB
|
Inflammation/Immunology
Cancer
|
|
Cusatuzumab (ARGX-110) is a selective competitive blocker targeting CD70 (with an equilibrium dissociation constant of 17 pM for binding to human CD70). Cusatuzumab also possesses enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) activity. It is a humanized IgG1 monoclonal antibody, artificially synthesized through humanization and genetic engineering modifications (CH2 region mutation to enhance effector function). Cusatuzumab has a dual mechanism of action: firstly, it competitively blocks the interaction between CD70 and CD27, inhibiting the CD27-NF-κB signaling pathway, reducing regulatory T cell (Treg) activation and tumor cell proliferation; secondly, by enhancing binding to FcγRIIIa, it mediates ADCC and antibody-dependent cellular phagocytosis (ADCP), directly lysing CD70-positive tumor cells. Cusatuzumab can efficiently eliminate leukemia stem cells (LSCs), induce tumor cell differentiation and apoptosis, restore immune surveillance, and target CD70-positive tumors. Cusatuzumab is used in the study of acute myeloid leukemia (AML) .
|
-

- HY-123996
-
|
|
IRE1
Apoptosis
Arrestin
CHIKV
|
Infection
Cancer
|
|
3-ethoxy-5,6-dibromosalicylaldehyde is a potent, non-competitive, selective IRE1 (including IRE1α) inhibitor (IC50s: ∼0.12 μM for hIRE1α-cyto; 6 μM for yeast Ire1). 3-ethoxy-5,6-dibromosalicylaldehyde inhibits XBP-1 splicing. 3-ethoxy-5,6-dibromosalicylaldehyde induces Apoptosis. 3-ethoxy-5,6-dibromosalicylaldehyde upregulates the mRNA expression level of TXNIP, while downregulating the expression level of TXN. 3-ethoxy-5,6-dibromosalicylaldehyde exhibits anticancer activity against pancreatic cancer. 3-ethoxy-5,6-dibromosalicylaldehyde significantly inhibits chikungunya virus replication .
|
-

- HY-123859
-
|
|
Casein Kinase
FLT3
CDK
|
Neurological Disease
Inflammation/Immunology
Cancer
|
|
SR-2890 is a highly selective, ATP-competitive inhibitor of casein kinase CK1δ and CK1ε, with IC50 values of 4 nM and 44 nM, respectively, and a Ki of 14 nM for CK1δ. SR-2890 exhibits antiproliferative effects. SR-2890 blocks the serine/threonine kinase activity of CK1δ and weakly inhibits a few off-target kinases such as FLT3, CDK4. SR-2890 has an oral bioavailability of 10% and a blood-brain barrier penetration rate of <1%. SR-2890 demonstrates stable in vitro metabolism and favorable in vivo pharmacokinetic properties, effectively inhibiting the growth of human A375 melanoma cells. SR-2890 can be used in melanoma research and is also a useful compound for studying CK1δ/ε-related diseases such as Alzheimer's disease .
|
-

- HY-118917
-
|
|
IMPDH
|
Inflammation/Immunology
|
|
VX-148 is an orally active immunosuppressant, which is a non-competitive inosine-5'-monophosphate dehydrogenase (IMPDH) inhibitor with Ki values for IMPDH Ⅱ and IMPDH Ⅰ of 6 and 14 nM respectively. VX-148 can significantly inhibit the proliferation of human peripheral blood mononuclear cells (PBMC) stimulated by T-cell mitogen (PHA) or B-cell mitogen (SPAS). VX-148 has high selectivity for lymphocytes (such as L1210, Jurkat T cells, and Raji B cells), but has no significant toxicity to non-lymphoid cells. VX-148 can inhibit antibody responses in mouse models and significantly prolong the survival time of transplanted skin in allogeneic skin transplantation models. VX-148 can be used in the research of autoimmune diseases (such as rheumatoid arthritis, psoriasis) and organ transplantation anti-rejection .
|
-

- HY-114118
-
Semaglutide
Maximum Cited Publications
35 Publications Verification
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-100607AR
-
|
ONO1101 hydrochloride (Standard)
|
Reference Standards
Adrenergic Receptor
Calcium Channel
Potassium Channel
Reactive Oxygen Species (ROS)
|
Cardiovascular Disease
Inflammation/Immunology
Endocrinology
|
|
Landiolol (ONO1101) hydrochloride (Standard) is the analytical standard of Landiolol hydrochloride (HY-100607A). This product is intended for research and analytical applications. Landiolol (ONO1101) hydrochloride is a highly selective, ultra-short-acting competitive inhibitor of β1 adrenergic receptors. Landiolol hydrochloride specifically blocks cardiac β1 receptors, reducing heart rate and myocardial oxygen consumption. Landiolol hydrochloride inhibits TNF-α-induced excessive mitochondrial oxygen consumption and reactive oxygen species production in a sepsis model, alleviating renal injury. Landiolol hydrochloride has little effect on cardiac ion channels (such as L-type calcium current and inward rectifier potassium current) and has a weak negative inotropic effect. Landiolol hydrochloride can be used for perioperative tachycardia control and protection studies of sepsis-related acute kidney injury .
|
-

- HY-114118B
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide acetate is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide acetate promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide acetate also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide acetate has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide acetate can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-120877A
-
|
|
MARK
Salt-inducible Kinase (SIK)
AMPK
Apoptosis
|
Cancer
|
|
(R)-MRT199665 is an isomer of MRT199665 (HY-120877). MRT199665 is a potent and ATP-competitive, selective MARK/SIK/AMPK inhibitor with IC50s of 2/2/3/2 nM, 10/10 nM, and 110/12/43 nM for MARK1/MARK2/MARK3/MARK14, AMPKα1/AMPKα2, and SIK1/SIK2/SIK3, respectively. MRT199665 causes apoptosis in MEF2C-activated human acute myeloid leukemias (AML) cells. MRT199665 inhibits the phosphorylation of SIK substrate CRTC3 at S370 .
|
-

- HY-176854
-
|
|
PI3K
mTOR
|
Cancer
|
|
PI3K/mTOR-IN-18 (Compound 12) is a highly selective dual PI3K/mTOR inhibitor. PI3K/mTOR-IN-18 shows antitumor effects via competitive binding to PI3Kα (Ki=0.130 nM) and mTOR (Ki=0.111 nM). PI3K/mTOR-IN-18 blocks the PI3K/AKT/mTOR pathway and inhibits tumor cell proliferation (IC50=144 nM). PI3K/mTOR-IN-18 is promising for research of solid tumors (e.g., breast, NSCLC) .
|
-

- HY-174425
-
|
|
Cytochrome P450
|
Cancer
|
CYP1B1-IN-9 is a highly selective and competitive CYP1B1 Inhibitor with IC50 values of 1.48 nM, > 100 μM, and > 80 μM for CYP1B1, CYP1A1, and CYP1A2, respectively. CYP1B1-IN-9 significantly inhibits the migration and invasion of A549/T cells. CYP1B1-IN-9 has the ability to resensitize Paclitaxel (HY-B0015)-resistant cells, and good metabolic stability and safety, and shows favorable pharmacokinetic parameters. CYP1B1-IN-9 can be used for the study of tumor-drug resistance .
|
-

- HY-114118C
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide sodium is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide sodium promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide sodium also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide sodium has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide sodium can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-P99144A
-
|
|
PD-1/PD-L1
|
Inflammation/Immunology
Cancer
|
|
Anti-Mouse PD-1 Antibody (S-5001) is a selective inhibitor targeting PD-1, blocking the PD-1/PD-L1 immune checkpoint axis through competitive binding to PD-1. Anti-Mouse PD-1 Antibody (S-5001) works by reversing the tumor immunosuppressive microenvironment and reactivating the anti-tumor activity of cytotoxic T lymphocytes. It can be used in research on tumors such as melanoma and HPV-positive oropharyngeal squamous cell carcinoma. Anti-Mouse PD-1 Antibody (S-5001) is often combined with photothermal therapy, chemotherapy, etc., to enhance efficacy .
|
-

- HY-B1194R
-
|
(±)-Tetramisole hydrochloride (Standard); DL-Tetramisole hydrochloride (Standard); R-829 (Standard)
|
Reference Standards
Potassium Channel
Parasite
PKA
|
Infection
Cardiovascular Disease
|
|
Tetramisole hydrochloride (Standard) is the analytical standard of Tetramisole (hydrochloride). This product is intended for research and analytical applications. Tetramisole hydrochloride is an orally active, selective inward rectifier potassium channel agonist with an EC50 of approximately 30 μM for the Kir2.1 subunit. Tetramisole hydrochloride is also an anti-nematode agent that blocks neuromuscular transmission by non-competitive depolarization. Tetramisole hydrochloride promotes the forward transport of Kir2.1 channels, hyperpolarizes the resting potential (RP), shortens the action potential duration (APD), inhibits intracellular calcium overload and the PKA signaling pathway, and exerts anti-arrhythmic and anti-myocardial remodeling activities. Tetramisole hydrochloride can be used in cardiac electrophysiology research and research related to myocardial ischemia and heart failure .
|
-

- HY-15574R
-
|
SB-207266 (Standard)
|
5-HT Receptor
Reference Standards
|
Cardiovascular Disease
|
|
Piboserod (Standard) is the analytical standard of Piboserod. This product is intended for research and analytical applications. Piboserod is an orally available selective antagonist of the 5-HT4 receptor, with a Ki value of approximately 0.1 nM for human 5-HT4 receptors. Piboserod can competitively bind to the 5-HT4 receptor and block the activation of the 5-HT4 receptor. Piboserod can inhibit the enhancing effect of 5-HT on the nerve-mediated contraction response of the human bladder detrusor muscle. Piboserod is mainly used in the research of urinary system diseases (such as overactive bladder) and cardiovascular diseases (such as chronic heart failure) .
|
-

- HY-125959R
-
|
|
Apoptosis
|
Cardiovascular Disease
Neurological Disease
|
|
Ucf-101 (Standard) is the analytical standard of Ucf-101. This product is intended for research and analytical applications. Ucf-101 is a selective and competitive inhibitor of pro-apoptotic protease Omi/HtrA2, with an IC50 of 9.5 μM for His-Omi. Ucf-101 exhibits very little activity against various other serine proteases (IC50>200 μM). Ucf-101 has a natural red fluorescence at 543 nm that is used to monitor its ability to enter mammalian cells. Ucf-101 has a significant cardioprotective effect against MI/R injury and also has certain neuroprotective effect .
|
-

- HY-136265
-
|
|
Aminoacyl-tRNA Synthetase
|
Cancer
|
|
BC-LI-0186 is a potent and selective inhibitor of Leucyl-tRNA synthetase (LRS; LeuRS) and Ras-related GTP-binding protein D (RagD) interaction (IC50=46.11 nM). BC-LI-0186 competitively binds to the RagD interacting site of LRS (Kd=42.1 nM) and has on effects on LRS-Vps34, LRS-EPRS, RagB-RagD association, mTORC1 complex formation or the activities of 12 kinases. BC-LI-0186 can effectively suppress the activity of cancer-associated?MTOR?mutants and the growth of rapamycin-resistant cancer cells.?BC-LI-0186 is a promising agent for lung cancer research .
|
-
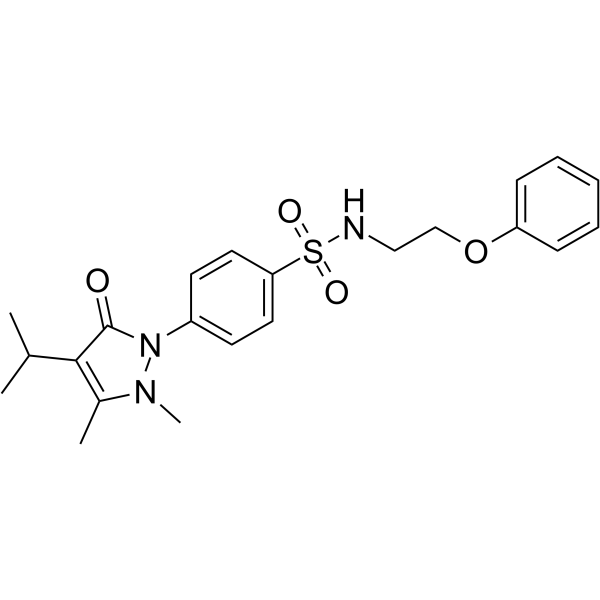
- HY-123597
-
|
DDUG; NCI C04808
|
Autophagy
Checkpoint Kinase (Chk)
|
Cancer
|
|
NSC 109555 is an ATP-competitive inhibitor of checkpoint kinase 2 (Chk2; IC50=200 nM in a cell-free kinase assay). It is selective for Chk2 over Chk1 and 16 kinases in a panel but does inhibit Brk, c-Met, IGFR, and LCK with IC50 values of 210, 6,000, 7,400, and 7,100 nM, respectively. NSC 109555 inhibits Chk2 autophosphorylation and phosphorylation of the Chk2 substrate histone H1 in vitro (IC50=240 nM). It inhibits the growth of, and induces autophagy in, L1210 leukemia cells in vitro.2 NSC 109555 (1,250 nM) potentiates gemcitabine-induced cytotoxicity in MIA PaCa-2, CFPAC-1, PANC-1, and BxPC-3 pancreatic cancer cells, as well as reduces gemcitabine-induced increases in Chk2 phosphorylation and enhances gemcitabine-induced production of reactive oxygen species (ROS) in MIA PaCa-2 cells.
|
-

- HY-178057
-
|
|
EGFR
Akt
Anaplastic lymphoma kinase (ALK)
Apoptosis
|
Cancer
|
|
EGFR-IN-176 is an orally active and ATP-competitive EGFR mutant inhibitor (particularly C797S-mediated EGFR triple mutant). EGFR-IN-176 effectively inhibits subsequent AKT signaling and induces apoptosis in Ba/F3 and PC-9 cells expressing EGFR 19del/T790M/C797S and EGFR L858R/T790M/C797S. EGFR-IN-176 selectively inhibits EGFR signaling in cell lines harboring EGFR triple mutation and shows no inhibitory effect against A431 cells that express wild-type EGFR. EGFR-IN-176 can effectively inhibit the enzymatic activity of ALK (IC50 < 0.5 nM). EGFR-IN-176 can be used for the study of non-small cell lung cancer (NSCLC) .
|
-

- HY-133178R
-
|
3,4,8,9-Tetrahydroxy urolithin (Standard)
|
Reference Standards
Ephrin Receptor
PPAR
AMPK
|
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
Urolithin D (Standard) (3,4,8,9-Tetrahydroxy urolithin (Standard)) is the analytical standard of Urolithin D (HY-133178). This product is intended for research and analytical applications. Urolithin D (3,4,8,9-Tetrahydroxy urolithin) is a colonic metabolite of Ellagitannins and a competitive, reversible, and selective antagonist of the EphA receptor. Urolithin D inhibits EphA2-ephrin-A1 binding with an IC50 of 0.9 μM. Urolithin D is also a potent antioxidant that scavenges free radicals and repairs oxidized DNA damage. Additionally, Urolithin D suppresses triglyceride accumulation and promotes fatty acid oxidation by activating the AMPK signaling pathway. Urolithin D can be used for research on tumors, metabolic, and inflammatory diseases .
|
-
- HY-B2132
-
|
3-(2-Aminoethyl)indole~2-(3-Indolyl)ethylamine
|
Endogenous Metabolite
5-HT Receptor
Aryl Hydrocarbon Receptor
|
Neurological Disease
Metabolic Disease
|
|
Tryptamine is a selective, blood-brain-penetrating 5-HT4 receptor agonist (EC50=1-3 mM) and an endogenous ligand of the aryl hydrocarbon receptor (AHR) (Kd=10-50 nM). Tryptamine promotes intestinal anion secretion and fluid transport by activating G protein-coupled receptors (GPCRs) and accelerates gastrointestinal motility. Tryptamine regulates Th17/Treg balance to inhibit neuroinflammation, competitively binds to 5-HT receptors to regulate central nervous system activity, and participates in temperature regulation and spinal reflex regulation as a neuromodulator. Tryptamine can be used to study intestinal motility disorders such as functional constipation, and has shown significant efficacy in multiple sclerosis models .
|
-

- HY-W414915R
-
|
CGP 48933 methyl ester (Standard)
|
Reference Standards
Drug Derivative
Angiotensin Receptor
|
Cardiovascular Disease
|
|
Valsartan (CGP 48933) methyl ester (Standard) is the analytical standard of Valsartan methyl ester (HY-W414915). This product is intended for research and analytical applications. Valsartan methyl ester is the methyl ester derivative of Valsartan (HY-18204). Valsartan is a selective angiotensin II type 1 (AT1) receptor blocker (ARB) with potent antihypertensive and cardioprotective effects. Valsartan competitively binds to AT1 receptors, inhibiting the binding of angiotensin II to AT1 receptors, thereby blocking angiotensin II-mediated vasoconstriction, sodium retention, and myocardial hypertrophy signaling pathways. Valsartan reduces systolic blood pressure in L-NAME-induced hypertensive rats. Valsartan can be used for the study and treatment of arterial hypertension, hypertensive heart disease, and heart failure .
|
-

- HY-135446
-
|
|
Endothelin Receptor
|
Cardiovascular Disease
Endocrinology
|
|
BQ-610 is a selective antagonist of the endothelin A receptor (ETA receptor). BQ-610 specifically blocks the ETA receptor, competitively inhibiting the binding of endothelin-1 (ET-1) (a vasoconstrictive peptide) to the receptor, thereby blocking the effects of ET-1 such as vascular smooth muscle contraction, cell mitosis, and inhibition of hormone secretion. BQ-610 significantly alleviates cerebral vasospasm in rabbits. BQ-610 blocks the bronchial epithelial and pulmonary vascular cell proliferation caused by cigarette smoke in rat models. BQ-610 can delay the natural luteal regression in the cow's uterus. BQ-610 can be used for research on vasospasm, abnormal cell proliferation, and reproductive endocrine disorders .
|
-

- HY-145836
-
|
|
FGFR
|
Cancer
|
|
FGFR4-IN-8 (Compound 7v) is an ATP-competitive, highly selective covalent inhibitor of wild-type and gatekeeper mutant FGFR4. FGFR4-IN-8 exhibits excellent potency against FGFR4, FGFR4 V550L, FGFR4 V550M and FGFR4 C552S with IC50s of 0.5, 0.25, 1.6, 931 nM, respectively. FGFR4-IN-8 exhibits potent antiproliferative activity against Hep3B hepatocellular carcinoma cells with the IC50 value of 29 nM. FGFR4-IN-8 demonstrates modest in vivo antitumor efficacy in nude mice bearing the Huh-7 xenograft model .
|
-
- HY-145939
-
|
BRD5846
|
Casein Kinase
|
Cancer
|
|
BAY-888 is a selective CK1α/CSNK1A1 (casein kinase 1α) ATP-competitive inhibitor (IC50: 4 nM@10 μM ATP; 63 nM@1 mM ATP). BAY-888 blocks the negative regulation of p53 and other signaling pathways by CK1α, induces apoptosis and inhibits proliferation of tumor cells. BAY-888 has shown inhibitory efficacy against cancers such as acute myeloid leukemia (AML) in PRISM barcoded cell line screening and can mimic the effects of shRNA-mediated CK1α knockdown. BAY-888 is primarily used for the development of anticancer drugs for p53 wild-type tumors and for the study of the mechanisms of CK1α-related signaling pathways .
|
-
- HY-114118S1
-
|
|
Isotope-Labeled Compounds
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide-d8 tetraTFA is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-114118A
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide TFA is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide TFA promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide TFA also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide TFA has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide TFA can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-114118CP
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide (crude) is the crude form of Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances Autophagy, inhibits oxidative stress and Apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-114118S
-
|
|
Isotope-Labeled Compounds
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide-d8 is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-176219
-
|
|
Bcl-2 Family
Apoptosis
Necroptosis
|
Cancer
|
|
Bcl-2-IN-23 (compound 5) is a selective inhibitor targeting Bcl-2. The IC50 of Bcl-2-IN-23 in HTB-140, HeLa and SW620 cells is 25.7-33.7 μM. Bcl-2-IN-23 can non-covalently competitively bind to Bcl-2 protein, significantly reduce its expression, and induce late apoptosis and necroptosis of cancer cells. Bcl-2-IN-23 enhances the sensitivity of cancer cells to apoptosis and reduces the release of IL-6 inflammatory factors by disrupting the Bcl-2-mediated mitochondrial apoptosis inhibition pathway. Bcl-2-IN-23 can be used for anti-apoptosis research of malignant tumors such as melanoma, cervical cancer, and colorectal cancer .
|
-

- HY-164090
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-

- HY-120006A
-
|
|
ERK
|
Cardiovascular Disease
|
|
(rel)-AR234960 is a selective and competitive agonist of the G protein-coupled receptor MAS. (rel)-AR234960 binds to the MAS receptor to activate the downstream ERK1/2 signaling pathway, inducing the expression of connective tissue growth factor (CTGF) and its downstream collagen subtype genes (such as COL1A1, COL3A1). (rel)-AR234960 promotes collagen synthesis in cardiac fibroblasts through the MAS-ERK1/2-CTGF pathway and aggravates extracellular matrix remodeling. (rel)-AR234960's in vitro effect can be blocked by the MAS inverse agonist AR244555 and MEK1 inhibitor. (rel)-AR234960 regulates the expression of cardiac fibrosis-related genes and can be used in the study of heart failure .
|
-
- HY-N9422
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate triethylamine, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-W250153
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-W250153A
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
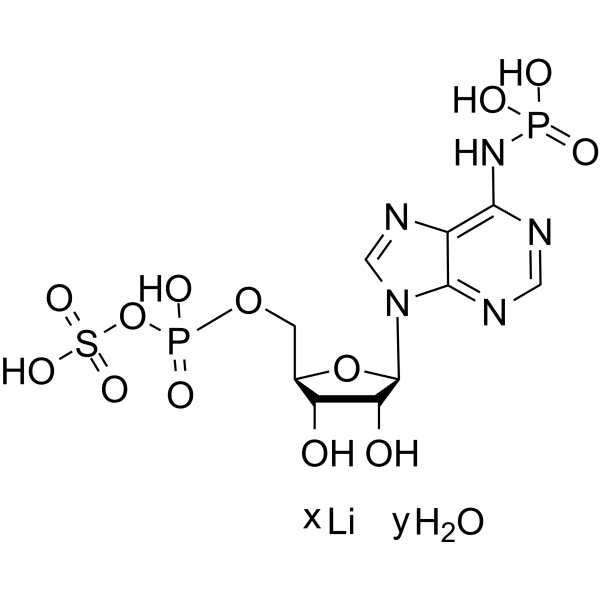
- HY-46286
-
|
N-(4-tert-butyl-1,3-thiazol-2-yl)-3-fluorobenzamide
|
5-HT Receptor
nAChR
GABA Receptor
Glycine Receptor (GlyR)
|
Neurological Disease
|
|
TTFB (N-(4-tert-butyl-1,3-thiazol-2-yl)-3-fluorobenzamide) is a selective, non-competitive zinc-activated channel (ZAC) antagonist. TTFB inhibits Zn 2+- and H +-induced ZAC currents with IC50 values of 3 μM and 8.5 μM, respectively, and has an IC50 of 4.7 μM against spontaneous activity. TTFB shows no significant agonistic, antagonistic or modulatory activity towards representative classical Cys-loop receptors including m5-HT3AR, hα3β4 nAChR, hα1β2γ2S GABAAR and hα1 GlyR. TTFB can be used to investigate the physiological and pathological functions of ZAC.
|
-

- HY-147294
-
|
ACT-539313
|
Orexin Receptor (OX Receptor)
Cytochrome P450
|
Neurological Disease
|
|
Nivasorexant (ACT-539313) is an orally active, blood-brain barrier penetrant, selective orexin OX1R inhibitor. Nivasorexant specifically blocks central OX1Rs without affecting OX2Rs, and exhibits competitive inhibitory activity against CYP2C8, CYP2C9, CYP2C19 and CYP3A4 (IC50 values are 25 μM, 8.6 μM, 1.6 μM, 19 μM/44 μM, respectively). Nivasorexant significantly reduces binge-like eating behavior of highly palatable food in rat models and has long-acting properties. Nivasorexant shows no relevant off-target activity against over 130 selected proteins, exhibits favorable safety profiles, and can be used for studies related to binge eating disorder .
|
-
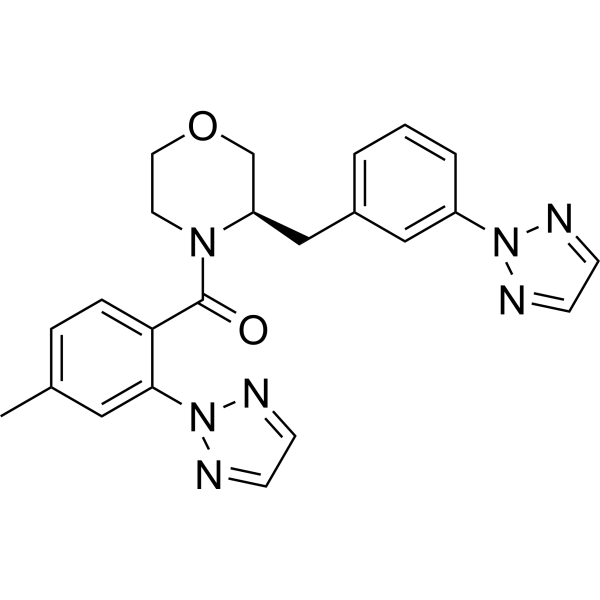
- HY-141539
-
|
|
Histone Methyltransferase
Akt
|
Cancer
|
|
SETDB1-TTD-IN-1 is a SETDB1 methyltransferase activator and SETDB1-TTD competitive inhibitor (Kd of 88 nM), and selectivity for SETDB1-TTD over other tudor and bromodomain proteins. SETDB1-TTD-IN-1 stimulates methyltransferase activity via increased catalytic activity, promotes Akt1 Lys64 methylation, Akt1 Thr308 phosphorylation and activation. SETDB1-TTD-IN-1 prevents SETDB1-TTD-histone H3 peptide association, induces global gene expression changes, exhibits cellular target engagement, and acts as a tool compound for SETDB1-TTD function exploration. SETDB1-TTD-IN-1 can be used for the research of breast cancer .
|
-
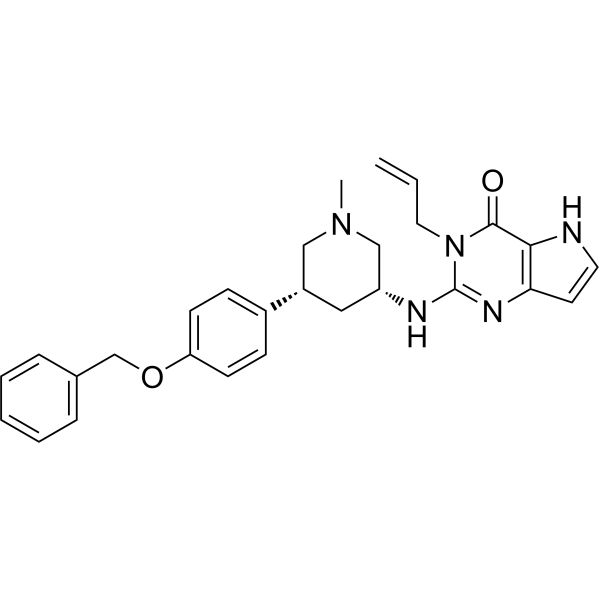
- HY-114118S3
-
|
|
Isotope-Labeled Compounds
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Metabolic Disease
|
|
Semaglutide- 13C6, 15N TFA is the 13C- and 15N-labeled Semaglutide TFA (HY-114118A). Semaglutide TFA is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide TFA promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide TFA also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide TFA has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide TFA can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-

- HY-108831
-
|
|
Integrin
|
Inflammation/Immunology
Cancer
|
|
Natalizumab (AN100226; BG00002) is a humanized monoclonal IgG4 antibody inhibitor that selectively targets α4 integrin (CD49d). It blocks the interaction of integrins such as α4β1 (VLA-4) with vascular cell adhesion molecule VCAM-1, intercellular adhesion molecule ICAM-1, and fibronectin by competitively binding to the α4 subunit. Natalizumab inhibits the adhesion, retention, and transendothelial migration of immune cells (such as CD4 + T cells), reducing the infiltration of inflammatory cells into the central nervous system or lesion sites, thus exerting anti-inflammatory and immunomodulatory activity. Natalizumab is used in the study of relapsing-remitting multiple sclerosis (RRMS) and also has applications in the study of autoimmune or inflammation-related diseases such as Crohn's disease, B-cell lymphoma, and non-infectious uveitis. Natalizumab can also prevent lymphocytes from entering the central nervous system, thereby preventing acute demyelinating relapses .
|
-

- HY-162080A
-
|
|
DNA Methyltransferase
Pyruvate Kinase
|
Cancer
|
|
METTL1-WDR4-IN-1 (Compound 1) TFA is a selective competitive inhibitor of the methyltransferase complex METTL1-WDR4 (IC50=144 μM). METTL1-WDR4-IN-1 TFA inhibits the m 7G methyltransferase activity of the METTL1-WDR4 complex, blocking the m 7G modification of PKM mRNA, reducing PKM2 protein expression, disrupting the METTL1/PKM2/H3K9la positive feedback loop, and simultaneously inhibiting PKM2 nuclear translocation-mediated CD155 transcriptional activation. METTL1-WDR4-IN-1 TFA can inhibit tumor cell proliferation, weaken glycolytic metabolism, reverse tumor immune evasion (restoring NK cell and CD8 + T cell function), and regulate RNA epigenetic modification and the tumor immune microenvironment. METTL1-WDR4-IN-1 TFA can be used in immunotherapy research for cancers such as colorectal cancer, and is particularly suitable for use in combination with PKM2 inhibitors to enhance anti-tumor treatment efficacy .
|
-

- HY-162080
-
|
|
DNA Methyltransferase
Pyruvate Kinase
|
Cancer
|
|
METTL1-WDR4-IN-1 (Compound 1) is a selective competitive inhibitor of the methyltransferase complex METTL1-WDR4 (IC50 = 144 μM). METTL1-WDR4-IN-1 inhibits the m 7G methyltransferase activity of the METTL1-WDR4 complex, blocking m 7G modification of PKM mRNA, reducing PKM2 protein expression, disrupting the METTL1/PKM2/H3K9la positive feedback loop, and simultaneously inhibiting PKM2 nuclear translocation-mediated CD155 transcriptional activation. METTL1-WDR4-IN-1 can inhibit tumor cell proliferation, weaken glycolytic metabolism, reverse tumor immune evasion (restoring NK cell and CD8 + T cell function), and regulate RNA epigenetic modification and the tumor immune microenvironment. METTL1-WDR4-IN-1 can be used in immunotherapy research for cancers such as colorectal cancer, and is particularly suitable for use in combination with PKM2 inhibitors to enhance anti-tumor treatment efficacy .
|
-
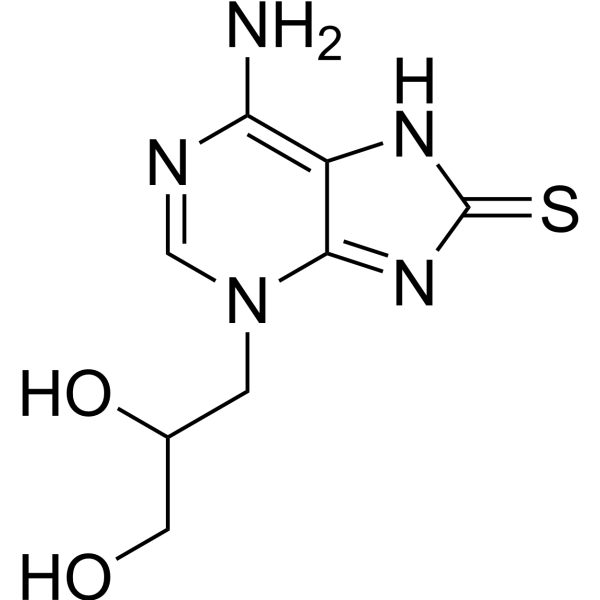
- HY-118156
-
|
|
Others
|
Others
|
|
L-699333 is a 5-lipoxygenase (5-LO) inhibitor belonging to the thieno[2,3,4-cd]indole class. This compound has a 2-ethoxybutyric acid side chain and is a potent inhibitor of the biosynthesis of 5-HPETE and LTB4 produced from human 5-LO, with ICm values of 22 nM, 7 nM, and 3.8 pM for human neutrophils and whole blood, respectively. L-699333 has shown anti-inflammatory and antiasthmatic effects in a variety of animal models, including rat pleurisy models, antigen-induced wheezing models, and awake macaque and sheep asthma models. Its inhibition of 5-LO is highly selective, with higher ICm values or stronger competitive inhibition in FLAP binding assays compared to inhibition of human 15-LO, porcine 12-LO, and ram epididymal cyclooxygenase. The racemic enantiomer 14g of L-699333 is the most potent enantiomer to date, with inhibitory effects similar to those of the known MK-0591, which has been shown in clinical trials to inhibit the biochemical effects of LTB4 biosynthesis in vitro and LTE4 excretion in urine.
|
-

- HY-W154247
-
|
|
Bacterial
|
Infection
|
IP6C is a specific inhibitor and phage sensitizer targeting type II Thoeris systems. IP6C competitively binds to histidine in the catalytic pocket of ThsB, blocks the production of the His-ADPR alarm signal and inhibits ThsA activation, thereby relieving bacterial stasis of phage replication. IP6C selectively resensitizes drug-resistant bacteria carrying type II Thoeris systems (such as Pseudomonas aeruginosa) to phage lysis, without affecting other bacteria, and shows no toxicity to mice and human cell lines. IP6C significantly improves the survival rate of infected mice, and can be used to overcome bacterial phage defense mechanisms and study Pseudomonas aeruginosa infections .
Thoeris system: (named after the Egyptian goddess of fertility and protection), is a widespread anti-phage immune defense system in bacteria and archaea. Thoeris system belongs to the "Abortion Infection (Abi)" mechanism of bacteria: when an individual bacterium detects phage invasion, it initiates a suicide program and dies, thereby blocking phage replication and spread, and protecting the surrounding bacterial population from infection.
|
-

- HY-114243
-
|
|
NF-κB
JNK
Caspase
Apoptosis
|
Neurological Disease
Cancer
|
|
DpC is a selective, orally active iron chelator with anticancer activity. DpC acts on signaling pathway-related targets such as JNK, NF-κB, and its activity is competitively inhibited by another iron chelator Dp44mT (HY-18973). By chelating intracellular iron and copper ions in tumor cells to form redox-active complexes, DpC induces oxidative stress, activates the JNK, NF-κB pathways and downregulates IκBα, upregulates the expressions of neuroglobin and cytoglobin, activates caspase 3/9 to induce tumor cell apoptosis. It also overcomes P-glycoprotein-mediated multidrug resistance through a lysosome-targeting mechanism, and exhibits broad-spectrum synergistic effects when combined with various chemotherapeutic agents. DpC inhibits tumor metastasis and increases TNF-α levels in the tumor microenvironment to enhance endogenous immune responses. DpC is applicable to the research of various malignancies including neuroblastoma, pancreatic cancer, prostate cancer, lung cancer, and breast cancer .
|
-
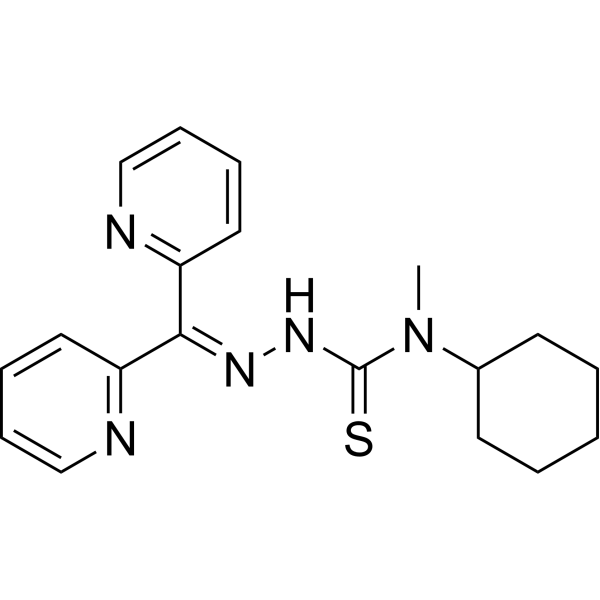
- HY-108831A
-
|
AN100226; BG00002
|
Integrin
|
Inflammation/Immunology
Cancer
|
|
Natalizumab (Anti-CD49d) (AN100226; BG00002) Solution is a humanized monoclonal IgG4 antibody inhibitor that selectively targets α4 integrin (CD49d), blocking the interaction of integrins such as α4β1 (VLA-4) with vascular cell adhesion molecule VCAM-1, intercellular adhesion molecule ICAM-1, and fibronectin by competitively binding to the α4 subunit. Natalizumab solution inhibits the adhesion, retention, and transendothelial migration of immune cells (such as CD4 + T cells), reducing the infiltration of inflammatory cells into the central nervous system or lesion sites, thereby exerting anti-inflammatory and immunomodulatory activity. Natalizumab (Anti-CD49d) solution is used in the study of relapsing-remitting multiple sclerosis (RRMS) and is also applied in the research of autoimmune or inflammation-related diseases such as Crohn's disease, B-cell lymphoma, and non-infectious uveitis. Natalizumab (Anti-CD49d) can also prevent lymphocytes from entering the central nervous system, thus preventing acute demyelinating relapses .
|
-
- HY-170524
-
|
|
SARS-CoV
DNA Methyltransferase
Cytochrome P450
|
Infection
|
|
TDI-015051 is a highly selective, orally active antiviral agent that targets the coronavirus NSP14 guanine-N7 methyltransferase. TDI-015051 binds to substrates in a non-competitive manner and forms a stable ternary complex, precisely blocking the capping and methylation processes of viral mRNA. TDI-015051 potently inhibits a variety of coronaviruses (including SARS-CoV-2 and MERS). By impairing viral replication and translation and inducing a moderate type I interferon-mediated immune response, it significantly reduces pulmonary viral load and exhibits a synergistic effect with Nirmatrelvir (HY-138687). In addition, TDI-015051 does not inhibit non-coronavirus methyltransferases, and the drug-resistant mutations it induces impair viral fitness, demonstrating excellent antiviral properties and safety. TDI-015051 can be used for research on COVID-19 and the replication mechanism of coronaviruses .The IC50 values of TDI-015051 against SARS-CoV-2, α-hCoV-NL63, α-hCoV-229E, β-hCoV-MERS are 0.15 nM, 1.7 nM, 2.6 nM and 3.6 nM, respectively, and the Ka value against SARS-CoV-2 is 0.061 nM .
|
-

- HY-18006
-
NKP608
1 Publications Verification
|
Neurokinin Receptor
Wnt
Bcl-2 Family
β-catenin
Cyclin G-associated Kinase (GAK)
VEGFR
Caspase
Cadherin
Apoptosis
|
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
NKP608 is a non-peptidic derivative of 4-aminopiperidine, a highly selective, orally active, neurokinin-1 (NK1) receptor antagonist with IC50 of 2.6 nM. NKP608 is active both in vitro and in vivo, showing extremely low affinity for NK2, NK3 receptors. NKP608 exerts its effects by blocking the NK₁ receptor, regulate cell proliferation and apoptosis, affect neurotransmitter functions and gastric mucosal repair mechanisms, and suppress the Wnt/β-catenin pathway in antitumor research. NKP608 is applicable to research related to various diseases, including cough, anxiety disorders, depression, gastric mucosal injury, and colorectal cancer .
|
-
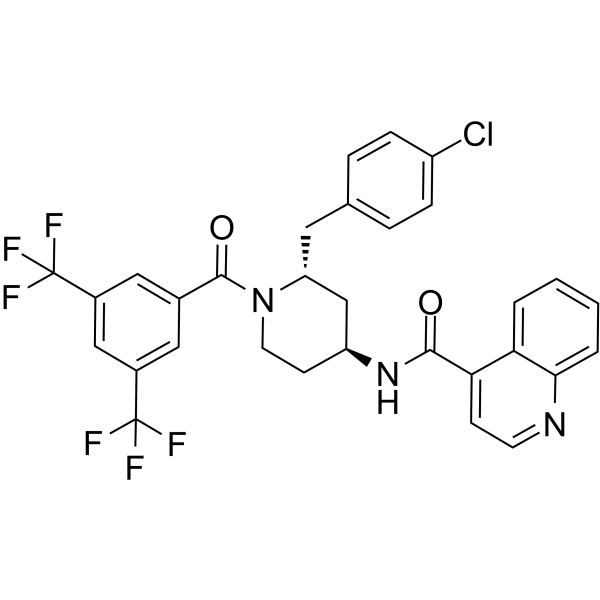
-
-
HY-L158
-
|
|
6,135 compounds
|
|
According to reports, most known kinase inhibitors exert their effects through competitive binding in highly conserved ATP pockets. Although genetic techniques such as RNA interference can inactivate specific genes, most kinases are multi domain proteins, each of which has an independent function. Highly selective inhibitors have higher efficiency than non-selective inhibitors, and the selectivity to the target is at least 100 times higher. Therefore, ensuring the validation of targets with the most selective inhibitors is crucial for a more thorough understanding of the pharmacology of the kinase field. The Highly Selective Inhibitors Library contains 6,135 compounds, covering multiple targets and subtypes, such as GPCR protein family, Ion channel, multiple kinases, etc. The Highly Selective Inhibitors Library is an effective tool for screening different phenotypes
|
| Cat. No. |
Product Name |
Type |
-
- HY-10254G
-
|
PD0325901; PD325901
|
Fluorescent Dyes
|
|
Mirdametinib (PD0325901) (GMP) is Mirdametinib (HY-10254) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Mirdametinib is an orally active, selective and non-ATP-competitive MEK inhibitor .
|
-
- HY-15141G
-
|
Antibiotic AM-2282; STS; AM-2282
|
Fluorescent Dyes
|
|
Staurosporine (AM-2282) (GMP) is Staurosporine (HY-15141) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases .
|
-
- HY-13418G
-
|
Compound C dihydrochloride; BML-275 dihydrochloride
|
Fluorescent Dyes
|
|
Dorsomorphin dihydrochloride (GMP) is the GMP level of Dorsomorphin dihydrochloride (HY-13418). GMP guidelines are used to produce Dorsomorphin dihydrochloride (GMP). GMP small molecules works appropriately as an auxiliary reagent for cell research manufacture. Dorsomorphin dihydrochloride (GMP) is a potent, selective and ATP-competitive AMPK inhibitor. Dorsomorphin dihydrochloride (GMP) can be used for the research of induced differentiation of pluripotent stem cells (PSCs) .
|
-
- HY-70044G
-
|
GSK-1070916A
|
Fluorescent Dyes
|
|
GSK-1070916 (GMP) is GSK-1070916 (HY-70044) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. GSK-1070916 is a potent and selective ATP-competitive inhibitor of aurora B and aurora C with Kis of 0.38 and 1.5 nM, respectively, and is >250- fold selective over Aurora A .
|
| Cat. No. |
Product Name |
Type |
-
- HY-W251428
-
|
Egg PG
|
Biochemical Assay Reagents
|
|
Phosphatidylglycerols (PG) is a selective inhibitor targeting the TLR4 accessory protein CD14/MD-2 complex, inhibiting LPS or virus (such as RSV)-mediated inflammatory signaling pathways through competitive binding. Phosphatidylglycerols directly bind to viral particles to block infection, inhibit COX-2 expression to reduce the release of inflammatory factors (IL-6, IL-8), and improve oxidative stress by regulating mitochondrial membrane phospholipid remodeling. Phosphatidylglycerols can be taken orally or by inhalation and can be used in the study of chronic inflammatory diseases (such as atherosclerosis) and respiratory viral infections (such as RSV) .
|
-
- HY-10254G
-
|
PD0325901; PD325901
|
Biochemical Assay Reagents
|
|
Mirdametinib (PD0325901) (GMP) is Mirdametinib (HY-10254) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Mirdametinib is an orally active, selective and non-ATP-competitive MEK inhibitor .
|
-
- HY-15141G
-
|
Antibiotic AM-2282; STS; AM-2282
|
Biochemical Assay Reagents
|
|
Staurosporine (AM-2282) (GMP) is Staurosporine (HY-15141) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases .
|
-
- HY-W250153
-
|
|
Biochemical Assay Reagents
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-13418G
-
|
Compound C dihydrochloride; BML-275 dihydrochloride
|
Biochemical Assay Reagents
|
|
Dorsomorphin dihydrochloride (GMP) is the GMP level of Dorsomorphin dihydrochloride (HY-13418). GMP guidelines are used to produce Dorsomorphin dihydrochloride (GMP). GMP small molecules works appropriately as an auxiliary reagent for cell research manufacture. Dorsomorphin dihydrochloride (GMP) is a potent, selective and ATP-competitive AMPK inhibitor. Dorsomorphin dihydrochloride (GMP) can be used for the research of induced differentiation of pluripotent stem cells (PSCs) .
|
-
- HY-W250153A
-
|
|
Biochemical Assay Reagents
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-70044G
-
|
GSK-1070916A
|
Biochemical Assay Reagents
|
|
GSK-1070916 (GMP) is GSK-1070916 (HY-70044) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. GSK-1070916 is a potent and selective ATP-competitive inhibitor of aurora B and aurora C with Kis of 0.38 and 1.5 nM, respectively, and is >250- fold selective over Aurora A .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-114118
-
Semaglutide
Maximum Cited Publications
35 Publications Verification
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-114118B
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide acetate is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide acetate promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide acetate also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide acetate has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide acetate can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-114118A
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide TFA is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide TFA promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide TFA also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide TFA has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide TFA can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-P1178
-
|
|
Trk Receptor
p38 MAPK
|
Neurological Disease
|
|
Cyclotraxin B is a BBB-penetrable and selective TrkB inhibitor. Cyclotraxin B inhibits BDNF-induced TrkB activity in a non-competitive manner, with an IC50 of 0.30 nM. Cyclotraxin B has analgesic and anxiolytic effects .
|
-
- HY-114118CP
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide (crude) is the crude form of Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances Autophagy, inhibits oxidative stress and Apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-P0097
-
|
Melanostatine-5
|
Melanocortin Receptor
|
Metabolic Disease
Endocrinology
Cancer
|
|
Nonapeptide-1 (Melanostatine-5), a peptide hormone, is a selective antagonist of MC1R (Ki: 40 nM). Nonapeptide-1 is a competitive α-MSH antagonist that potently inhibits intracellular cAMP and melanosome dispersion induced by α-MSH in melanocytes (IC50: 2.5 nM and 11 nM, respectively). Nonapeptide-1 inhibits melanin synthesis, and can be used in the research of skin pigmentation and regulation of steroid production in the adrenal gland, skin cancer .
|
-
- HY-P1291
-
|
|
PKA
Epigenetic Reader Domain
Flavivirus
|
Infection
Neurological Disease
|
|
PKI 14-22 amide, myristoylated is a selective, cAMP-dependent, competitive PKA inhibitor with Ki=~36 nM. The myristoylation modification of PKI 14-22 amide, myristoylated makes it more permeable to cell membranes and blood-brain barriers than the precursor molecule. PKI 14-22 amide, myristoylated can block the phosphorylation of cAMP-dependent downstream targets (such as CREB). PKI 14-22 amide, myristoylated can prevent the development of morphine analgesic tolerance in mice, and also inhibits protein translation and negative-strand RNA synthesis of Zika virus. PKI 14-22 amide, myristoylated can be used in research fields such as opioid tolerance mechanisms and antiviral drugs .
|
-
- HY-P1137
-
10Panx
1 Publications Verification
|
Gap Junction Protein
|
Others
|
|
10Panx is a competitive inhibitor of selective Pannexin 1 (PANX1) channels. 10Panx blocks the opening of PANX1 channels, inhibits ATP release and downstream P2X7 receptor-mediated signaling pathways, thereby reducing cell death and inflammatory responses. 10Panx can be used in the study of diseases such as neuropathic pain, inflammatory bowel disease, and Clostridioides difficile infection. 10Panx can effectively reduce mechanical hyperalgesia and enhanced C-reflexes, and inhibit the expression of pro-inflammatory factors such as IL-6[1][2][3].
|
-
- HY-13634B
-
-
- HY-114118S1
-
|
|
Isotope-Labeled Compounds
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide-d8 tetraTFA is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-114118S
-
|
|
Isotope-Labeled Compounds
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide-d8 is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
- HY-P2271
-
|
|
Apelin Receptor (APJ)
|
Cancer
|
|
MM 54 (compound 5) is a competitive antagonist at APJ, with an IC50 of 93 nM. MM 54 behaves as a potent and selective inhibitor of apelin binding and APLNR activation .
|
-
- HY-P0097A
-
|
Melanostatine-5 acetate salt
|
Melanocortin Receptor
|
Metabolic Disease
Endocrinology
Cancer
|
|
Nonapeptide-1 (Melanostatine-5) acetate salt, a peptide hormone, is a selective antagonist of MC1R (Ki: 40 nM). Nonapeptide-1 acetate salt is a competitive α-MSH antagonist that potently inhibits intracellular cAMP and melanosome dispersion induced by α-MSH in melanocytes (IC50: 2.5 nM and 11 nM, respectively). Nonapeptide-1 acetate salt inhibits melanin synthesis, and can be used in the research of skin pigmentation and regulation of steroid production in the adrenal gland, skin cancer .
|
-
- HY-P1291A
-
|
|
PKA
Epigenetic Reader Domain
Flavivirus
|
Infection
Neurological Disease
|
|
PKI 14-22 amide, myristoylated TFA is a selective, cAMP-dependent, competitive PKA inhibitor with Ki=~36 nM. The myristoylation modification of PKI 14-22 amide, myristoylated TFA makes it more permeable to cell membranes and blood-brain barriers than the precursor molecule. PKI 14-22 amide, myristoylated TFA can block the phosphorylation of cAMP-dependent downstream targets (such as CREB). PKI 14-22 amide, myristoylated TFA can prevent the development of analgesic tolerance in mice, and also inhibits protein translation and negative-strand RNA synthesis of Zika virus. PKI 14-22 amide, myristoylated TFA can be used in research fields such as opioid tolerance mechanisms and antiviral drugs .
|
-
- HY-P5542
-
|
SB-01; Peniel 2000
|
Factor Xa
TGF-beta/Smad
|
Inflammation/Immunology
|
|
Vicatertide (SB-01, Peniel 2000) is a polypeptide with both competitive inhibitory activity against TGF-β1 and selective inhibitory activity against human factor XIa (hFXIa, with a Ka of 80 nM for hFXIa). Vicatertide binds allosterically to the two binding sites of dimeric hFXI/hFXIa, while directly binding to activated TGF-β1, selectively blocking the Smad1/5/8 pathway and maintaining low-level activation of the Smad2 pathway to enhance the synthesis of type Ⅱ collagen and aggrecan. Vicatertide inhibits thrombus formation in arteriovenous thrombosis models, and also reduces thrombus weight and thrombus incidence in mouse lung cancer models. Vicatertide can be used for research on degenerative disc disease and thrombosis-related diseases .
|
-
- HY-P1173
-
|
Myristoylated L 803; GSK-3β inhibitor XIII
|
GSK-3
Amyloid-β
|
Neurological Disease
|
|
L803-mts (Myristoylated L 803) is a selective and substrate-competitive GSK-3 peptide inhibitor (IC50: 40 μM). L803-mts also reduces Aβ deposits and ameliorates cognitive deficits in 5XFAD mice. L803-mts shows antidepressive effect in the forced swimming test .
|
-
- HY-P10392B
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R TFA, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R TFA inhibits competitively the binding of β-catenin to TCF4. aStAx-35R TFA selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-
- HY-P1293
-
|
|
iGluR
|
Neurological Disease
|
|
Conantokin G, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G has neuroprotective properties .
|
-
- HY-P10392
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R inhibits competitively the binding of β-catenin to TCF4. aStAx-35R selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-
- HY-P5158
-
|
|
Adrenergic Receptor
|
Others
|
|
Conopeptide rho-TIA is a peptide derived from the venom contained in the predatory sea snail Conus tulipa, has highly selective and noncompetitive inhibitor at human α1B-Adrenergic Receptor. Conopeptide rho-TIA acts a competitive inhibitor at human α1A-Adrenergic Receptor and α1D-Adrenergic Receptor. Conopeptide rho-TIA binds to each subtype and may provide useful information for the development of novel α1-Adrenergic Receptor subtype-selective drugs .
|
-
- HY-P5756
-
|
|
Opioid Receptor
|
Neurological Disease
|
|
CSD-CH2(1,8)-NH2 is a selective and competitive KOR antagonist (Ki: 6.8 nM). CSD-CH2(1,8)-NH2 inhibits calcium mobilization in DRG neurons. CH2(1,8)-NH2 antagonizes the antinociceptive effect of U50,488. CSD-CH2(1,8)-NH2 can be used for research of neuropsychiatric disorders .
|
-
- HY-P1178A
-
|
|
Trk Receptor
|
Neurological Disease
|
|
Cyclotraxin B TFA is a BBB-penetrable and selective TrkB inhibitor. Cyclotraxin B TFA inhibits BDNF-induced TrkB activity in a non-competitive manner, with an IC50 of 0.30 nM. Cyclotraxin B TFA has analgesic and anxiolytic effects .
|
-
- HY-P1694
-
|
|
Bradykinin Receptor
|
Cardiovascular Disease
|
|
B4148 is a selective competitive bradykinin (BK) antagonist that significantly inhibits BK-induced hypotension in rats. In a rat model of endotoxin shock induced by Escherichia coli lipopolysaccharide, B4148 significantly attenuated the decrease in mean arterial blood pressure compared with the control group .
|
-
- HY-P1293A
-
|
|
iGluR
|
Neurological Disease
|
|
Conantokin G TFA, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G TFA inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G TFA has neuroprotective properties .
|
-
- HY-P11019
-
|
|
CXCR
Calcium Channel
|
Cancer
|
|
IS4 is a selective CXCR4 competitive antagonist, with an IC50 of 0.65 nM in THP-1 cells and 38.75 nM in Jurkat cells. IS4 is stable in serum and non-cytotoxic. IS4 competitively binds to CXCR4 with CXCL12, thereby inhibiting CXCL12-induced intracellular Ca 2+ release and cancer cell migration. IS4 can be used in the research on the prevention of metastasis of breast cancer, prostate cancer, leukemia, and other diseases .
|
-
- HY-P2055
-
|
|
Endogenous Metabolite
|
Endocrinology
|
|
A-57696 is a cholecystokinin antagonist with selective activity at cortical CCK-B receptors (IC50 = 25 nM). A-57696 behaves as a competitive antagonist in reversing CCK8-stimulated pancreatic alpha-amylase secretion and phosphatidylinositol degradation. A-57696 fails to induce gallbladder contraction and inhibits CCK8-induced contraction. A-57696 behaves as a partial agonist at CCK-B/gastrin receptors on NCI-H345 cells, achieving 80% of the maximal CCK8 response. A-57696 and CCK8 inhibit each other in a calcium mobilization assay .
|
-
- HY-114118C
-
|
|
GLP Receptor
Insulin Receptor
α-synuclein
Apoptosis
p38 MAPK
Autophagy
Bcl-2 Family
|
Neurological Disease
Metabolic Disease
Cancer
|
|
Semaglutide sodium is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide sodium promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide sodium also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide sodium has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide sodium can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
| Cat. No. |
Product Name |
Target |
Research Area |
Image |
-
- HY-P99014
-
|
ARGX-110
|
Fc Receptor (FcR)
NF-κB
|
Inflammation/Immunology
Cancer
|
|
Cusatuzumab (ARGX-110) is a selective competitive blocker targeting CD70 (with an equilibrium dissociation constant of 17 pM for binding to human CD70). Cusatuzumab also possesses enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) activity. It is a humanized IgG1 monoclonal antibody, artificially synthesized through humanization and genetic engineering modifications (CH2 region mutation to enhance effector function). Cusatuzumab has a dual mechanism of action: firstly, it competitively blocks the interaction between CD70 and CD27, inhibiting the CD27-NF-κB signaling pathway, reducing regulatory T cell (Treg) activation and tumor cell proliferation; secondly, by enhancing binding to FcγRIIIa, it mediates ADCC and antibody-dependent cellular phagocytosis (ADCP), directly lysing CD70-positive tumor cells. Cusatuzumab can efficiently eliminate leukemia stem cells (LSCs), induce tumor cell differentiation and apoptosis, restore immune surveillance, and target CD70-positive tumors. Cusatuzumab is used in the study of acute myeloid leukemia (AML) .
|
-

(5)
-
- HY-P99144A
-
|
|
PD-1/PD-L1
|
Inflammation/Immunology
Cancer
|
|
Anti-Mouse PD-1 Antibody (S-5001) is a selective inhibitor targeting PD-1, blocking the PD-1/PD-L1 immune checkpoint axis through competitive binding to PD-1. Anti-Mouse PD-1 Antibody (S-5001) works by reversing the tumor immunosuppressive microenvironment and reactivating the anti-tumor activity of cytotoxic T lymphocytes. It can be used in research on tumors such as melanoma and HPV-positive oropharyngeal squamous cell carcinoma. Anti-Mouse PD-1 Antibody (S-5001) is often combined with photothermal therapy, chemotherapy, etc., to enhance efficacy .
|
-
(5)
-
- HY-108831A
-
|
AN100226; BG00002
|
Integrin
|
Inflammation/Immunology
Cancer
|
|
Natalizumab (Anti-CD49d) (AN100226; BG00002) Solution is a humanized monoclonal IgG4 antibody inhibitor that selectively targets α4 integrin (CD49d), blocking the interaction of integrins such as α4β1 (VLA-4) with vascular cell adhesion molecule VCAM-1, intercellular adhesion molecule ICAM-1, and fibronectin by competitively binding to the α4 subunit. Natalizumab solution inhibits the adhesion, retention, and transendothelial migration of immune cells (such as CD4 + T cells), reducing the infiltration of inflammatory cells into the central nervous system or lesion sites, thereby exerting anti-inflammatory and immunomodulatory activity. Natalizumab (Anti-CD49d) solution is used in the study of relapsing-remitting multiple sclerosis (RRMS) and is also applied in the research of autoimmune or inflammation-related diseases such as Crohn's disease, B-cell lymphoma, and non-infectious uveitis. Natalizumab (Anti-CD49d) can also prevent lymphocytes from entering the central nervous system, thus preventing acute demyelinating relapses .
|
-
(5)
-
- HY-108831
-
|
|
Integrin
|
Inflammation/Immunology
Cancer
|
|
Natalizumab (AN100226; BG00002) is a humanized monoclonal IgG4 antibody inhibitor that selectively targets α4 integrin (CD49d). It blocks the interaction of integrins such as α4β1 (VLA-4) with vascular cell adhesion molecule VCAM-1, intercellular adhesion molecule ICAM-1, and fibronectin by competitively binding to the α4 subunit. Natalizumab inhibits the adhesion, retention, and transendothelial migration of immune cells (such as CD4 + T cells), reducing the infiltration of inflammatory cells into the central nervous system or lesion sites, thus exerting anti-inflammatory and immunomodulatory activity. Natalizumab is used in the study of relapsing-remitting multiple sclerosis (RRMS) and also has applications in the study of autoimmune or inflammation-related diseases such as Crohn's disease, B-cell lymphoma, and non-infectious uveitis. Natalizumab can also prevent lymphocytes from entering the central nervous system, thereby preventing acute demyelinating relapses .
|
-
(5)
-
- HY-P99463
-
|
AVB-500; AVB-S6-500
|
TAM Receptor
PI3K
Akt
p38 MAPK
|
Cancer
|
|
Batiraxcept (AVB-500; AVB-S6-500) is a selective, soluble AXL receptor and GAS6 inhibitor that targets the GAS6-AXL signaling axis. Batiraxcept is orally inactive and does not cross the blood-brain barrier. Batiraxcept competitively binds to GAS6 ((KD <1 nM), preventing its interaction with the AXL receptor tyrosine kinase, thereby inhibiting downstream PI3K/AKT and MAPK signaling pathways, reducing tumor cell glycolysis, angiogenesis, and metastatic potential. Batiraxcept has demonstrated antitumor activity in preclinical models of endometrial, cholangiocarcinoma, and ovarian cancer by inhibiting tumor growth, invasion, and metastasis .
|
-
(5)
-
- HY-P991869
-
|
AQmabAM; rAb-53
|
Aquaporin
|
Neurological Disease
|
|
Aquaporumab (AQmabAM) is an engineered human monoclonal IgG antibody. Aquaporumab is a selective inhibitor of aquaporin 4 (AQP4), binding tightly to AQP4 and competitively displacing AQP4-IgG from serum [1][2]. Aquaporumab significantly reduces neuromyelitis optica lesions in spinal cord slice cultures and in mice receiving intracerebral injection of AQP4-IgG and complement [1][2]. Aquaporumab can be used for research on neuromyelitis optica spectrum disorders [1][2]."}
|
-
(5)
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-15141
-
-
-
- HY-B2132
-
|
3-(2-Aminoethyl)indole~2-(3-Indolyl)ethylamine
|
Alkaloids
Microorganisms
Classification of Application Fields
Other Diseases
Endogenous metabolite
Disease Research Fields
Indole Alkaloids
|
Endogenous Metabolite
5-HT Receptor
Aryl Hydrocarbon Receptor
|
|
Tryptamine is a selective, blood-brain-penetrating 5-HT4 receptor agonist (EC50=1-3 mM) and an endogenous ligand of the aryl hydrocarbon receptor (AHR) (Kd=10-50 nM). Tryptamine promotes intestinal anion secretion and fluid transport by activating G protein-coupled receptors (GPCRs) and accelerates gastrointestinal motility. Tryptamine regulates Th17/Treg balance to inhibit neuroinflammation, competitively binds to 5-HT receptors to regulate central nervous system activity, and participates in temperature regulation and spinal reflex regulation as a neuromodulator. Tryptamine can be used to study intestinal motility disorders such as functional constipation, and has shown significant efficacy in multiple sclerosis models .
|
-
-
- HY-B0442
-
-
-
- HY-18732A
-
-
-
- HY-B0140
-
-
-
- HY-B0290
-
-
-
- HY-B0442A
-
-
-
- HY-I1070
-
|
(R)-Isoleucine
|
Microorganisms
Source Classification
|
ASCT
|
|
D-Isoleucine is a selective competitive activator of the Asc-1 antiporter (Ki=0.98 mM). D-Isoleucine promotes the release of D-serine and glycine by binding to the Asc-1 protein on the neuronal cell membrane, and enhances NMDA receptor-dependent synaptic plasticity. D-Isoleucine can be used in the study of neurodegenerative diseases (such as Alzheimer's disease and schizophrenia). D-Isoleucine also acts as a non-classical D-amino acid, interferes with bacterial peptidoglycan synthesis, and inhibits the formation of Staphylococcus aureus biofilm, and has potential antibacterial application value[1][2].
|
-
-
- HY-126752
-
-
-
- HY-A0009
-
|
Galantamine hydrobromide
|
Alkaloids
Other Alkaloids
Plants
Amaryllidaceae
|
Cholinesterase (ChE)
nAChR
|
|
Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 µM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
-
- HY-15237
-
-
-
- HY-I0096
-
|
|
Ketones, Aldehydes, Acids
Endogenous metabolite
Source Classification
|
iGluR
HIV
HIV Integrase
|
|
Indole-2-carboxylic acid (I2CA) is a competitive antagonist of the glycine site of the NMDA receptor (Ki=15 μM, 5-fluoro-I2CA) and an inhibitor of HIV-1 integrase. Indole-2-carboxylic acid is selective for the glycine site of the NMDA receptor and blocks the enhancement of NMDA receptor by competitively inhibiting the binding of glycine to the NMDA receptor. Indole-2-carboxylic acid can also inhibit the strand transfer activity of HIV-1 integrase by chelating Mg 2+ at the active site of integrase and interacting with the hydrophobic cavity. Indole-2-carboxylic acid can be used in the study of neurological diseases (such as stroke, epilepsy) and HIV-1 infection .
|
-
-
- HY-B0442B
-
-
-
- HY-110399
-
-
-
- HY-N2554
-
|
Ostenol
|
Coumarins
Phenols
Polyphenols
Phenylpropanoids
Kleinia odora (Forssk.) DC.
Umbelliferae
Plants
Source Classification
|
Monoamine Oxidase
PI3K
Akt
Apoptosis
|
|
Osthenol (Ostenol) is a reversible, selective, competitive inhibitor of hMAO-A (IC50=0.74 μM, Ki=0.26 μM), with antifungal and antibacterial activity. Osthenol inhibits the oxidative deamination of hMAO-A and regulates the metabolism of monoamine neurotransmitters. Osthenol also inhibits the PI3K/AKT signaling pathway to induce apoptosis of colon cancer cells, arrest the cell cycle at the G1 phase, and inhibit cell proliferation. Osthenol is mainly used in the study of neurological diseases and cancer, especially depression-related MAO-A targeted intervention and colon cancer .
|
-
-
- HY-B0927
-
-
-
- HY-18963
-
-
-
- HY-N6057
-
-
-
- HY-119413
-
-
-
- HY-133178
-
|
3,4,8,9-Tetrahydroxy urolithin
|
Microorganisms
Classification of Application Fields
Phenols
Polyphenols
Disease Research Fields
Source Classification
Cancer
|
Ephrin Receptor
PPAR
AMPK
|
|
Urolithin D (3,4,8,9-Tetrahydroxy urolithin) is a colonic metabolite of Ellagitannins and a competitive, reversible, and selective antagonist of the EphA receptor. Urolithin D inhibits EphA2-ephrin-A1 binding with an IC50 of 0.9 μM. Urolithin D is also a potent antioxidant that scavenges free radicals and repairs oxidized DNA damage. Additionally, Urolithin D suppresses triglyceride accumulation and promotes fatty acid oxidation by activating the AMPK signaling pathway. Urolithin D can be used for research on tumors, metabolic, and inflammatory diseases .
|
-
-
- HY-B0290A
-
-
-
- HY-130199
-
-
-
- HY-15141R
-
-
-
- HY-B0290R
-
-
-
- HY-N8540
-
|
|
Natural Products
Microorganisms
Source Classification
|
Phosphoglycerate Kinase (PGK)
Fungal
Apoptosis
|
|
Ilicicolin H is a selective and non-ATP-competitive phosphoglycerate kinase 1 (PGK1) (IC50 = 9.02 μM) and mitochondrial cytochrome bc1 reductase (IC50 = 2-3 ng/mL) inhibitor. Ilicicolin H directly binds to PGK1 with KD of 60 μM .Ilicicolin H can inhibit cell proliferation and induce apoptosis. Ilicicolin H can inhibit the lactate production and glucose uptake of hepatocellular carcinoma (HCC) cells. Ilicicolin H has a broad antifungal spectrum including C. albicans, Cryptococcus and A. fumigatus. Ilicicolin H can be used for the researches of cancer and infection, such as hepatocellular carcinoma (HCC) and C. albicans infection .
|
-
-
- HY-N10488
-
-
-
- HY-122369
-
-
-
- HY-100513
-
|
|
Structural Classification
Microorganisms
Antibiotics
Source Classification
|
DNA/RNA Synthesis
Apoptosis
Antibiotic
|
|
(±)-Dehydroaltenusin, an antibiotic, is a selective eukaryotic DNA polymerase α (pol α) inhibitor with an IC50 of 0.68 μM. (±)-Dehydroaltenusin can be isolated from fungus Alternaria tenuis. (±)-Dehydroaltenusin competitively inhibits the DNA template primer (Ki: 0.23 μM) and non-competitively suppresses the 2'-deoxyribonucleoside 5'-triphosphate substrate (Ki: 0.18 μM). (±)-Dehydroaltenusin induces the cancer cell S-phase cycle arrest and apoptosis. (±)-Dehydroaltenusin can be used for cancers like human adenocarcinoma research .
|
-
-
- HY-N7536
-
-
-
- HY-W661499
-
|
|
Structural Classification
Natural Products
Microorganisms
Source Classification
|
Phosphatase
Apoptosis
Reactive Oxygen Species (ROS)
Caspase
|
|
Orellanine, a nephrotoxic alkaloid found in Cortinarius orellanus, is an orally active and selective non-competitive inhibitor of alkaline phosphatase. Orellanine chelates iron, generates reactive oxygen species (ROS), induces DNA scission, forms ortho-semiquinone radicals, downregulates antioxidant defenses, and inhibits mitochondrial function. Orellanine induces caspase 8/9-mediated apoptosis. Orellanine inhibits synthesis of proteins, RNA, DNA, and mitochondrial protein synthesis, with metabolic activation required for cell-free protein synthesis inhibition. Orellanine can be used for the research of metastatic clear cell renal cell carcinoma, acute renal failure, chronic renal insufficiency, and kidney damage .
|
-
-
- HY-N1151
-
|
|
Structural Classification
Hydrangeaceae
Coumarins
Phenylpropanoids
Plants
Source Classification
|
Bacterial
Cholinesterase (ChE)
MMP
TNF Receptor
|
|
Thunberginol C is an orally active, selective, and non-competitive inhibitor of AChE and BChE, with IC50 values of 41.96 and 42.36 μM, respectively. Thunberginol C exerts cytoprotective, pro-collagen type I restorative, MMP-1 inhibitory, hyaluronic acid restorative, anti-photoaging effects in skin cells. Thunberginol C exerts neuroprotective, anxiolytic, TNF-α inhibitory, neuroinflammation inhibitory, and oxidative stress inhibitory effects. Thunberginol C can be used for the research of Alzheimer’s disease, UVB-induced skin photoaging, allergic reactions, oral bacterial infections, and stress-induced anxiety .
|
-
-
- HY-P1293
-
|
|
Natural Products
Animals
Source Classification
|
iGluR
|
|
Conantokin G, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G has neuroprotective properties .
|
-
-
- HY-B0290AR
-
-
-
- HY-A0009R
-
|
Galantamine hydrobromide (Standard)
|
Structural Classification
Alkaloids
Other Alkaloids
Plants
Amaryllidaceae
Source Classification
|
nAChR
Cholinesterase (ChE)
Reference Standards
|
|
Galanthamine (hydrobromide) (Standard) is the analytical standard of Galanthamine (hydrobromide). This product is intended for research and analytical applications. Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 µM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
-
- HY-110399R
-
-
-
- HY-18963R
-
-
-
- HY-P1293A
-
|
|
Natural Products
Animals
Source Classification
|
iGluR
|
|
Conantokin G TFA, a 17-amino-acid peptide, is a potent, selective and competitive antagonist of N-methyl-D-aspartate (NMDA) receptors. Conantokin G TFA inhibits NMDA-evoked currents in murine cortical neurons with an IC50 of 480 nM. Conantokin G TFA has neuroprotective properties .
|
-
-
- HY-113015R
-
-
-
- HY-N13917
-
|
|
Natural Products
Microorganisms
Source Classification
|
Proteasome
Bacterial
|
|
Argyrin B, a natural product cyclic peptide, is a reversible, non-competitive immunoproteasome inhibitor. Argyrin B shows selective inhibition of the β5i and β1i sites of the immunoproteasome over the β5c and β1c sites of the constitutive proteasome with nearly 20-fold selective inhibition of β1i over the homologous β1c. Argyrin B has antibacterial effects .
|
-
-
- HY-B0442C
-
-
-
- HY-18732AR
-
|
Tilarginine acetate (Standard); Methylarginine acetate (Standard)
|
Structural Classification
Natural Products
Endogenous metabolite
Source Classification
|
Reference Standards
NO Synthase
|
|
L-NMMA (Tilarginine) acetate (Standard) is the analytical standard of L-NMMA acetate (HY-18732A). This product is intended for research and analytical applications. L-NMMA (Tilarginine) acetate is a non-selective and competitive inhibitor of nitric oxide synthase. L-NMMA acetate inhibits three subtypes, namely nNOS, eNOS, and iNOS, and reduces NO production . L-NMMA acetate alleviates mechanical allodynia, thermal hyperalgesia, and choroidal fibrosis. L-NMMA acetate is applicable to research related to nociception, bone cancer pain, and myopia .
|
-
-
- HY-B0442CR
-
|
|
Structural Classification
Natural Products
Endogenous metabolite
Source Classification
|
Reference Standards
Endogenous Metabolite
Phosphodiesterase (PDE)
|
|
Vardenafil (dihydrochloride) (Standard) is the analytical standard of Vardenafil (dihydrochloride). This product is intended for research and analytical applications. Vardenafil dihydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil dihydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM respectively, while IC50s are >1000 nM for PDE3 and PDE4. Vardenafil dihydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil dihydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-
-
- HY-B0442AR
-
|
|
Structural Classification
Natural Products
Endogenous metabolite
Source Classification
|
Phosphodiesterase (PDE)
Endogenous Metabolite
Reference Standards
|
|
Vardenafil (hydrochloride) (Standard) is the analytical standard of Vardenafil (hydrochloride). This product is intended for research and analytical applications. Vardenafil hydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-
-
- HY-B0442R
-
-
-
- HY-W049735R
-
|
|
Monophenols
Phenols
Endogenous metabolite
Source Classification
|
Drug Metabolite
Reference Standards
Endogenous Metabolite
|
|
Vardenafil (dihydrochloride) (Standard) is the analytical standard of Vardenafil (dihydrochloride). This product is intended for research and analytical applications. Vardenafil dihydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil dihydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM respectively, while IC50s are >1000 nM for PDE3 and PDE4. Vardenafil dihydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil dihydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-
-
- HY-B0442BR
-
|
|
Structural Classification
Natural Products
Endogenous metabolite
Source Classification
|
Reference Standards
Endogenous Metabolite
Phosphodiesterase (PDE)
|
|
Vardenafil (hydrochloride trihydrate) (Standard) is the analytical standard of Vardenafil (hydrochloride trihydrate). This product is intended for research and analytical applications. Vardenafil hydrochloride trihydrate is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride trihydrate shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride trihydrate competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride trihydrate can be used for the research of erectile dysfunction, hepatitis, diabetes - .
|
-
-
- HY-B0927R
-
|
(-)-β-Hydrastine (Standard); (1R,9S)-β-Hydrastine (Standard)
|
Structural Classification
Alkaloids
Piperidine Alkaloids
Ranunculaceae
Plants
|
Reference Standards
Tyrosine Hydroxylase
Dopamine Receptor
OAT
|
|
Hydrastine (Standard) is the analytical standard of Hydrastine (HY-B0927). This product is intended for research and analytical applications. Hydrastine ((-)-β-Hydrastine; (1R,9S)-β-Hydrastine) is a selective competitive inhibitor of tyrosine hydroxylase (TH), inhibiting dopamine biosynthesis (IC50=20.7 μM, PC12 cells). Hydrastine also inhibits the organic cation transporter OCT1 (IC50=6.6 μM). Hydrastine may cause neuronal toxicity through mitochondrial dysfunction rather than oxidative stress damage, and can aggravate cell apoptosis when combined with L-DOPA. Hydrastine can be used to study Parkinson's disease-related dopaminergic neuronal damage .
|
-
-
- HY-133178R
-
|
3,4,8,9-Tetrahydroxy urolithin (Standard)
|
Microorganisms
Phenols
Polyphenols
Source Classification
|
Reference Standards
Ephrin Receptor
PPAR
AMPK
|
|
Urolithin D (Standard) (3,4,8,9-Tetrahydroxy urolithin (Standard)) is the analytical standard of Urolithin D (HY-133178). This product is intended for research and analytical applications. Urolithin D (3,4,8,9-Tetrahydroxy urolithin) is a colonic metabolite of Ellagitannins and a competitive, reversible, and selective antagonist of the EphA receptor. Urolithin D inhibits EphA2-ephrin-A1 binding with an IC50 of 0.9 μM. Urolithin D is also a potent antioxidant that scavenges free radicals and repairs oxidized DNA damage. Additionally, Urolithin D suppresses triglyceride accumulation and promotes fatty acid oxidation by activating the AMPK signaling pathway. Urolithin D can be used for research on tumors, metabolic, and inflammatory diseases .
|
-
-
- HY-N14540
-
-
-
- HY-N18289
-
-
- HY-N14094
-
|
|
Structural Classification
Alkaloids
Other Alkaloids
Rubiaceae
Plants
Pogonopus tubulosus (A.Rich. ex DC.) K.Schum.
Source Classification
|
JAK
Apoptosis
Autophagy
|
|
Tubulosine is an alkaloid. Tubulosine can be isolated from Pogonopus tubulosus (DC.) Schumann. Tubulosine is an ATP-competitive, selective JAK3 inhibitor with an IC50 value of 9.9 nM. Tubulosine also inhibits the kinase activities of other JAK family members, the extent of inhibition is less than that of JAK3, with IC50 values of 69.5, 84.9 and 76.3 nM for JAK1, JAK2 and TYK2, respectively. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. Tubulosine induces apoptotic and necrotic/autophagic cell death. Tubulosine inhibits the process of peptide chain elongation by eukaryotic polysomes by, specifically preventing the elongation-factor-2-dependent step of translocation. Tubulosine exhibits anticancer activity in breast cancer cells .
|
-
- HY-180420
-
-
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-114118S3
-
|
|
|
Semaglutide- 13C6, 15N TFA is the 13C- and 15N-labeled Semaglutide TFA (HY-114118A). Semaglutide TFA is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide TFA promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide TFA also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide TFA has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide TFA can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
-
- HY-114118S1
-
|
|
|
Semaglutide-d8 tetraTFA is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
-
- HY-114118S
-
|
|
|
Semaglutide-d8 is the deuterium labeled Semaglutide (HY-114118). Semaglutide is a long-acting, selective, competitive GLP-1R agonist that can penetrate the blood-brain barrier. After activating GLP-1R, Semaglutide promotes insulin secretion, inhibits gastric emptying and appetite, and at the same time enhances autophagy, inhibits oxidative stress and apoptosis. Semaglutide also regulates mitochondrial function and lipid metabolism (such as reducing de novo lipogenesis in the liver). Semaglutide has activities such as lowering blood sugar, reducing weight, neuroprotection (such as improving motor function in Parkinson's disease models, reducing α-synuclein aggregation) and improving hepatic steatosis. Semaglutide can be used for the study of neurodegenerative diseases and liver diseases such as type 2 diabetes, obesity, Parkinson's disease, metabolic associated fatty liver disease (MASLD), and cancer .
|
-
-
- HY-13011S
-
|
|
|
Alectinib-d8 is the deuterium labeled Alectinib. Alectinib (CH5424802) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
-
- HY-16985S
-
|
|
|
Darolutamide-d4 (ODM-201-d4) is deuterium labeled Darolutamide (HY-16985). Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-
-
- HY-B0442AS
-
|
|
|
Vardenafil-d5 hydrochloride is deuterated labeled Vardenafil hydrochloride (HY-B0442A). Vardenafil hydrochloride is a selective and orally active inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil hydrochloride shows inhibitory towards PDE1, PDE6 with IC50s of 180 nM, and 11 nM, while IC50s are >1000 nM for PDE3 and PDE4 . Vardenafil hydrochloride competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels . Vardenafil hydrochloride can be used for the research of erectile dysfunction, hepatitis, diabetes [1]-[6].
|
-
-
- HY-17623S
-
|
|
|
Tegoprazan (CJ-12420; RQ-00000004), a potassium-competitive acid blocker, is a reversible, oral active and highly selective inhibitor of gastric H+/K+-ATPase that could control gastric acid secretion and motility, with IC50 values ranging from 0.29-0.52 μM for porcine, canine, and human H +/K +-ATPases in vitro. Tegoprazan significantly improves colitis in mice and enhances the intestinal epithelial barrier function. Tegoprazan is promising for research of Inflammatory bowel, gastric acid-related, motilityimpaired diseases .
|
-
-
- HY-15656S
-
|
|
|
Ceritinib (LDK378)-d7 is a deuterium labeled Ceritinib (HY-15656). Ceritinib is a selective, orally bioavailable and ATP-competitive ALK tyrosine kinase inhibitor . Ceritinib is a selective, orally bioavailable, and ATP-competitive ALK tyrosine kinase inhibitor with an IC50 of 200 pM. Ceritinib also inhibits IGF-1R, InsR, and STK22D with IC50 values of 8, 7, and 23 nM, respectively. Ceritinib shows great antitumor potency .
|
-
-
- HY-10195BS
-
|
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
-
- HY-13011S1
-
|
|
|
Alectinib-d6 is deuterium labeled Alectinib. Alectinib (CH5424802) is a potent, selective, and orally available ALK inhibitor with an IC50 of 1.9 nM and a Kd value of 2.4 nM (in an ATP-competitive manner), and also inhibits ALK F1174L and ALK R1275Q with IC50s of 1 nM and 3.5 nM, respectively . Alectinib demonstrates effective central nervous system (CNS) penetration .
|
-
-
- HY-50878AS
-
|
|
|
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
-
- HY-W700452
-
|
|
|
Y-27632-d4 hydrochloride hydrate is the deuterium labeled Y-27632 hydrochloride hydrate (HY-10071A). Y-27632 hydrochloride hydrate is an orally active, ATP-competitive inhibitor of ROCK-I and ROCK-II, with Kis of 220 and 300 nM, respectively. Y-27632 hydrochloride hydrate attenuates Doxorubicin-induced apoptosis of human cardiac stem cells. Y-27632 hydrochloride hydrate also suppresses dissociation-induced apoptosis of murine prostate stem/progenitor cells. Y-27632 hydrochloride hydrate primes human induced pluripotent stem cells (hIPSCs) to selectively differentiate towards mesendodermal lineage via epithelial-mesenchymal transition-like modulation .
|
-
-
- HY-10512S
-
|
|
|
AR-A014418-d3 is the deuterium labeled AR-A014418. AR-A014418 is a potent, selective, and ATP-competitive GSK3β inhibitor (IC50=104 nM; Ki=38 nM) .
|
-
-
- HY-10917S
-
|
|
|
GW2580-d6 is the deuterium labeled GW2580. GW2580 is an orally bioavailable and selective inhibitor of c-Fms kinase which completely inhibits human cFMS kinase in vitro at 0.06 μM. GW2580 acts as a competitive inhibitor of ATP binding to the cFMS kinase and inhibits colony-stimulating-factor-1 signaling .
|
-
-
- HY-B0442S
-
|
|
|
Vardenafil-d5 is deuterium labeled Vardenafil. Vardenafil is a selective, orally active, potent inhibitor of phosphodiesterase-5 (PDE5), with an IC50 of 0.7 nM. Vardenafil shows selectivity over PDE1 (180 nM), PDE6 (11 nM), PDE2, PDE3, and PDE4 (>1000 nM). Vardenafil competitively inhibits cyclic guanosine monophosphate (cGMP) hydrolysis and thus increases cGMP levels. Vardenafil can be used for the research of erectile dysfunction .
|
-
-
- HY-10285S2
-
|
|
|
Saxagliptin- 13C2 (BMS-477118- 13C2) is 13C labeled Saxagliptin. Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-
-
- HY-10119S
-
|
|
|
Vorapaxar-d5 is deuterated labeled Vorapaxar (HY-10119). Vorapaxar (SCH 530348), an antiplatelet agent, is a selective, orally active, and competitive thrombin receptor protease-activated receptor (PAR-1) antagonist (Ki=8.1 nM). Vorapaxar (SCH 530348) inhibits thrombin receptor-activating peptide (TRAP)-induced platelet aggregation in a dose-dependent manner .
|
-
-
- HY-13204AS
-
|
|
|
Biperiden-d5 (KL 373-d5) is deuterium labeled Biperiden. Biperiden (KL 373) is a non-selective muscarinic receptor antagonist that competitively binds to M1 muscarinic receptors, thereby inhibiting acetylcholine and enhancing dopamine signaling in the central nervous system. Biperiden has the potential for the research of Parkinson's disease and other related psychiatric disorders .
|
-
-
- HY-10409S
-
|
|
|
Fedratinib-d9 (TG-101348-d9) is deuterium labeled Fedratinib. Fedratinib (TG-101348) is a potent, selective, ATP-competitive and orally active JAK2 inhibitor with IC50s of 3 nM for both JAK2 and JAK2V617F kinase. Fedratinib shows 35- and 334-fold selectivity over JAK1 and JAK3, respectively. Fedratinib induces cancer cell apoptosis and has the potential for myeloproliferative disorders research .
|
-
-
- HY-B0290S1
-
|
|
|
Pranlukast-d4 is deuterium labeled Pranlukast. Pranlukast is a highly potent, selective and competitive antagonist of peptide leukotrienes. Pranlukast inhibits [3H]LTE4, [3H]LTD4, and [3H]LTC4 bindings to lung membranes with Kis of 0.63±0.11, 0.99±0.19, and 5640±680 nM, respectively.
|
-
-
- HY-18174S
-
|
|
|
Prexasertib-d4 (LY2606368-d4) is the deuterium labeled Prexasertib (HY-18174). Prexasertib (LY2606368) is a selective, ATP-competitive second-generation checkpoint kinase 1 (CHK1) inhibitor with a Ki of 0.9 nM and an IC50 of <1 nM. Prexasertib inhibits CHK2 (IC50=8 nM) and RSK1 (IC50=9 nM). Prexasertib causes double-stranded DNA breakage and replication catastrophe resulting in apoptosis. Prexasertib shows potent anti-tumor activity .
|
-
-
- HY-A0009S
-
|
|
|
Galanthamine-d3 (hydrobromide) is deuterium labeled Galanthamine (hydrobromide). Galanthamine hydrobromide (Galantamine hydrobromide) is a selective, reversible, competitive, alkaloid AChE inhibitor, with an IC50 of 0.35 μM. Galanthamine hydrobromide is a potent allosteric potentiating ligand (APL) of human α3β4, α4β2, α6β4 nicotinic receptors ( nAChRs). Galanthamine hydrobromide is developed for the research of Alzheimer's disease (AD) .
|
-
-
- HY-10285S
-
|
|
|
Saxagliptin-15N,d2 Hydrochloride (BMS-477118-15N,d2 Hydrochloride) is the 15N and deuterium labeled isotope of Saxagliptin (HY-10285). Saxagliptin (BMS-477118) is a potent, selective, reversible, competitive and orally active dipeptidyl peptidase-4 (DPP-4) (Ki = 0.6-1.3 nM) inhibitor. Saxagliptin has the peotential for type 2 diabetes mellitus research .
|
-
-
- HY-118835S
-
|
|
|
Zimeldine-d6 is the deuterium labeled Zimeldine (HY-118835) . Zimelidine is an orally active selective serotonin reuptake inhibitor. Zimelidine competitively inhibits central 5-HT uptake and desensitizes 5-HT autoreceptors in dorsal raphe nucleus. Zimelidine time-dependently modulates 5-HT neuronal firing and hippocampal CA3 responses. Zimelidine strengthens central serotonergic neurotransmission and produces related behavioral changes. Zimelidine exerts anxiolytic, analgesic, feeding-suppressive and tolerance-attenuating effects. Zimelidine is used for the study of depressive disorders and analgesic tolerance .
|
-
-
- HY-136270S
-
|
|
|
Gartisertib-d8 (VX-803-d8) is the deuterium labeled Gartisertib (HY-136270). Gartisertib (VX-803) is an ATP-competitive, orally active, and selective ATR inhibitor, with a Ki of <150 pM. Gartisertib potently inhibits ATR-driven phosphorylated checkpoint kinase-1 (Chk1) phosphorylation with an IC50 of 8 nM. Antitumor activity .
|
-
-
- HY-10280S
-
|
|
|
YM-60828-d3 is the deuterium labeled YM-60828 (HY-10280). YM-60828 is an orally active, selective and competitive factor Xa inhibitor with a Ki of 1.3 nM and an IC50 of 2.3 nM. YM-60828 inhibits thrombus formation and platelet aggregation. YM-60828 can be used for the research of venous thrombosis, arterial thrombosis, and thromboembolic disorders .
|
-
| Cat. No. |
Product Name |
|
Classification |
-
- HY-23460
-
|
4-Ethynyl-L-phenylalanine
|
|
Alkynes
|
|
p-Ethynylphenylalanine (4-Ethynyl-L-phenylalanine) is a potent, selective, reversible and competitive inhibitor of tryptophan hydroxylase (TPH), with a Ki of 32.6 μM . p-Ethynylphenylalanine is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-23460A
-
|
4-Ethynyl-L-phenylalanine hydrochloride
|
|
Alkynes
|
|
p-Ethynylphenylalanine hydrochloride (4-Ethynyl-L-phenylalanine hydrochloride) is a potent, selective, reversible and competitive inhibitor of tryptophan hydroxylase (TPH), with a Ki of 32.6 μM . p-Ethynylphenylalanine (hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-104066
-
|
Xiliertinib; HMPL-309
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-146691
-
|
|
|
Alkynes
|
|
hMAO-B-IN-2 (compound 6j) is an orally active, potent, selective and BBB penetrated and competitive reversible hMAO-B inhibitor, with an IC50 of 4 nM. hMAO-B-IN-2 shows low toxicity and good neuroprotective effects in SH-SY5Y cell. hMAO-B-IN-2 can be used for alzheimer’s disease research . hMAO-B-IN-2 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
- HY-104066A
-
|
Xiliertinib tartrate; HMPL-309 tartrate
|
|
Alkynes
|
|
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
| Cat. No. |
Product Name |
|
Classification |
-
- HY-12854
-
|
GRN163L
|
|
Antisense Oligonucleotides
|
|
Imetelstat (GRN163L) is a 13-mer oligonucleotide and competitive Telomerase inhibitor. Imetelstat binds with high affinity to the template region of the RNA component of human telomerase. Imetelstat induces Apoptosis. Imetelstat is capable of selectively eliminating myelofibrosis hematopoietic stem cells. Imetelstat leads to the loss of a cancer cell's ability to maintain telomere length, resulting in the inhibition of cell proliferation .
|
-
- HY-W250153
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-164090
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-W250153A
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-10254G
-
|
PD0325901; PD325901
|
MEK
|
Cancer
|
|
Mirdametinib (PD0325901) (GMP) is Mirdametinib (HY-10254) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Mirdametinib is an orally active, selective and non-ATP-competitive MEK inhibitor .
|
-
-
- HY-70044G
-
|
GSK-1070916A
|
Apoptosis
Aurora Kinase
|
Cancer
|
|
GSK-1070916 (GMP) is GSK-1070916 (HY-70044) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. GSK-1070916 is a potent and selective ATP-competitive inhibitor of aurora B and aurora C with Kis of 0.38 and 1.5 nM, respectively, and is >250- fold selective over Aurora A .
|
-
-
- HY-15141G
-
|
Antibiotic AM-2282; STS; AM-2282
|
PKC
|
Infection
Cancer
|
|
Staurosporine (AM-2282) (GMP) is Staurosporine (HY-15141) produced by using GMP guidelines. GMP small molecules works appropriately as an auxiliary reagent for cell therapy manufacture. Staurosporine is a potent, ATP-competitive and non-selective inhibitor of protein kinases .
|
-
-
- HY-13418G
-
|
Compound C dihydrochloride; BML-275 dihydrochloride
|
AMPK
|
Neurological Disease
Cancer
|
|
Dorsomorphin dihydrochloride (GMP) is the GMP level of Dorsomorphin dihydrochloride (HY-13418). GMP guidelines are used to produce Dorsomorphin dihydrochloride (GMP). GMP small molecules works appropriately as an auxiliary reagent for cell research manufacture. Dorsomorphin dihydrochloride (GMP) is a potent, selective and ATP-competitive AMPK inhibitor. Dorsomorphin dihydrochloride (GMP) can be used for the research of induced differentiation of pluripotent stem cells (PSCs) .
|
-
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent: