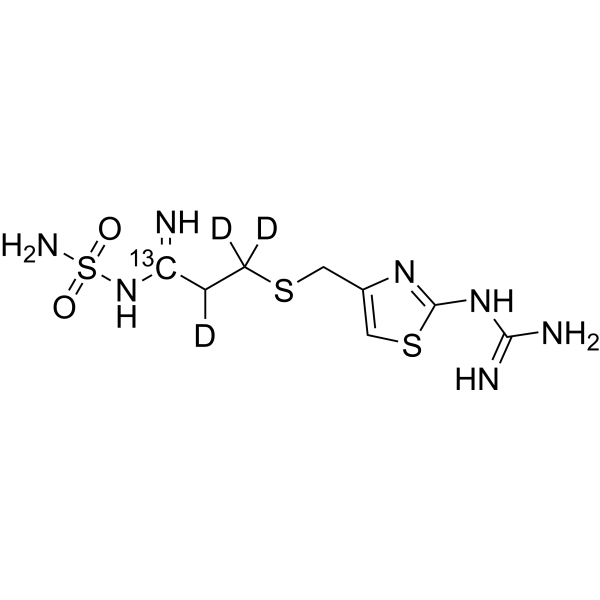Search Result
Results for "
Competitive inhibition
" in MedChemExpress (MCE) Product Catalog:
3
Biochemical Assay Reagents
15
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
- HY-50878
-
|
PF-02341066
|
Ligands for Target Protein for PROTAC
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
|
Cancer
|
|
Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
-
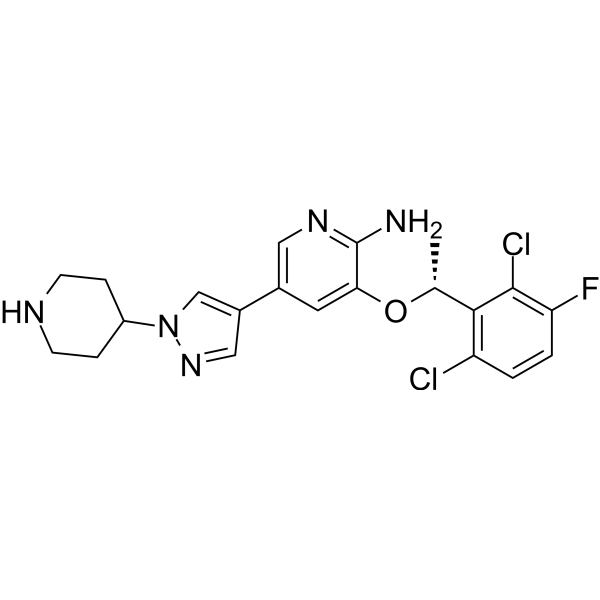
- HY-17000
-
|
OPC-41061
|
Vasopressin Receptor
Autophagy
|
Cardiovascular Disease
Endocrinology
Cancer
|
|
Tolvaptan is a selective, competitive and orally active vasopressin receptor 2 (V2R) antagonist with an IC50 of 1.28 μM for the inhibition of arginine vasopressin (AVP)-induced platelet aggregation. Tolvaptan induces cell apoposis and affects cell cycle. Tolvaptan can be used for the research of hyponatremia .
|
-
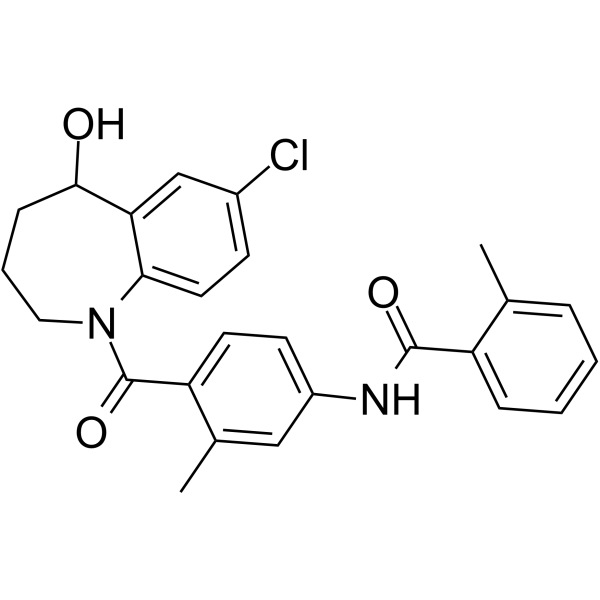
-
- HY-B0377
-
-
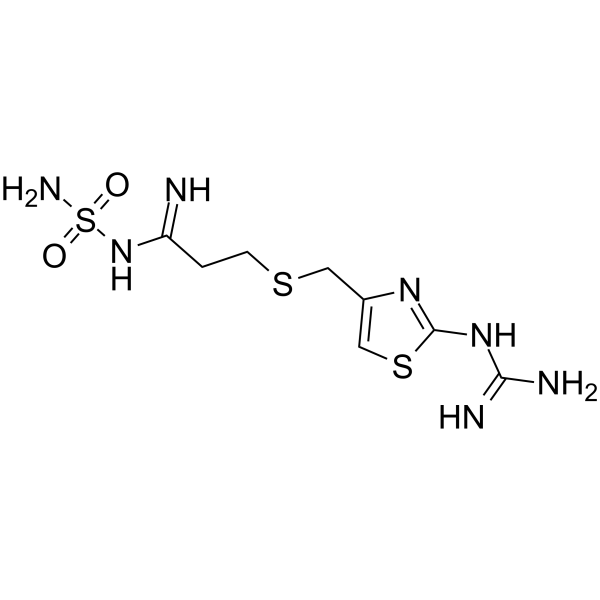
-
- HY-12481
-
|
|
PI3K
Autophagy
|
Cancer
|
|
SAR405 is a first-in-class, selective, and ATP-competitive PI3K class III (PIK3C3) isoform Vps34 inhibitor (IC50=1.2 nM; Kd=1.5 nM). SAR405 inhibits autophagy induced either by starvation or by mTOR inhibition. Anticancer activity .
|
-
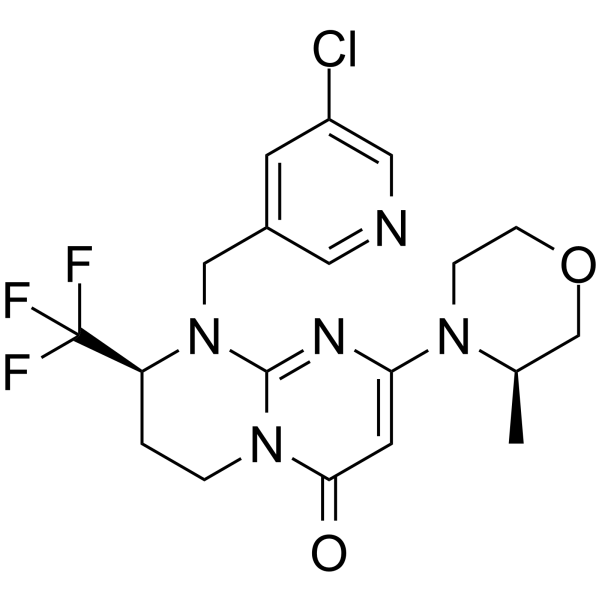
-
- HY-108913
-
|
|
Beta-lactamase
Antibiotic
Bacterial
|
Infection
|
|
Nitrocefin is a highly activated, chromogenic cephalosporin derivative. Nitrocefin is a chromogenic β-lactamase substrate. Nitrocefin undergoes a distinctive color change from yellow to red as the amide bond in the β-lactam ring is hydrolyzed by β-lactamase. Nitrocefin is used in competitive inhibition studies in developmental work on β-lactamase-resistant antibiotics .
|
-
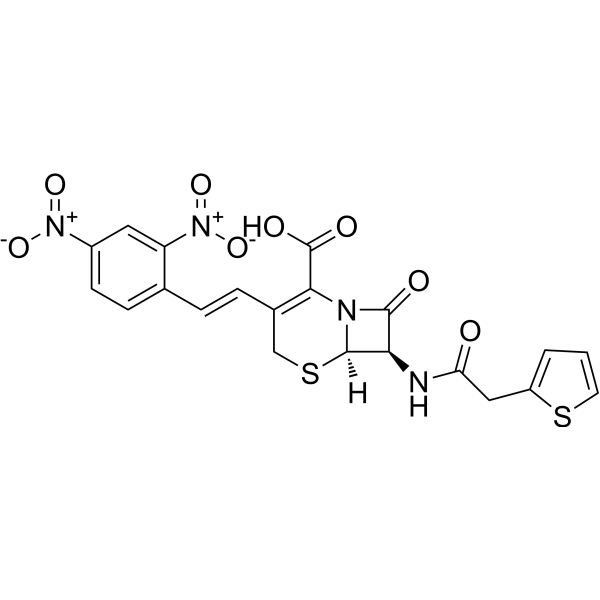
-
- HY-12037A
-
|
ON-01910
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib (ON-01910) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3 kinase/Akt pathway, promots the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
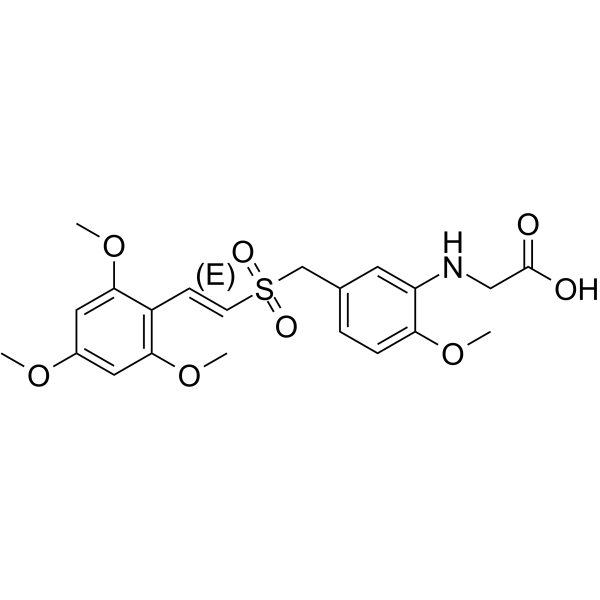
-
- HY-402410
-
TETi76
1 Publications Verification
|
TET Protein
|
Cancer
|
|
TETi76 is an orally active TET family inhibitor with IC50 values ??of 1.5, 9.4 and 8.8 μM for TET1, TET2 and TET3, respectively. TETi76 competitively binds to the active site of TET enzymes, reduces cytosine hydroxymethylation and restricts clonal growth of TET2 mutants in vitro and in vivo, but does not affect the growth of normal hematopoietic precursor cells. TETi76 can be used for leukemia research .
|
-

-
- HY-15663
-
|
|
PAK
|
Cancer
|
|
IPA-3 is a selective non-ATP competitive PAK1 inhibitor with IC50 of 2.5 μM, and shows no inhibition to group II PAKs (PAKs 4-6).
|
-
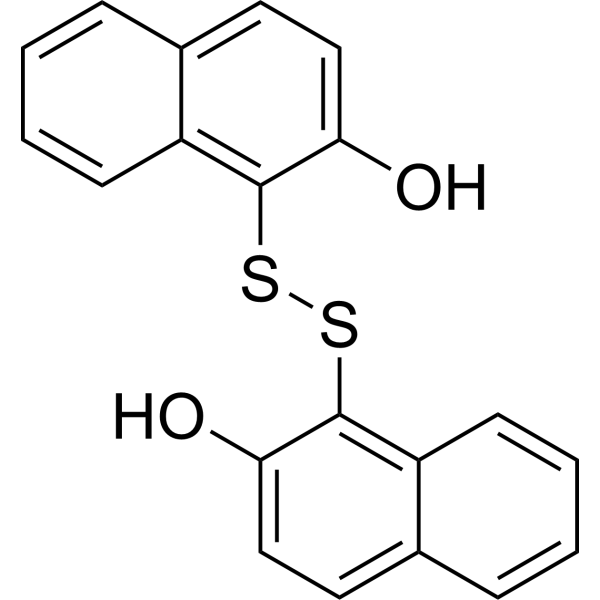
-
- HY-13007
-
|
PF-03758309
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
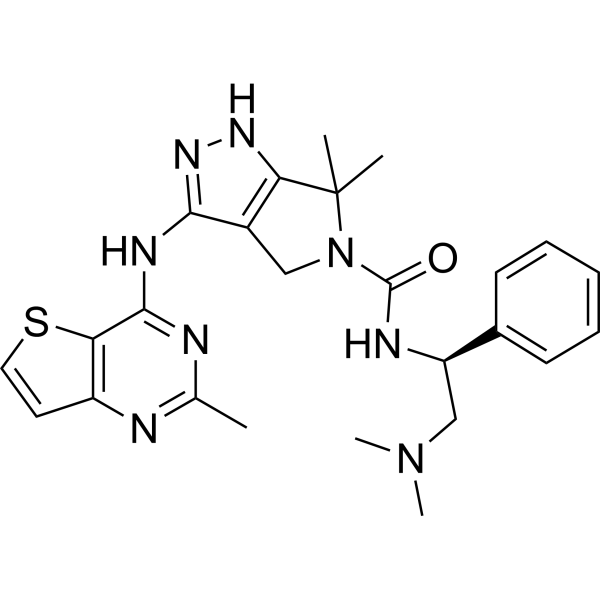
-
- HY-13965
-
|
ML161
|
Protease Activated Receptor (PAR)
|
Cardiovascular Disease
|
|
Parmodulin 2 (ML161) is an allosteric inhibitor of protease-activated receptor 1 (PAR1) with an IC50 of 0.26 μM . Parmodulin 2 is a potent and non-competitive inhibitor of SFLLRN-induced P-selectin expression leading to inhibition of platelet aggregation in vitro and platelet thrombus formation in vivo .
|
-
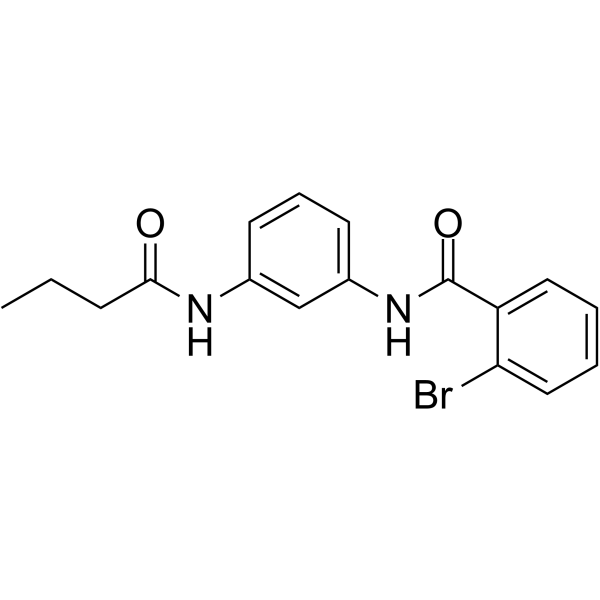
-
- HY-10195B
-
|
LY333531 hydrochloride
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
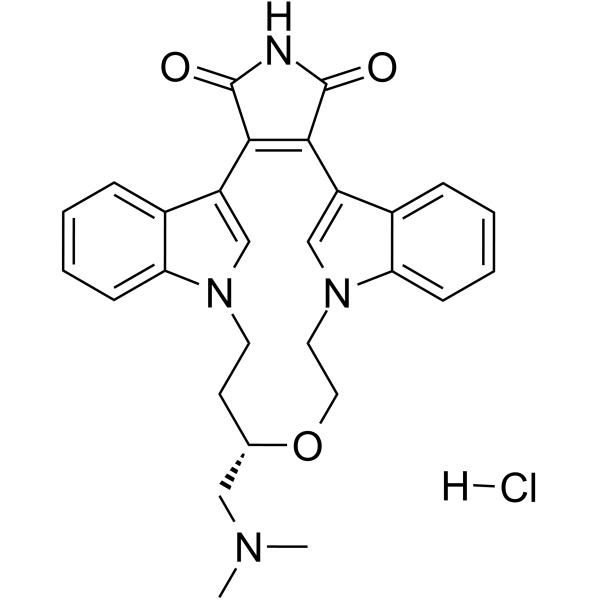
-
- HY-10195
-
|
LY333531
|
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (LY333531) is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
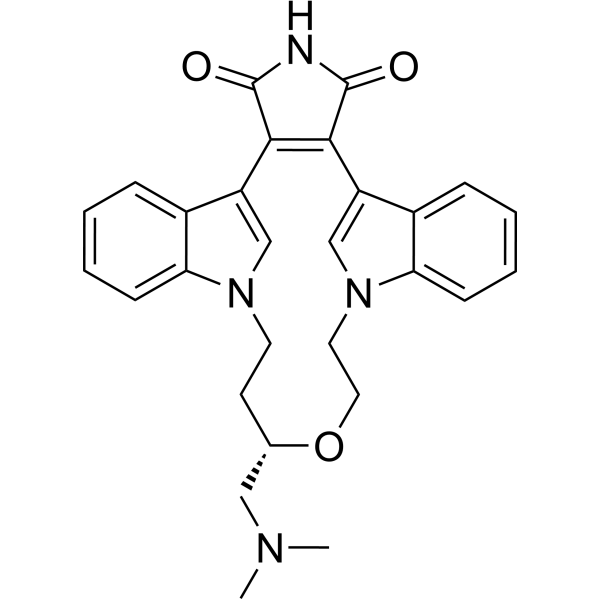
-
- HY-100414
-
|
BYK61359
|
Proton Pump
|
Metabolic Disease
|
|
Soraprazan (BYK61359) is a selective, reversible K-competitive inhibitor of the H,K-ATPase (Ki=6.4 nM), with an IC50 of 0.19 μM in gastric glands. Soraprazan binds to the H,K-ATPase with a Kd of 28.27 nM. Soraprazan shows immediate inhibition of acid secretion and is more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases .
|
-
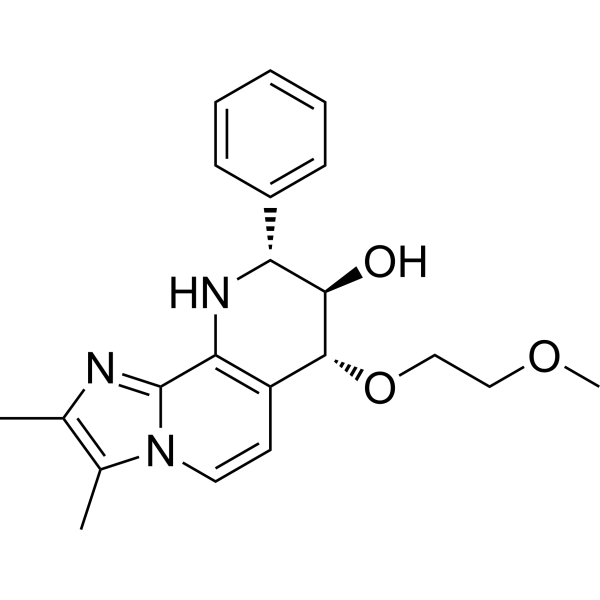
-
- HY-113763
-
|
|
TNF Receptor
|
Others
|
|
L524-0366 is a specific, dose-dependent TWEAK-Fn14 inhibitor. L524-0366 specifically binds to the Fn14 surface with a KD of 7.12 μM, competitively inhibiting the binding of Fn14 to TWEAK (inhibition rate reaching 16%). L524-0366 inhibits TWEAK-induced glioma cell migration without potential cytotoxic effects .
|
-

-
- HY-N0598
-
|
20(S)-Ginsenoside F1
|
Cytochrome P450
Endogenous Metabolite
|
Cancer
|
|
Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
|
-
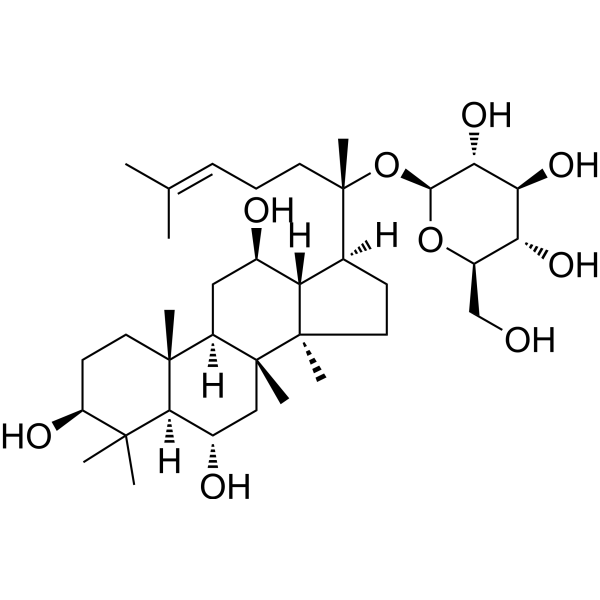
-
- HY-15687A
-
|
|
ROCK
|
Metabolic Disease
|
|
SAR407899 is a selective, potent and ATP-competitive ROCK inhibitor, with an IC50 of 135 nM for ROCK-2, and Kis of 36 nM and 41 nM for human and rat ROCK-2, respectively. SAR407899 shows stable inhibition of migrasome formation.
|
-
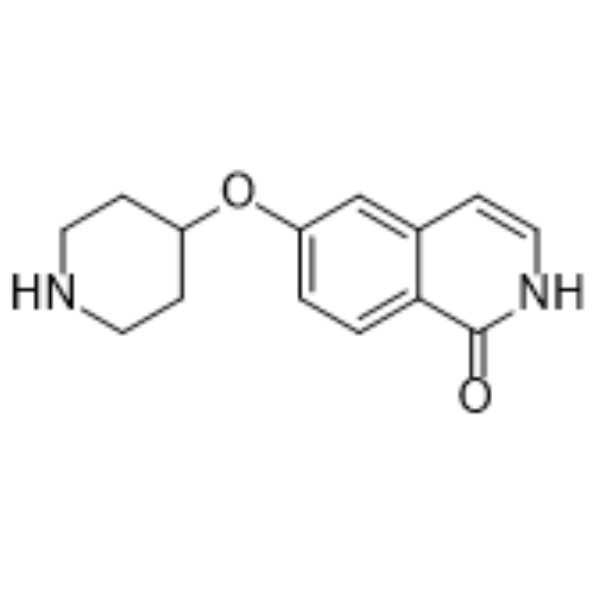
-
- HY-113268
-
|
|
Endogenous Metabolite
|
Metabolic Disease
|
|
Biotin sulfone, a structural analog and metabolite of Biotin (HY-B0511), exerts competitive inhibition against Biotin in Lactobacillus arabinosus 17-5 .
|
-
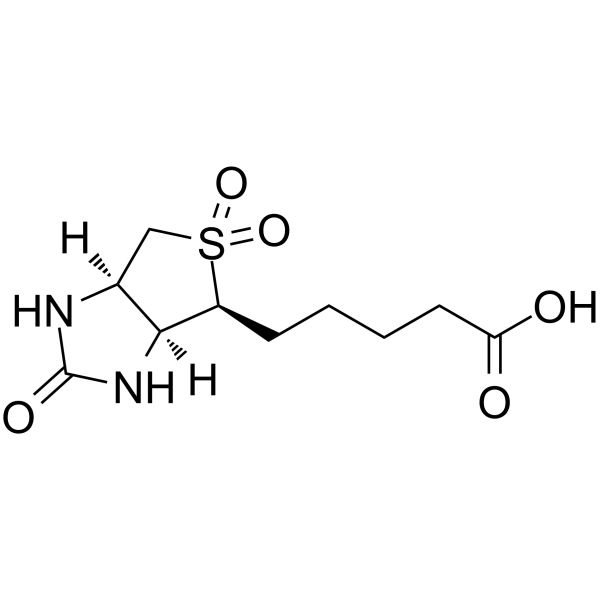
-
- HY-117769
-
|
|
Fatty Acid Synthase (FASN)
|
Metabolic Disease
|
|
GSK837149A is a selective inhibitor of human Fatty Acid Synthase (FASN) targeting the KR domain. GSK837149A has reversible inhibition effect on FASN and selectivity for type I FASN (Ki=30 nM). GSK837149A is also a competitive inhibitor of NADPH and a non-competitive inhibitor of acetoacetyl-CoA. GSK837149A can be used for the research of obesity and breast cancer .
|
-
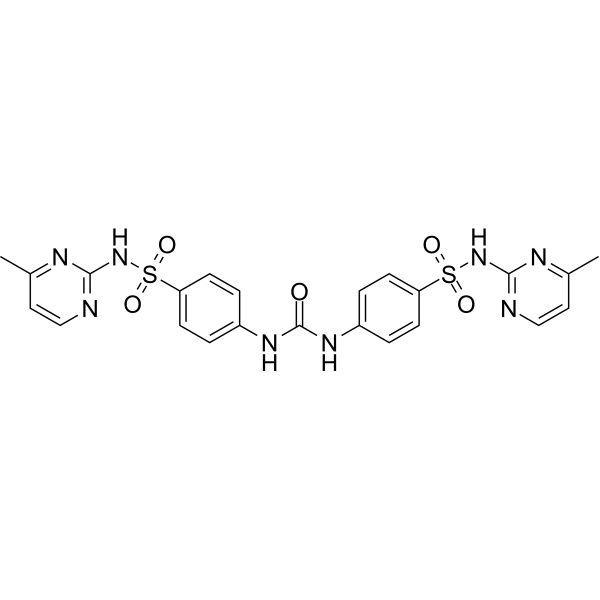
-
- HY-137506
-
-
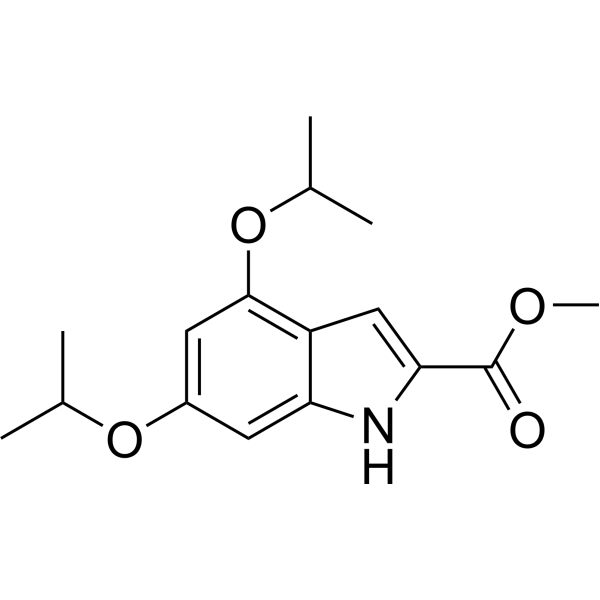
-
- HY-112179
-
GSK180
2 Publications Verification
|
KMO
|
Inflammation/Immunology
|
|
GSK180 is a selective, competitive, and potent inhibitor of kynurenine-3-monooxygenase (KMO), a key enzyme of tryptophan metabolism (IC50, ~6 nM), but shows negligible activity against other enzymes on the tryptophan pathway. GSK180 rapidly changes levels of kynurenine pathway metabolites, and acts as a useful tool to probe the therapeutic potential of KMO inhibition .
|
-
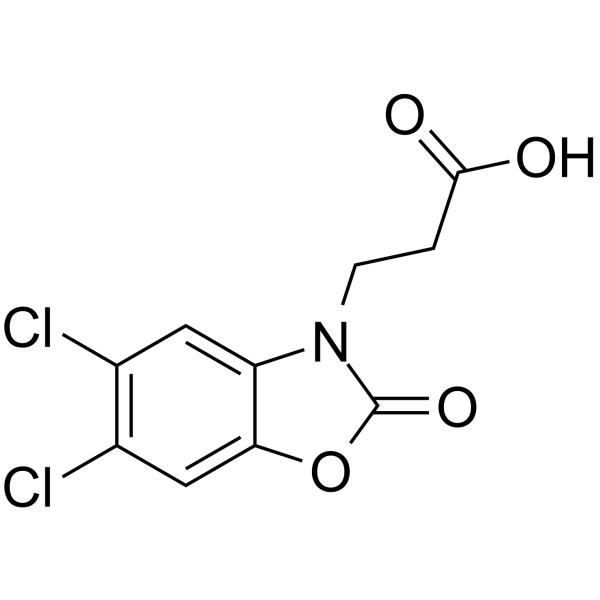
-
- HY-12854
-
|
GRN163L
|
Telomerase
Apoptosis
|
Cardiovascular Disease
Cancer
|
|
Imetelstat (GRN163L) is a 13-mer oligonucleotide and competitive Telomerase inhibitor. Imetelstat binds with high affinity to the template region of the RNA component of human telomerase. Imetelstat induces Apoptosis. Imetelstat is capable of selectively eliminating myelofibrosis hematopoietic stem cells. Imetelstat leads to the loss of a cancer cell's ability to maintain telomere length, resulting in the inhibition of cell proliferation .
|
-

-
- HY-12037
-
|
ON-01910 sodium
|
Polo-like Kinase (PLK)
PI3K
Apoptosis
|
Cancer
|
|
Rigosertib sodium (ON-01910 sodium) is a multi-kinase inhibitor and a selective anti-cancer agent, which induces apoptosis by inhibition the PI3K/Akt pathway, promotes the phosphorylation of histone H2AX and induces G2/M arrest in cell cycle . Rigosertib sodium is a selective and non-ATP-competitive inhibitor of PLK1 with an IC50 of 9 nM .
|
-
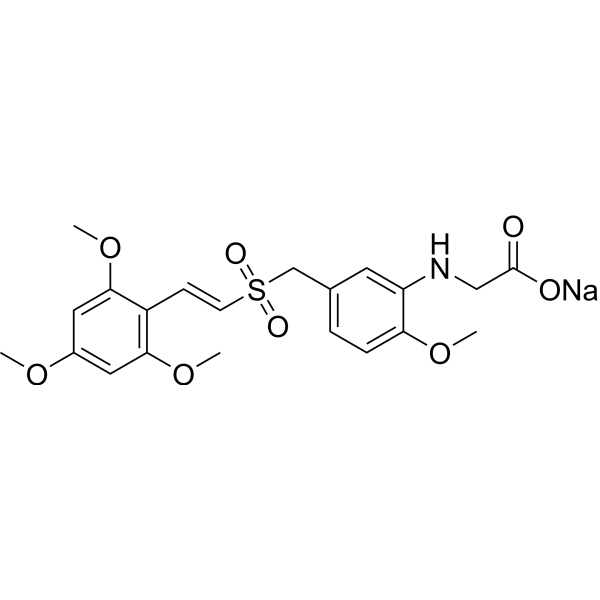
-
- HY-N2511
-
|
|
Cholinesterase (ChE)
Phosphatase
|
Infection
Neurological Disease
|
|
Trimyristin is an orally active compound. Trimyristin can be isolated from the seeds of nutmeg (Myristica fragrans). Trimyristin inhibits the activities of acetylcholinesterase (AChE), ACP and ALP, with IC50 values of 0.11, 0.16 and 0.18 mM, respectively. Trimyristin exerts competitive-noncompetitive inhibition on acetylcholinesterase, uncompetitive inhibition on ACP, and competitive/noncompetitive inhibition on ALP. Trimyristin restores the downregulated acetylcholinesterase concentration in the cerebral cortex of rats exposed to sodium arsenite. Trimyristin can be used in studies related to fascioliasis and neurotoxicity .
|
-

-
- HY-W719041
-
|
|
HIV
Reverse Transcriptase
|
Infection
|
|
1,6-Di-O-galloyl-β-D-glucose is a compound found in the fruit of Phyllanthus emblica. 1,6-Di-O-galloyl-β-D-glucose has HIV-1 reverse transcriptase (RT) inhibitory activity with an IC50 value of 270 μM. The inhibitory mechanism of 1,6-Di-O-galloyl-β-D-glucose is competitive inhibition of the template primer and non-competitive inhibition of the substrate (dTTP). 1,6-Di-O-galloyl-β-D-glucose can be used in anti-HIV research .
|
-

-
- HY-N7389A
-
|
|
Endogenous Metabolite
Drug Intermediate
|
Metabolic Disease
|
|
GDP-D-mannose disodium consists of GDP-α-D-mannose (HY-N7389B) and GDP-β-D-mannose. GDP-α-D-mannose serves as a donor substrate for mannosyltransferases and acts as a precursor of GDP-β-L-fucose. GDP-α-D-mannose exerts competitive inhibition on GTP (with a Ki value of 14.7 μM) and non-competitive inhibition on mannose-1-P (with a Ki value of 115 μM). GDP-D-mannose disodium is metabolized to GDP-L-fucose (HY-134433) via GMDS (Gmd) and TSTA3 (WcaG) .
|
-
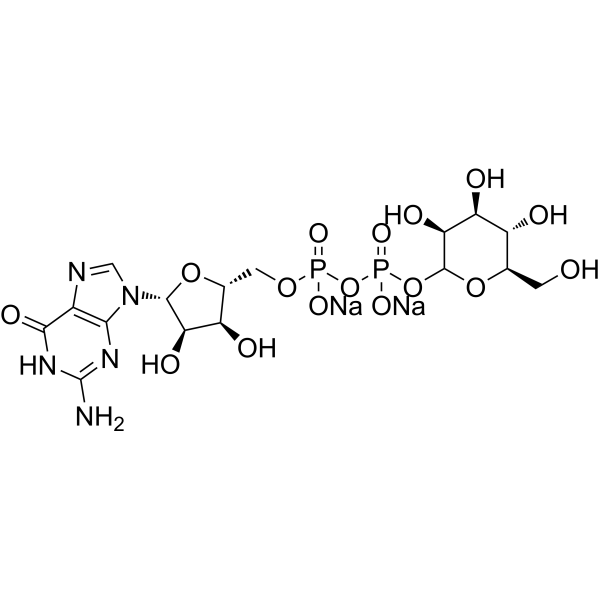
-
- HY-114858
-
|
|
Casein Kinase
|
Cancer
|
|
Epiblastin A is an ATP competitive casein kinase 1 (CK1) inhibitor with IC50s of 8.9, 0.5, and 4.7 µM for CK1α, CK1δ, and CK1 ɛ, respectively. Epiblastin A induces reprogramming of epiblast stem cells into embryonic stem cells by inhibition of CK1 .
|
-
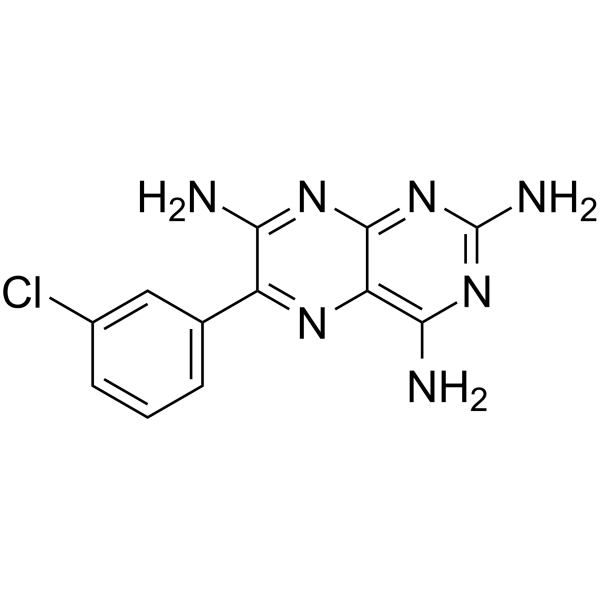
-
- HY-15003
-
|
|
FLT3
Apoptosis
|
Cancer
|
|
ATH686 is a potent, selective and ATP-competitive FLT3 inhibitor. ATH686 target mutant FLT3 protein kinase activity and inhibit the proliferation of cells harboring FLT3 mutants via induction of apoptosis and cell cycle inhibition. ATH686 has antileukemic effects .
|
-
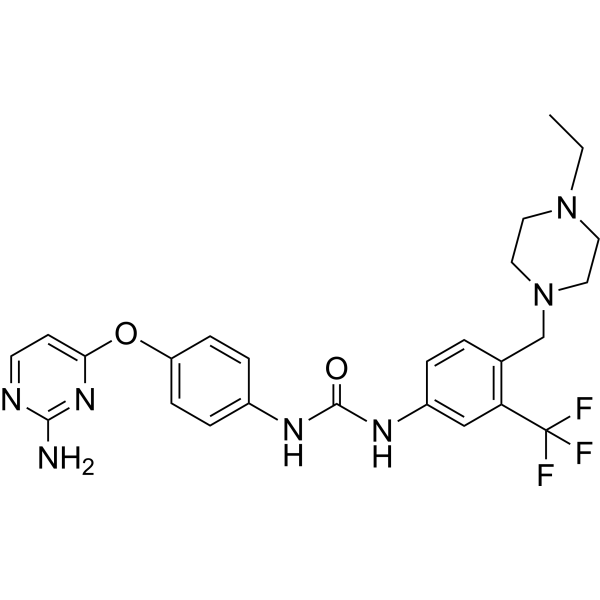
-
- HY-114333
-
|
1-Octyl 2-oxopentanedioate
|
HIF/HIF Prolyl-Hydroxylase
|
Metabolic Disease
Cancer
|
|
Octyl-α-ketoglutarate (1-Octyl 2-oxopentanedioate) is a stable, cell-permeable form of α-ketoglutarate which accumulates rapidly in HEK293 cells with a dysfunctional tricarboxylic acid (TCA) cycle, stimulating prolyl hydroxylase (PHD) activity. In addition, Octyl-α-ketoglutarate competitively blocks succinate- or fumarate-mediated inhibition of PHD .
|
-

-
- HY-145702
-
|
|
MEK
ERK
|
Cancer
|
|
MAP855 is a highly potent, selective, ATP-competitive and orally active MEK1/2 kinase inhibitor (MEK1 ERK2 cascade IC50=3 nM, pERK EC50=5 nM). MAP855 shows equipotent inhibition of wild-type and mutant MEK1/2 .
|
-
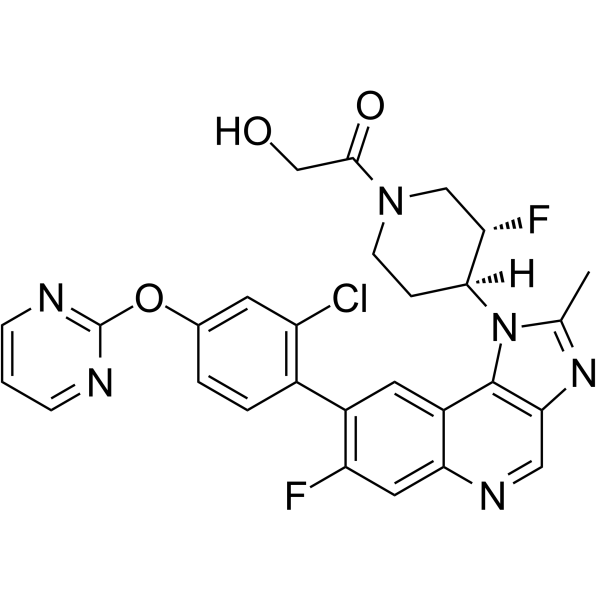
-
- HY-15814
-
|
|
Bcr-Abl
PDGFR
c-Kit
Src
JAK
Apoptosis
|
Cancer
|
|
HG-7-85-01 is a type II ATP competitive inhibitor of wild-type and gatekeeper mutations forms of Bcr-Abl, PDGFRα, Kit, and Src kinases. HG-7-85-01 inhibits T315I mutant Bcr-Abl kinase, KDR and RET with IC50s of 3 nM, 20 nM and 30 nM, and is only weak or no inhibition of other kinases (IC50>2 μM). HG-7-85-01 inhibits the cell proliferation, which is mediated by the induction of apoptosis, and inhibition of cell-cycle progression .
|
-
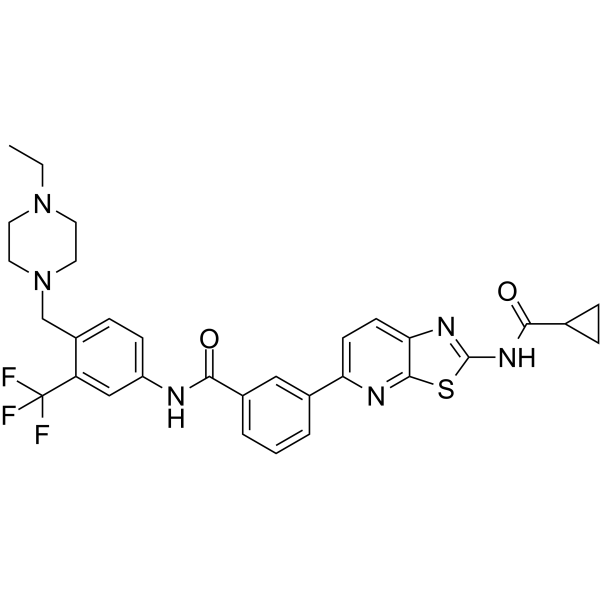
-
- HY-130199
-
|
Parellic acid
|
Others
|
Metabolic Disease
|
|
Psoromic acid is a potent and selective RabGGTase (Rab geranylgeranyl transferase) inhibitor with an IC50 value of 1.3 µM. Psoromic acid is an antioxidative agent. Psoromic acid exhibits a competitive type of HMGR inhibition and mixed type of ACE (angiotensin converting enzyme) inhibition .
|
-
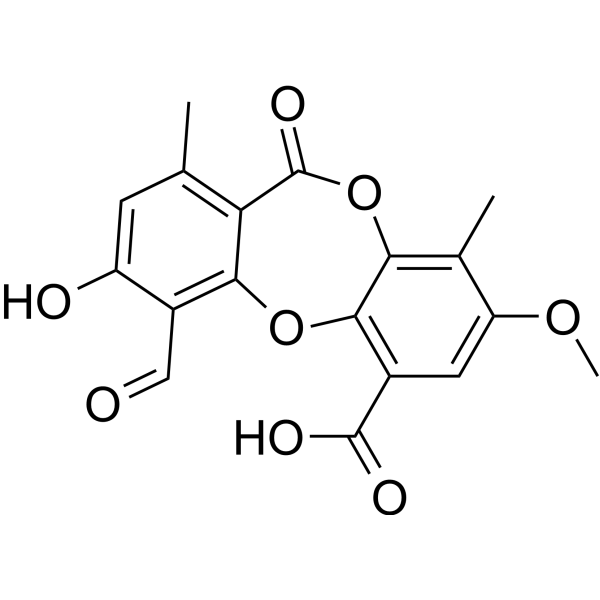
-
- HY-N3394
-
|
|
Others
|
Infection
|
|
Lecanoric acid is a histidine-decarboxylase inhibitor isolated from fungus. The inhibition by lecanoric acid is competitive with histidineand noncompetitive with pyridoxal phosphate. Lecanoric acid did not inhibit aromatic amino acid decarboxylase .
|
-
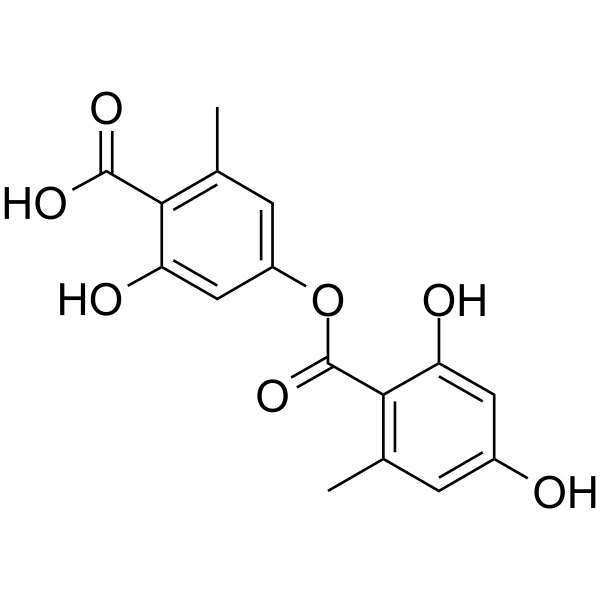
-
- HY-120566
-
-

-
- HY-111173
-
|
|
Dipeptidyl Peptidase
|
Others
|
|
Diprotin B is a dipeptidyl aminopeptidase IV (DPP IV) inhibitor. The apparent competitive inhibition of DPP-IV by the diprotins is a kinetic artifact, derived from the substrate-like nature of tripeptides containing a penultimate proline residue .
|
-
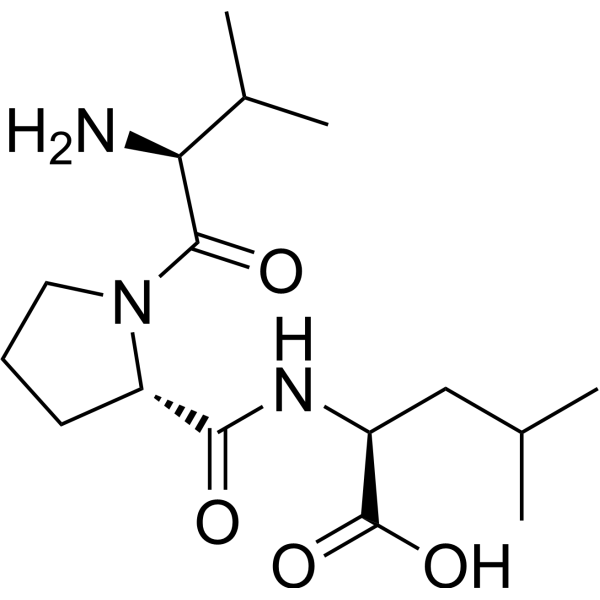
-
- HY-P10392B
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R TFA, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R TFA inhibits competitively the binding of β-catenin to TCF4. aStAx-35R TFA selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-

-
- HY-122369
-
|
|
Carboxypeptidase
|
Metabolic Disease
|
|
Histargin is a selective carboxypeptidase B inhibitor with an IC50 of 17 μg/mL and a Ki of 30 μM. Histargin exerts competitive inhibition with substrate, with inhibitory activity abolished by metal cations. Histargin shows no significant inhibitory activity against carboxypeptidase A, aminopeptidase A, or aminopeptidase B .
|
-

-
- HY-163174
-
|
|
Amine N-methyltransferase
|
Cardiovascular Disease
Neurological Disease
Metabolic Disease
Cancer
|
|
II399 is a potent, selective NNMT bisubstrate inhibitor containing an unconventional SAM mimic, with a Ki of 5.9 nM. II399 exhibits an explicit pattern of competitive inhibition for NAM. II399 occupies both the substrate and cofactor binding pockets. II399 has the potential for the research of cancers, metabolic, cardiovascular, and neurodegenerative diseases .
|
-
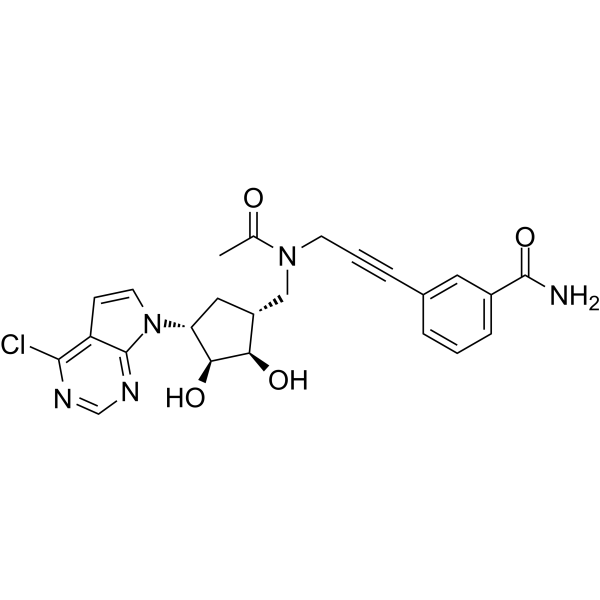
-
- HY-N7536
-
|
|
TRP Channel
|
Others
|
|
Voacangine is an antagonist for TRPV1 and TRPM8 but as an agonist for TRPA1 (EC50=8 μM). Voacangine competitively blockes capsaicin binding to TRPV1 (IC50=50 μM). Voacangine competitively inhibits the binding of menthol to TRPM8 (IC50=9 μM) and it shows noncompetitive inhibition against icilin (IC50=7 μM). Voacangine selectively abrogates chemical agonist-induced TRPM8 activation and did not affect cold-induced activation. Voacangine is an alkaloid isolated from the root bark of Voacanga africana .
|
-

-
- HY-18569AR
-
|
Indole-3-acetic acid sodium (Standard); 3-IAA sodium (Standard)
|
Reference Standards
Endogenous Metabolite
|
Metabolic Disease
|
|
Ginsenoside F1 (Standard) is the analytical standard of Ginsenoside F1. This product is intended for research and analytical applications. Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
|
-

-
- HY-123129
-
|
|
5-HT Receptor
Monoamine Oxidase
|
Neurological Disease
|
|
RS 2232 is an orally active, competitive 5-HT deamination inhibitor with a Ki of 0.054 μM. RS-2232 exhibits reversible and specific inhibition of type A monoamine oxidase. RS 2232 can be used in brain research .
|
-

-
- HY-157220
-
|
|
Vasopressin Receptor
|
Cardiovascular Disease
|
|
Tolvaptan phosphate ester sodium, a prodrug of Tolvaptan (HY-17000), can be used in the study of cardiac edema. Tolvaptan is a selective, competitive and orally active vasopressin receptor 2 (V2R) antagonist with an IC50 of 1.28 μM for the inhibition of arginine vasopressin (AVP)-induced platelet aggregation .
|
-
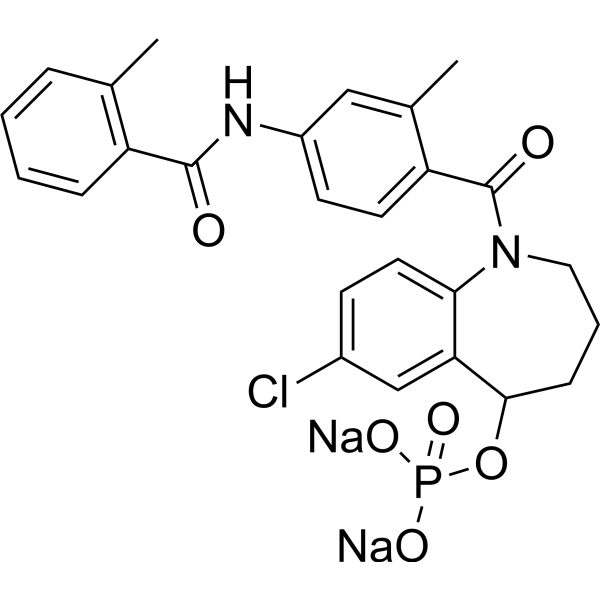
-
- HY-W142169
-
|
Formyl-L-histidine
|
Aminoacyl-tRNA Synthetase
|
Others
|
|
N-Formyl-L-histidine shows binding affinity to histidyl-tRNA synthetase with a Ki value of 4.6 μM. N-Formyl-L-histidine shows a competitive inhibition against L-histidine ammonia-lyase, inhibits urocanic acid formation from L-histidine with a Ki value of 4.26 mM .
|
-
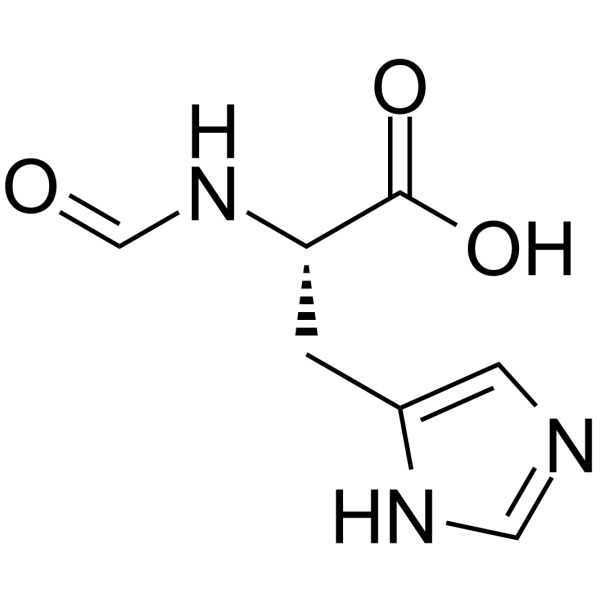
-
- HY-165498
-
|
|
Na+/K+ ATPase
|
Metabolic Disease
|
|
AU-461 is an orally active and reversible inhibitor of the gastric H⁺/K⁺ ATPase with IC₅₀ values for rabbit-derived and pig-derived enzymes are 12.15 μM and 4.20 μM respectively. AU-461 competes with activated cationic K⁺ (Kᵢ = 1.64 μM). AU-461 reduces both histamine-stimulated gastric acid secretion and basal gastric acid secretion in rats. AU-461 inhibits ulcer formation caused by ethanol or sodium hydroxide, and restores the plasma gastrin level to normal. AU-461 can be used for the study of peptic ulcers .
|
-

-
- HY-N7389B
-
|
|
Endogenous Metabolite
|
Metabolic Disease
|
|
GDP-α-D-mannose disodium is the donor substrate for mannosyltransferases and the precursor of GDP-β-L-fucose. GDP-α-D-mannose disodium gives a competitive inhibition with respect to GTP (Ki 14.7 μM) and an uncompetitive inhibition with respect to mannose-1-P (Ki 115 μM) .
|
-
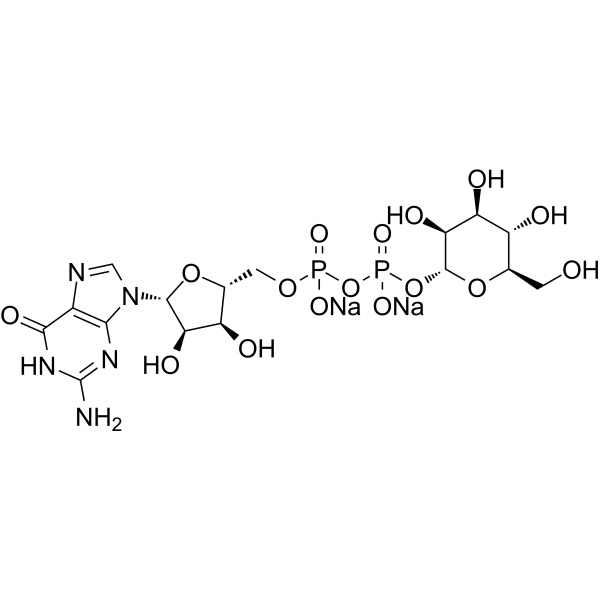
-
- HY-13007A
-
|
PF-03758309 hydrochloride
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) hydrochloride is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 hydrochloride has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
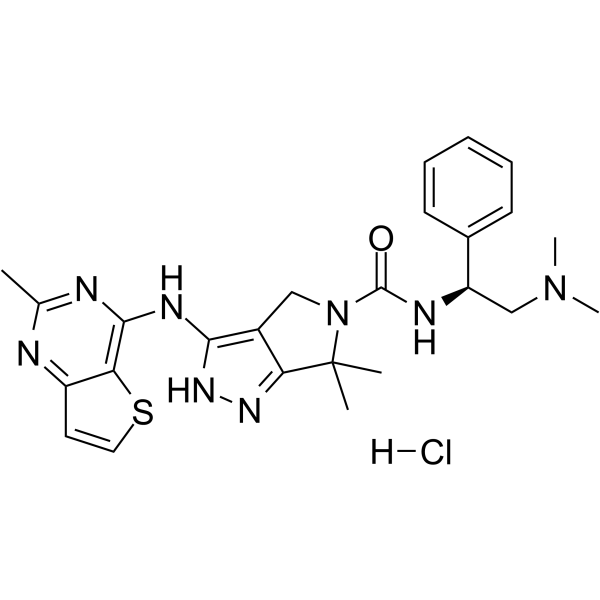
-
- HY-13007B
-
|
PF-03758309 dihydrochloride
|
PAK
Apoptosis
|
Cancer
|
|
PF-3758309 (PF-03758309) dihydrochloride is a potent, orally available, and reversible ATP-competitive inhibitor of PAK4 (Kd= 2.7 nM; Ki=18.7 nM). PF-3758309 dihydrochloride has the expected cellular functions of a PAK4 inhibitor: inhibition of anchorage-independent growth, induction of apoptosis, cytoskeletal remodeling, and inhibition of proliferation .
|
-
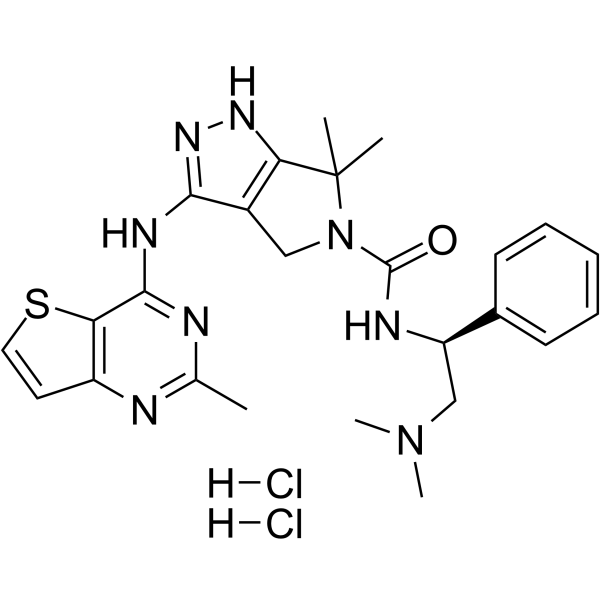
-
- HY-B0690
-
-
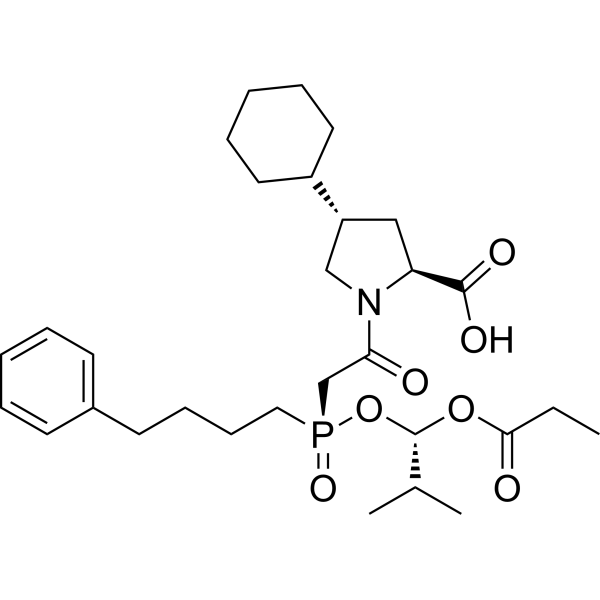
-
- HY-B0377S
-
-
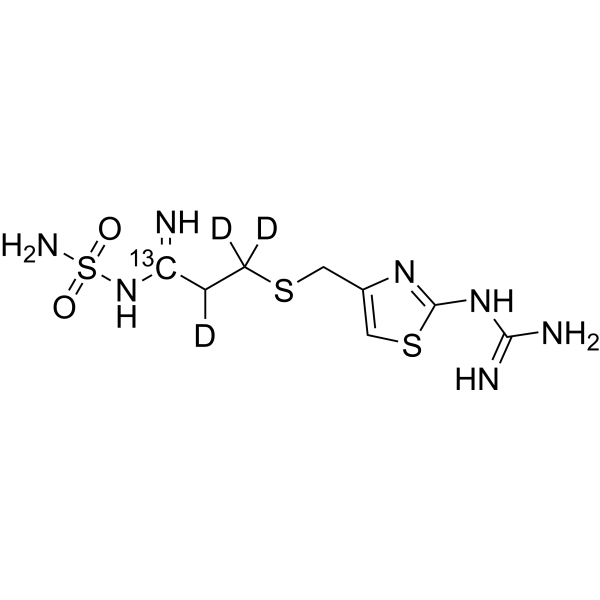
-
- HY-146142
-
|
|
Cholinesterase (ChE)
Amyloid-β
|
Neurological Disease
|
|
AChE/BuChE-IN-2 (Compound 5f) is an orally active AChE and BuChE inhibitor with IC50 values of 0.72 μM and 0.16 μM, respectively. AChE/BuChE-IN-2 shows a non-competitive inhibition with AChE and shows potent self-induced β-amyloid (Aβ) aggregation inhibition with an IC50 of 62.52 μM. AChE/BuChE-IN-2 can cross the BBB .
|
-
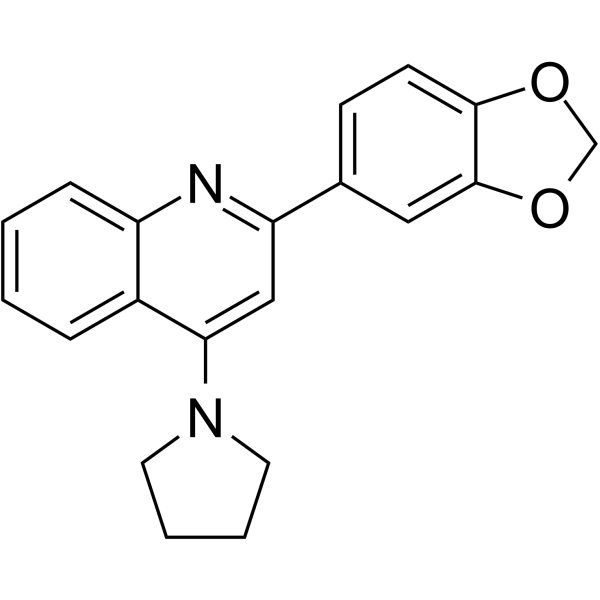
-
- HY-W707517
-
-

- HY-15663R
-
|
|
PAK
|
Cancer
|
|
IPA-3 (Standard) is the analytical standard of IPA-3. This product is intended for research and analytical applications. IPA-3 is a selective non-ATP competitive PAK1 inhibitor with IC50 of 2.5 μM, and shows no inhibition to group II PAKs (PAKs 4-6).
|
-

- HY-139226
-
|
2-Guanidine-4-methylquinazoline
|
GABA Receptor
|
Inflammation/Immunology
|
|
GMQ is an acid-sensing ion channel modulator, competitive GABAAR antagonist. GMQ preferentially, potently, competitively inhibits GABAARs. GMQ inhibits α1β2, α1β2γ2, α4β2γ2 and α5β2γ2 GABAARs. GMQ enhances neuronal excitation through inhibition of GABAergic transmission. GMQ has anti-histamine effects in the enteric system, inhibiting gastric acid secretion .
|
-

- HY-135111
-
|
|
Drug Metabolite
|
Infection
Metabolic Disease
Cancer
|
|
4-Desmethoxy Omeprazole is the active metabolite of Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .
|
-
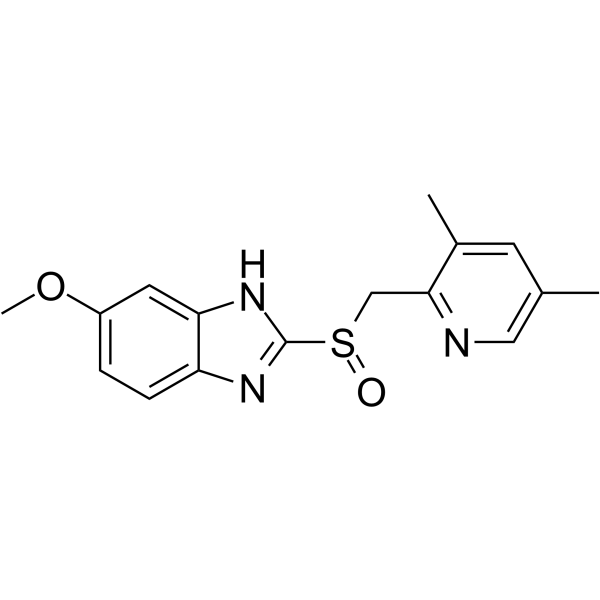
- HY-10195BS
-
|
|
Isotope-Labeled Compounds
PKC
|
Metabolic Disease
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
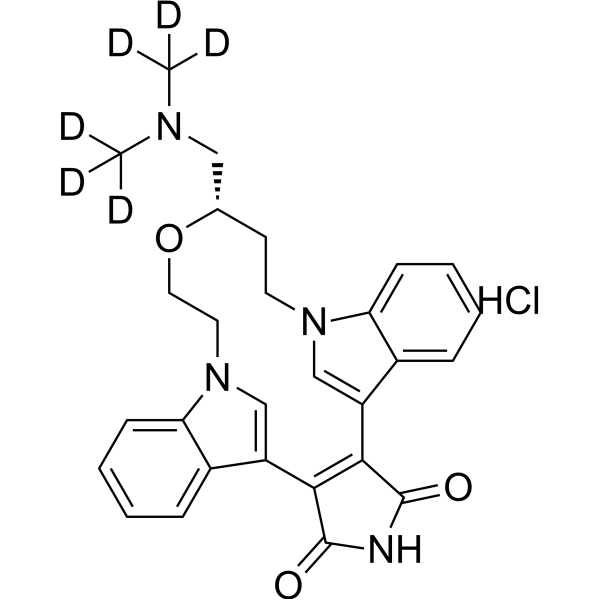
- HY-P10392
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R inhibits competitively the binding of β-catenin to TCF4. aStAx-35R selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-

- HY-171835
-
|
|
HIV
HIV Protease
|
Infection
|
|
DPC 684 is a potent and selective HIV-1 protease inhibitor (IC90 = 5.7-40 nM, Ki = 0.021 nM). DPC 684 competitively inhibits HIV-1 protease and blocks viral polyprotein cleavage. DPC 684 has low protein binding and broad-spectrum inhibition against a variety of wild-type and mutant HIV-1 proteases. DPC 684 has low protein binding and broad-spectrum inhibition (IC90 = 1.9-6.3 nM). DPC 684 has research significance for HIV .
|
-

- HY-10572A
-
|
(R)-DMP 266; (R)-EFV; (R)-L-743726
|
Reverse Transcriptase
|
Infection
|
|
(R)-Efavirenz ((R)-DMP 266) is a non-nucleoside reverse transcriptase inhibitor that acts by non-competitive inhibition of the viral enzyme. (R)-Efavirenz can be metabolized by CYP2B6 to 8-hydroxyefavirenz in a highly stereoselective manner. (R)-Efavirenz is promising to be a useful probe for the CYP2B6 active site and catalytic mechanisms .
|
-

- HY-17000R
-
|
OPC-41061 (Standard)
|
Reference Standards
Vasopressin Receptor
Autophagy
|
Cardiovascular Disease
Endocrinology
Cancer
|
|
Tolvaptan (Standard) is the analytical standard of Tolvaptan. This product is intended for research and analytical applications. Tolvaptan is a selective, competitive and orally active vasopressin receptor 2 (V2R) antagonist with an IC50 of 1.28 μM for the inhibition of arginine vasopressin (AVP)-induced platelet aggregation. Tolvaptan induces cell apoposis and affects cell cycle. Tolvaptan can be used for the research of hyponatremia .
|
-

- HY-19885
-
|
|
Bacterial
Dihydrofolate reductase (DHFR)
|
Infection
|
|
AR-102 has inhibitory activity towards Staphylococcus aureus. AR-102 exhibits a competitive potent inhibition of the F98Y mutant DHFR (Ki = 0.22 nM). AR-102 has been determined as ternary complex with NADPH in both wild-type S. aureus DHFR and the TMP-resistant F98Y mutant enzyme .
|
-

- HY-108913R
-
|
|
Beta-lactamase
Reference Standards
Antibiotic
|
Infection
|
|
Nitrocefin (Standard) is the analytical standard of Nitrocefin. This product is intended for research and analytical applications. Nitrocefin is a chromogenic β-lactamase substrate that undergoes a distinctive color change from yellow to red as the amide bond in the β-lactam ring is hydrolyzed by β-lactamase. Nitrocefin is used in competitive inhibition studies in developmental work on β-lactamase-resistant antibiotics .
|
-

- HY-129278
-
|
|
Parasite
|
Infection
Cardiovascular Disease
Neurological Disease
|
|
Lunarine is an alkaloid, which can be isolated from the seeds of kale (Lunaria annua). Lunarine lowers the blood pressure, stimulates small intestinal motility, inhibits spontaneous contractions of uterus and nervous system, and exhibits an acute toxicity with LD50 of 62.3 mg/kg in dogs and rabbits. Lunarine exhibits antiparasitic activity through a competitive inhibition of protozoan oxidoreductase trypanothione reductase (TryR) with Ki of 304 μM .
|
-

- HY-174231
-
|
|
EGFR
Apoptosis
|
Cancer
|
|
EGFR-IN-163 (Compound 13) is a competitive epidermal growth factor receptor (EGFR) inhibitor (IC50=0.079 μM, selective for HER-2 inhibition). EGFR-IN-163 induces tumor cell apoptosis and cell cycle arrest at G₂/M phase. EGFR-IN-163 is promising for research of estrogen receptor-positive (ER+) breast cancer .
|
-

- HY-110333
-
|
|
EGFR
|
Cancer
|
|
BMS-599626 dihydrochloride is a small molecule pan-HER (human epidermal growth factor receptor) kinase inhibitor. BMS-599626 dihydrochloride primarily targets HER1 (IC50=20 nmol/L) and HER2 (IC50=30 nmol/L) kinase activity in the HER family. BMS-599626 inhibits the kinase activity of HER1 and HER2 by competing with their ATP-binding sites, and can inhibit the downstream signaling pathway by blocking the heterodimer formation of HER1 and HER2. BMS-599626 dihydrochloride can be used to study the antitumor effects of multiple HER1 or HER2 overexpressed tumor models .
|
-

- HY-161507
-
|
|
Carbonic Anhydrase
Cholinesterase (ChE)
|
Metabolic Disease
|
|
hCAI/II-IN-8 (Compound 8) is a hydrazide derivative based on 4-hydroxybenzaldehyde. hCAI/II-IN-8 primarily targets human carbonic anhydrase isomerase I (hCA I) and II (hCA II) for inhibition (IC50 = 21.35 ± 0.39 nM (hCA I); 7.12 ± 0.12 nM (hCA II)). hCAI/II-IN-8 inhibits AChE and BChE as well(IC50 = 46.27 ±0.75 nM (AChE); 43.38 ± 0.83 nM (BChE)). . .
|
-

- HY-N12427
-
|
|
Tyrosinase
|
Cancer
|
|
Litchinol B (compound 2) is a non-competitive tyrosinase inhibitor with an inhibition constant of 5.70 μM .
|
-
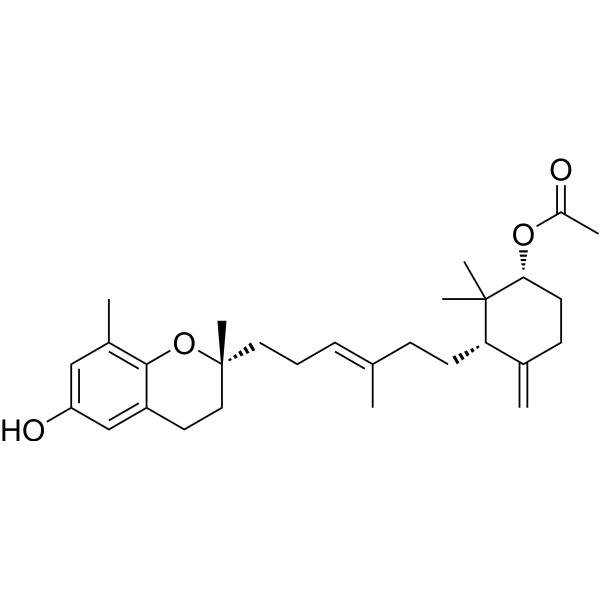
- HY-P10552
-
|
|
CXCR
|
Inflammation/Immunology
|
|
pCXCL8-1aa is an anti-inflammatory peptide. pCXCL8-1aa competitively inhibits the binding of CXCL8 to glycosaminoglycans such as heparin sulfate (HS) by binding with high affinity. This reduces the presentation of CXCL8 on the surface of vascular endothelial cells, thereby inhibiting neutrophil migration and inflammatory responses. pCXCL8-1aa can be used to study inflammatory diseases such as rheumatoid arthritis .
|
-

- HY-N0598R
-
|
20(S)-Ginsenoside F1 (Standard)
|
Reference Standards
Cytochrome P450
Endogenous Metabolite
|
Cancer
|
|
Ginsenoside F1 (Standard) is the analytical standard of Ginsenoside F1. This product is intended for research and analytical applications. Ginsenoside F1, an enzymatically modified derivative of Ginsenoside Rg1, demonstrates competitive inhibition of CYP3A4 activity and weaker inhibition of CYP2D6 activity.
|
-

- HY-N3136
-
|
|
Histamine Receptor
|
Others
|
|
Onitin is a natural product, that can be isolated from Onychium siliculosum. Onitin is also a non-competitive antagonist of histamine. Onitin shows activity in blocking the peristaltic reflex of the guinea-pig ileum, in inhibition of the responses of guinea-pig ileum to histamine and of inhibition of the responses of guinea-pig tracheal muscle to histamine .
|
-
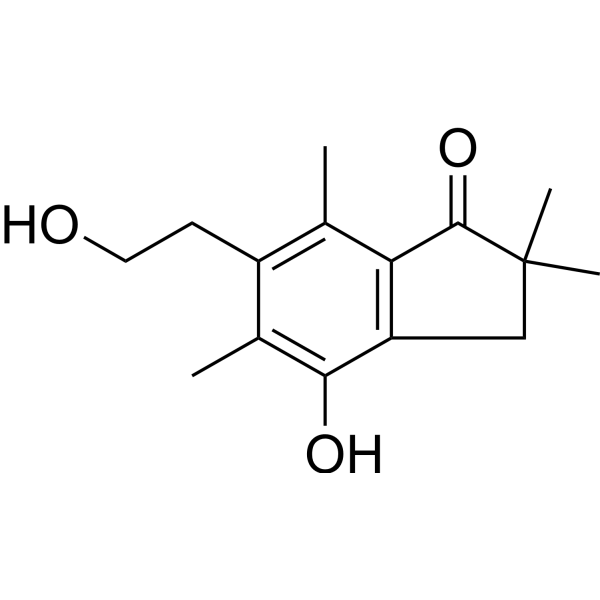
- HY-103562
-
|
3,3'-Dimethoxybenzaldazine
|
mGluR
|
Neurological Disease
|
|
DMeOB (3,3'-Dimethoxybenzaldazine) is a mGluR5 receptor negative allosteric modulator with an IC50 of 3 μM. DMeOB displays reversible non-competitive inhibition of mGlu5-mediated responses .
|
-
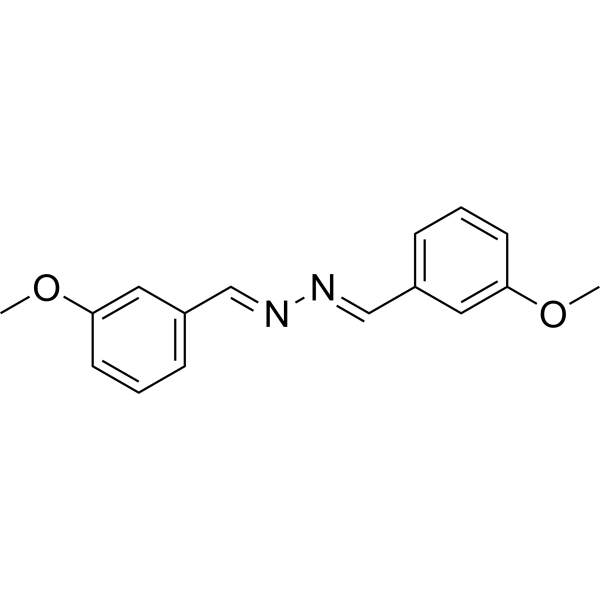
- HY-17000S
-
-
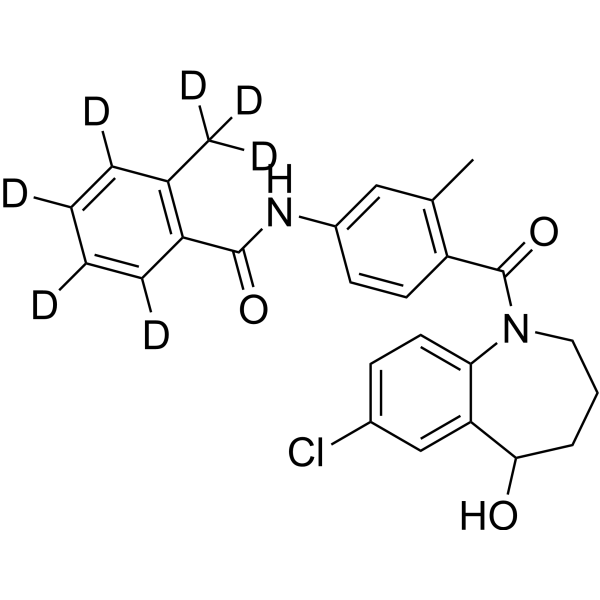
- HY-B0377R
-
|
MK-208 (Standard)
|
Reference Standards
Histamine Receptor
|
Metabolic Disease
Endocrinology
Cancer
|
|
Famotidine (Standard) is the analytical standard of Famotidine. This product is intended for research and analytical applications. Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion.
|
-

- HY-121787
-
|
OLC20
|
Olfactory Receptor
|
Others
|
|
OX1a (OLC20) is an Orco antagonist with non-competitive activity that inhibits the activation of oxygen odor receptors (ORs). OX1a is able to reduce the activation of ORs by competitively inhibiting the effects of Orco agonists. OX1a also shows non-competitive inhibition of odor molecules, which may affect the olfactory-mediated behavior of insects. Through structural optimization, OX1a analogs have shown higher antagonistic potency, indicating that this type of compound may have application potential in a wide range of insect species .
|
-

- HY-B0377S1
-
-

- HY-145701
-
|
|
MEK
|
Cancer
|
|
MEK1/2-IN-2 is a potent ATP-competitive MEK1/2 inhibitor and shows equipotent inhibition of WT MEK1/2 and a panel of MEK1/2 mutant cell lines .
|
-
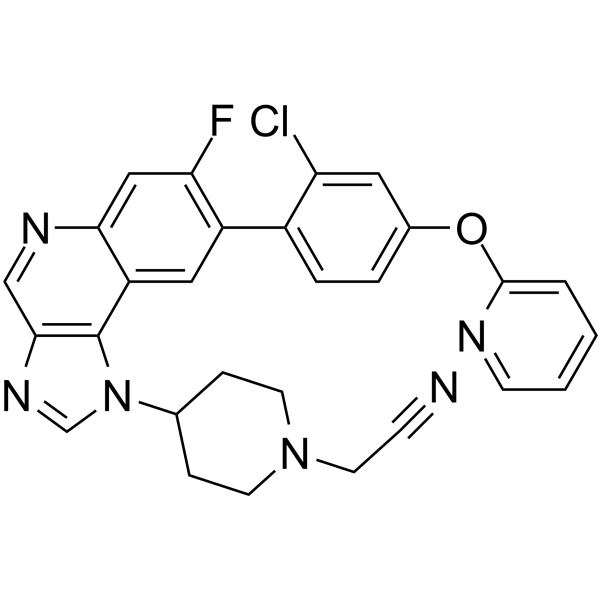
- HY-170599
-
-

- HY-161421
-
|
|
Glycosidase
|
Metabolic Disease
|
α-Glucosidase-IN-61 (Compd 1j), a competitive α-glucosidase inhibitor, demonstrates excellent inhibition with an IC50 of 0.73 μM. α-Glucosidase-IN-61 (Compd 1j) is used for the research of type 2 diabetes mellitus .
|
-
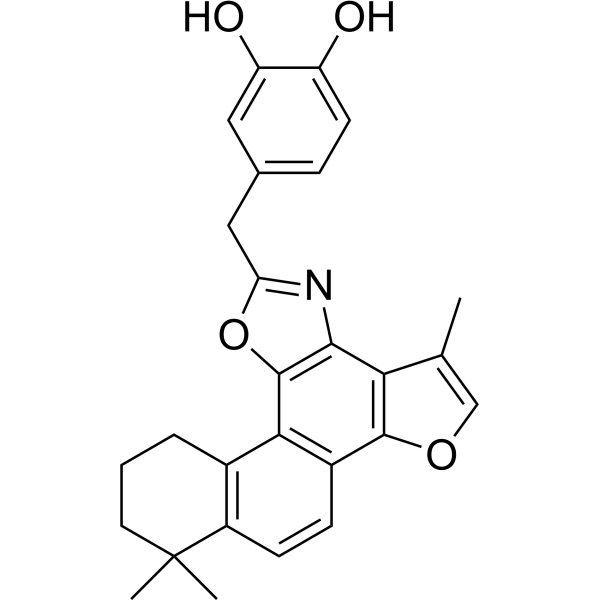
- HY-113268R
-
|
|
Reference Standards
Endogenous Metabolite
|
Metabolic Disease
|
|
Biotin sulfone (Standard) is the analytical standard of Biotin sulfone (HY-113268). This product is intended for research and analytical applications. Biotin sulfone, a structural analog and metabolite of Biotin (HY-B0511), exerts competitive inhibition against Biotin in Lactobacillus arabinosus 17-5 .
|
-

- HY-129907
-
|
2-Aminoethyl methylphosphonate
|
GABA Receptor
|
Neurological Disease
|
|
2-AEMP (2-Aminoethyl methylphosphonate) is an anti-GABA(A)-ρ1 receptor compound with antagonist activity. 2-AEMP exhibits competitive inhibition compared to TPMPA with an IC(50) value of 18 μM, compared to 7 μM for TPMPA. The release rate of inhibition at termination of 2-AEMP is significantly higher than that of TPMPA. The preincubation time required for the onset of inhibition of 2-AEMP is much shorter than that of TPMPA. Some analogs of 2-AEMP, especially those with benzyl or n-butyl substituents, show lower potency in terms of biological activity .
|
-

- HY-P10346
-
|
Smooth-Muscle Myosin Light-Chain Kinase (796-815)
|
Myosin
|
Cardiovascular Disease
Inflammation/Immunology
|
|
smMLCK peptide is a specific inhibitor of smooth muscle myosin light chain kinase (smMLCK). The smMLCK peptide mimics the substrate and competitively inhibits the binding of the actual substrate to the enzyme, thereby inhibiting the kinase activity. This inhibition prevents the phosphorylation of the myosin light chain, thus inhibiting muscle contraction .
|
-

- HY-W777002
-
-

- HY-P1115
-
|
|
Akt
|
Others
|
|
AKTide-2T is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-

- HY-N13917
-
|
|
Proteasome
Bacterial
|
Infection
Cancer
|
|
Argyrin B, a natural product cyclic peptide, is a reversible, non-competitive immunoproteasome inhibitor. Argyrin B shows selective inhibition of the β5i and β1i sites of the immunoproteasome over the β5c and β1c sites of the constitutive proteasome with nearly 20-fold selective inhibition of β1i over the homologous β1c. Argyrin B has antibacterial effects .
|
-

- HY-P1115A
-
|
|
Akt
|
Others
|
|
AKTide-2T TFA is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T TFA mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-
- HY-107727
-
|
|
Neuropeptide Y Receptor
|
Neurological Disease
|
|
BMS-193885 (L-Lactic acid) is a potent, selective, and brain-penetrant neuropeptide Y1 receptor antagonist. BMS-193885 has a Ki value of 3.3 nM for the neuropeptide Y1 receptor, competitively acts on the neuropeptide Y binding site, and can reduce food intake and body weight through central Y1 inhibition .
|
-
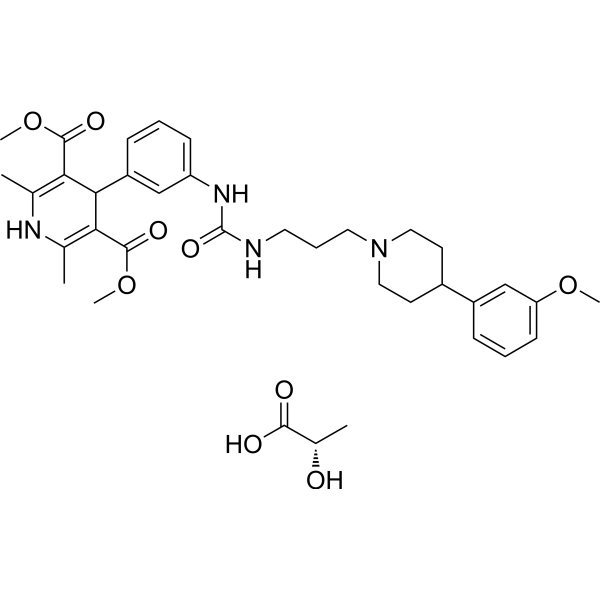
- HY-116138
-
|
|
EGFR
|
Others
|
|
RG-14467 is an epidermal growth factor receptor tyrosine kinase inhibitor with activity that inhibits enzyme activity. RG-14467 has similar inhibition kinetics to Lavendustin-A, with a dissociation constant of 3.4μM for the initial rapidly formed complex and an overall dissociation constant estimated to be less than or equal to 30nM, and is a partially competitive inhibitor for ATP.
|
-

- HY-B0690S
-
-

- HY-10195R
-
|
LY333531 (Standard)
|
Reference Standards
PKC
|
Metabolic Disease
|
|
Ruboxistaurin (Standard) is the analytical standard of Ruboxistaurin. This product is intended for research and analytical applications. Ruboxistaurin (LY333531) is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin inhibits PKC beta II with an IC50 of 5.9 nM .
|
-

- HY-10287A
-
|
|
Dipeptidyl Peptidase
|
Metabolic Disease
|
|
Gosogliptin hydrochloride is the hydrochloride of Gosogliptin (HY-10287). Gosogliptin (PF-00734200) is a potent, orally active, selective, and competitive inhibitor of DPP-IV, the enzyme mainly responsible for the degradation of the incretin peptides GLP-1 and glucose-dependent insulinotropic polypeptide. Gosogliptin demonstrates rapid and reversible inhibition of plasma DPP-4 activity. Gosogliptin stimulates insulin secretion and improves glucose tolerance .
|
-

- HY-18953
-
|
|
mTOR
|
Cancer
|
|
mTOR inhibitor-23 (compound DHM25) is a selective, competitive, irreversible and covalent inhibitor of mTOR. mTOR inhibitor-23 has the mechanism of inhibition occurs mainly through its capacity to covalently interact with a nucleophilic amino acid inside the ATP pocket. mTOR inhibitor-23 exerts potent antitumor activity against triple-negative breast tumor cell lines .
|
-

- HY-116973
-
|
|
HIV
|
Infection
|
|
L-738372 is a non-competitive reversible inhibitor of HIV-1 reverse transcriptase (RT) with an inhibition constant (Ki) of 140 nM against dTTP. When combined with any nucleoside analogs (such as azidothymidine triphosphate, didexoyinosine triphosphate, or didexoycytidine triphosphate), L-738372 exhibits synergistic inhibition of RT activity. L-738372 has 2-3 times more inhibitory potency against the azidothymidine-resistant RT (D67N, K70R, T215Y, K219Q) compared to the wild-type RT. L-738372 holds promise for research in the field of HIV virus treatment .
|
-

- HY-50878B
-
|
PF-02341066 acetate
|
Anaplastic lymphoma kinase (ALK)
ROS Kinase
c-Met/HGFR
|
Cancer
|
|
Crizotinib (PF-02341066) acetate is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib acetate inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib acetate is also a ROS1 inhibitor. Crizotinib acetate has effective tumor growth inhibition .
|
-
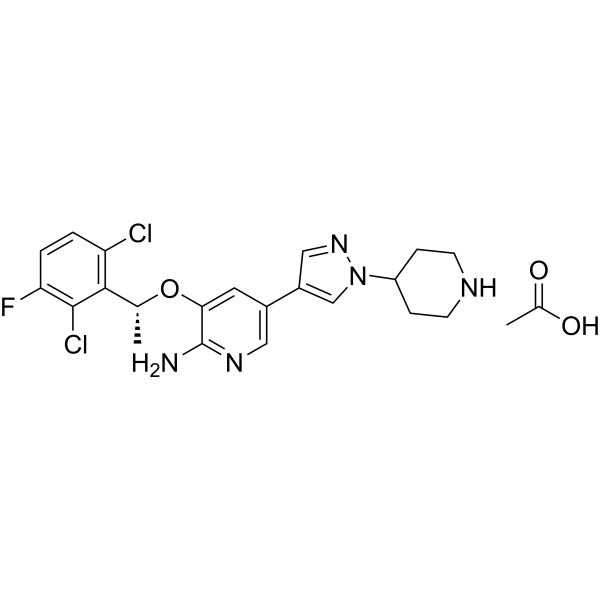
- HY-114923
-
|
|
DNA-PK
PI3K
|
Cancer
|
|
SU-11752 is an inhibitor for DNA-dependent protein kinase (DNA-PK) with an IC50 of 0.13 μM. SU-11752 inhibits PI3K p110γ kinase with IC50 of 1.1 μM. SU-11752 binds competitively for ATP-site in DNA-PK, results in inhibition of intracellular DNA double-strand break repair and increases the sensitivity of cells to radiotherapy .
|
-

- HY-162480
-
|
|
Glycosidase
Phosphatase
|
Endocrinology
|
PTP1B-IN-27 (Compound 7i) is an inhibitor of protein tyrosine phosphatase 1-B (PTP‐1B)(IC50=8.2 µM). PTP1B-IN-27 also inhibits α-Glucosidase (IC50=120 µM) and shows competitive inhibition (Ki=118 µM) .
|
-
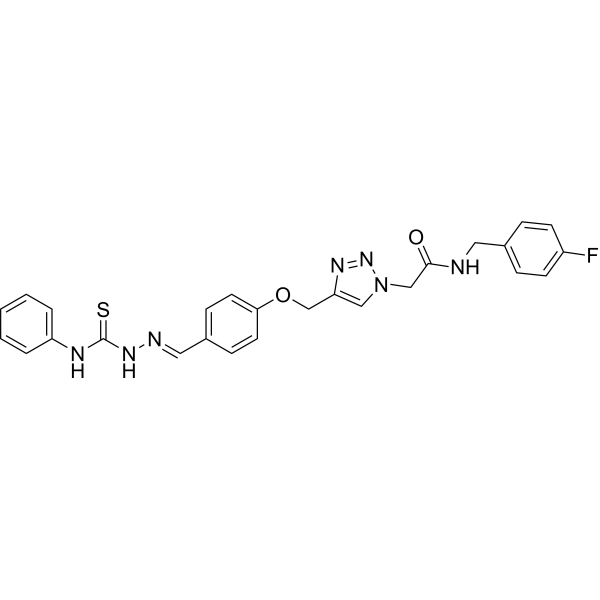
- HY-179598
-
-

- HY-137921
-
|
|
DNA/RNA Synthesis
|
Cancer
|
|
Zelpolib is a non-competitive Pol δ inhibitor. Zelpolib shows robust inhibition of Pol δ activity against dTTP with a Ki of 4.3 μM. Zelpolib inhibits cellular DNA replication. Zelpolib shows antiproliferative properties .
|
-

- HY-165443
-
|
Nervocidine
|
Na+/K+ ATPase
|
Cardiovascular Disease
|
|
Cassaine (Nervocidine) is a Na +/K +-ATPase inhibitor. Cassaine stabilizes phosphorylated intermediates, slows spontaneous dephosphorylation and blocks potassium-stimulated dephosphorylation. Cassaine exhibits non-competitive inhibition with respect to K +, shows inhibition enhanced by Pi and antagonized by Na +. Cassaine induces positive inotropic effects in perfused hearts with faster inotropy offset. Cassaine can be used for research on cardiac arrhythmias .
|
-

- HY-105101
-
|
|
Histamine Receptor
|
Inflammation/Immunology
|
|
Osutidine is a selective histamine H2 receptor antagonist, can effectively inhibit histamine-stimulated gastric acid secretion. Osutidine does not affect [ 14C]aminopyrine accumulation stimulated by carbachol or dibutyryl-cAMP. Osutidine is insurmountable and includes non-competitive inhibition. Osutidine can be used for the study of gastric mucosal injury .
|
-

- HY-10195BR
-
|
LY333531 hydrochloride (Standard)
|
Reference Standards
PKC
|
Metabolic Disease
|
|
Ruboxistaurin hydrochloride (Standard) is the analytical standard of Ruboxistaurin hydrochloride (HY-10195B). This product is intended for research and analytical applications. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-

- HY-123965
-
|
|
MAP4K
TNF Receptor
|
Metabolic Disease
Cancer
|
|
PF-06745013 (Compound 37) is a MAP4K4 inhibitor without time-dependent inhibition (TDI) risk of CYP3A4 (IC50 of 0.4 nM for MAP4K4). PF-06745013 has no accumulation CNS-impaired and non-ATP competitive activities in mouse models. PF-06745013 can be used for inflammatory diseases like diabetes and cancers research .
|
-

- HY-137744
-
|
|
Adenylate Cyclase
Nucleoside Antimetabolite/Analog
|
Infection
|
|
MANT-GppNHp is a competitive adenyl cyclase (AC) inhibitor. MANT-GppNHp is a fluorescently labeled GTP (HY-113225) analogue. MANT-GppNHp interacts with the hydrophobic pocket near the AC catalytic site through its MANT group, thereby directly blocking the binding of the substrate ATP. MANT-GppNHp can be used to study diseases related to the increased activity of AC (such as cholera) .
|
-

- HY-103430
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 hydrobromide is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for the research of parkinson’s disease and nicotine craving alleviation .
|
-
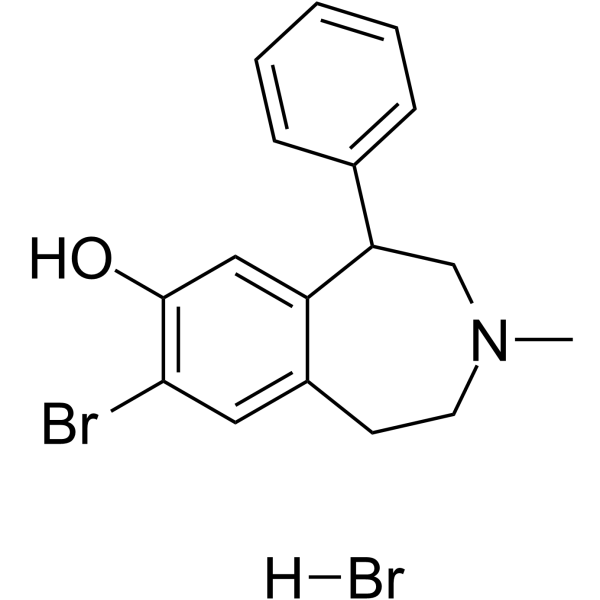
- HY-10780
-
|
|
Endogenous Metabolite
|
Cardiovascular Disease
|
|
JTV-803 mesylate is a human factor Xa inhibitor with oral anticoagulant activity. JTV-803 exhibits competitive inhibition of human factor Xa, with a Ki value of 0.019μM and IC50Value is 0.081μM. JTV-803 is 100 times more selective at inhibiting human factor Xa than its comparator. JTV-803 is an effective oral anticoagulant for the prevention of thrombosis .
|
-

- HY-120214
-
|
|
Syk
RET
|
Inflammation/Immunology
|
|
TAS05567 is a potent, highly selective, ATP-competitive and orally active Syk inhibitor with an IC50 of 0.37 nM. In a panel of 192 kinases, TAS05567 only shows >70% inhibition of Syk and 4 other kinases (FLT3, JAK2, KDR and RET with IC50s of 10 nM, 4.8 nM, 600 nM and 29 nM, respectively). TAS05567 can be used for humoral immune-mediated inflammatory conditions such as autoimmune and allergic diseases .
|
-
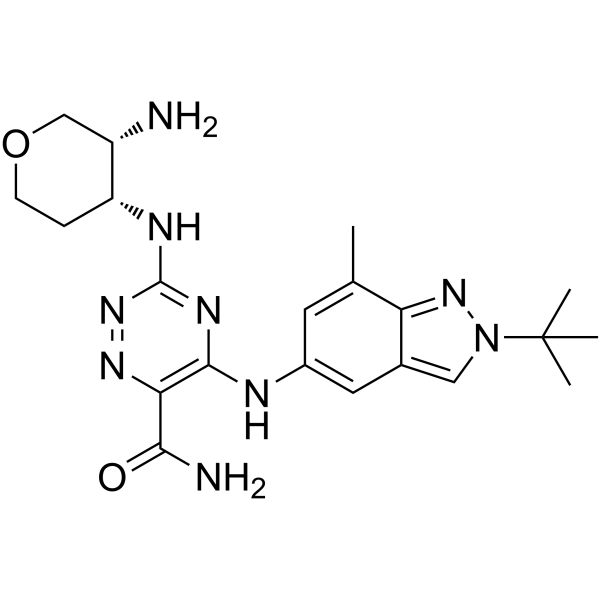
- HY-118061
-
|
|
Bacterial
|
Infection
|
|
VCC234718 is a molecule with mycobacterial growth inhibitory activity, specifically targeting Mycobacterium tuberculosis (Mtb). The primary molecular target of VCC234718 is inosine monophosphate dehydrogenase (GuaB2), and it inhibits the growth of Mtb by affecting the function of this enzyme. VCC234718 inhibits GuaB2 with a K value of 100 nM and exhibits non-competitive inhibition with IMP and NAD+. VCC234718 exerts its inhibitory effect by directly interacting with IMP and binding at the NAD+ site .
|
-

- HY-W040220
-
|
|
Bacterial
|
Infection
|
|
N-(3-Hydroxyoctanoyl)-DL-homoserine lactone (Compound 40) is a competitive inhibitor of the quorum sensing receptor LuxR with an IC50 value of 4 μM. N-(3-Hydroxyoctanoyl)-DL-homoserine lactone shows selective inhibition toward quorum sensing systems in Gram-negative bacteria like Pseudomonas aeruginosa. N-(3-Hydroxyoctanoyl)-DL-homoserine lactone is promising for research of bacterial infections .
|
-

- HY-105409
-
|
NVP-XAA-296; XAA 296
|
Microtubule/Tubulin
Apoptosis
|
Cancer
|
|
Discodermolide (NVP-XAA-296) is a potent microtubule-stabilizing agent with a Ki of 0.4 μM. Discodermolide stabilizes microtubules, induces G2 or M phase cell cycle arrest and apoptosis, leading to inhibition of cancer cell proliferation. Discodermolide competitively inhibits the binding of Paclitaxel (HY-B0015) to tubulin polymers, and inhibits the growth of Paclitaxel-resistant cells. Discodermolide can be used for breast and colon cancer research .
|
-

- HY-115570
-
|
GW108X
|
Kinesin
ULK
Autophagy
|
Cancer
|
|
GW406108X is a specific Kif15 (Kinesin-12) inhibitor with an IC50 of 0.82 uM in ATPase assays. GW406108X, a potent autophagy inhibitor, shows ATP competitive inhibition against ULK1 with a pIC50 of 6.37 (427 nM). GW406108X inhibits ULK1 kinase activity and blocks autophagic flux, without affecting the upstream signaling kinases mTORC1 and AMPK .
|
-
- HY-183249
-
|
|
Acetyl-CoA synthetase
Fungal
|
Infection
|
|
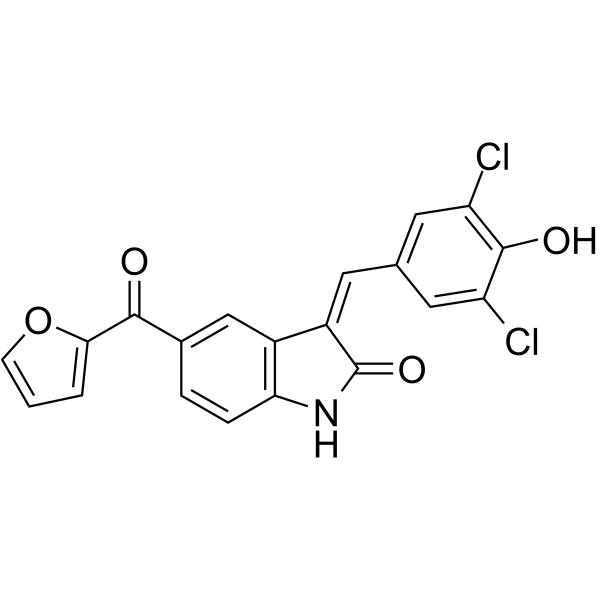
Ac-CoA Synthase-IN-2 is an Ac-CoA Synthase (ACS) inhibitor and antifungal agent. Ac-CoA Synthase-IN-2 binds in the ATP/acetyl-AMP pocket of fungal and human ACS enzymes to exert competitive inhibition with ATP, and inhibits Cryptococcus neoformans CnKbc1-mediated acetoacetate-to-aceto-acetyl CoA conversion. Ac-CoA Synthase-IN-2 can be used for the research of fungal infections .
|
-

- HY-135111S
-
|
|
Isotope-Labeled Compounds
Drug Metabolite
|
Infection
Metabolic Disease
Cancer
|
|
4-Desmethoxy Omeprazole-d3 is the deuterium labeled 4-Desmethoxy Omeprazole. 4-Desmethoxy Omeprazole is the active metabolite of Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Kiof 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .
|
-

- HY-15186
-
|
GDC-0068; RG7440
|
Organoid
Akt
Apoptosis
|
Cancer
|
|
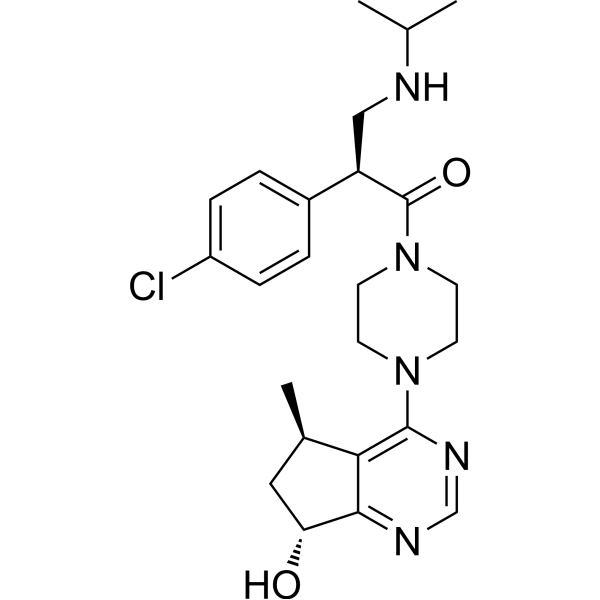
Ipatasertib (GDC-0068) is an orally active, highly selective and ATP-competitive pan-Akt inhibitor with IC50 values of 5, 18, 8 nM for Akt1/2/3, respectively. Ipatasertib synchronously activates FoxO3a and NF-κB through inhibition of Akt leading to p53-independent activation of PUMA. Ipatasertib also induces apoptosis in cancer cells and inhibits tumor growth in xenograft mouse models .
|
-
- HY-15186C
-
|
GDC-0068 tosylate; RG7440 tosylate
|
Organoid
Akt
Apoptosis
|
Cancer
|
|
Ipatasertib (GDC-0068) tosylate is an orally active, highly selective and ATP-competitive pan-Akt inhibitor with IC50 values of 5, 18, 8 nM for Akt1/2/3, respectively. Ipatasertib tosylate synchronously activates FoxO3a and NF-κB through inhibition of Akt leading to p53-independent activation of PUMA. Ipatasertib tosylate also induces apoptosis in cancer cells and inhibits tumor growth in xenograft mouse models .
|
-

- HY-50878S
-
|
PF-02341066-d5
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
- HY-131339
-
SP-96
2 Publications Verification
|
Aurora Kinase
|
Cancer
|
|
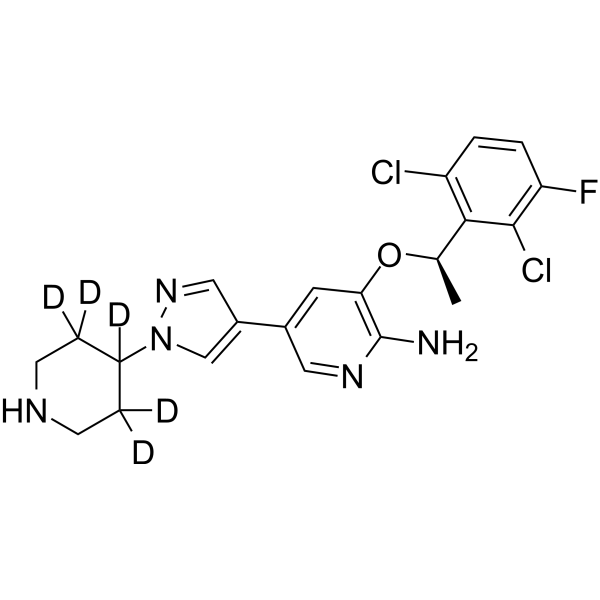
SP-96 is a highly potent, selective and non-ATP-competitive Aurora B (IC50=0.316 nM) inhibitor and shows >2000 fold selectivity against FLT3 and KIT. SP-96 shows selective growth inhibition in NCI60 screening, incluing MDA-MD-468 (GI50=107 nM). SP-96 can be used for the research of triple negative breast cancer (TNBC) .
|
-
- HY-113371R
-
|
Methylcitric acid (Standard)
|
Reference Standards
Endogenous Metabolite
|
Metabolic Disease
|
|
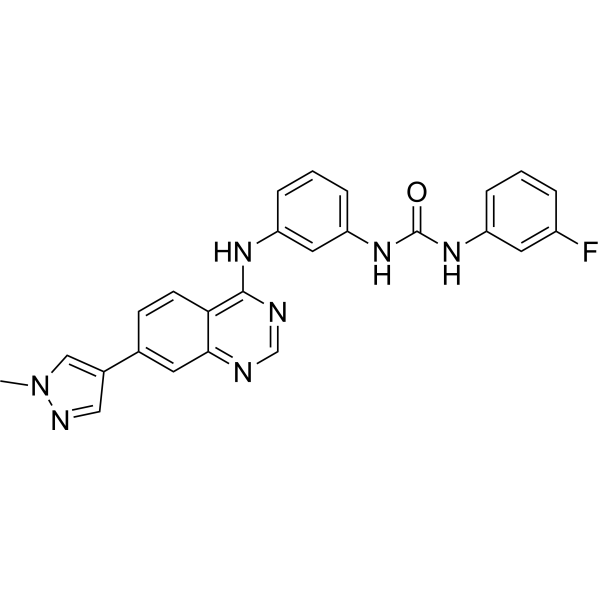
Omeprazole (sodium) (Standard) is the analytical standard of Omeprazole (sodium). This product is intended for research and analytical applications. Omeprazole sodium (H 16868 sodium), a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole sodium shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole sodium also inhibits growth of Gram-positive and Gram-negative bacteria . Omeprazole is a potent brain penetrant neutral sphingomyelinase (N-SMase) inhibitor (exosome inhibitor) .
|
-

- HY-16525
-
|
|
Monoamine Transporter
Adrenergic Receptor
|
Neurological Disease
|
|
XEN-2174 is a noradrenaline transporter (NET) inhibitor. XEN-2174 inhibits the reuptake of noradrenaline through non-competitive inhibition, increasing the concentration of noradrenaline in the synaptic cleft, thereby activating α2-adrenergic receptor at the spinal level and exerting analgesic effects. XEN-2174 exhibits long-lasting analgesic effects in models of neuropathic pain and postoperative pain in rats. XEN-2174 can be used in pain research .
|
-

- HY-144636
-
|
|
Atg4
Cathepsin
Phospholipase
Autophagy
|
Cancer
|
|
Atg4B-IN-2 is a potent competitive Atg4B inhibitor with Ki value of 3.1 μM, also possesses declining PLA2 inhibitory potency, IC50s of 11 μM and 3.5 μM for Atg4B and PLA2, respectively. Atg4B-IN-2 enhances the anticancer activity of anti-castration-resistant prostate cancer agents via autophagy inhibition .
|
-
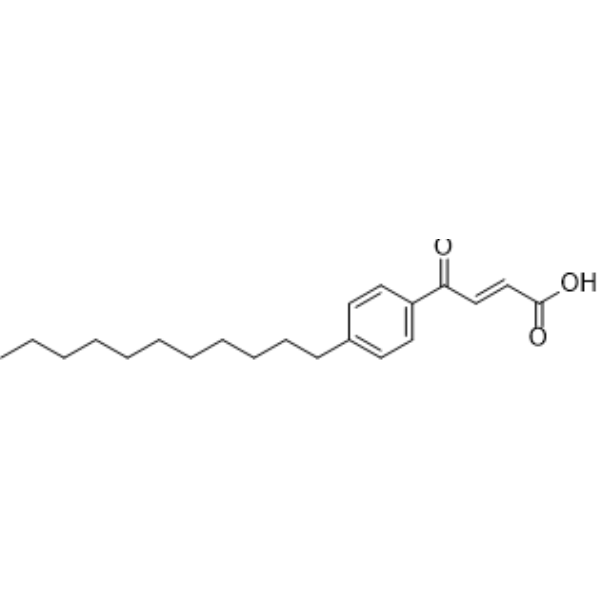
- HY-135111R
-
|
|
Reference Standards
Drug Metabolite
|
Infection
Metabolic Disease
Cancer
|
|
4-Desmethoxy Omeprazole (Standard) is the analytical standard of 4-Desmethoxy Omeprazole. This product is intended for research and analytical applications. 4-Desmethoxy Omeprazole is the active metabolite of Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Ki of 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .
|
-

- HY-162485
-
|
|
Dengue Virus
Virus Protease
|
Infection
|
|
DV-B-120 is a competitive inhibitor for dengue virus (DENV) with IC50s of 5.35, 7.39, 10.49 and 8.58 μM, for DENV-1, DENV-2, DENV-3 and DENV-4, respectively, by inhibiting NS2B-NS3 protease. DV-B-120 exhibits antiviral activity through inhibition of DENV replication .
|
-

- HY-103430A
-
|
|
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for research of parkinson’s disease and nicotine craving alleviation .
|
-
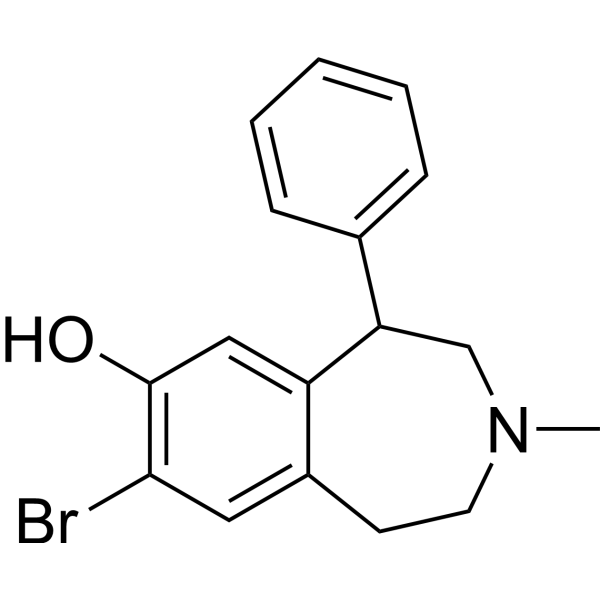
- HY-117747
-
|
JCR 424; XM 323
|
HIV Protease
|
Infection
|
|
DMP 323 is a potent, nonpeptide cyclic urea inhibitor of HIV protease, effective against both HIV type 1 and type 2. Designed using structural information and database searching, it competitively inhibits the cleavage of both peptide and HIV-1 gag polyprotein substrates. DMP 323 shows comparable potency to other highly effective HIV protease inhibitors like A-80987 and Ro-31-8959. Importantly, its efficacy against HIV protease remains unaffected by human plasma or serum, suggesting low affinity for plasma proteins. Furthermore, DMP 323 demonstrates minimal inhibition of various mammalian proteases at concentrations much higher than those needed for HIV protease inhibition, highlighting its specificity for viral targets .
|
-

- HY-50878A
-
|
PF-02341066 hydrochloride
|
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
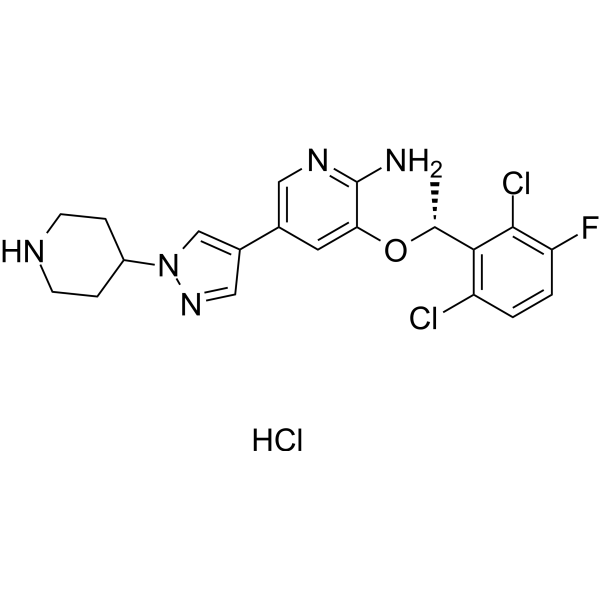
- HY-N15587
-
|
Gostatine
|
Aminotransferases (Transaminases)
|
Metabolic Disease
|
|
Gostatin is an inhibitor of aspartate aminotransferase (GOT). Gostatin is found in Streptomyces sumanensis nov. sp. NK-23. Gostatin has a strong inhibitory effect on pig heart GOT, a weak inhibitory effect on wheat germ GOT and GPT, and no significant effect on glutamate dehydrogenase and glutamine synthetase. The inhibitory mechanism of gostatin is similar to substrate competitive inhibition, and aspartate has a protective effect on its inhibitory effect. Gostatin can be used to study the catalytic mechanism of GOT and its role in nitrogen metabolism .
|
-

- HY-N1584
-
Halofuginone
Maximum Cited Publications
20 Publications Verification
RU-19110
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (RU-19110), a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
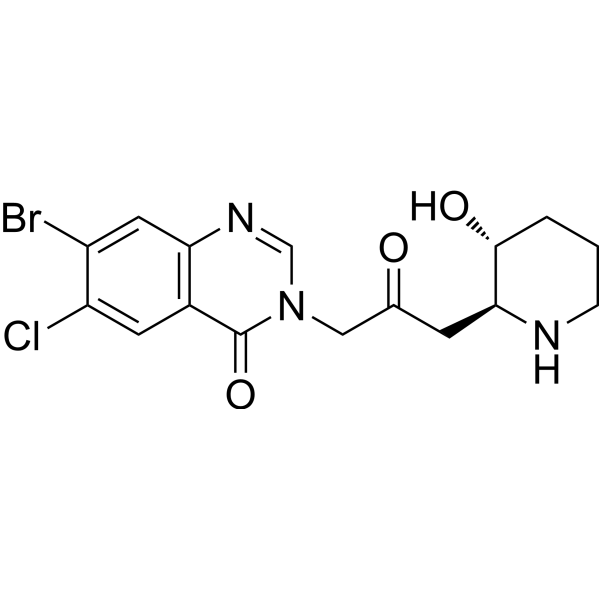
- HY-N1584C
-
|
RU-19110 lactate
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone lactate, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone lactate is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone lactate is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone lactate has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-

- HY-50878R
-
|
PF-02341066 (Standard)
|
Reference Standards
Anaplastic lymphoma kinase (ALK)
c-Met/HGFR
ROS Kinase
Autophagy
|
Cancer
|
|
Crizotinib (Standard) is the analytical standard of Crizotinib. This product is intended for research and analytical applications. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-

- HY-100414R
-
|
BYK61359 (Standard)
|
Proton Pump
Reference Standards
|
Metabolic Disease
|
|
Soraprazan (Standard) is the analytical standard of Soraprazan (HY-100414). This product is intended for research and analytical applications. Soraprazan (BYK61359) is a selective, reversible K-competitive inhibitor of the H,K-ATPase (Ki=6.4 nM), with an IC50 of 0.19 μM in gastric glands. Soraprazan binds to the H,K-ATPase with a Kd of 28.27 nM. Soraprazan shows immediate inhibition of acid secretion and is more than 2000-fold selective for H,K-ATPase over Na,K- and Ca-ATPases .
|
-

- HY-103430AR
-
|
|
Reference Standards
Dopamine Receptor
5-HT Receptor
Adenylate Cyclase
|
Neurological Disease
|
|
SKF-83566 (Standard) is the analytical standard of SKF-83566 (HY-103430A). This product is intended for research and analytical applications. SKF-83566 is a potent, blood-brain permeable and orally active D1-like dopamine receptor (D1DR) antagonist and a weaker competitive antagonist at the vascular 5-HT2 receptor (Ki=11 nM) . SKF-83566 is a competitive DAT (dopamine transporter) inhibitor with an IC50 of 5.7 μM . SKF-83566 also shows selective inhibition for adenylyl cyclase 2 (AC2) over AC1 and AC5 in the isolated rabbit thoracic aorta . SKF-83566 can be used for research of parkinson’s disease and nicotine craving alleviation .
|
-

- HY-110079
-
|
|
IPK Superfamily
|
Cancer
|
|
TNP is a competitive, reversible inhibitor of IP6K1 and IP3K, with IC50s of 0.55 μM and 10.2 μM for IP6K1 and IP3K, respectively. TNP competitively binds to the ATP binding site of IP6K, inhibits the generation of 5-IP7, and thus relieves the inhibition of 5-IP7 on the AKT signaling pathway. TNP can enhance insulin sensitivity and promote thermogenesis in adipose tissue. TNP cannot effectively pass through the blood-brain barrier and is mainly used in the study of obesity, type 2 diabetes, and metabolic syndrome. However, TNP also inhibits CYP3A4 and may need further optimization[1][2][3].
|
-
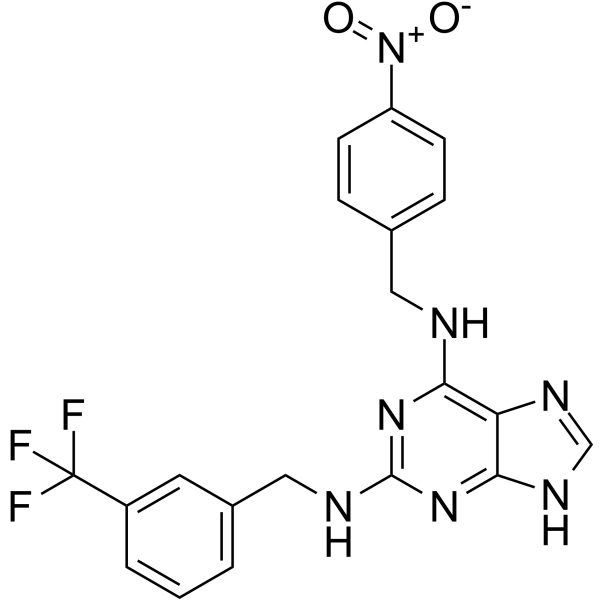
- HY-50878S1
-
|
PF-02341066-d8
|
Isotope-Labeled Compounds
ROS Kinase
Autophagy
c-Met/HGFR
Anaplastic lymphoma kinase (ALK)
|
Cancer
|
|
Crizotinib-d8 (PF-02341066-d8) is deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-

- HY-N1584B
-
|
RU-19110 hydrochloride
|
Calcium Channel
DNA/RNA Synthesis
Parasite
Sodium Channel
TGF-beta/Smad
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM. Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity. Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
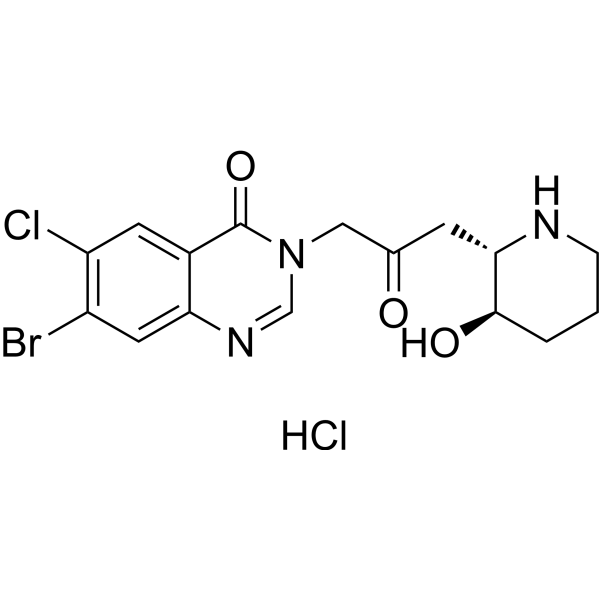
- HY-P5272
-
|
|
Bacterial
|
Inflammation/Immunology
|
|
Histatin-3 TFA, a 32 amino acid peptide, possesses powerful antimicrobial properties. Histatin-3 TFA behaves as a substrate for proprotein convertase 1 (PC1), being cleaved by this endoprotease primarily at a site carboxy terminal to the single Arg25 residue (HRGYR decrease SN). Histatin-3 TFA is a moderately potent, reversible and competitive inhibitor of the furin-mediated cleavage of the pentapeptide pGlu-Arg-Thr-Lys-Arg-MCA fluorogenic substrate, with an estimated inhibition constant Ki of 1.98 μM .
|
-

- HY-50878S2
-
|
PF-02341066-d9
|
Isotope-Labeled Compounds
|
Cancer
|
|
Crizotinib-d9 (PF-02341066-d9) is deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-

- HY-137697
-
|
|
Nucleoside Antimetabolite/Analog
DNA/RNA Synthesis
HIV Protease
HIV
Drug Metabolite
|
Infection
|
|
ddCTP is a type of chain-terminating deoxynucleotide. ddCTP can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-

- HY-N1584A
-
|
RU-19110 hydrobromide
|
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
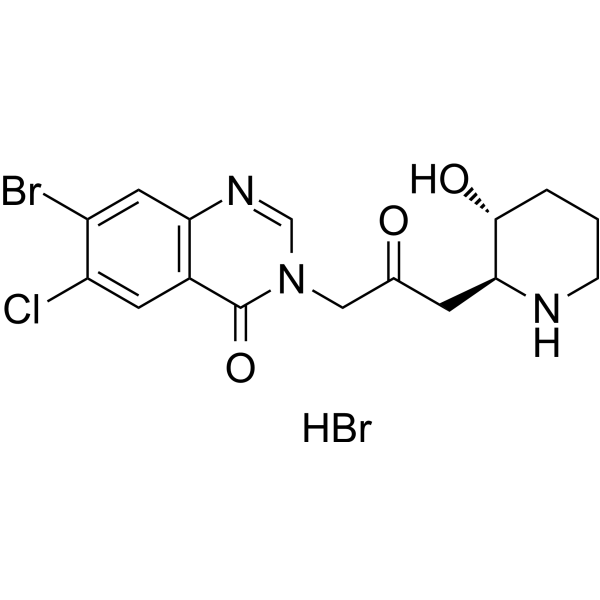
- HY-17422S1
-
|
Aciclovir-d4; Acycloguanosine-d4
|
Isotope-Labeled Compounds
HSV
Bacterial
Apoptosis
Antibiotic
|
Infection
Cancer
|
|
Acyclovir-d4 is the deuterium labeled Acyclovir. Acyclovir (Aciclovir) is a guanosine analogue and an orally active antiviral agent. Acyclovir inhibits HSV-1 (IC50 of 0.85 μM), HSV-2 (IC50 of 0.86 μM) and varicella-zoster virus. Acyclovir can be phosphorylated by viral thymidine kinase (TK), and Acyclovir triphosphate interferes with viral DNA polymerization through competitive inhibition with guanosine triphosphate and obligatory chain termination . Acyclovir prevents bacterial infections during induction therapy for acute leukaemia .
|
-
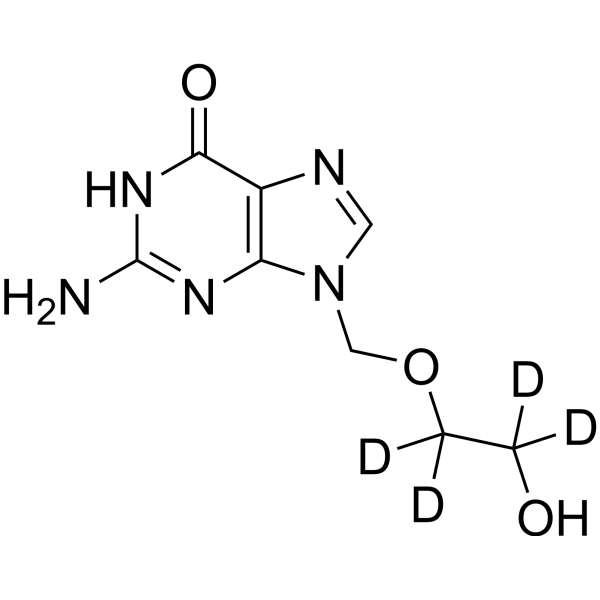
- HY-137697A
-
|
|
Drug Metabolite
HIV
Nucleoside Antimetabolite/Analog
HIV Protease
DNA/RNA Synthesis
|
Infection
|
|
ddCTP tetrasodium is a type of chain-terminating deoxynucleotide. ddCTP tetrasodium can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP tetrasodium can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP tetrasodium is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-

- HY-W105272R
-
|
|
Reference Standards
Endogenous Metabolite
|
Metabolic Disease
|
|
Halofuginone (Standard) is the analytical standard of Halofuginone. This product is intended for research and analytical applications. Halofuginone (RU-19110), a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca2+ channels. Halofuginone has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-

- HY-137697B
-
|
|
HIV
DNA/RNA Synthesis
Nucleoside Antimetabolite/Analog
Drug Metabolite
HIV Protease
|
Infection
|
|
ddCTP trilithium is a type of chain-terminating deoxynucleotide. ddCTP trilithium can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP trilithium can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP trilithium is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-
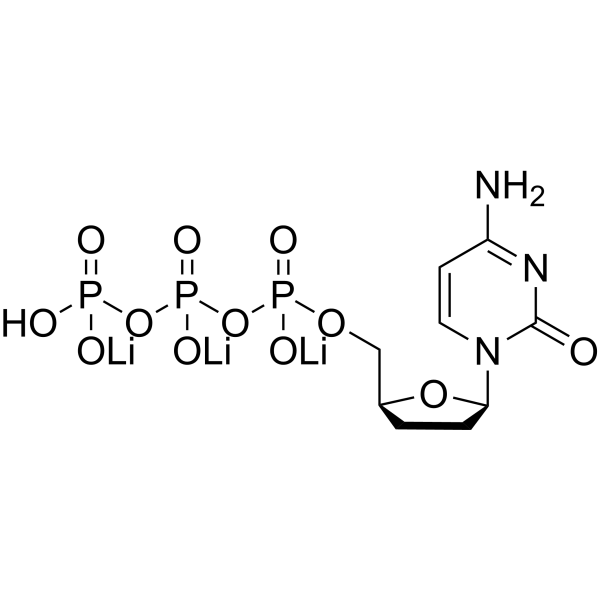
- HY-N1584R
-
|
RU-19110 (Standard)
|
Reference Standards
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (Standard) is the analytical standard of Halofuginone. This product is intended for research and analytical applications. Halofuginone (RU-19110), a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca2+ channels. Halofuginone has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-

- HY-118156
-
|
|
Others
|
Others
|
|
L-699333 is a 5-lipoxygenase (5-LO) inhibitor belonging to the thieno[2,3,4-cd]indole class. This compound has a 2-ethoxybutyric acid side chain and is a potent inhibitor of the biosynthesis of 5-HPETE and LTB4 produced from human 5-LO, with ICm values of 22 nM, 7 nM, and 3.8 pM for human neutrophils and whole blood, respectively. L-699333 has shown anti-inflammatory and antiasthmatic effects in a variety of animal models, including rat pleurisy models, antigen-induced wheezing models, and awake macaque and sheep asthma models. Its inhibition of 5-LO is highly selective, with higher ICm values or stronger competitive inhibition in FLAP binding assays compared to inhibition of human 15-LO, porcine 12-LO, and ram epididymal cyclooxygenase. The racemic enantiomer 14g of L-699333 is the most potent enantiomer to date, with inhibitory effects similar to those of the known MK-0591, which has been shown in clinical trials to inhibit the biochemical effects of LTB4 biosynthesis in vitro and LTE4 excretion in urine.
|
-

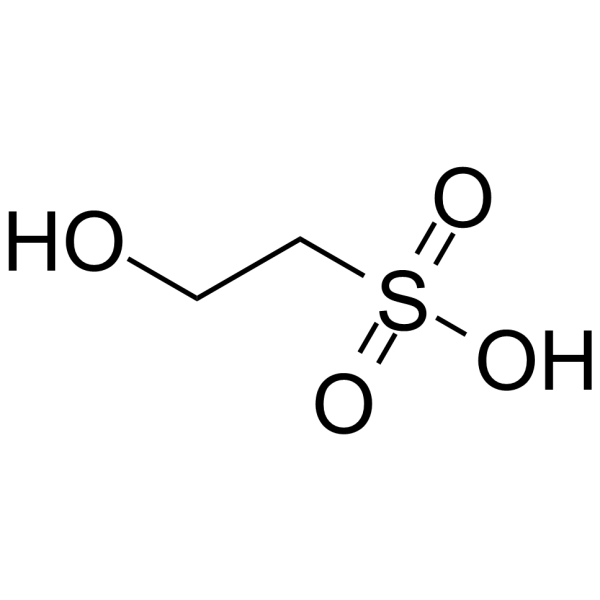
- HY-Y0095
-
|
2-Hydroxyethanesulfonic acid
|
Environmental Pollutants
Mitochondrial Metabolism
Parasite
|
Metabolic Disease
|
|
Isethionic acid is a calcium binder and anionic detergent that enhances mitochondrial calcium binding capacity by competitively binding to calcium binding sites on the outer mitochondrial membrane. Isethionic acid can inhibit calcium-activated mitochondrial respiration. Isethionic acid inhibits barnacle (Balanus amphitrite) larvae with LC50s of 23 μg/mL (24 h) and 17 μg/mL (48 h), respectively. Isethionic acid can inhibit the attachment of barnacle larvae (complete inhibition at 10 μg/mL) and regulate mitochondrial calcium transport, and can enhance ATP-dependent calcium uptake at high calcium concentrations. Isethionic acid can be used to study the mechanism of mitochondrial calcium metabolism.
|
-
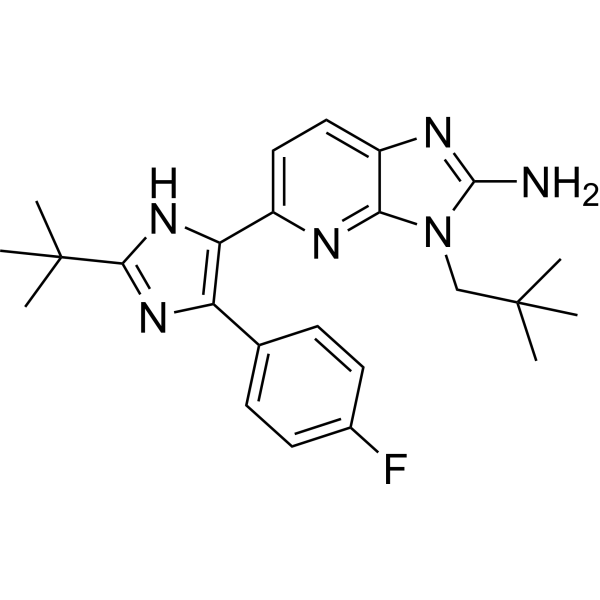
- HY-13241A
-
|
LY2228820
|
p38 MAPK
Autophagy
|
Cancer
|
|
Ralimetinib is an ATP-competitive p38α and p38β MAPK inhibitor with an IC50 of 5.3 nmol/L against human p38α and an IC50 of 3.2 nmol/L against human p38β. Ralimetinib slows tumor growth in preclinical in vivo cancer models, exhibits oral bioavailability in mice, and achieves sustained target inhibition for 4 to 8 h. Ralimetinib is applicable for research on melanoma, non-small cell lung cancer, ovarian cancer, glioma, multiple myeloma, breast cancer, renal cancer, and head and neck squamous cell carcinoma .
|
-
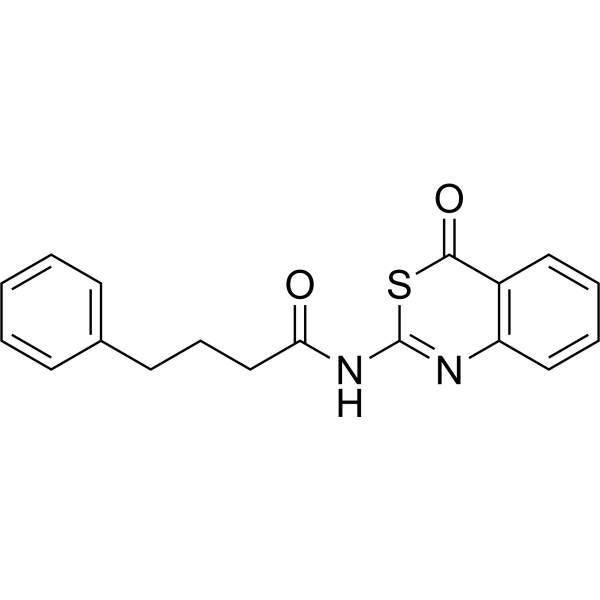
- HY-180506
-
|
|
Tyrosinase
Glycosidase
Cholinesterase (ChE)
|
Others
|
|
Tyrosinase-IN-47 (compound 6a) is a potent competitive tyrosinase inhibitor with an IC50 of 1.43 µM and a Ki of 0.1142 μM. Tyrosinase-IN-47 also shows inhibition activity in α-Glucosidase (IC50 = 36.26 μM) and acetylcholinesterase (IC50 = 8.26 μM). Tyrosinase-IN-47 exhibits in vitro antioxidant activity, with good scavenging ability for DPPH (IC50 = 4.75 μM) and ABTS (IC50 = 0.04 μM). Tyrosinase-IN-47 displays anti-browning effect on freshly cut potatoes. Tyrosinase-IN-47 can be used for pharmaceutical research .
|
-

- HY-N1584BR
-
|
RU-19110 hydrochloride (Standard)
|
Reference Standards
Calcium Channel
DNA/RNA Synthesis
Parasite
Sodium Channel
TGF-beta/Smad
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (hydrochloride) (Standard) is the analytical standard of Halofuginone (hydrochloride). This product is intended for research and analytical applications. Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM. Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity. Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects [4] .
|
-

- HY-120826
-
|
|
Monoamine Oxidase
Adenosine Receptor
|
Neurological Disease
|
|
A2AAR/hMAO-B-IN-1 (compoudn 17) is a non-xanthine dual-target inhibitor targeting the A2A adenosine receptor (A2AAR) (IC50: 34.9 nM) andMAO-B (Ki: 39.5 nM, human). A2AAR/hMAO-B-IN-1 inhibits A2AAR-induced cAMP accumulation and exhibits competitive, reversible inhibition of MAO-B. A2AAR/hMAO-B-IN-1 can be used in the study of neurodegenerative diseases such as Parkinson's disease (PD) .
|
-
- HY-50878AS
-
|
PF-02341066-d9 hydrochloride
|
c-Met/HGFR
Autophagy
Anaplastic lymphoma kinase (ALK)
ROS Kinase
Isotope-Labeled Compounds
|
Cancer
|
|
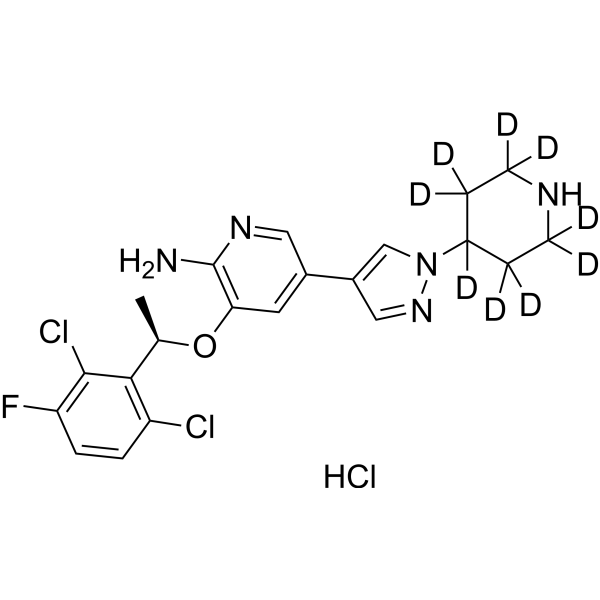
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
- HY-110137A
-
|
DB75; NSC 305831
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine (DB75) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine is also an antiparasite agent .
|
-
- HY-110137
-
|
DB75 dihydrochloride; NSC 305831 dihydrochloride
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine dihydrochloride (DB75 dihydrochloride) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine dihydrochloride is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 µM, 283 µM, and >400 µM, respectively). Furamidine dihydrochloride is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine dihydrochloride is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine dihydrochloride is also an antiparasite agent .
|
-
- HY-N1584AR
-
|
RU-19110 hydrobromide (Standard)
|
Reference Standards
DNA/RNA Synthesis
TGF-beta/Smad
Parasite
Sodium Channel
Calcium Channel
|
Infection
Cardiovascular Disease
Inflammation/Immunology
Cancer
|
|
Halofuginone (hydrobromide) (Standard) is the analytical standard of Halofuginone (hydrobromide). This product is intended for research and analytical applications. Halofuginone (RU-19110) hydrobromid, a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone hydrobromid is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone hydrobromid is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca 2+ channels. Halofuginone hydrobromid has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-

- HY-121998
-
|
|
Aurora Kinase
|
Others
|
|
Binucleine 2 is an isoform-specific and ATP-competitive inhibitor of Drosophila Aurora B kinase (Ki=0.36 μM), a kinase involved in cell division. It is specific for Drosophila Aurora B kinase, inhibiting it in a dose-dependent manner, with minimal inhibition of human or X. laevis Aurora B kinases at concentrations up to 100 μM. Binucleine 2 induces mitotic and cytokinesis defects in Drosophila Kc167 cells. It prevents Drosophila S2 cells from assembling a contractile ring during cell division when used at a concentration of 40 μM but does not affect ring ingression, suggesting that Aurora B kinase activity is not required for that step.
|
-

- HY-15076
-
|
NS-1209 sodium
|
iGluR
|
Neurological Disease
|
|
SPD-502 sodium is a novel glutamate antagonist with potential neuroprotective properties, particularly in brain ischemia. It selectively targets the AMPA receptor, showing high affinity (IC50 = 0.043 μM) and competitive inhibition of AMPA-induced effects in rat cortical membranes and cultured mouse cortical neurons. In vivo, SPD-502 sodium effectively blocks AMPA-evoked spike activity in the hippocampus after intravenous administration, significantly increasing the seizure threshold in mice and demonstrating robust protection against ischemia-induced damage to hippocampal neurons in gerbils. These findings suggest SPD-502 sodium may be promising for studying neurodegenerative conditions associated with glutamate excitotoxicity .
|
-

- HY-15074
-
|
NS-1209
|
iGluR
|
Neurological Disease
|
|
SPD-502 is a novel glutamate antagonist with potential neuroprotective properties, particularly in brain ischemia. It selectively targets the AMPA receptor, showing high affinity (IC50 = 0.043 μM) and competitive inhibition of AMPA-induced effects in rat cortical membranes and cultured mouse cortical neurons. In vivo, SPD-502 effectively blocks AMPA-evoked spike activity in the hippocampus after intravenous administration, significantly increasing the seizure threshold in mice and demonstrating robust protection against ischemia-induced damage to hippocampal neurons in gerbils. These findings suggest SPD-502 may be promising for studying neurodegenerative conditions associated with glutamate excitotoxicity .
|
-

- HY-137697D
-
|
|
HIV
DNA/RNA Synthesis
Nucleoside Antimetabolite/Analog
Drug Metabolite
HIV Protease
|
Infection
|
|
ddCTP trilithium solution (100 mM) is a chain-terminating dideoxynucleotide. ddCTP trilithium is a type of chain-terminating deoxynucleotide. ddCTP trilithium can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP trilithium can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP trilithium is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-
- HY-W657887
-
|
|
Histone Methyltransferase
GSK-3
Tau Protein
Amyloid-β
Glucocorticoid Receptor
Androgen Receptor
Adrenergic Receptor
|
Neurological Disease
|
|
GSK-3β/G9a-IN-1 (Compound T2) is an orally active, selective, blood-brain-barrier permeable, competitive G9a (substrate-competitive, IC50: 1.1 μM) and GSK-3β (ATP competitive, IC50: 0.8 μM) inhibitor. GSK-3β/G9a-IN-1 is a potent H3K9me2 inhibitor that reshapes chromatin landscape. GSK-3β/G9a-IN-1 lowers tau phosphorylation, reduces Aβ aggregation. GSK-3β/G9a-IN-1 displays inhibition toward glucocorticoid receptor, androgen receptor, and alpha-2A adrenergic receptor. GSK-3β/G9a-IN-1 also upregulates SAGA complex members such as Eny2 and Sgf29. GSK-3β/G9a-IN-1 markedly improves memory, restores social behaviors, and increases synaptic complexity in late-onset Alzheimer’s disease .
|
-

- HY-177944
-
|
|
Phosphoglycerate Kinase (PGK)
Keap1-Nrf2
Interleukin Related
|
Inflammation/Immunology
|
|
DC-PGKI is an orally active ATP-competitive PGK1 inhibitor(IC50 = 0.16 Μm, Kd = 99.08 nM). DC-PGKI stabilizes PGK1 in vitro and in vivo, and suppresses both glycolytic activity and the kinase function of PGK1. DC-PGKI-mediated inhibition of PGK1 leads to the accumulation of NRF2 (nuclear factor-erythroid factor 2-related factor 2, NFE2L2), which then translocates to the nucleus, binds to the proximal regions of IL-1β and IL-6 genes, and suppresses the LPS-induced expression of these genes. DC-PGKI can be used for the study of colitis .
|
-

- HY-116142
-
|
|
iGluR
|
Neurological Disease
|
|
CP-283097 is an orally active and conformationally restricted and NR2B subtype-selective NMDA antagonist. CP-283097 efficiently competitively inhibits the binding of [³H]CP-101,606 to the rat meninges, with an IC50 value of 18 nM. CP-283097 exhibits nearly complete inhibition of the current mediated by the NR2B receptor (IC50 = 206 nM), while the inhibitory effect on the NR2A or NR2C receptors is very weak. CP-283097 demonstrates excellent central nervous system permeability and in vivo efficacy in animal models. CP-283097 can be used for neurological diseases related to excessive activation of NMDA receptors .
|
-

- HY-10256
-
Adezmapimod
Maximum Cited Publications
601 Publications Verification
SB 203580; RWJ 64809
|
Organoid
p38 MAPK
Autophagy
Mitophagy
HSP
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (SB 203580) is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod can inhibit p38 MAPK and lead to the inhibition of downstream HSP27 phosphorylation. Adezmapimod does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-
- HY-W661499
-
|
|
Phosphatase
Apoptosis
Reactive Oxygen Species (ROS)
Caspase
|
Cancer
|
|
Orellanine, a nephrotoxic alkaloid found in Cortinarius orellanus, is an orally active and selective non-competitive inhibitor of alkaline phosphatase. Orellanine chelates iron, generates reactive oxygen species (ROS), induces DNA scission, forms ortho-semiquinone radicals, downregulates antioxidant defenses, and inhibits mitochondrial function. Orellanine induces caspase 8/9-mediated apoptosis. Orellanine inhibits synthesis of proteins, RNA, DNA, and mitochondrial protein synthesis, with metabolic activation required for cell-free protein synthesis inhibition. Orellanine can be used for the research of metastatic clear cell renal cell carcinoma, acute renal failure, chronic renal insufficiency, and kidney damage .
|
-
- HY-110137R
-
|
DB75 dihydrochloride (Standard); NSC 305831 dihydrochloride (Standard)
|
Histone Methyltransferase
Phosphodiesterase (PDE)
Parasite
Reference Standards
|
Infection
Inflammation/Immunology
Cancer
|
|
Furamidine (dihydrochloride) (Standard) is the analytical standard of Furamidine (dihydrochloride). This product is intended for research and analytical applications. Furamidine dihydrochloride (DB75 dihydrochloride) is a selective protein arginine methyltransferase 1 (PRMT1) inhibitor with an IC50 of 9.4 μM. Furamidine dihydrochloride is selective for PRMT1 over PRMT5, PRMT6, and PRMT4 (CARM1) (IC50s of 166 μM, 283 μM, and >400 μM, respectively). Furamidine dihydrochloride is a potent, reversible and competitive tyrosyl-DNA phosphodiesterase 1 (TDP-1) inhibitor. Inhibition of TDP-1 by Furamidine dihydrochloride is effective both with single- and double-stranded DNA substrates but is slightly stronger with the duplex DNA. Furamidine dihydrochloride is also an antiparasite agent .
|
-

- HY-W015490R
-
|
|
Reference Standards
DNA/RNA Synthesis
NF-κB
Monoamine Oxidase
TNF Receptor
Bacterial
|
Infection
Inflammation/Immunology
Cancer
|
|
1,4-Naphthoquinone is an inhibitor with broad-spectrum inhibitory activity targeting DNA polymerase, NF-κB and monoamine oxidase (MAO-A/B), with antibacterial and anti-biofilm efficacy. 1,4-Naphthoquinone is a competitive inhibitor of MAO-B (Ki=1.4 μM) and a non-competitive inhibitor of MAO-A (Ki=7.7 μM). 1,4-Naphthoquinone inhibits DNA polymerase pol α, β, γ, δ, ε, λ with IC50 ranging from 5.57-128 μM. 1,4-Naphthoquinone inhibits tumor cell proliferation, induces apoptosis and necrosis, and has anti-angiogenic and anti-inflammatory activities by inducing oxidative stress, depleting glutathione (GSH), inhibiting DNA polymerase-mediated DNA synthesis and blocking NF-κB nuclear translocation. 1,4-Naphthoquinone can be used in anti-bacterial , anti-tumor and anti-inflammatory studies, including inhibition of melanoma and colon cancer cell growth and endothelial cell function, as well as LPS-induced inflammation models .
|
-

- HY-W015490
-
|
|
Environmental Pollutants
DNA/RNA Synthesis
NF-κB
Monoamine Oxidase
TNF Receptor
Bacterial
|
Infection
Inflammation/Immunology
Cancer
|
|
1,4-Naphthoquinone is an inhibitor with broad-spectrum inhibitory activity targeting DNA polymerase, NF-κB and monoamine oxidase (MAO-A/B), with antibacterial and anti-biofilm efficacy. 1,4-Naphthoquinone is a competitive inhibitor of MAO-B (Ki=1.4 μM) and a non-competitive inhibitor of MAO-A (Ki=7.7 μM). 1,4-Naphthoquinone inhibits DNA polymerase pol α, β, γ, δ, ε, λ with IC50 ranging from 5.57-128 μM. 1,4-Naphthoquinone inhibits tumor cell proliferation, induces apoptosis and necrosis, and has anti-angiogenic and anti-inflammatory activities by inducing oxidative stress, depleting glutathione (GSH), inhibiting DNA polymerase-mediated DNA synthesis and blocking NF-κB nuclear translocation. 1,4-Naphthoquinone can be used in anti-bacterial , anti-tumor and anti-inflammatory studies, including inhibition of melanoma and colon cancer cell growth and endothelial cell function, as well as LPS-induced inflammation models .
|
-
- HY-403733A
-
|
|
Androgen Receptor
|
Cancer
|
|
(-)-JJ-450 is a non-competitive antagonist targeting the androgen receptor (AR). (-)-JJ-450 is more potent than (+)-JJ-450 (HY-403733B) in inhibiting androgen-induced AR activity, and the mechanism of AR inhibition by (+)-JJ-450 is different from that of Enzalutamide (MDV3100) (HY-70003), which may target the ligand binding domain (LBD) of AR. (-)-JJ-450 inhibits the transcriptional activity of wild-type AR and mutant AR F876L by inhibiting AR nuclear translocation and promoting nuclear degradation of unbound AR. (-)-JJ-450 can be used in the study of castration-resistant prostate cancer (CRPC) resistant to Enzalutamide .
|
-

- HY-182513
-
|
|
Prostaglandin Receptor
|
Others
Inflammation/Immunology
|
|
ONO-8539 is an orally active prostanoid EP1 receptor antagonist with competitive, insurmountable binding and slow receptor dissociation for long-duration inhibition. ONO-8539 modulates afferent nerve function related to bladder activity, inhibits detrusor overactivity-related contractions, decreases nonvoiding contractions and voiding duration, and increases uroflow rate with ATP (HY-B2176)-induced detrusor overactivity. ONO-8539 attenuates acid-induced heartburn symptoms, extends time to first heartburn sensation, and prevents primary hypersensitivity after distal esophageal acidification. ONO-8539 can be used for the research of overactive bladder and gastroesophageal reflux disease .
|
-

- HY-P0299
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) can readily crosse the blood-brain barrier .
|
-
- HY-P0299A
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) TFA is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) TFA can readily crosse the blood-brain barrier .
|
-
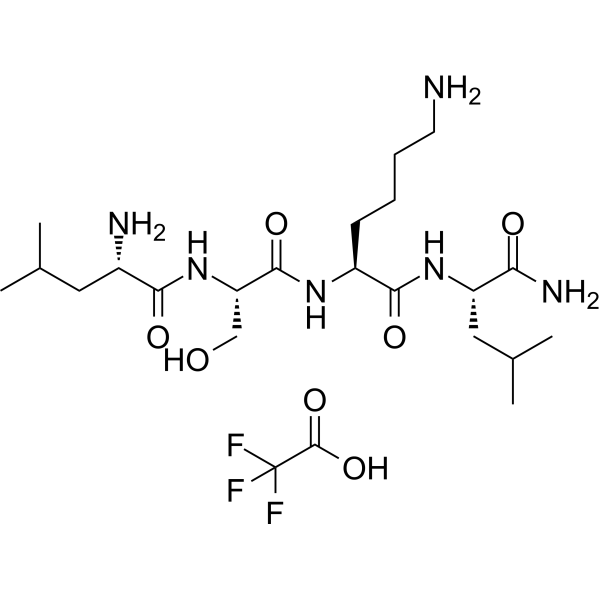
- HY-10256R
-
|
SB 203580 (Standard); RWJ 64809 (Standard)
|
Organoid
Reference Standards
p38 MAPK
Autophagy
Mitophagy
HSP
|
Inflammation/Immunology
Cancer
|
|
Adezmapimod (Standard) is the analytical standard of Adezmapimod. This product is intended for research and analytical applications. Adezmapimod (SB 203580) is a selective and ATP-competitive p38 MAPK inhibitor with IC50s of 50 nM and 500 nM for SAPK2a/p38 and SAPK2b/p38β2, respectively. Adezmapimod inhibits LCK, GSK3β and PKBα with IC50s of 100-500-fold higher than that for SAPK2a/p38. Adezmapimod can inhibit p38 MAPK and lead to the inhibition of downstream HSP27 phosphorylation. Adezmapimod does not disrupt JNK activity and is an autophagy and mitophagy activator .
|
-

- HY-135446
-
|
|
Endothelin Receptor
|
Cardiovascular Disease
Endocrinology
|
|
BQ-610 is a selective antagonist of the endothelin A receptor (ETA receptor). BQ-610 specifically blocks the ETA receptor, competitively inhibiting the binding of endothelin-1 (ET-1) (a vasoconstrictive peptide) to the receptor, thereby blocking the effects of ET-1 such as vascular smooth muscle contraction, cell mitosis, and inhibition of hormone secretion. BQ-610 significantly alleviates cerebral vasospasm in rabbits. BQ-610 blocks the bronchial epithelial and pulmonary vascular cell proliferation caused by cigarette smoke in rat models. BQ-610 can delay the natural luteal regression in the cow's uterus. BQ-610 can be used for research on vasospasm, abnormal cell proliferation, and reproductive endocrine disorders .
|
-

- HY-B1341
-
|
Enidrel; SC-4642; NSC 15432
|
Endogenous Metabolite
Progesterone Receptor
|
Cancer
|
|
Norethynodrel (Enidrel; SC-4642) is an orally active progestogen analog that reduces estrogen-like effects and enhances progestogen-like responses in endometrial stromal cells. Norethynodrel also promotes cell maturation and predecidual cell formation by inducing organelle hyperplasia and glycogen accumulation. Norethynodrel competitively inhibits drug-metabolizing enzymes in rat liver microsomes, thereby prolonging Pentobarbital sleep time, while exhibiting multiple effects including reduced body weight gain, attenuated heart rate elevation and ovulation inhibition. In mouse models, Norethynodrel significantly increases the incidence of mammary adenocarcinoma, cervical cancer and pituitary tumors. Norethynodrel can be used for mechanism research on related diseases such as mammary adenocarcinoma, cervical cancer, ovarian tubular adenoma and pituitary adenoma .
|
-
- HY-182407
-
|
|
Angiotensin-converting Enzyme (ACE)
|
Cardiovascular Disease
|
|
CV 5975 is an orally active angiotensin converting enzyme (ACE) competitive inhibitor with a rabbit lung ACE IC50 of 3.1 nM and Ki values of 2.6 nM. CV 5975 inhibits ACE in plasma, aorta, kidney, and brain, intensifying inhibition with repeated administration. CV 5975 inhibits Angiotensin I (HY-P1032)-induced pressor responses and ileum contraction, and augments bradykinin-induced ileum contraction and depressor responses. CV 5975 reduces blood pressure via ACE-independent mechanisms, with sustained action across multiple hypertensive and normotensive animal models, intensified by repeated dosing or Hydrochlorothiazide (HY-B0252) co-administration. CV 5975 can be used for the research of hypertension .
|
-

- HY-W015490S
-
|
|
Isotope-Labeled Compounds
DNA/RNA Synthesis
NF-κB
Monoamine Oxidase
TNF Receptor
Bacterial
|
Infection
Inflammation/Immunology
Cancer
|
|
1,4-Naphthoquinone-d6 is the deuterium labeled 1,4-Naphthoquinone. 1,4-Naphthoquinone is an inhibitor with broad-spectrum inhibitory activity targeting DNA polymerase, NF-κB and monoamine oxidase (MAO-A/B), with antibacterial and anti-biofilm efficacy. 1,4-Naphthoquinone is a competitive inhibitor of MAO-B (Ki=1.4 μM) and a non-competitive inhibitor of MAO-A (Ki=7.7 μM). 1,4-Naphthoquinone inhibits DNA polymerase pol α, β, γ, δ, ε, λ with IC50 ranging from 5.57-128 μM. 1,4-Naphthoquinone inhibits tumor cell proliferation, induces apoptosis and necrosis, and has anti-angiogenic and anti-inflammatory activities by inducing oxidative stress, depleting glutathione (GSH), inhibiting DNA polymerase-mediated DNA synthesis and blocking NF-κB nuclear translocation. 1,4-Naphthoquinone can be used in anti-bacterial , anti-tumor and anti-inflammatory studies, including inhibition of melanoma and colon cancer cell growth and endothelial cell function, as well as LPS-induced inflammation models .
|
-
- HY-176219
-
|
|
Bcl-2 Family
Apoptosis
Necroptosis
|
Cancer
|
|
Bcl-2-IN-23 (compound 5) is a selective inhibitor targeting Bcl-2. The IC50 of Bcl-2-IN-23 in HTB-140, HeLa and SW620 cells is 25.7-33.7 μM. Bcl-2-IN-23 can non-covalently competitively bind to Bcl-2 protein, significantly reduce its expression, and induce late apoptosis and necroptosis of cancer cells. Bcl-2-IN-23 enhances the sensitivity of cancer cells to apoptosis and reduces the release of IL-6 inflammatory factors by disrupting the Bcl-2-mediated mitochondrial apoptosis inhibition pathway. Bcl-2-IN-23 can be used for anti-apoptosis research of malignant tumors such as melanoma, cervical cancer, and colorectal cancer .
|
-

- HY-164090
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-

- HY-N14094
-
|
|
JAK
Apoptosis
Autophagy
|
Cancer
|
|
Tubulosine is an alkaloid. Tubulosine can be isolated from Pogonopus tubulosus (DC.) Schumann. Tubulosine is an ATP-competitive, selective JAK3 inhibitor with an IC50 value of 9.9 nM. Tubulosine also inhibits the kinase activities of other JAK family members, the extent of inhibition is less than that of JAK3, with IC50 values of 69.5, 84.9 and 76.3 nM for JAK1, JAK2 and TYK2, respectively. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. Tubulosine induces apoptotic and necrotic/autophagic cell death. Tubulosine inhibits the process of peptide chain elongation by eukaryotic polysomes by, specifically preventing the elongation-factor-2-dependent step of translocation. Tubulosine exhibits anticancer activity in breast cancer cells .
|
-

- HY-179581
-
|
|
Amino acid Transporter
|
Metabolic Disease
|
|
SLC6A19-IN-4 is an allosteric-competitive and orally active B 0AT1 inhibitor. SLC6A19-IN-4 inhibits both human and mouse B 0AT1 with IC50 values of 513 nM and 295 nM, respectively. SLC6A19-IN-4 exhibits excellent metabolic stability. SLC6A19-IN-4 significantly increases urinary phenylalanine (Phe) excretion and reduces plasma Phe levels through dual inhibition of B 0AT1 in both the intestine (reducing absorption) and kidney (promoting excretion) in vivo. SLC6A19-IN-4 can be used for phenylketonuria (PKU) and other disorders involving SLC6-family transporters research .
|
-

- HY-N9422
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate triethylamine, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate triethylamine can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-W250153
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
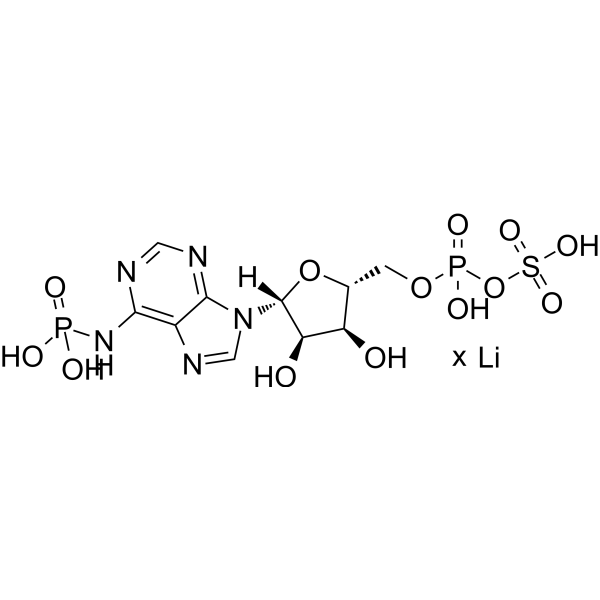
- HY-W250153A
-
|
|
P2Y Receptor
|
Cardiovascular Disease
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
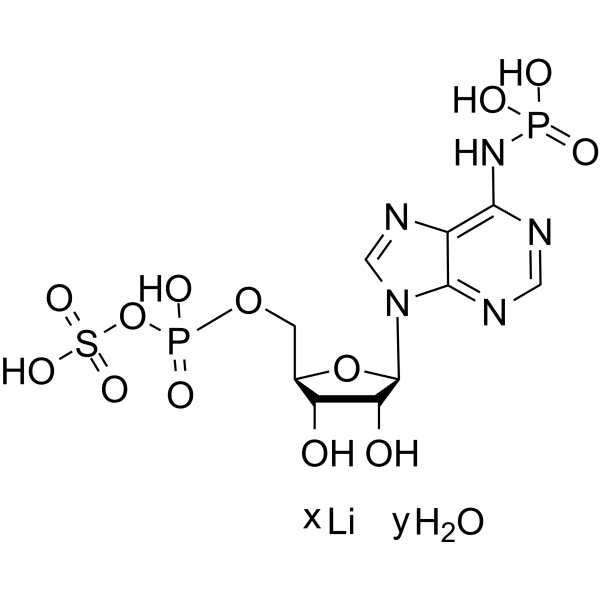
- HY-161449
-
|
|
11β-HSD
|
Metabolic Disease
|
|
JTT-654 is an orally active, potent and selective11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor. The IC50 of JTT-654 for 11β-HSD1 is 4.65, 0.97, and 0.74 nM in human, rat, and mouse recombinant enzymes, respectively. JTT-654 showed competitive inhibition against human recombinant enzyme. The IC50 value for human 11β-HSD2 is > 30 μM (human 11β-HSD2 is responsible for the reverse reaction against human 11β-HSD1). JTT-654 ameliorates insulin resistance and non-obese type 2 diabetes by inhibiting adipose tissue and liver 11β-HSD1 .
|
-

- HY-A0139
-
|
NSC 108165; Navan; Navane
|
Sigma Receptor
mAChR
Histamine Receptor
Dopamine Receptor
Adrenergic Receptor
|
Others
|
|
Thiothixene is a typical antipsychotic. It selectively binds to dopamine D2 over D1, D3, and D4 receptors (Kis=0.417, 338, 186.2, and 363.1 nM, respectively). Thiothixene also binds to various serotonin (5-HT), histamine H1, α1- and α2-adrenergic, muscarinic acetylcholine, and sigma receptors (Kis=15-5,754 nM) as well as the dopamine, norepinephrine, and serotonin transporters (Kis=3.16-30 μM). In vivo, thiothixene reduces spontaneous and amphetamine-induced locomotor activity in rats. It enhances latent inhibition, as measured by a decreased lick latency in response to light and foot shock stimuli, which is a measure of selective attention in rats.3 Thiothixene also increases competitive behavior in submissive mice, indicating antidepressant-like behavior.
|
-

- HY-W748758
-
|
NSC 108165-d8; Navan-d8; Navane-d8
|
Isotope-Labeled Compounds
Dopamine Receptor
Histamine Receptor
mAChR
Adrenergic Receptor
Sigma Receptor
|
Others
|
|
(Z)-Thiothixene-d8 (NSC 108165-d8; Navan-d8; Navane-d8) is the deuterium labeled Thiothixene (HY-A0139). Thiothixene is a typical antipsychotic. It selectively binds to dopamine D2 over D1, D3, and D4 receptors (Kis=0.417, 338, 186.2, and 363.1 nM, respectively). Thiothixene also binds to various serotonin (5-HT), histamine H1, α1- and α2-adrenergic, muscarinic acetylcholine, and sigma receptors (Kis=15-5,754 nM) as well as the dopamine, norepinephrine, and serotonin transporters (Kis=3.16-30 μM). In vivo, thiothixene reduces spontaneous and amphetamine-induced locomotor activity in rats. It enhances latent inhibition, as measured by a decreased lick latency in response to light and foot shock stimuli, which is a measure of selective attention in rats.3 Thiothixene also increases competitive behavior in submissive mice, indicating antidepressant-like behavior.
|
-

| Cat. No. |
Product Name |
Type |
-
- HY-W250153
-
|
|
Biochemical Assay Reagents
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-137697
-
|
|
Biochemical Assay Reagents
|
|
ddCTP is a type of chain-terminating deoxynucleotide. ddCTP can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-
- HY-W250153A
-
|
|
Biochemical Assay Reagents
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P0299
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) can readily crosse the blood-brain barrier .
|
-
- HY-P0299A
-
|
|
TGF-β Receptor
|
Cancer
|
|
LSKL, Inhibitor of Thrombospondin (TSP-1) TFA is a latency-associated protein (LAP)-TGFβ derived tetrapeptide and a competitive TGF-β1 antagonist. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA inhibits the binding of TSP-1 to LAP and alleviates renal interstitial fibrosis and hepatic fibrosis. LSKL, Inhibitor of Thrombospondin (TSP-1) TFA suppresses subarachnoid fibrosis via inhibition of TSP-1-mediated TGF-β1 activity, prevents the development of chronic hydrocephalus and improves long-term neurocognitive defects following subarachnoid hemorrhage (SAH). LSKL, Inhibitor of Thrombospondin (TSP-1) TFA can readily crosse the blood-brain barrier .
|
-
- HY-111173
-
|
|
Dipeptidyl Peptidase
|
Others
|
|
Diprotin B is a dipeptidyl aminopeptidase IV (DPP IV) inhibitor. The apparent competitive inhibition of DPP-IV by the diprotins is a kinetic artifact, derived from the substrate-like nature of tripeptides containing a penultimate proline residue .
|
-
- HY-P10392B
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R TFA, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R TFA inhibits competitively the binding of β-catenin to TCF4. aStAx-35R TFA selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-
- HY-P5272
-
|
|
Bacterial
|
Inflammation/Immunology
|
|
Histatin-3 TFA, a 32 amino acid peptide, possesses powerful antimicrobial properties. Histatin-3 TFA behaves as a substrate for proprotein convertase 1 (PC1), being cleaved by this endoprotease primarily at a site carboxy terminal to the single Arg25 residue (HRGYR decrease SN). Histatin-3 TFA is a moderately potent, reversible and competitive inhibitor of the furin-mediated cleavage of the pentapeptide pGlu-Arg-Thr-Lys-Arg-MCA fluorogenic substrate, with an estimated inhibition constant Ki of 1.98 μM .
|
-
- HY-W142169
-
|
Formyl-L-histidine
|
Aminoacyl-tRNA Synthetase
|
Others
|
|
N-Formyl-L-histidine shows binding affinity to histidyl-tRNA synthetase with a Ki value of 4.6 μM. N-Formyl-L-histidine shows a competitive inhibition against L-histidine ammonia-lyase, inhibits urocanic acid formation from L-histidine with a Ki value of 4.26 mM .
|
-
- HY-P10392
-
|
|
β-catenin
Wnt
|
Cancer
|
|
aStAx-35R, a stapled peptide, antagonizes nuclear form of β-catenin and inhibits Wnt signaling. aStAx-35R inhibits competitively the binding of β-catenin to TCF4. aStAx-35R selectively induces growth inhibition of Wnt-dependent cancer cells .
|
-
- HY-16525
-
|
|
Monoamine Transporter
Adrenergic Receptor
|
Neurological Disease
|
|
XEN-2174 is a noradrenaline transporter (NET) inhibitor. XEN-2174 inhibits the reuptake of noradrenaline through non-competitive inhibition, increasing the concentration of noradrenaline in the synaptic cleft, thereby activating α2-adrenergic receptor at the spinal level and exerting analgesic effects. XEN-2174 exhibits long-lasting analgesic effects in models of neuropathic pain and postoperative pain in rats. XEN-2174 can be used in pain research .
|
-
- HY-P10552
-
|
|
CXCR
|
Inflammation/Immunology
|
|
pCXCL8-1aa is an anti-inflammatory peptide. pCXCL8-1aa competitively inhibits the binding of CXCL8 to glycosaminoglycans such as heparin sulfate (HS) by binding with high affinity. This reduces the presentation of CXCL8 on the surface of vascular endothelial cells, thereby inhibiting neutrophil migration and inflammatory responses. pCXCL8-1aa can be used to study inflammatory diseases such as rheumatoid arthritis .
|
-
- HY-P10346
-
|
Smooth-Muscle Myosin Light-Chain Kinase (796-815)
|
Myosin
|
Cardiovascular Disease
Inflammation/Immunology
|
|
smMLCK peptide is a specific inhibitor of smooth muscle myosin light chain kinase (smMLCK). The smMLCK peptide mimics the substrate and competitively inhibits the binding of the actual substrate to the enzyme, thereby inhibiting the kinase activity. This inhibition prevents the phosphorylation of the myosin light chain, thus inhibiting muscle contraction .
|
-
- HY-P1115
-
|
|
Akt
|
Others
|
|
AKTide-2T is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
-
- HY-P1115A
-
|
|
Akt
|
Others
|
|
AKTide-2T TFA is an excellent in vitro substrate for AKT and shows competitive inhibition of histone H2B phosphorylation with a Ki of 12 nM. AKTide-2T TFA mimics the optimal phosphorylation sequence of Akt and is an inhibitory peptide with the wildtype AKTide lacking Thr in the S22 position .
|
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-N1584
-
-
-
- HY-N1584A
-
-
-
- HY-Y0095
-
-
-
- HY-N0598
-
-
-
- HY-113268
-
-
-
- HY-N2511
-
-
-
- HY-W719041
-
-
-
- HY-N7389A
-
-
-
- HY-130199
-
-
-
- HY-N3394
-
-
-
- HY-B1341
-
|
Enidrel; SC-4642; NSC 15432
|
Structural Classification
Classification of Application Fields
Endogenous metabolite
Disease Research Fields
Endocrinology
Steroids
Source Classification
|
Endogenous Metabolite
Progesterone Receptor
|
|
Norethynodrel (Enidrel; SC-4642) is an orally active progestogen analog that reduces estrogen-like effects and enhances progestogen-like responses in endometrial stromal cells. Norethynodrel also promotes cell maturation and predecidual cell formation by inducing organelle hyperplasia and glycogen accumulation. Norethynodrel competitively inhibits drug-metabolizing enzymes in rat liver microsomes, thereby prolonging Pentobarbital sleep time, while exhibiting multiple effects including reduced body weight gain, attenuated heart rate elevation and ovulation inhibition. In mouse models, Norethynodrel significantly increases the incidence of mammary adenocarcinoma, cervical cancer and pituitary tumors. Norethynodrel can be used for mechanism research on related diseases such as mammary adenocarcinoma, cervical cancer, ovarian tubular adenoma and pituitary adenoma .
|
-
-
- HY-122369
-
-
-
- HY-N7536
-
-
-
- HY-N1584R
-
-
-
- HY-N1584AR
-
-
-
- HY-W661499
-
|
|
Structural Classification
Natural Products
Microorganisms
Source Classification
|
Phosphatase
Apoptosis
Reactive Oxygen Species (ROS)
Caspase
|
|
Orellanine, a nephrotoxic alkaloid found in Cortinarius orellanus, is an orally active and selective non-competitive inhibitor of alkaline phosphatase. Orellanine chelates iron, generates reactive oxygen species (ROS), induces DNA scission, forms ortho-semiquinone radicals, downregulates antioxidant defenses, and inhibits mitochondrial function. Orellanine induces caspase 8/9-mediated apoptosis. Orellanine inhibits synthesis of proteins, RNA, DNA, and mitochondrial protein synthesis, with metabolic activation required for cell-free protein synthesis inhibition. Orellanine can be used for the research of metastatic clear cell renal cell carcinoma, acute renal failure, chronic renal insufficiency, and kidney damage .
|
-
-
- HY-18569AR
-
-
-
- HY-N7389B
-
-
-
- HY-129278
-
|
|
Alkaloids
Lunaria annua L.
Other Alkaloids
Plants
Brassicaceae
Source Classification
|
Parasite
|
|
Lunarine is an alkaloid, which can be isolated from the seeds of kale (Lunaria annua). Lunarine lowers the blood pressure, stimulates small intestinal motility, inhibits spontaneous contractions of uterus and nervous system, and exhibits an acute toxicity with LD50 of 62.3 mg/kg in dogs and rabbits. Lunarine exhibits antiparasitic activity through a competitive inhibition of protozoan oxidoreductase trypanothione reductase (TryR) with Ki of 304 μM .
|
-
-
- HY-N12427
-
-
-
- HY-N0598R
-
-
-
- HY-N3136
-
-
-
- HY-113268R
-
-
-
- HY-N13917
-
|
|
Natural Products
Microorganisms
Source Classification
|
Proteasome
Bacterial
|
|
Argyrin B, a natural product cyclic peptide, is a reversible, non-competitive immunoproteasome inhibitor. Argyrin B shows selective inhibition of the β5i and β1i sites of the immunoproteasome over the β5c and β1c sites of the constitutive proteasome with nearly 20-fold selective inhibition of β1i over the homologous β1c. Argyrin B has antibacterial effects .
|
-
-
- HY-113371R
-
-
-
- HY-N15587
-
|
Gostatine
|
Structural Classification
Microorganisms
Ketones, Aldehydes, Acids
Source Classification
|
Aminotransferases (Transaminases)
|
|
Gostatin is an inhibitor of aspartate aminotransferase (GOT). Gostatin is found in Streptomyces sumanensis nov. sp. NK-23. Gostatin has a strong inhibitory effect on pig heart GOT, a weak inhibitory effect on wheat germ GOT and GPT, and no significant effect on glutamate dehydrogenase and glutamine synthetase. The inhibitory mechanism of gostatin is similar to substrate competitive inhibition, and aspartate has a protective effect on its inhibitory effect. Gostatin can be used to study the catalytic mechanism of GOT and its role in nitrogen metabolism .
|
-
-
- HY-W105272R
-
|
|
Structural Classification
Natural Products
Endogenous metabolite
Source Classification
|
Reference Standards
Endogenous Metabolite
|
|
Halofuginone (Standard) is the analytical standard of Halofuginone. This product is intended for research and analytical applications. Halofuginone (RU-19110), a Febrifugine derivative, is a competitive prolyl-tRNA synthetase inhibitor with a Ki of 18.3 nM . Halofuginone is a specific inhibitor of type-I collagen synthesis and attenuates osteoarthritis (OA) by inhibition of TGF-β activity . Halofuginone is also a potent pulmonary vasodilator by activating Kv channels and blocking voltage-gated, receptor-operated and store-operated Ca2+ channels. Halofuginone has anti-malaria, anti-inflammatory, anti-cancer, anti-fibrosis effects .
|
-
-
- HY-165443
-
-
-
- HY-N14094
-
|
|
Structural Classification
Alkaloids
Other Alkaloids
Rubiaceae
Plants
Pogonopus tubulosus (A.Rich. ex DC.) K.Schum.
Source Classification
|
JAK
Apoptosis
Autophagy
|
|
Tubulosine is an alkaloid. Tubulosine can be isolated from Pogonopus tubulosus (DC.) Schumann. Tubulosine is an ATP-competitive, selective JAK3 inhibitor with an IC50 value of 9.9 nM. Tubulosine also inhibits the kinase activities of other JAK family members, the extent of inhibition is less than that of JAK3, with IC50 values of 69.5, 84.9 and 76.3 nM for JAK1, JAK2 and TYK2, respectively. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. Tubulosine induces apoptotic and necrotic/autophagic cell death. Tubulosine inhibits the process of peptide chain elongation by eukaryotic polysomes by, specifically preventing the elongation-factor-2-dependent step of translocation. Tubulosine exhibits anticancer activity in breast cancer cells .
|
-
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-17422S1
-
|
|
|
Acyclovir-d4 is the deuterium labeled Acyclovir. Acyclovir (Aciclovir) is a guanosine analogue and an orally active antiviral agent. Acyclovir inhibits HSV-1 (IC50 of 0.85 μM), HSV-2 (IC50 of 0.86 μM) and varicella-zoster virus. Acyclovir can be phosphorylated by viral thymidine kinase (TK), and Acyclovir triphosphate interferes with viral DNA polymerization through competitive inhibition with guanosine triphosphate and obligatory chain termination . Acyclovir prevents bacterial infections during induction therapy for acute leukaemia .
|
-
-
- HY-W015490S
-
|
|
|
1,4-Naphthoquinone-d6 is the deuterium labeled 1,4-Naphthoquinone. 1,4-Naphthoquinone is an inhibitor with broad-spectrum inhibitory activity targeting DNA polymerase, NF-κB and monoamine oxidase (MAO-A/B), with antibacterial and anti-biofilm efficacy. 1,4-Naphthoquinone is a competitive inhibitor of MAO-B (Ki=1.4 μM) and a non-competitive inhibitor of MAO-A (Ki=7.7 μM). 1,4-Naphthoquinone inhibits DNA polymerase pol α, β, γ, δ, ε, λ with IC50 ranging from 5.57-128 μM. 1,4-Naphthoquinone inhibits tumor cell proliferation, induces apoptosis and necrosis, and has anti-angiogenic and anti-inflammatory activities by inducing oxidative stress, depleting glutathione (GSH), inhibiting DNA polymerase-mediated DNA synthesis and blocking NF-κB nuclear translocation. 1,4-Naphthoquinone can be used in anti-bacterial , anti-tumor and anti-inflammatory studies, including inhibition of melanoma and colon cancer cell growth and endothelial cell function, as well as LPS-induced inflammation models .
|
-
-
- HY-50878S
-
|
|
|
Crizotinib-d5 is the deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
-
- HY-B0377S
-
|
|
|
Famotidine- 13C,d3 is the 13C- and deuterium labeled Famotidine. Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion.
|
-
-
- HY-W707517
-
|
|
|
Famotidine-d4 (MK-208-d4) is deuterium labeled Famotidine. Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion .
|
-
-
- HY-10195BS
-
|
|
|
Ruboxistaurin-d6 (hydrochloride) is the deuterium labeled Ruboxistaurin hydrochloride. Ruboxistaurin (LY333531) hydrochloride is an orally active, selective PKC beta inhibitor (Ki=2 nM). Ruboxistaurin hydrochloride exhibits ATP dependent competitive inhibition of PKC beta I with an IC50 of 4.7 nM. Ruboxistaurin hydrochloride inhibits PKC beta II with an IC50 of 5.9 nM .
|
-
-
- HY-50878AS
-
|
|
|
Crizotinib-d9 hydrochloride is deuterated labeled Crizotinib hydrochloride (HY-50878A). Crizotinib hydrochloride (PF-02341066 hydrochloride) is an orally bioavailable, selective, and ATP-competitive dual ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib hydrochloride (PF-02341066 hydrochloride) inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. It is also a ROS proto-oncogene 1 (ROS1) inhibitor. Crizotinib hydrochloride (PF-02341066 hydrochloride) has effective tumor growth inhibition .
|
-
-
- HY-17000S
-
|
|
|
Tolvaptan-d7 is the deuterium labeled Tolvaptan. Tolvaptan is a selective, competitive arginine vasopressin receptor 2 antagonist with an IC50 of 1.28μM for the inhibition of AVP-induced platelet aggregation .
|
-
-
- HY-B0377S1
-
|
|
|
Famotidine- 13C (MK-208- 13C) is 13C labeled Famotidine. Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion.
|
-
-
- HY-W777002
-
|
|
|
Famotidine- 13C3 (MK-208- 13C3) is the 13C-labeled Famotidine (HY-B0377). Famotidine (MK-208) is a competitive histamine H2-receptor antagonist. Its main pharmacodynamic effect is the inhibition of gastric secretion.
|
-
-
- HY-B0690S
-
|
|
|
Fosinopril-d5 (SQ28555-d5 (free acid)) is deuterium labeled Fosinopril. Fosinopril (SQ28555 free acid) is the ester proagent of angiotensin-converting enzyme (ACE) inhibitor with an IC50 value of 0.18 μM. Fosinopril demonstrates a non-competitive inhibition effect on ACE activity with an Ki value of 1.675 μM .
|
-
-
- HY-135111S
-
|
|
|
4-Desmethoxy Omeprazole-d3 is the deuterium labeled 4-Desmethoxy Omeprazole. 4-Desmethoxy Omeprazole is the active metabolite of Omeprazole. Omeprazole, a proton pump inhibitor (PPI), is available for treatment of acid-related gastrointestinal disorders. Omeprazole shows competitive inhibition of CYP2C19 activity with a Kiof 2 to 6 μM . Omeprazole also inhibits growth of Gram-positive and Gram-negative bacteria .
|
-
-
- HY-50878S1
-
|
|
|
Crizotinib-d8 (PF-02341066-d8) is deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
-
- HY-50878S2
-
|
|
|
Crizotinib-d9 (PF-02341066-d9) is deuterium labeled Crizotinib. Crizotinib (PF-02341066) is an orally bioavailable, ATP-competitive ALK and c-Met inhibitor with IC50s of 20 and 8 nM, respectively. Crizotinib inhibits tyrosine phosphorylation of NPM-ALK and tyrosine phosphorylation of c-Met with IC50s of 24 and 11 nM in cell-based assays, respectively. Crizotinib is also a ROS1 inhibitor. Crizotinib has effective tumor growth inhibition .
|
-
-
- HY-W748758
-
|
|
|
(Z)-Thiothixene-d8 (NSC 108165-d8; Navan-d8; Navane-d8) is the deuterium labeled Thiothixene (HY-A0139). Thiothixene is a typical antipsychotic. It selectively binds to dopamine D2 over D1, D3, and D4 receptors (Kis=0.417, 338, 186.2, and 363.1 nM, respectively). Thiothixene also binds to various serotonin (5-HT), histamine H1, α1- and α2-adrenergic, muscarinic acetylcholine, and sigma receptors (Kis=15-5,754 nM) as well as the dopamine, norepinephrine, and serotonin transporters (Kis=3.16-30 μM). In vivo, thiothixene reduces spontaneous and amphetamine-induced locomotor activity in rats. It enhances latent inhibition, as measured by a decreased lick latency in response to light and foot shock stimuli, which is a measure of selective attention in rats.3 Thiothixene also increases competitive behavior in submissive mice, indicating antidepressant-like behavior.
|
-
| Cat. No. |
Product Name |
|
Classification |
-
- HY-B1341
-
|
Enidrel; SC-4642; NSC 15432
|
|
Alkynes
|
|
Norethynodrel (Enidrel; SC-4642) is an orally active progestogen analog that reduces estrogen-like effects and enhances progestogen-like responses in endometrial stromal cells. Norethynodrel also promotes cell maturation and predecidual cell formation by inducing organelle hyperplasia and glycogen accumulation. Norethynodrel competitively inhibits drug-metabolizing enzymes in rat liver microsomes, thereby prolonging Pentobarbital sleep time, while exhibiting multiple effects including reduced body weight gain, attenuated heart rate elevation and ovulation inhibition. In mouse models, Norethynodrel significantly increases the incidence of mammary adenocarcinoma, cervical cancer and pituitary tumors. Norethynodrel can be used for mechanism research on related diseases such as mammary adenocarcinoma, cervical cancer, ovarian tubular adenoma and pituitary adenoma .
|
| Cat. No. |
Product Name |
|
Classification |
-
- HY-12854
-
|
GRN163L
|
|
Antisense Oligonucleotides
|
|
Imetelstat (GRN163L) is a 13-mer oligonucleotide and competitive Telomerase inhibitor. Imetelstat binds with high affinity to the template region of the RNA component of human telomerase. Imetelstat induces Apoptosis. Imetelstat is capable of selectively eliminating myelofibrosis hematopoietic stem cells. Imetelstat leads to the loss of a cancer cell's ability to maintain telomere length, resulting in the inhibition of cell proliferation .
|
-
- HY-137697D
-
|
|
|
Nucleotide Analogs
|
|
ddCTP trilithium solution (100 mM) is a chain-terminating dideoxynucleotide. ddCTP trilithium is a type of chain-terminating deoxynucleotide. ddCTP trilithium can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP trilithium can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP trilithium is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-
- HY-W250153
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-164090
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
-
- HY-137697
-
|
|
|
Nucleotide Analogs
|
|
ddCTP is a type of chain-terminating deoxynucleotide. ddCTP can be incorporated into the extension primer chain that lacks the 3'-hydroxyl group, thereby terminating primer extension, viral genome replication, and DNA synthesis. ddCTP can distinguish almost identical RNA through distinguishable extension products in primer extension inhibition experiments. ddCTP is the active metabolite of Zalcitabine (HY-17392), which can competitively inhibit HIV reverse transcriptase, terminate the synthesis of viral DNA chains, and thereby inhibit HIV replication .
|
-
- HY-W250153A
-
|
|
|
Nucleotide Analogs
Adenine Nucleotide
|
|
Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate, an adenine nucleotide derivative, is a selective P2Y1 antagonist with no effect on P2Y2, P2Y4, or P2Y6 receptors. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can competitive inhibit ADP-induced platelet aggregation, as well as the ability of ADP to cause shape change and increases in Ca 2+ in platelets, but had no effect on the inhibition of stimulated adenylate cyclase by ADP. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate is a co-substrate used for the sulfonation of glycans. Adenosine 3'-phosphate 5'-phosphosulfate lithium hydrate can be used for Golgi-resident PAP-specific 3'-phosphatase-coupled sulfotransferase assays, which as donor substrate to transfer a sulfonate group .
|
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent: