Search Result
Results for "
wild-type receptor
" in MedChemExpress (MCE) Product Catalog:
1
Biochemical Assay Reagents
4
Isotope-Labeled Compounds
| Cat. No. |
Product Name |
Target |
Research Areas |
Chemical Structure |
-
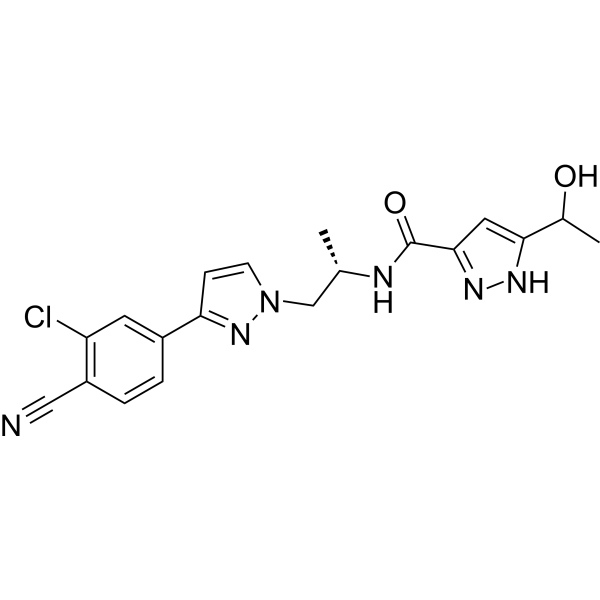
- HY-16985
-
|
ODM-201; BAY-1841788
|
Androgen Receptor
|
Cancer
|
|
Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist. Darolutamide has a Ki of 11 nM for rat wild-type AR (wtAR) and IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-
-
- HY-13223
-
Crenolanib
Maximum Cited Publications
17 Publications Verification
CP-868596
|
FLT3
PDGFR
Autophagy
|
Cancer
|
|
Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-
-
- HY-W587486
-
|
|
Bacterial
Endogenous Metabolite
TGF-β Receptor
|
Metabolic Disease
|
|
N‑acetyltaurine is an orally active endogenous sulfonate that is synthesized from taurine and acetate in the renal cortex. N‑acetyltaurine supports bacterial growth as a sole fixed nitrogen or carbon source. N‑acetyltaurine buffers acetyl moieties of mitochondrial acetyl‑CoA in skeletal muscle. N‑acetyltaurine reduces food intake and body weight in obese and lean wild‑type mice in a GFRAL‑dependent manner. N‑acetyltaurine can be used for the research of diet‑induced obesity, hyperacetatemia and diabetes .
|
-

-
- HY-P99384
-
|
B-701; MFGR-1877S; RG-7444
|
FGFR
|
Cancer
|
|
Vofatamab (B-701) is an anti-FGFR3 monoclonal antibody (mAb). Vofatamab blocks activation of both the wildtype and genetically activated receptor. Vofatamab can be used in the research of metastatic urothelial carcinoma (mUC). Recommend Isotype Controls: Human IgG1 kappa, Isotype Control (HY-P99001) .
|
-

-
- HY-19815
-
|
CK-101; RX518
|
EGFR
|
Cancer
|
|
Olafertinib (CK-101) is an orally available, third generation and irreversible epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). Olafertinib selectively inhibits both EGFR-TKI-sensitizing and resistance mutations with minimal activity on wild-type EGFR. Olafertinib can be used in research for non-small cell lung cancer (NSCLC) with EGFR mutations and other advanced malignancies .
|
-
-
- HY-112256
-
|
|
Androgen Receptor
|
Inflammation/Immunology
|
|
ACP-105 is an orally available, selective amd potent androgen receptor modulator (SARM), with pEC50s of 9.0 and 9.3 for AR wild type and T877A mutant, respectively.
|
-
-
- HY-100131
-
GSK481
1 Publications Verification
|
RIP kinase
|
Inflammation/Immunology
|
|
GSK481 is a highly potent, selective, and specific receptor interacting protein 1 (RIP1) kinase inhibitor with an IC50 of 1.3 nM, which inhibits Ser 166 phosphorylation in wild-type human RIP1 (IC50=2.8 nM). GSK481 also exhibits excellent translation in the U937 cellular assay with an IC50 of 10 nM .
|
-
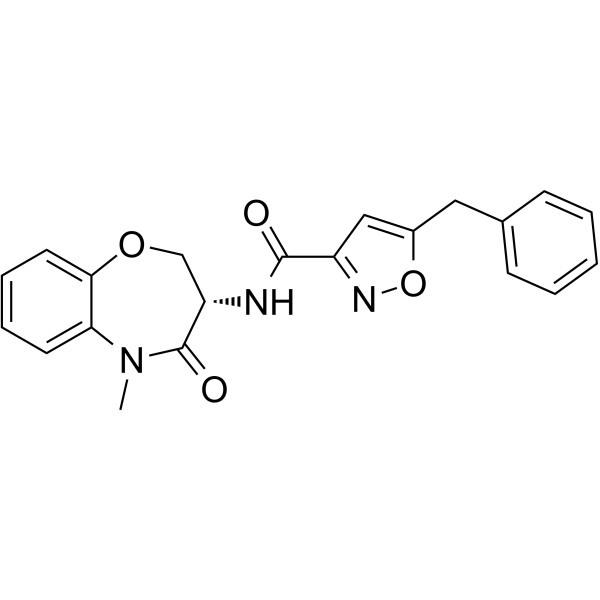
-
- HY-112611
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
H3B-5942 is a selective, irreversible and orally active estrogen receptor covalent antagonist, inactivates both wild-type and mutant ERα by targeting Cys530, with Kis of 1 nM and 0.41 nM, respectively. H3B-5942 reduces ERα target gene GREB1, shows potent antitumor activity both in multiple cell lines or animals bearing ERα WT or ERα mutations .
|
-
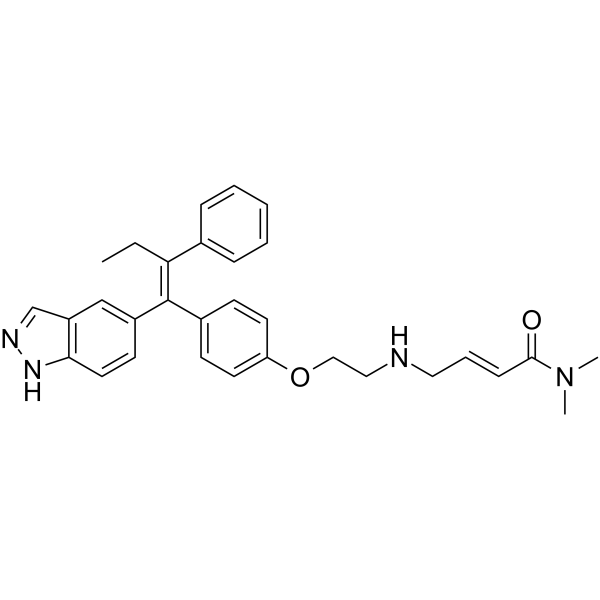
-
- HY-136242
-
|
|
Estrogen Receptor/ERR
|
Endocrinology
Cancer
|
|
UT-34 is a potent, selective, orally bioactive second-generation pan-androgen receptor (AR) antagonist and degrader, with IC50 values of 211.7 nM, 262.4 nM, and 215.7 nM for wild-type AR, F876L-AR, and W741L-AR, respectively. UT-34 binds to the ligand-binding domain (LBD) and functional 1 (AF-1) domain of AR and requires the ubiquitin-proteasome pathway for AR degradation. UT-34 has anti-prostate cancer effects.
|
-
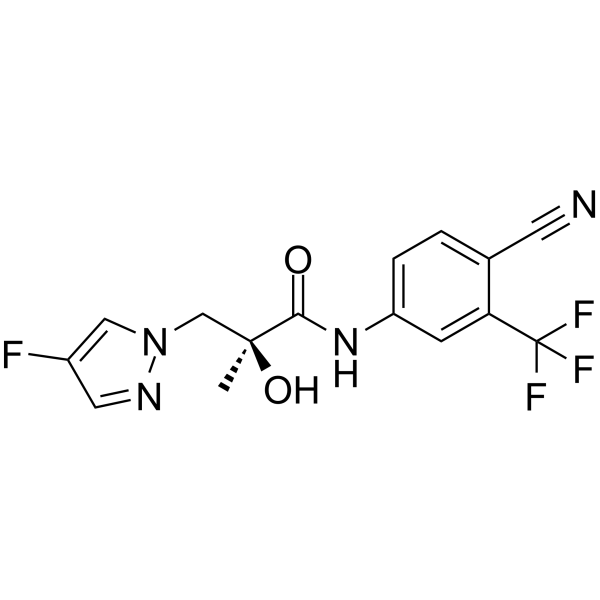
-
- HY-19337
-
|
BAY 1896953; ORM-15341
|
Androgen Receptor
|
Cancer
|
|
Ketodarolutamide (ORM-15341) is a potent, high-affinity nonsteroidal competitive full antagonist of androgen receptor (AR). Ketodarolutamide displays a Ki value of 8 nM for rat wild-type AR and an IC50 value of 38 nM in AR-HEK293 cells . Ketodarolutamide inhibits testosterone-induced nuclear translocation of the AR and antagonizes both overexpressed and mutant ARs . Ketodarolutamide specifically suppresses the proliferation of AR-dependent prostate cancer cells and exhibits antitumor activity in models of castration-resistant prostate cancer (CRPC) . Ketodarolutamide is suitable for the mechanistic and therapeutic research of prostate cancer .
|
-
-
- HY-W010162
-
|
L-Alanyl-L-alanine; Ala-Ala
|
Amino Acid Derivatives
|
Others
|
H-Ala-Ala-OH is a substrate of human intestinal oligopeptide transporter (PEPT1/SLC15A1), with an IC50 of 0.25 mM and an EC50 of 0.08 mM against human PEPT1. When used as a dipeptide linker in antibody-drug conjugates (ADCs) loaded with glucocorticoid receptor regulator payloads, H-Ala-Ala-OH enables efficient intracellular payload release .
|
-

-
- HY-160142
-
|
|
PROTACs
Btk
Syk
MEK
ERK
|
Cancer
|
|
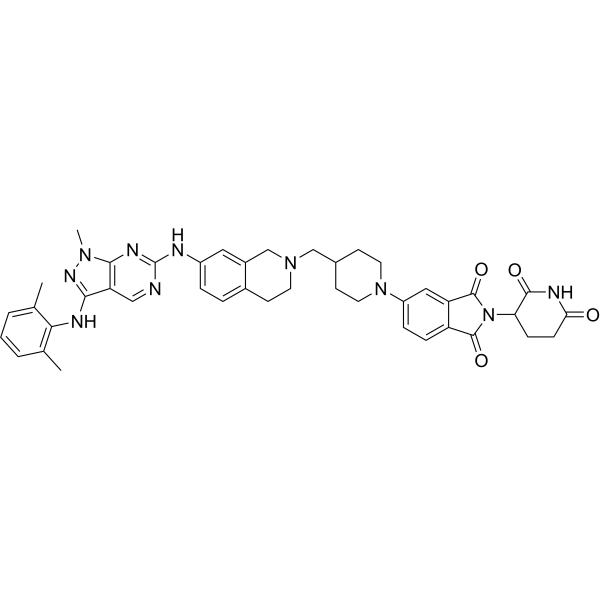
UBX-382 is an orally active BTK PROTAC degrader with a DC50 of 4.56 nM. UBX-382 inhibits B-cell receptor signaling by targeting BTK. UBX-382 shows superior degradation activity for wild-type and mutant BTK proteins. UBX-382 inhibits tumor growth in murine xenograft models harboring wild-type or C481S mutant BTK-expressing TMD-8 cells. UBX-382 can be used for the study of B-cell-related blood cancers .
|
-
-
- HY-N0475
-
|
Hypolide; (+)-Triptophenolide
|
Androgen Receptor
Pyroptosis
Caspase
Bcl-2 Family
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
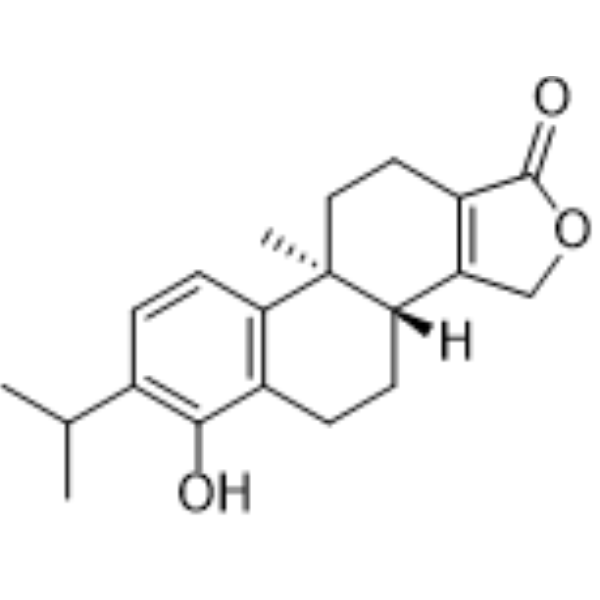
Triptophenolide (Hypolide) is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
|
-
-
- HY-115282A
-
|
TRC-253
|
Androgen Receptor
|
Cancer
|
|
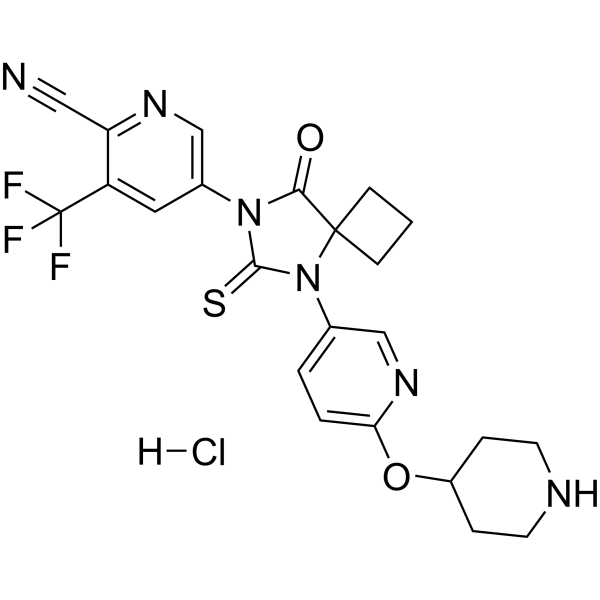
JNJ-63576253 (TRC-253) is a potent and orally active full antagonist of androgen receptor (AR), with IC50s of 37 and 54 nM for F877L mutant AR and wild-type AR in LNCaP cells. JNJ-63576253 can be used for the research of castration-resistant prostate cancer (CRPC) .
|
-
-
- HY-103320A
-
|
|
CaSR
|
Metabolic Disease
|
|
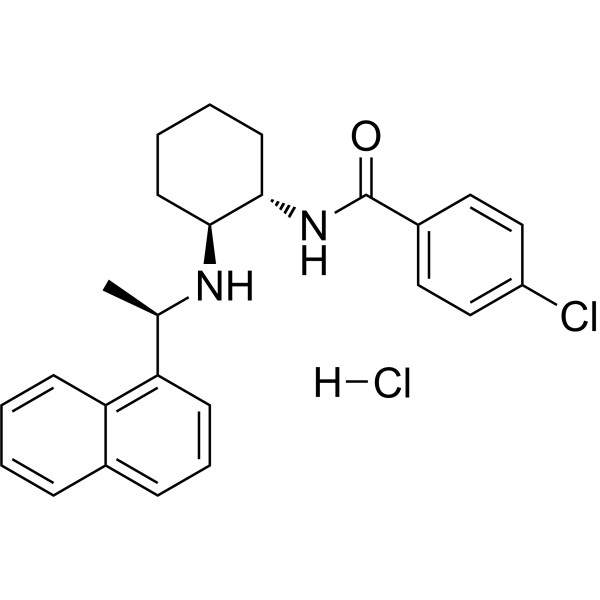
Calhex 231 hydrochloride is a potent negative allosteric modulator that blocks (IC50 = 0.39 μM) increases in [ 3H]inositol phosphates elicited by activating the human wild-type CaSR transiently Ca 2+-sensing receptor. Calhex 231 hydrochloride can be used in the study of traumatic hemorrhagic shock (THS) and diabetic cardiomyopathy (DCM) .
|
-
-
- HY-117764
-
|
|
mGluR
|
Neurological Disease
|
|
LSP4-2022 is a potent and brain-penetrant mGlu4-selective orthosteric agonist, with an EC50 of 0.11 μM. LSP4-2022 inhibits neurotransmission in cerebellar slices from wild-type but not mGlu4 receptor-knockout mice. LSP4-2022 shows pro-depressant activity .
|
-
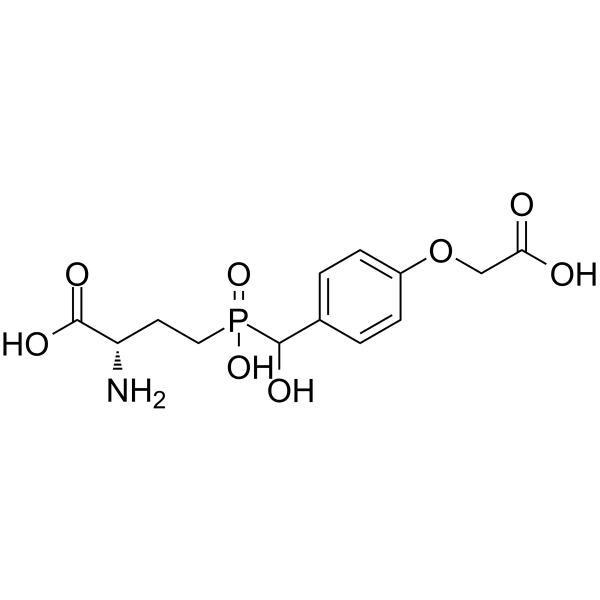
-
- HY-110094
-
BIMU 8
1 Publications Verification
|
5-HT Receptor
|
Neurological Disease
|
|
BIMU 8 is a potent and selective 5-HT4 agonist with EC50s of 18 nM, 77 nM, and 540 nM for wild type 5HT4 receptor, T3.36A, and W6.48A mutant 5-HT4 receptors .
|
-
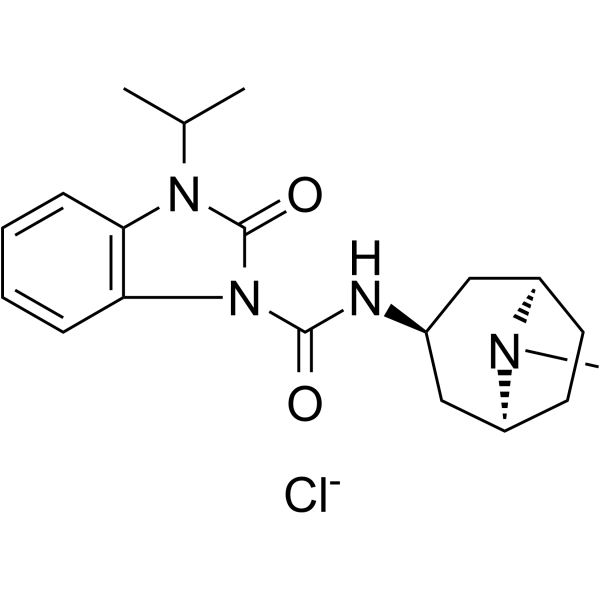
-
- HY-122578
-
|
|
MDM-2/p53
|
Cancer
|
|
P53R3 is a potent p53 reactivator and restores sequence-specific DNA binding of p53 hot spot mutants, including p53 R175H, p53 R248W and p53 R273H. P53R3 induces p53-dependent antiproliferative effects with much higher specificity than PRIMA-1. P53R3 enhances the recruitment of wild-type p53 and p53 M237I to several target gene promoters. P53R3 strongly enhances the mRNA, total protein and cell surface expression of the death receptor death receptor 5 (DR5). P53R3 is used for cancer research .
|
-
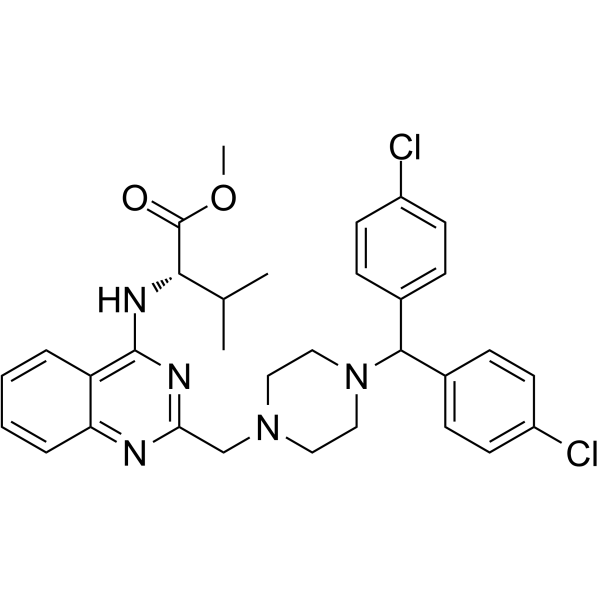
-
- HY-123450
-
|
|
Bcr-Abl
Apoptosis
PDGFR
|
Cancer
|
|
S116836, a potent, orally active BCR-ABL tyrosine kinase inhibitor, blocks both wild-type as well as T315I Bcr-Abl. S116836 arrests the cells in the G0/G1 phase of cell cycle, induces apoptosis, increases ROS production, and decreases GSH production in BaF3/WT and BaF3/T315I cells. S116836 also inhibits SRC, LYN, HCK, LCK and BLK, and receptor tyrosine kinases such as FLT3, TIE2, KIT, PDGFR-β. Antitumor activies . S116836 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
|
-
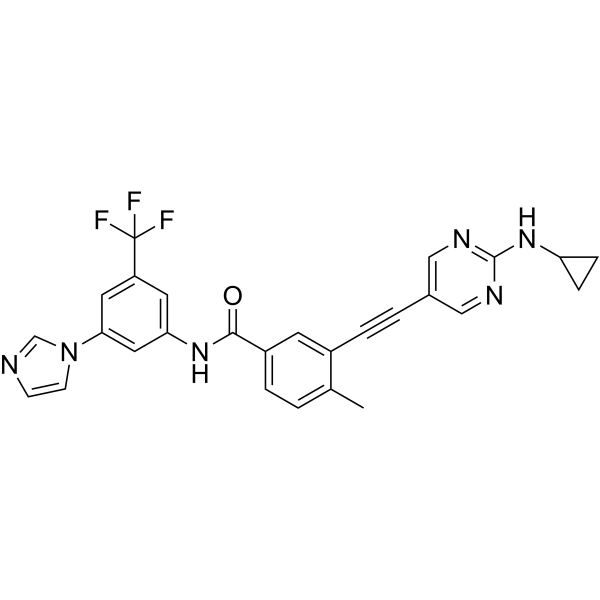
-
- HY-162735
-
|
|
GPR65
|
Inflammation/Immunology
|
|
BRD5080 is a GPR65 agonist. BRD5080 activates GPR65 to enhance the cAMP signaling pathway, reduce the expression of inflammatory cytokines and chemokines in dendritic cells, enhance the activity of human wild-type, mouse wild-type, and human GPR65 I231L variant receptors in a pH-dependent manner, and mediate the recruitment of Gαs protein. BRD5080 can be used in the research of inflammatory bowel disease .
|
-

-
- HY-121392
-
|
|
5-HT Receptor
|
Cardiovascular Disease
Neurological Disease
|
|
GR 125743 is a selective 5-HT1B/1D receptor antagonist, with pKis of 8.85 and 8.31 for wild-type h5-HT1B and wild-type h5-HT1D, respectively. GR 125743 is used for the research of Parkinson's disease and cardiovascular diseases .
|
-
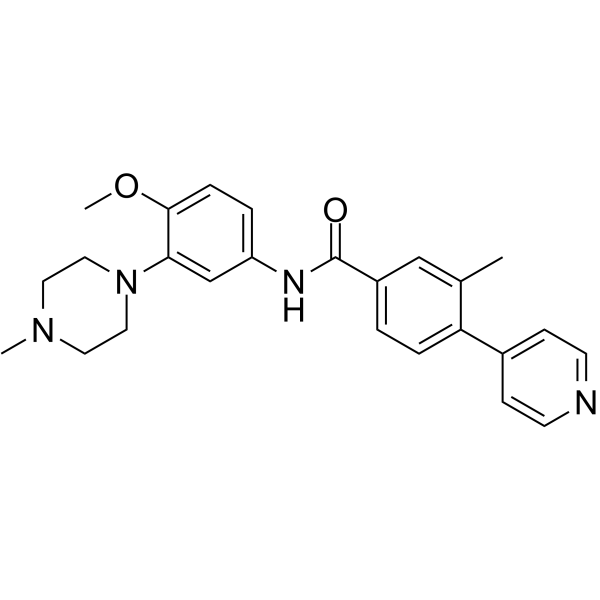
-
- HY-13223A
-
|
CP 868596-26
|
Autophagy
PDGFR
FLT3
|
Cancer
|
|
Crenolanib benzenesulfonate is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

-
- HY-159613
-
|
|
Apoptosis
Estrogen Receptor/ERR
|
Inflammation/Immunology
Cancer
|
|
PELP1-IN-1 is a PELP1 inhibitor and an apoptosis inducer with no cytotoxic activity against non-cancer cell lines. PELP1-IN-1 targets wild-type, mutant and drug-resistant ER + breast cancer, and promotes PELP1 degradation through the proteasome pathway. As an analog of SMIP34 (HY-169903), PELP1-IN-1 is applicable to the research of estrogen receptor α-positive breast cancer .
|
-

-
- HY-16985S
-
|
ODM-201-d4; BAY-1841788-d4
|
Isotope-Labeled Compounds
Androgen Receptor
|
Cancer
|
|
Darolutamide-d4 (ODM-201-d4) is deuterium labeled Darolutamide (HY-16985). Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-

-
- HY-19985A
-
|
|
EGFR
|
Cancer
|
|
(3S, 4S)-PF-06459988 is the S enantiomer of PF-06459988 with less active. PF-06459988 is a potent irreversible inhibitor of T790M mutant epidermal growth factor receptor (EGFR). PF-06459988 has excellent selectivity against EGFR wild-type while possessing a minimally reactive electrophile that reduces the propensity of off-target labeling .
|
-
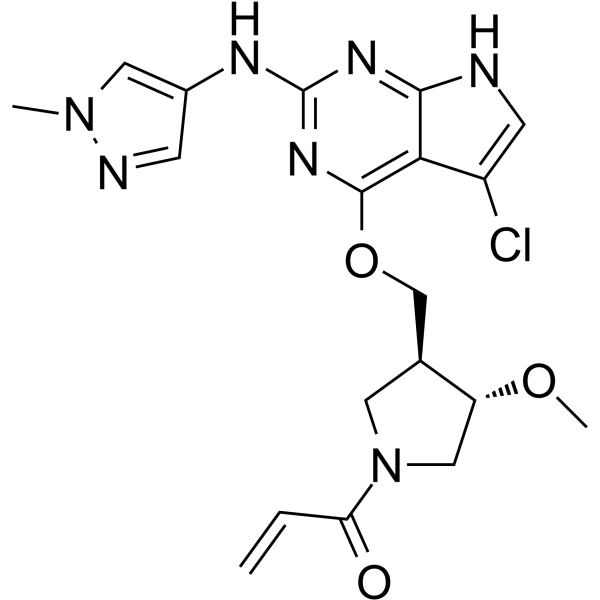
-
- HY-175652
-
|
|
PROTACs
Adenosine Receptor
|
Cancer
|
|
AZD9750 is an orally active, Cereblon (CRBN)-recruiting, androgen receptor (AR)-targeted PROTAC degrader. AZD9750 induces the proteasome-dependent degradation of both wild-type AR and the drug-resistant mutant AR L702H, thereby inhibiting the AR signaling pathway. AZD9750 suppresses tumor growth in mouse prostate cancer xenograft models and has been utilized in prostate cancer research .
|
-

-
- HY-136881
-
|
|
CCR
|
Inflammation/Immunology
|
|
CH0076989 is a specific CCR3 agonist. CH0076989 activates eosinophils and transfectants expressing both wild-type CCR3 and a CCR1:CCR3 chimaeric receptor lacking the CCR3 amino-terminus. CH0076989 has a direct interaction with the transmembrane helices of CCR3, supported by the complete loss of its activity due to mutations of the residues Y41, Y113 and E287. CH0076989 can be used for the study of inflammation and allergic diseases (such as asthma) .
|
-

-
- HY-161633
-
|
|
PROTACs
EGFR
FAK
|
Cancer
|
|
PROTAC EGFR degrader 11 (Compound B71) is a PROTAC degrader for epidermal growth factor receptor (EGFR), with DC50 <100 nM. PROTAC EGFR degrader 11 binds CRBN-DDB1 with a Ki of 36 nM. PROTAC EGFR degrader 11 degrades EGFR, focal adhesion kinase (FAK), and RSK1, inhibits the proliferation of BaF3 wild type and EGFR mutants, with IC50 <100 nM.
|
-

-
- HY-18006
-
NKP608
1 Publications Verification
|
Neurokinin Receptor
Wnt
Bcl-2 Family
β-catenin
Cyclin G-associated Kinase (GAK)
VEGFR
Caspase
Cadherin
Apoptosis
|
Neurological Disease
Metabolic Disease
Inflammation/Immunology
Cancer
|
|
NKP608 is a non-peptidic derivative of 4-aminopiperidine, a highly selective, orally active, neurokinin-1 (NK1) receptor antagonist with IC50 of 2.6 nM. NKP608 is active both in vitro and in vivo, showing extremely low affinity for NK2, NK3 receptors. NKP608 exerts its effects by blocking the NK₁ receptor, regulate cell proliferation and apoptosis, affect neurotransmitter functions and gastric mucosal repair mechanisms, and suppress the Wnt/β-catenin pathway in antitumor research. NKP608 is applicable to research related to various diseases, including cough, anxiety disorders, depression, gastric mucosal injury, and colorectal cancer .
|
-
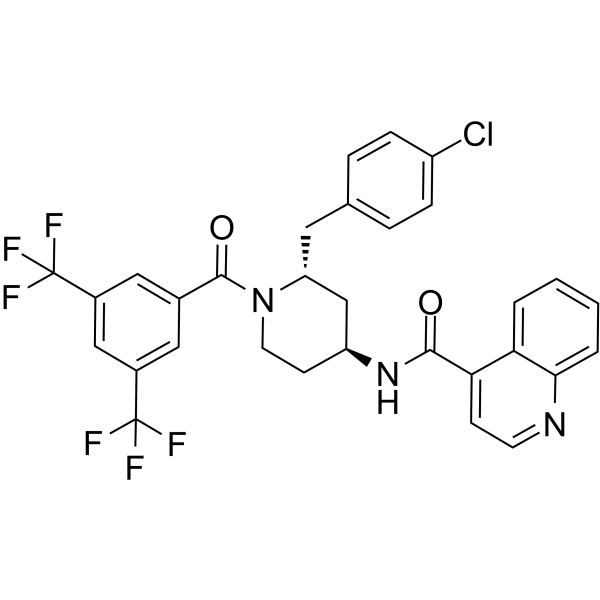
-
- HY-16985R
-
|
ODM-201 (Standard); BAY-1841788 (Standard)
|
Reference Standards
Androgen Receptor
|
Cancer
|
|
Darolutamide (Standard) is the analytical standard of Darolutamide (HY-16985). This product is intended for research and analytical applications. Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-

-
- HY-W050122
-
|
|
GABA Receptor
|
Neurological Disease
|
|
S-(+)-GABOB is an endogenous ligand with antiepileptic activity. S-(+)-GABOB is a metabolite of GABA and may function as a neurotransmitter. S-(+)-GABOB behaves as a full agonist when bound to the ρ(1) wild-type receptor. S-(+)-GABOB acts as a competitive antagonist in the ρ(1) T244S mutant receptor .
|
-

-
- HY-13223R
-
|
CP-868596 (Standard)
|
FLT3
PDGFR
Autophagy
Reference Standards
|
Cancer
|
|
Crenolanib (Standard) is the analytical standard of Crenolanib. This product is intended for research and analytical applications. Crenolanib is a potent and selective inhibitor of wild-type and mutant isoforms of the class III receptor tyrosine kinases FLT3 and PDGFRα/β with Kds of 0.74 nM and 2.1 nM/3.2 nM, respectively.
|
-

-
- HY-P991221
-
|
|
Melanocortin Receptor
|
Metabolic Disease
|
|
ALD1613 is a potent neutralizing monoclonal antibody against adrenocorticotropic hormone (ACTH). ALD1613 neutralizes ACTH-induced signaling and inhibits ACTH-induced cyclic AMP accumulation in a mouse adrenal cell line (Y1) via all five melanocortin receptors. ALD1613 significantly reduces plasma corticosterone levels in wild-type rats. ALD1613 can be used in the study of diseases associated with elevated ACTH levels .
|
-

-
- HY-124089
-
|
|
Cannabinoid Receptor
|
Metabolic Disease
|
|
Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-

-
- HY-114636
-
|
|
Insecticide
|
Others
|
|
RG-102240 is a ligand for insect ecdysone receptor (EcR), with IC50 of 85 and 13 nM, in wildtype G:CfE(DEF) and its A110P mutant. RG-102240 induces the reporter gene activity of wild-type CfEcR and A110P mutant .
|
-

-
- HY-118806A
-
|
|
mAChR
|
Neurological Disease
|
|
AC-42 hydrochloride is the hydrochloride salt form of AC-42 (HY-118806). AC-42 hydrochloride is an allosteric agonist for muscarinic M1 receptor with EC50s of 805 nM and 220 nM for human wild-type and Y381A mutated M1 receptors, respectively. AC-42 hydrochloride stimulates the inositol phosphate (IP)-accumulation and calcium mobilization in CHO cells .
|
-

-
- HY-126720
-
|
|
Endogenous Metabolite
|
Metabolic Disease
|
|
N-Lignoceroyl Taurine is an arachidonoyl amino acid and taurine conjugate with a fatty acid that can be isolated from bovine brain. N-Lignoceroyl Taurine is one of several novel taurine-conjugated fatty acids discovered during mass spectrometry lipidomic analysis of the brain and spinal cord of wild-type and fatty acid amide hydrolase (FAAH) knockout mice. N-Lignoceroyl Taurine levels were 23-26-fold higher in FAAH -/- mice compared to wild-type mice, suggesting that FAAH utilizes N-Lignoceroyl Taurine as a substrate. However, in vitro experiments with purified FAAH showed that N-Lignoceroyl Taurine was hydrolyzed 2,000-fold slower in FAAH compared to oleoylethanolamide. N-Acyl Taurines with polyunsaturated acyl chains can activate members of the transient receptor potential (TRP) calcium channel family, including TRPV1 and TRPV4.
|
-

-
- HY-101522
-
|
|
EGFR
BMX Kinase
Btk
MEK
|
Cancer
|
|
CHMFL-EGFR-202 is a potent, irreversible inhibitor of epidermal growth factor receptor (EGFR) mutant kinase, with IC50s of 5.3 nM and 8.3 nM for drug-resistant mutant EGFR T790M and WT EGFR kinases, respectively. CHMFL-EGFR-202 exhibits ~10-fold selectivity for EGFR L858R/T790M against the EGFR wild-type in cells. CHMFL-EGFR-202 adopts a covalent “DFG-in-C-helix-out” inactive binding conformation with EGFR, with strong antiproliferative effects against EGFR mutant-driven nonsmall-cell lung cancer (NSCLC) cell lines .
|
-
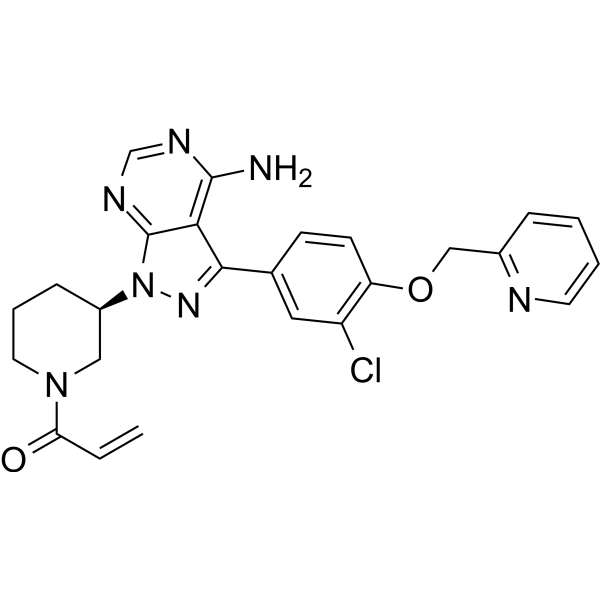
-
- HY-105506
-
|
|
Angiotensin Receptor
|
Cardiovascular Disease
|
|
EXP-7711 is a non peptide angiotensin II (Ang II) AT1 receptor antagonist. EXP-7711’s affinity for wild-type AT1 receptor (Ki =180 nM) is lower than peptide antagonists, but higher than losartan (Ki = 12 nM). EXP-7711 can be used for research on cardiovascular conditions .
|
-

-
- HY-103320
-
|
|
CaSR
|
Metabolic Disease
|
|
Calhex 231 is a potent negative allosteric modulator that blocks (IC50 = 0.39 μM) increases in [ 3H]inositol phosphates elicited by activating the human wild-type CaSR transiently Ca 2+-sensing receptor. Calhex 231 can be used in the study of traumatic hemorrhagic shock (THS) and diabetic cardiomyopathy (DCM) .
|
-
-
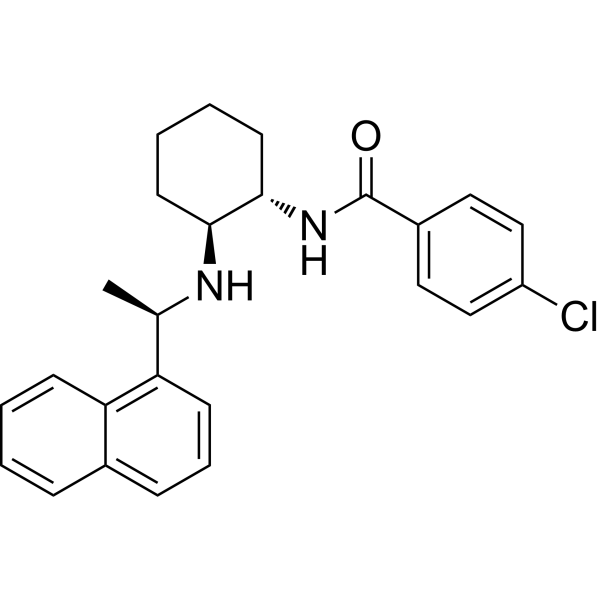
- HY-P991641
-
|
LY3012218
|
FLT3
p38 MAPK
STAT
PI3K
Akt
|
Cancer
|
|
IMC-EB10 (LY3012218) is an anti-FLT3 monoclonal antibody. IMC-EB10 binds to FLT3 with high affinity (Kd = 158 pM) and blocks the binding of FLT3 ligand to FLT3 (IC50 ≈ 10 nM), thereby inhibiting MAPK, STAT5, and PI3K/Akt signaling in leukemia cells. IMC-EB10 can enhance the anti-leukemic effect of Methotrexate (HY-14519) and inhibit leukemias expressing wild-type or ITD-mutated FLT3 receptors. IMC-EB10 prolongs the survival of acute lymphoblastic leukemia (ALL) cells and primary leukemia samples and reduces engraftment in non-obese diabetic/severe combined immunodeficiency patients. IMC-EB10 is indicated for leukemia research .
|
-

-
- HY-161632
-
|
|
PROTACs
EGFR
FAK
|
Cancer
|
|
PROTAC EGFR degrader 10 (Compound B56) is a PROTAC degrader for epidermal growth factor receptor (EGFR), with DC50 <100 nM. PROTAC EGFR degrader 10 binds CRBN-DDB1 with a Ki of 37 nM. PROTAC EGFR degrader 10 degrades EGFR, focal adhesion kinase (FAK), and RSK1, inhibits the proliferation of BaF3 wild type and EGFR mutants, with IC50 <150 nM .
|
-

-
- HY-171906A
-
|
1-Arachidonoyl-sn-glycerol 3-phosphate sodium; 1-Arachidonoyl LPA sodium; LPA (20:4) sodium
|
LPL Receptor
|
Cancer
|
|
1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) (1-Arachidonoyl-sn-glycerol 3-phosphate (sodium)) is a phospholipid with arachifonic acid at the sn-1 position. 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) binds to the LPA2/EDG4 receptor (EC50 of 10 nM). 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) can be found in rat brain as 37% of the arachidoinic acid-containing lysophosphatidic acid species. 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) is the precursor to 1-arachidonoyl glycerol. 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) can prevent TNF-α and IL-6 secretion in wild type LPS-stimulated dendritic cells. 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA (sodium) reduces differentiation of HT-29 colon carcinoma cells into goblet cells when sodium butyrate is present .
|
-

-
- HY-118806
-
|
|
mAChR
|
Neurological Disease
|
|
AC-42 is a poent M1 muscarinic selective allosteric agonist with EC50s of 805 nM and 220 nM for human wild-type and Y381A mutated M1 receptors, respectively. AC-42 stimulates the IP-accumulation and calcium mobilization in CHO cells .
|
-
-
- HY-138842
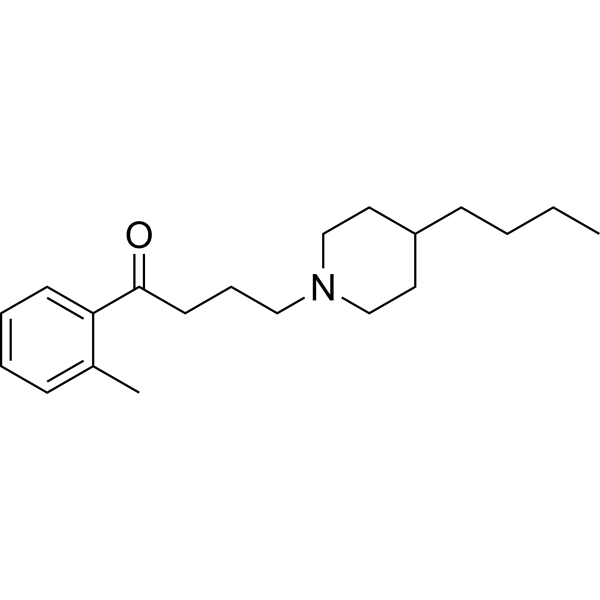
-
|
|
Insulin Receptor
Akt
ERK
|
Metabolic Disease
|
|
DDN is a selective insulin receptor (Insulin Receptor) activator, an insulin sensitizer, and a glucose-lowering insulin mimetic with oral bioavailability. DDN can directly bind to the receptor kinase domain and induce Akt and ERK phosphorylation, and it can also enhance insulin's effect on glucose uptake. DDN significantly reduces blood glucose levels in wild-type and diabetic ob/ob and db/db mice .
|
-

-
- HY-115282
-
|
TRC-253 free base
|
Androgen Receptor
|
Cancer
|
|
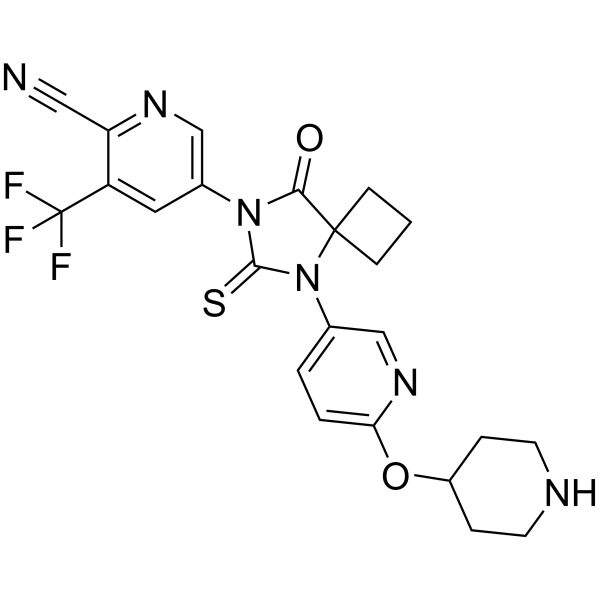
JNJ-63576253 (TRC-253) free base is a potent and orally active full antagonist of androgen receptor (AR), with IC50s of 37 and 54 nM for F877L mutant AR and wild-type AR in LNCaP cells. JNJ-63576253 free base can be used for the research of castration-resistant prostate cancer (CRPC) .
|
-
-
- HY-15041
-
|
|
Bradykinin Receptor
|
Inflammation/Immunology
|
|
NVP-SAA164 is an orally active nonpeptide kinin B1 receptor antagonist. NVP-SAA164 reverses CFA (Complete Freund's adjuvant) (HY-153808)-induced mechanical hyperalgesia and desArg10KD-induced hyperalgesia in hB1-KI mice, and is inactive in a model of inflammatory pain in wild-type mice .
|
-

-
- HY-145866
-
|
|
Drug Intermediate
|
Cancer
|
|
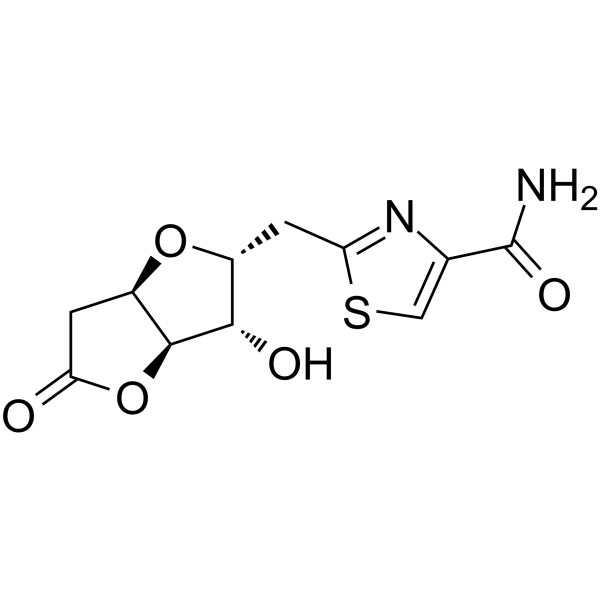
Antiproliferative agent-3 is an antiproliferative agent. Antiproliferative agent-3 exhibits anticancer activity against a variety of cancers, including estrogen receptor-positive breast cancer. Antiproliferative agent-3 shows no toxicity to wild-type zebrafish embryos. Antiproliferative agent-3 can be used in the research of chronic myeloid leukemia, promyelocytic leukemia, Burkitt's lymphoma, estrogen receptor-positive mammary adenocarcinoma, cervical cancer, and lung cancer .
|
-
-
- HY-114253
-
|
|
EGFR
|
Cancer
|
|
ODS-2004436 is a small molecule radiotracer that uses positron emission tomography (PET) imaging to measure the activity of the epidermal growth factor receptor (EGFR) in tumors. ODS-2004436 labeled with the radioactive isotope fluorine-18 shows significantly higher uptake in the xenograft models of lung cancer with EGFR mutations compared to the wild-type models, and could be used to distinguish the mutation status. ODS-2004436 can be used to identify EGFR-positive tumors and predict their response to certain reagent treatments, especially for non-small cell lung cancer (NSCLC) .
|
-

-
- HY-P11092
-
|
|
Endogenous Metabolite
|
Neurological Disease
|
|
TLQP-62 (mouse,rat) is a secreted C-terminal peptide that can be derived from protein VGF. TLQP-62 activates the BDNF-TrkB signaling pathway, inducing acute, transient phosphorylation of TrkB receptor and downstream CREB (Ser133) phosphorylation. TLQP-62 demonstrates excellent efficacy in promoting long-term fear memory formationin wild-type mice and reversing memory impairment in VGF heterozygous knock-out mice. TLQP-62 can be used for the study of memory-related neurological disorders (e.g., Alzheimer’s disease, frontotemporal dementia) .
|
-

- HY-403733A
-
|
|
Androgen Receptor
|
Cancer
|
|
(-)-JJ-450 is a non-competitive antagonist targeting the androgen receptor (AR). (-)-JJ-450 is more potent than (+)-JJ-450 (HY-403733B) in inhibiting androgen-induced AR activity, and the mechanism of AR inhibition by (+)-JJ-450 is different from that of Enzalutamide (MDV3100) (HY-70003), which may target the ligand binding domain (LBD) of AR. (-)-JJ-450 inhibits the transcriptional activity of wild-type AR and mutant AR F876L by inhibiting AR nuclear translocation and promoting nuclear degradation of unbound AR. (-)-JJ-450 can be used in the study of castration-resistant prostate cancer (CRPC) resistant to Enzalutamide .
|
-

- HY-171906
-
|
1-Arachidonoyl-sn-glycerol 3-phosphate; 1-Arachidonoyl LPA; LPA (20:4)
|
LPL Receptor
|
Inflammation/Immunology
|
|
1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA is a phospholipid containing arachidonic acid at the sn-1 position. It is a precursor to 1-arachidonoyl glycerol (1-Monoarachidin; HY-130567) . 1-Arachidonoyl-2-hydroxy-sn-glycero-3-PA binds to the LPA2/EDG4 receptor with an EC50 value of approximately 10 nM . It prevents TNF-α and IL-6 secretion in wild-type but not Lpa2-/- dendritic cells stimulated by LPS . It also decreases differentiation of HT-29 human colon carcinoma cells to goblet cells in the presence of sodium butyrate .
|
-

- HY-403733C
-
|
|
Androgen Receptor
|
Cancer
|
|
JJ-450 is a non-competitive antagonist androgen receptor (AR) that inhibits the transcriptional activity of wild-type AR and mutant AR F876L. JJ-450 has an IC50 of approximately 1-10 μM in inhibiting AR transcriptional activity in PC3 cells. It is selective for AR binding and does not compete with androgens for binding to the ligand binding domain (LBD) of AR. JJ-450 inhibits the transcriptional activity of AR and its splice variants (such as AR F876L) by inhibiting AR nuclear translocation and promoting the degradation of unliganded AR in the nucleus. JJ-450 can be used in castration-resistant prostate cancer (CRPC) studies that are resistant to Enzalutamide (MDV3100) (HY-70003) .
|
-

- HY-N0475R
-
|
Hypolide (Standard); (+)-Triptophenolide (Standard)
|
Reference Standards
Androgen Receptor
Pyroptosis
Caspase
Bcl-2 Family
Apoptosis
|
Inflammation/Immunology
Cancer
|
|
Triptophenolide (Standard) (Hypolide) is the analytical standard of Triptophenolide (HY-N0475). This product is intended for research and analytical applications. Triptophenolide is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
|
-

- HY-P11075
-
|
|
Neurokinin Receptor
|
Metabolic Disease
|
|
[Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) (ID 305) is a selective NK2R agonist with an EC50 of 3.7 nM for hNK2R. [Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) significantly inhibits weight loss in diet-induced obese (DIO) mice model and improves Loperamide (HY-156131)-induced dysfunctional voiding in wild-type mice. [Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) can be used for neurokinin receptor mediated disorders, such as obesity, insulin resistance and type 2 diabetes research .
|
-
![[Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10)](//file.medchemexpress.com/product_pic/hy-p11075.gif)
- HY-124089S
-
|
|
Isotope-Labeled Compounds
Cannabinoid Receptor
|
Metabolic Disease
|
|
Eicosapentaenoyl ethanolamide-d4 is the deuterium labeled Eicosapentaenoyl ethanolamide. Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-

- HY-168384
-
|
|
STING
TNF Receptor
Interleukin Related
CD28
|
Inflammation/Immunology
|
|
M04 is an agonist of STING. It induces the expression of the IFN reporter gene in HEK293T cells expressing wild-type human STING, but does not induce this expression in HEK293T cells expressing the R71H-G230A-R293Q (HAQ) STING variant or in mouse RAW 264.7 cells, indicating that its activity is dependent on allelic and species variations. M04 induces the production of TNF-α, IL-10, IL-1β, and IL-12p70 in human peripheral blood mononuclear cells (PBMCs). At a concentration of 50 µM, M04 stimulates dendritic cells isolated from PBMCs to express the MHC class II cell surface receptor HLA-DR and co-stimulatory molecules CD40, CD80, and CD86, and also enhances their ability to activate T cells in an ex vivo assay. M04 can be used in research on inflammatory immune diseases .
|
-

- HY-170438
-
|
|
EGFR
|
Cancer
|
|
EGFR-IN-139 (compound PD 18) is an epidermal growth factor receptor (EGFR) inhibitor, with IC50s of 12.88 (wild type), 10.84 (L858R/T790M), 42.68 (L858R/T790M/C797S) nM, respectively. EGFR-IN-139 displays strong anticancer activity against A549 and H1975 cancer cell lines, which are highly expressed EGFR. EGFR-IN-139 has a strong selectivity to cancer cells. EGFR-IN-139 can be used for nonsmall cell lung cancer (NSCLC) research[1].
|
-

- HY-125094
-
|
|
Angiotensin Receptor
|
Cardiovascular Disease
|
|
L-162782 is a high affinity AT1 receptor ligand for rat and human wild-type AT1 with IC50 values of 28.5 and 24.6 nM, respectively. L-162782 acts as a partial agonist (EC50 ≈ 30 nM) and insurmountable antagonist (IC50 = 6.5 μM) on wild-type rat AT1 receptors in COS-7 cells. L-162782 reduces Angiotensin II (HY-13948)-induced phosphatidylinositol turnover. L-162782 can be used for hypertension research .
|
-

- HY-W699451
-
|
|
Sodium Channel
|
Others
|
|
MTSET chloride is a membrane-impermeable, thiol-specific reagent that primarily acts as an inhibitor of the Nav1.5 sodium channel. MTSET chloride reduces the peak sodium current amplitude of most mutant Nav1.5 channels, induces a hyperpolarizing shift in steady-state inactivation, and slows the recovery process, but shows no activity against wild-type and non-inactivating Nav1.5 channel mutants. MTSET chloride reacts covalently with T336C mutant P2X2 receptors, partially reversibly blocks ATP-induced cation currents, and exerts no effect on wild-type P2X2 receptors .
|
-

- HY-163679
-
|
|
Estrogen Receptor/ERR
Cytochrome P450
PROTACs
Apoptosis
|
Cancer
|
|
PROTAC ERα Degrader-9 (Compound 18c) is a dual-targeting PROTAC degrader, which degrades estrogen receptor α (ERα) and aromatase (ARO). PROTAC ERα Degrader-9 binds to ERα with a Ki of 0.25 μM, inhibits ARO with an IC50 of 4.6 μM. PROTAC ERα Degrader-9 inhibits the proliferation of MCF-7 wildtype (IC50=0.54 μM) and ERα mutants MCF-7 EGFR (IC50=0.075 μM), MCF-7 D538G (IC50=0.31 μM), MCF-7 Y537S (IC50=2.3 μM), downregulates the expressions of ERS1 and MYC. PROTAC ERα Degrader-9 arrests the cell cycle at G2/M, induces apoptosis in MCF-7. PROTAC ERα Degrader-9 exhibits antitumor efficacy in mouse models. (Pink: ligand for target protein (HY-163680); Black: linker (HY-W007559); Blue: ligand for E3 ligase (HY-112078))
|
-

- HY-183667
-
|
|
Androgen Receptor
Kallikrein
|
Cancer
|
|
JNJ-pan-AR is an orally active androgen receptor (AR) inhibitor with an IC50 of 19 nM and a Ki of 8.4 nM against human wild-type AR. JNJ-pan-AR abolishes androgen-induced KLK2 and KLK3 mRNA expression and reduces androgen-dependent colony formation in prostate cancer cells. JNJ-pan-AR blocks AR nuclear translocation, inhibits PSA protein expression, and represses the growth of AR-dependent tumor cells and ARF877L-driven tumor xenografts. JNJ-pan-AR blocks transactivation and signaling of wild-type AR and various mutant AR variants. JNJ-pan-AR is applicable for research on castration-resistant prostate cancer .
|
-

- HY-182809
-
|
|
Androgen Receptor
HSP
|
Neurological Disease
Cancer
|
|
Androgen receptor degrader-6 is a blood-brain barrier-permeable AR degrader. Androgen receptor degrader-6 inhibits the chaperone activity of HSP27 and disrupts the HSP27-AR complex. Androgen receptor degrader-6 promotes the degradation of wild-type and mutant AR, reduces AR protein levels, and inhibits the growth of glioblastoma cells. Androgen receptor degrader-6 can be used in glioblastoma research .
|
-

- HY-176995
-
|
|
GHSR
|
Metabolic Disease
|
|
AZ-GHS-38 (compound 38) is an orally active CNS-penetrant inverse agonist of growth hormone secretagogue receptor (GHS-R1a) with an IC50 of 6.7 nM. AZ-GHS-38 results in acute reduction of food intake in wild-type mice. AZ-GHS-38 can be used for anti-obesity research .
|
-

- HY-125675
-
|
|
Estrogen Receptor/ERR
|
Cancer
|
|
VPC-16606 is a selective inhibitor targeting the activation function 2 (AF2) domain of estrogen receptor α (ERα). VPC-16606 blocks the interaction between ERα and coactivators, inhibits the activity of both wild-type and clinically drug-resistant mutant ERα, and exerts inhibitory effects on hormone-resistant breast cancer cells. VPC-16606 can be used in breast cancer research .
|
-

- HY-103320AR
-
|
|
Reference Standards
CaSR
|
Metabolic Disease
|
|
Calhex 231 hydrochloride (Standard) is the analytical standard of Calhex 231 hydrochloride (HY-103320A). This product is intended for research and analytical applications. Calhex 231 hydrochloride is a potent negative allosteric modulator that blocks (IC50 = 0.39 μM) increases in [3H]inositol phosphates elicited by activating the human wild-type CaSR transiently Ca2+-sensing receptor. Calhex 231 hydrochloride can be used in the study of traumatic hemorrhagic shock (THS) and diabetic cardiomyopathy (DCM) .
|
-

- HY-100131R
-
|
|
RIP kinase
Reference Standards
|
Inflammation/Immunology
|
|
GSK481 (Standard) is the analytical standard of GSK481 (HY-100131). This product is intended for research and analytical applications. GSK481 is a highly potent, selective, and specific receptor interacting protein 1 (RIP1) kinase inhibitor with an IC50 of 1.3 nM, which inhibits Ser166 phosphorylation in wild-type human RIP1 (IC50=2.8 nM). GSK481 also exhibits excellent translation in the U937 cellular assay with an IC50 of 10 nM .
|
-

- HY-19337S1
-
|
BAY 1896953-d6; ORM-15341-d6
|
Isotope-Labeled Compounds
Androgen Receptor
|
Cancer
|
|
Ketodarolutamide-d6 (BAY 1896953-d6) is the deuterium labeled Ketodarolutamide (HY-19337). Ketodarolutamide (ORM-15341) is a potent, high-affinity nonsteroidal competitive full antagonist of androgen receptor (AR). Ketodarolutamide displays a Ki value of 8 nM for rat wild-type AR and an IC50 value of 38 nM in AR-HEK293 cells . Ketodarolutamide inhibits testosterone-induced nuclear translocation of the AR and antagonizes both overexpressed and mutant ARs . Ketodarolutamide specifically suppresses the proliferation of AR-dependent prostate cancer cells and exhibits antitumor activity in models of castration-resistant prostate cancer (CRPC) . Ketodarolutamide is suitable for the mechanistic and therapeutic research of prostate cancer .
|
-

- HY-19337S
-
|
BAY 1896953-d3; ORM-15341-d3
|
Isotope-Labeled Compounds
Androgen Receptor
|
Cancer
|
|
Ketodarolutamide-d3 (BAY 1896953-d3) is the deuterium labeled Ketodarolutamide (HY-19337). Ketodarolutamide (ORM-15341) is a potent, high-affinity nonsteroidal competitive full antagonist of androgen receptor (AR). Ketodarolutamide displays a Ki value of 8 nM for rat wild-type AR and an IC50 value of 38 nM in AR-HEK293 cells . Ketodarolutamide inhibits testosterone-induced nuclear translocation of the AR and antagonizes both overexpressed and mutant ARs . Ketodarolutamide specifically suppresses the proliferation of AR-dependent prostate cancer cells and exhibits antitumor activity in models of castration-resistant prostate cancer (CRPC) . Ketodarolutamide is suitable for the mechanistic and therapeutic research of prostate cancer .
|
-

- HY-165357
-
|
(R)-Alprenolol; D-Alprenolol
|
5-HT Receptor
Adrenergic Receptor
|
Cardiovascular Disease
|
|
(+)-Alprenolol ((R)-Alprenolol; D-Alprenolol) is a 5-HT1A receptor antagonist with a Ki value of 5400 nM. (+)-Alprenolol also acts as a non-selective β-receptor (β-receptor) blocker. (+)-Alprenolol can be used in the research of cardiovascular and cerebrovascular diseases .
|
-

- HY-181649
-
|
|
Sigma Receptor
|
Neurological Disease
|
|
S1R agonist 3 is a potent, selective and brain-penetrant sigma-1 receptor (S1R) agonist with a Ki of 1.5 nM. S1R agonist 3 reduces hyperlocomotion in wfs1abKO zebrafish larvae without affecting locomotion in wildtype wfs1abWT zebrafish larvae. S1R agonist 3 reverses Aβ25-35 (HY-P0128)-induced learning and memory impairments. S1R ligand 1 can be used for the research of Alzheimer’s disease .
|
-

- HY-W587486A
-
|
|
Bacterial
Endogenous Metabolite
TGF-β Receptor
|
Metabolic Disease
|
|
N-Acetyltaurine hemimagnesium is an orally active endogenous sulfonate that is synthesized from taurine and acetate in the renal cortex. N-Acetyltaurine hemimagnesium supports bacterial growth as a sole fixed nitrogen or carbon source. N-Acetyltaurine hemimagnesium buffers acetyl moieties of mitochondrial acetyl‑CoA in skeletal muscle. N-Acetyltaurine hemimagnesium reduces food intake and body weight in obese and lean wild‑type mice in a GFRAL‑dependent manner. N-Acetyltaurine hemimagnesium can be used for the research of diet‑induced obesity, hyperacetatemia and diabetes .
|
-

- HY-186122
-
|
|
Cholecystokinin Receptor
|
Neurological Disease
Inflammation/Immunology
|
|
CCK2R agonist-1 (Compound 2S) is a CCK2R agonist, with an IC50 of 48 nM against wild-type CCK-2R and an IC50 of 450 nM against mutant CCK-2R (N353L). CCK2R agonist-1 stimulates the production of inositol phosphate. The changes in pH and HDC induced by CCK2R agonist-1 in mice are comparable to those induced by the full-length peptide agonist Gastrin. CCK2R agonist-1 can be used in studies of gastric diseases and pain .
|
-

- HY-182642
-
|
|
P2X Receptor
Nucleoside Antimetabolite/Analog
|
Cardiovascular Disease
|
|
MRS2339 is a ribose-modified nucleotide and a nucleotidase-resistant P2 receptor agonist. MRS2339 activates P2X4R. MRS2339 induces ionic currents via P2X receptors, reduces cardiomyocyte cross-sectional area and heart weight/body weight ratio, lacks vasodilatory activity, and extends the lifespan of mice with cardiomyopathy. MRS2339 can be used in research related to heart failure and cardiomyopathy .
|
-

- HY-18101
-
|
|
Sigma Receptor
TRP Channel
|
Neurological Disease
|
|
BD-1063 is a selective σ-1 receptor antagonist with inhibitory activity against TRPC5 and TRPM3. BD-1063 exerts anti-hyperalgesic and anti-allodynic effects by inhibiting sustained calcium influx mediated by TRPC5 and TRPM3, and reverses the effects of Carrageenan (HY-125474). BD-1063 also significantly reduces excessive ethanol self-administration behavior. BD-1063 is widely used in studies on the mechanisms underlying neuropathic pain, inflammatory hyperalgesia, and alcohol abuse and dependence .
|
-

| Cat. No. |
Product Name |
Type |
-
- HY-W699451
-
|
|
Fluorescent Dyes
|
|
MTSET chloride is a membrane-impermeable, thiol-specific reagent that primarily acts as an inhibitor of the Nav1.5 sodium channel. MTSET chloride reduces the peak sodium current amplitude of most mutant Nav1.5 channels, induces a hyperpolarizing shift in steady-state inactivation, and slows the recovery process, but shows no activity against wild-type and non-inactivating Nav1.5 channel mutants. MTSET chloride reacts covalently with T336C mutant P2X2 receptors, partially reversibly blocks ATP-induced cation currents, and exerts no effect on wild-type P2X2 receptors .
|
| Cat. No. |
Product Name |
Type |
-
- HY-W010162
-
|
L-Alanyl-L-alanine; Ala-Ala
|
Biochemical Assay Reagents
|
H-Ala-Ala-OH is a substrate of human intestinal oligopeptide transporter (PEPT1/SLC15A1), with an IC50 of 0.25 mM and an EC50 of 0.08 mM against human PEPT1. When used as a dipeptide linker in antibody-drug conjugates (ADCs) loaded with glucocorticoid receptor regulator payloads, H-Ala-Ala-OH enables efficient intracellular payload release .
|
| Cat. No. |
Product Name |
Target |
Research Area |
-
- HY-P11092
-
|
|
Endogenous Metabolite
|
Neurological Disease
|
|
TLQP-62 (mouse,rat) is a secreted C-terminal peptide that can be derived from protein VGF. TLQP-62 activates the BDNF-TrkB signaling pathway, inducing acute, transient phosphorylation of TrkB receptor and downstream CREB (Ser133) phosphorylation. TLQP-62 demonstrates excellent efficacy in promoting long-term fear memory formationin wild-type mice and reversing memory impairment in VGF heterozygous knock-out mice. TLQP-62 can be used for the study of memory-related neurological disorders (e.g., Alzheimer’s disease, frontotemporal dementia) .
|
-
- HY-P11075
-
|
|
Neurokinin Receptor
|
Metabolic Disease
|
|
[Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) (ID 305) is a selective NK2R agonist with an EC50 of 3.7 nM for hNK2R. [Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) significantly inhibits weight loss in diet-induced obese (DIO) mice model and improves Loperamide (HY-156131)-induced dysfunctional voiding in wild-type mice. [Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10) can be used for neurokinin receptor mediated disorders, such as obesity, insulin resistance and type 2 diabetes research .
|
| Cat. No. |
Product Name |
Target |
Research Area |
Image |
-
- HY-P99384
-
|
B-701; MFGR-1877S; RG-7444
|
FGFR
|
Cancer
|
|
Vofatamab (B-701) is an anti-FGFR3 monoclonal antibody (mAb). Vofatamab blocks activation of both the wildtype and genetically activated receptor. Vofatamab can be used in the research of metastatic urothelial carcinoma (mUC). Recommend Isotype Controls: Human IgG1 kappa, Isotype Control (HY-P99001) .
|
-

(5)
-
- HY-P991221
-
|
|
Melanocortin Receptor
|
Metabolic Disease
|
|
ALD1613 is a potent neutralizing monoclonal antibody against adrenocorticotropic hormone (ACTH). ALD1613 neutralizes ACTH-induced signaling and inhibits ACTH-induced cyclic AMP accumulation in a mouse adrenal cell line (Y1) via all five melanocortin receptors. ALD1613 significantly reduces plasma corticosterone levels in wild-type rats. ALD1613 can be used in the study of diseases associated with elevated ACTH levels .
|
-
(5)
-
- HY-P991641
-
|
LY3012218
|
FLT3
p38 MAPK
STAT
PI3K
Akt
|
Cancer
|
|
IMC-EB10 (LY3012218) is an anti-FLT3 monoclonal antibody. IMC-EB10 binds to FLT3 with high affinity (Kd = 158 pM) and blocks the binding of FLT3 ligand to FLT3 (IC50 ≈ 10 nM), thereby inhibiting MAPK, STAT5, and PI3K/Akt signaling in leukemia cells. IMC-EB10 can enhance the anti-leukemic effect of Methotrexate (HY-14519) and inhibit leukemias expressing wild-type or ITD-mutated FLT3 receptors. IMC-EB10 prolongs the survival of acute lymphoblastic leukemia (ALL) cells and primary leukemia samples and reduces engraftment in non-obese diabetic/severe combined immunodeficiency patients. IMC-EB10 is indicated for leukemia research .
|
-
(5)
| Cat. No. |
Product Name |
Category |
Target |
Chemical Structure |
-
- HY-N0475
-
-
-
- HY-N0475R
-
|
Hypolide (Standard); (+)-Triptophenolide (Standard)
|
Structural Classification
Monophenols
Terpenoids
Celastraceae
Phenols
Diterpenoids
Tripterygium wilfordii Hook. f.
Plants
Source Classification
|
Reference Standards
Androgen Receptor
Pyroptosis
Caspase
Bcl-2 Family
Apoptosis
|
|
Triptophenolide (Standard) (Hypolide) is the analytical standard of Triptophenolide (HY-N0475). This product is intended for research and analytical applications. Triptophenolide is a colorless crystal isolated from the ethyl acetate extract of Tripterygium wilfordii. Triptophenolide is an orally active pan‑antagonist of the androgen receptor (AR) with an IC50 of 467 nM against human wild‑type AR. Triptophenolide reduces AR expression, inhibits AR nuclear translocation, downregulates prostate‑specific antigen mRNA levels, and suppresses the growth of AR‑positive prostate cancer cells. Triptophenolide shows anti-tumor effects against breast cancer by inhibiting cell proliferation and migration, inducing G1-phase arrest and apoptosis, repressing xenograft tumor growth. Triptophenolide inhibits pyroptosis, alleviates tissue inflammation, and ameliorates synovial injury. Triptophenolide can be used for the study of prostate cancer, rheumatoid arthritis and breast cancer .
|
-
| Cat. No. |
Product Name |
Chemical Structure |
-
- HY-16985S
-
|
|
|
Darolutamide-d4 (ODM-201-d4) is deuterium labeled Darolutamide (HY-16985). Darolutamide (ODM-201) is an orally active competitive androgen receptor (AR) antagonist, with a Ki of 11 nM for rat wild-type AR (wtAR) and an IC50 of 26 nM for human wild-type AR (hAR)-mediated transcriptional activation . Darolutamide inhibits testosterone-induced AR nuclear translocation and transcriptional activation . Darolutamide exerts selective effects on AR-positive cells by inhibiting AR-dependent signaling pathways, and its active metabolite retains full antagonistic activity against AR mutants . Darolutamide can be used for the research of prostate cancer, including androgen receptor-dependent prostate cancer .
|
-
-
- HY-124089S
-
|
|
|
Eicosapentaenoyl ethanolamide-d4 is the deuterium labeled Eicosapentaenoyl ethanolamide. Eicosapentaenoyl ethanolamide, an omega-3 fatty acid, is one of N-acylethanolamines (NAEs). Eicosapentaenoyl ethanolamide is cannabinoid CB1/CB2 receptor agonist. Eicosapentaenoyl ethanolamide acts as a metabolic signal. Eicosapentaenoyl ethanolamide inhibits dietary restriction (DR)-induced lifespan extension in wild type animals and suppresses lifespan extension in a TOR pathway mutant .
|
-
-
- HY-19337S1
-
|
|
|
Ketodarolutamide-d6 (BAY 1896953-d6) is the deuterium labeled Ketodarolutamide (HY-19337). Ketodarolutamide (ORM-15341) is a potent, high-affinity nonsteroidal competitive full antagonist of androgen receptor (AR). Ketodarolutamide displays a Ki value of 8 nM for rat wild-type AR and an IC50 value of 38 nM in AR-HEK293 cells . Ketodarolutamide inhibits testosterone-induced nuclear translocation of the AR and antagonizes both overexpressed and mutant ARs . Ketodarolutamide specifically suppresses the proliferation of AR-dependent prostate cancer cells and exhibits antitumor activity in models of castration-resistant prostate cancer (CRPC) . Ketodarolutamide is suitable for the mechanistic and therapeutic research of prostate cancer .
|
-
-
- HY-19337S
-
|
|
|
Ketodarolutamide-d3 (BAY 1896953-d3) is the deuterium labeled Ketodarolutamide (HY-19337). Ketodarolutamide (ORM-15341) is a potent, high-affinity nonsteroidal competitive full antagonist of androgen receptor (AR). Ketodarolutamide displays a Ki value of 8 nM for rat wild-type AR and an IC50 value of 38 nM in AR-HEK293 cells . Ketodarolutamide inhibits testosterone-induced nuclear translocation of the AR and antagonizes both overexpressed and mutant ARs . Ketodarolutamide specifically suppresses the proliferation of AR-dependent prostate cancer cells and exhibits antitumor activity in models of castration-resistant prostate cancer (CRPC) . Ketodarolutamide is suitable for the mechanistic and therapeutic research of prostate cancer .
|
-
Your information is safe with us. * Required Fields.
Inquiry Information
- Product Name:
- Cat. No.:
- Quantity:
- MCE Japan Authorized Agent:































































![[Lys5,Tyr6,mLeu9,Nle10]-Neurokinin A (4-10)](http://file.medchemexpress.com/product_pic/hy-p11075.gif)



































