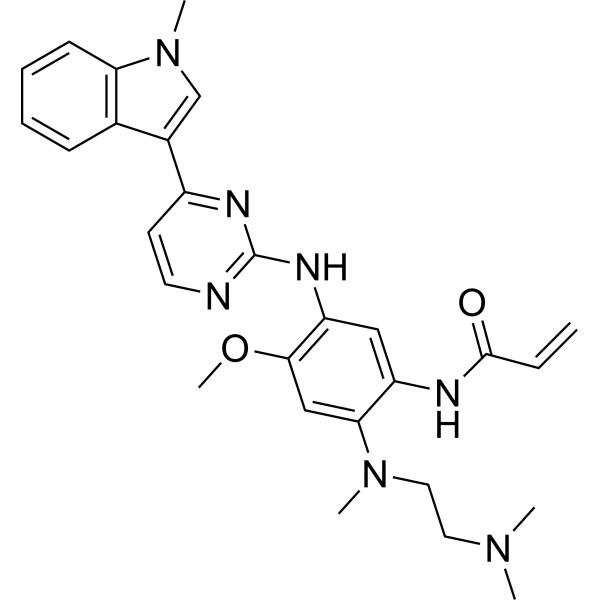
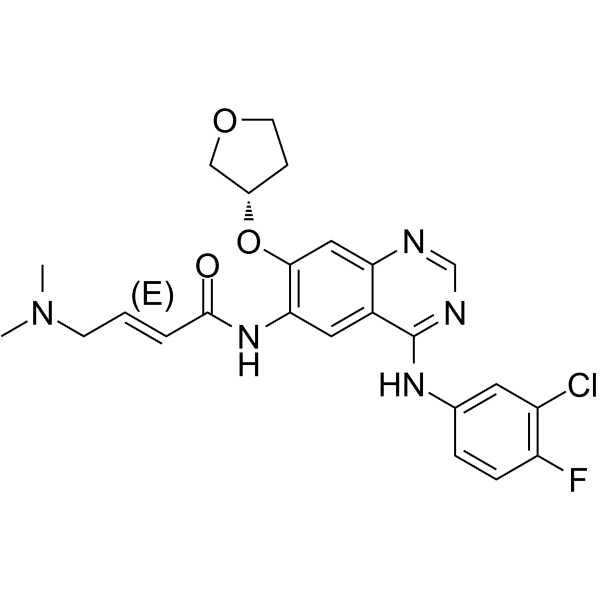
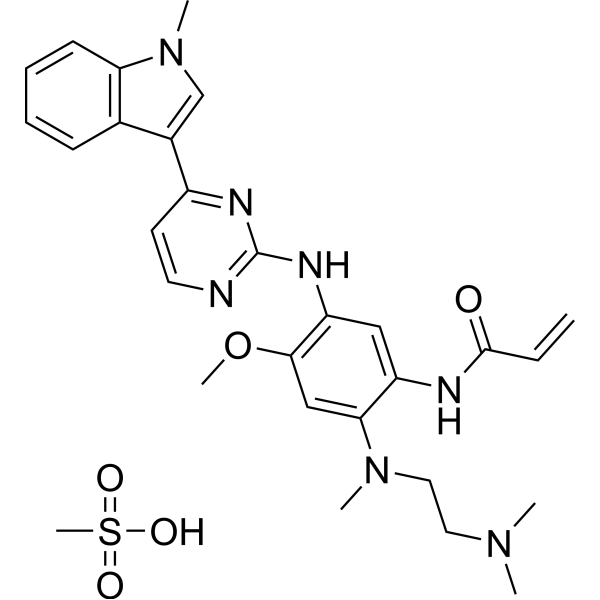
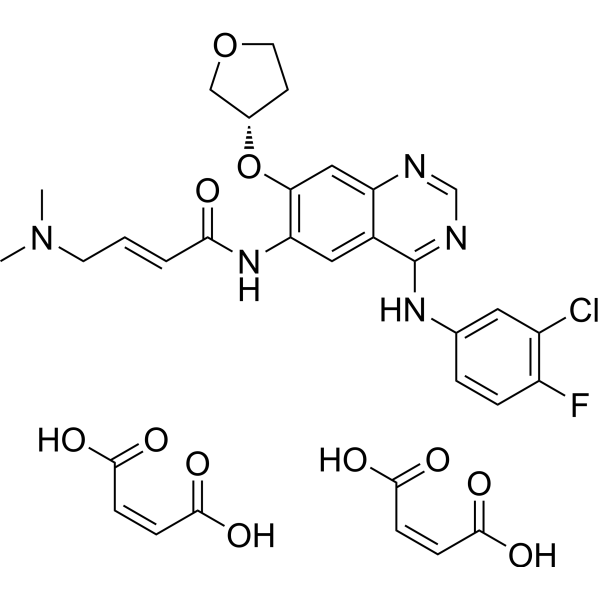
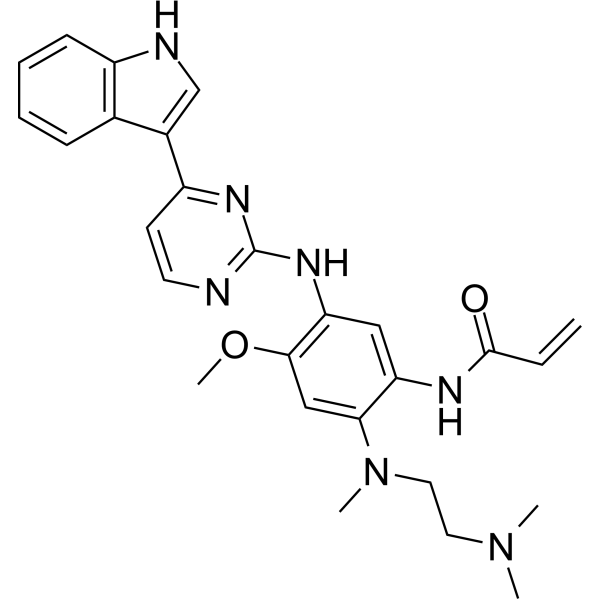
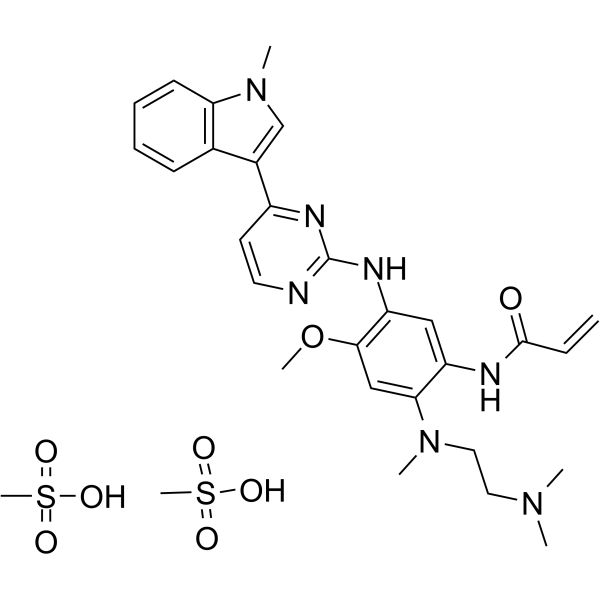
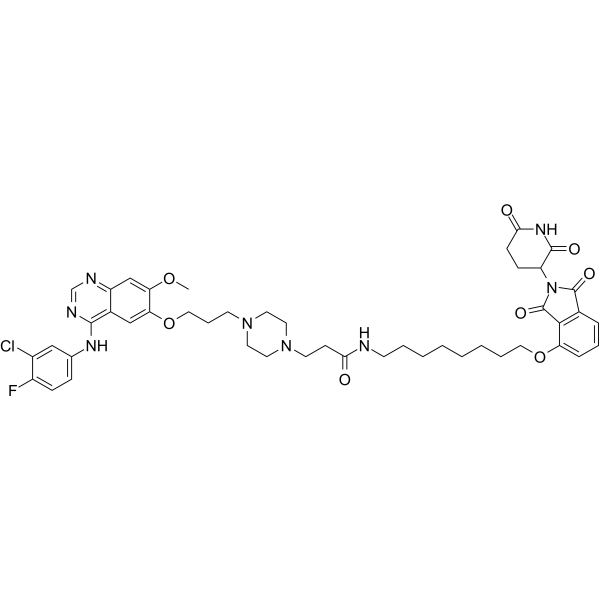
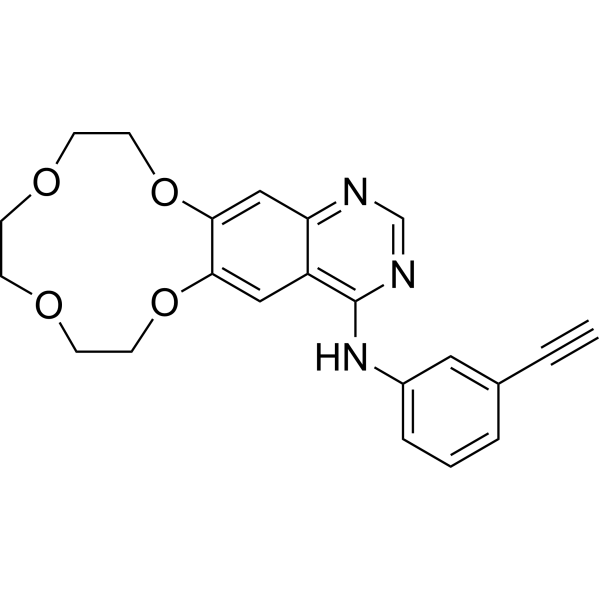
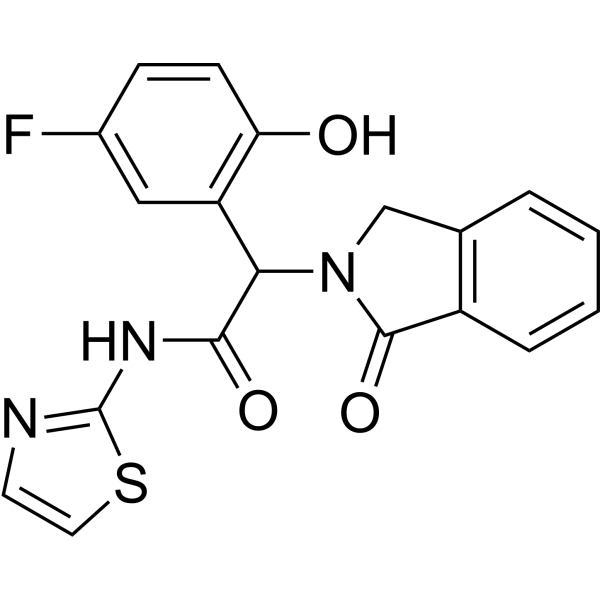
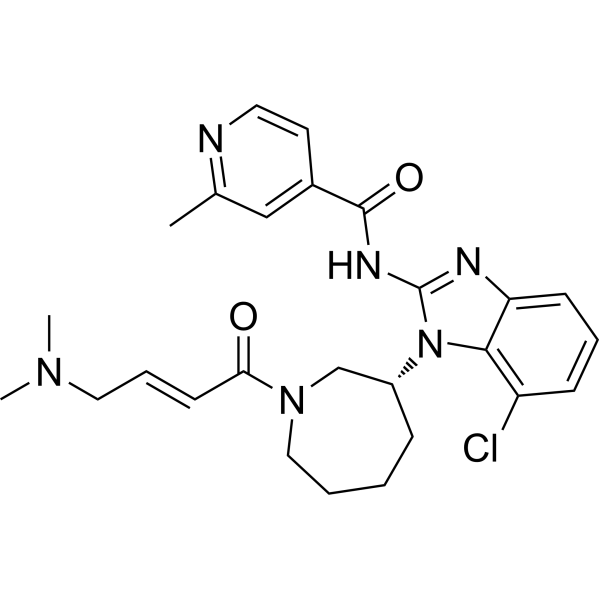
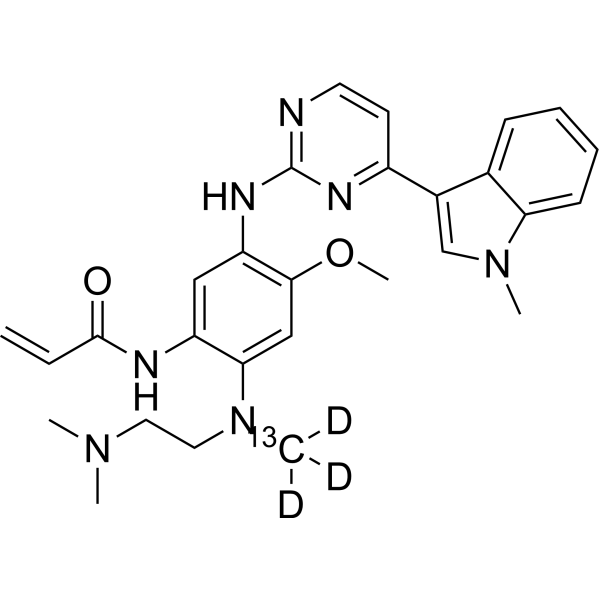
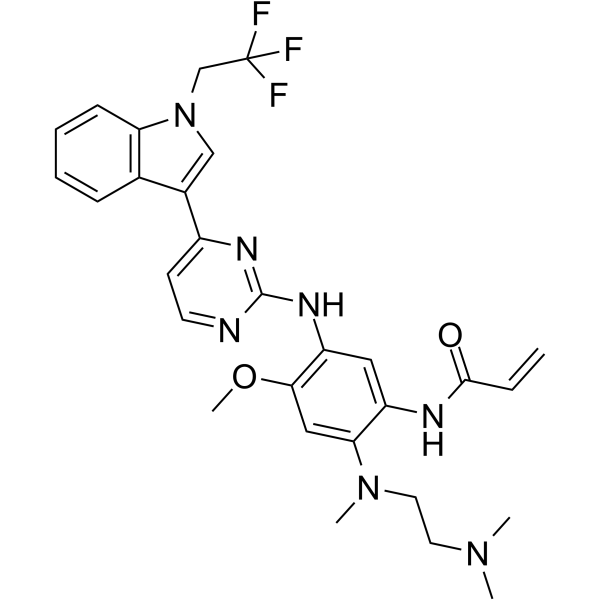
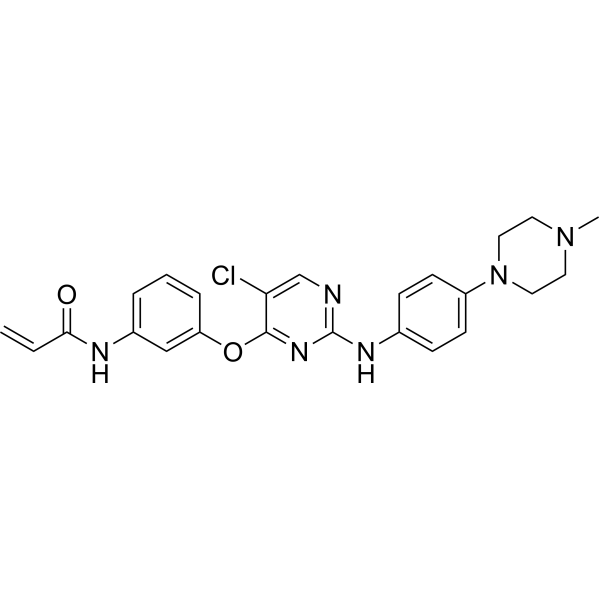
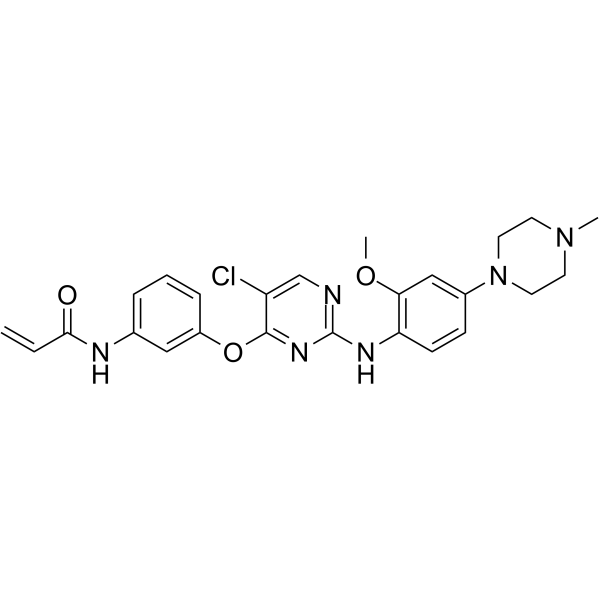
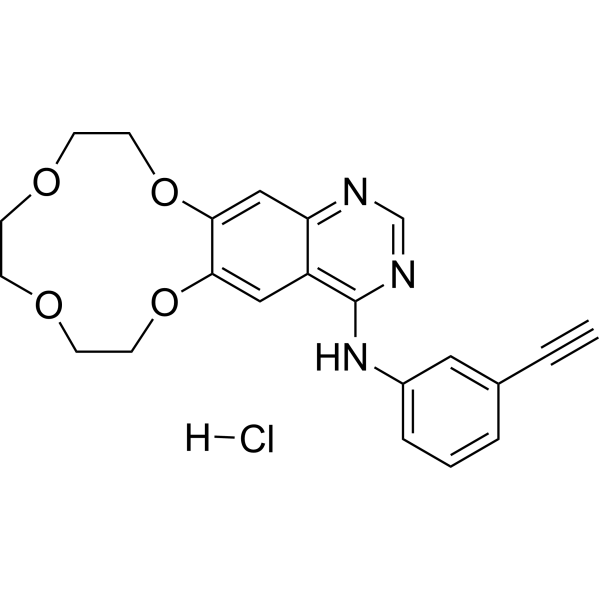
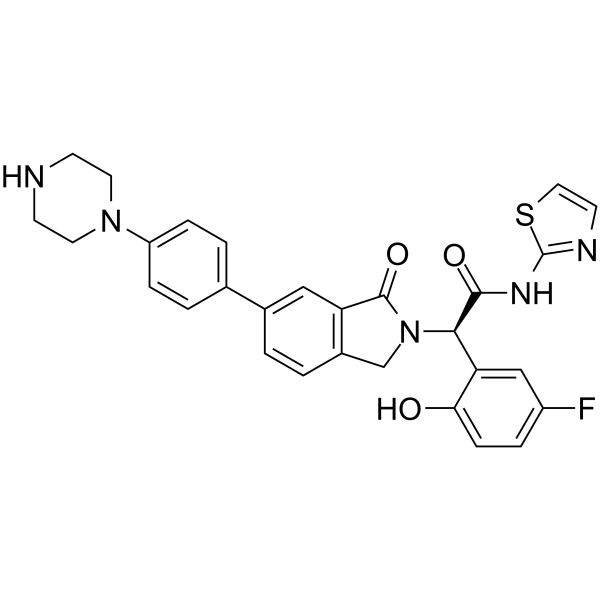
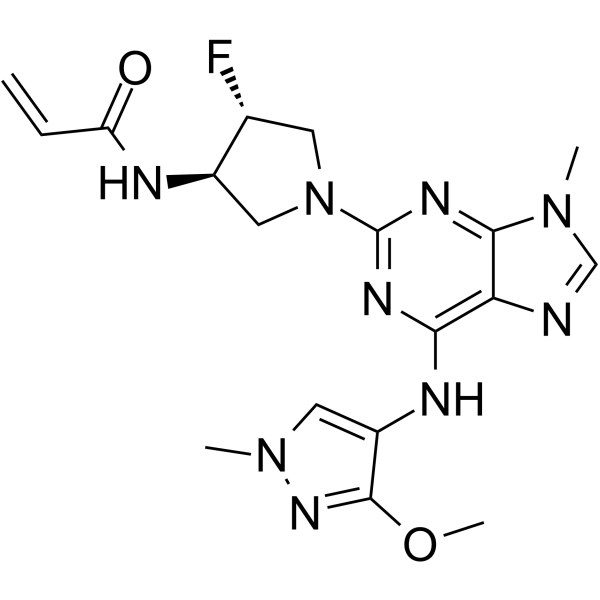
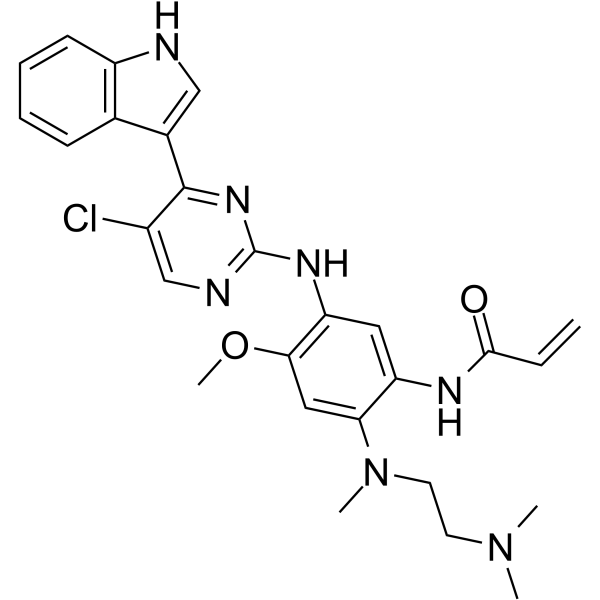
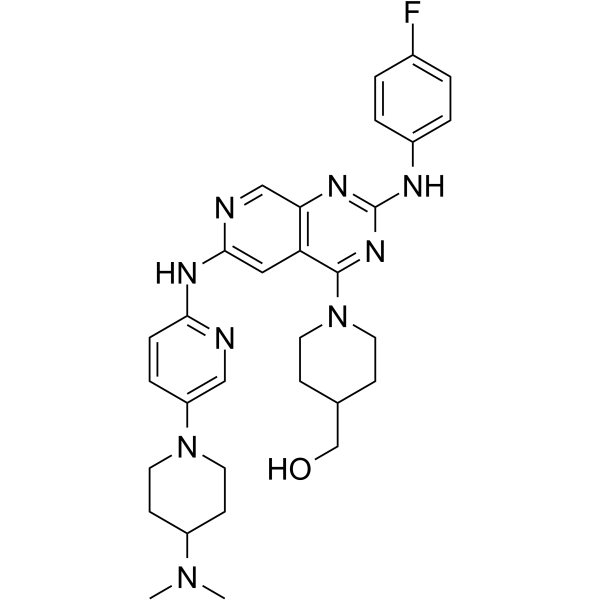
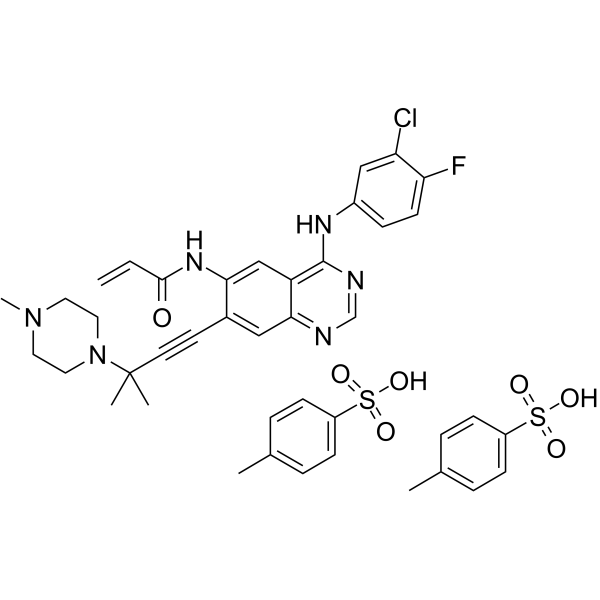
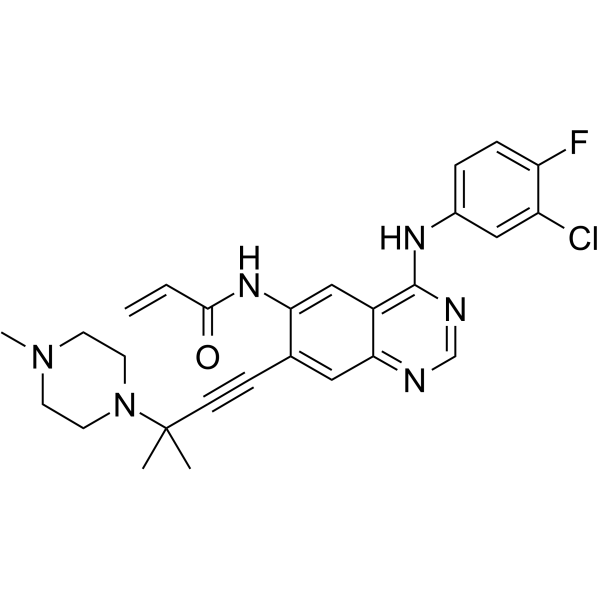
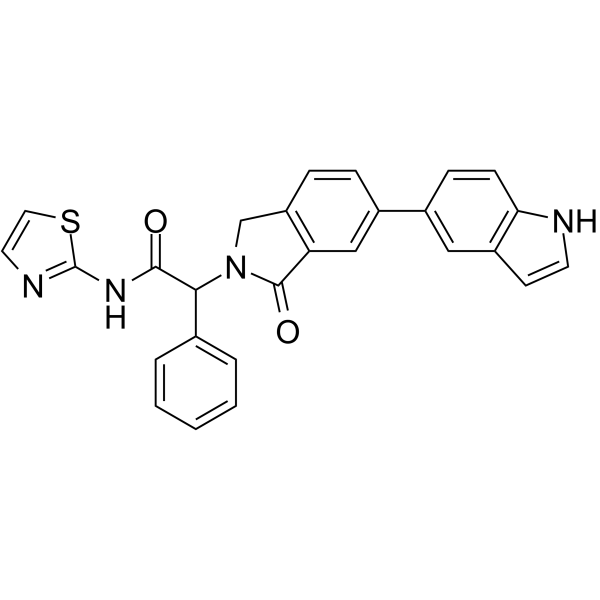
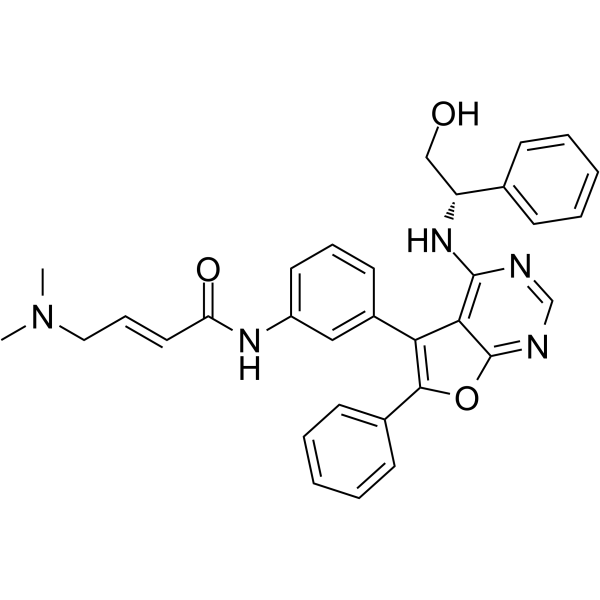
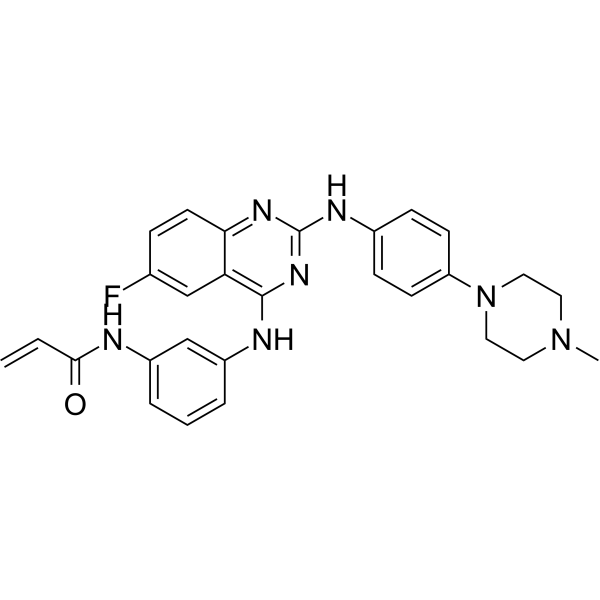
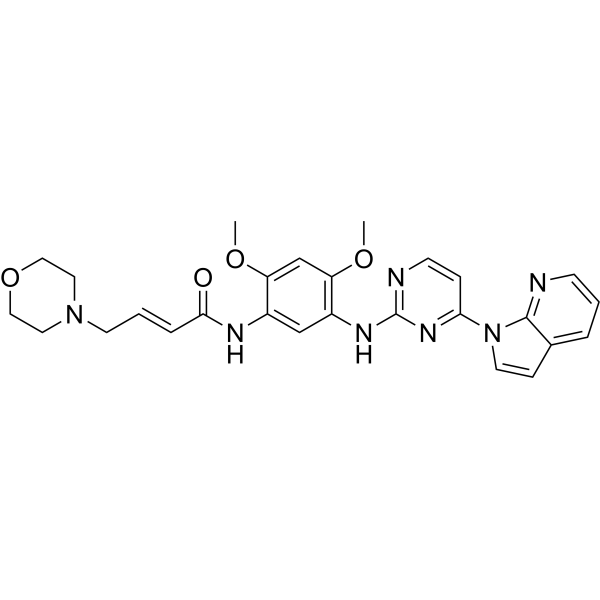
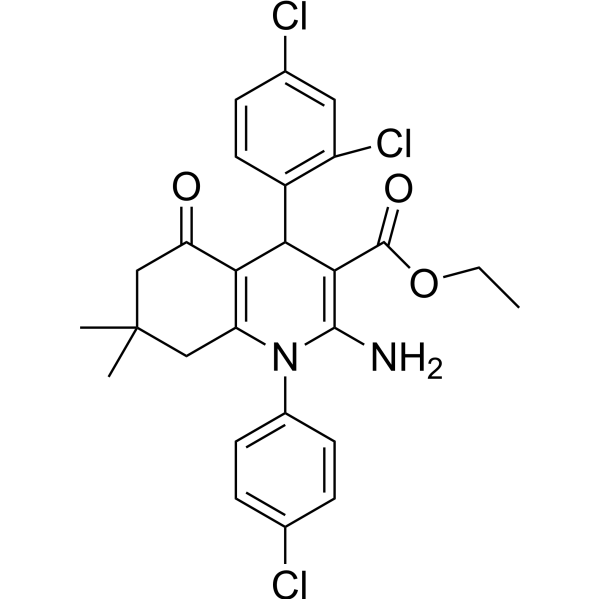
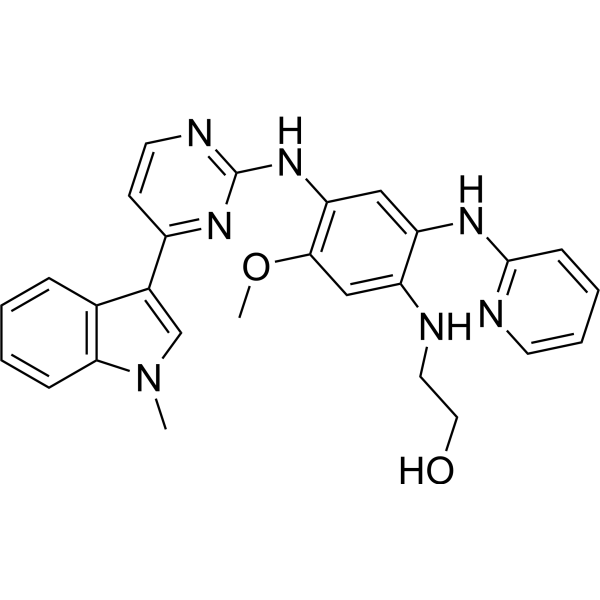
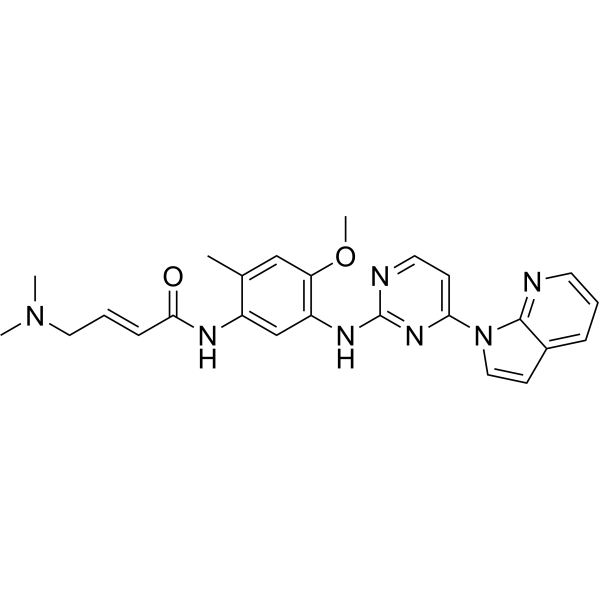
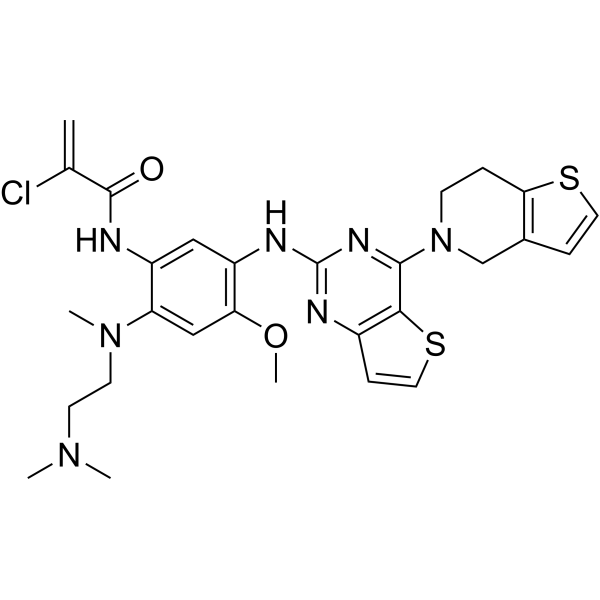
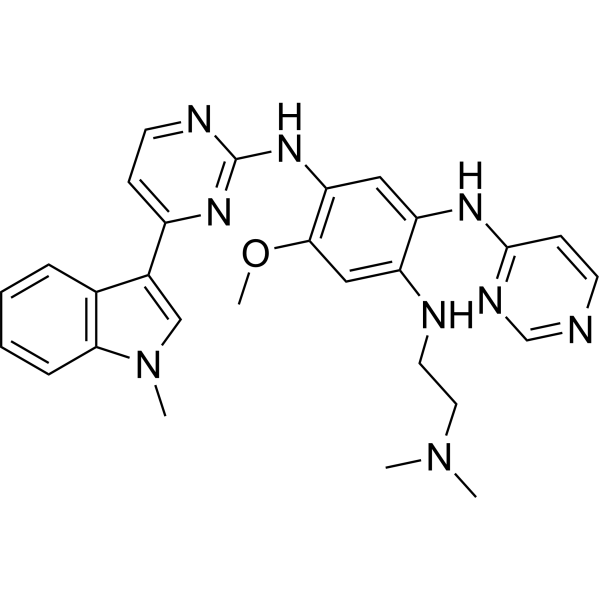
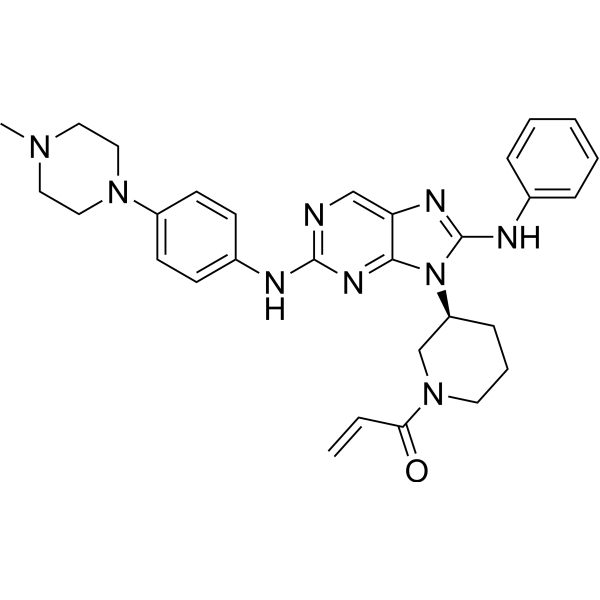
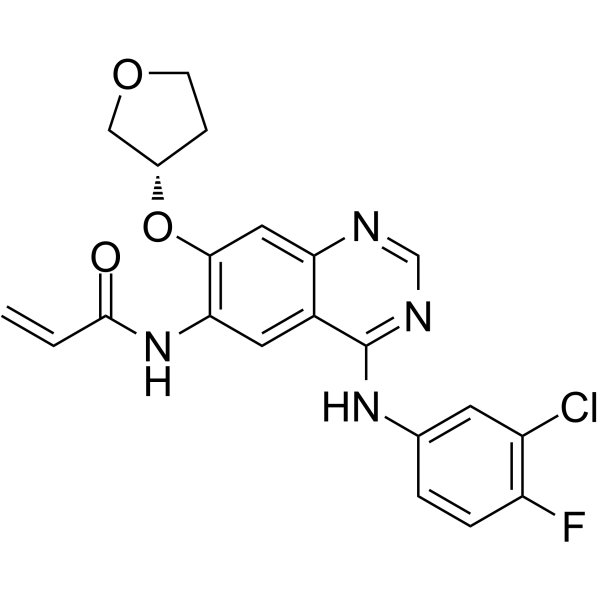
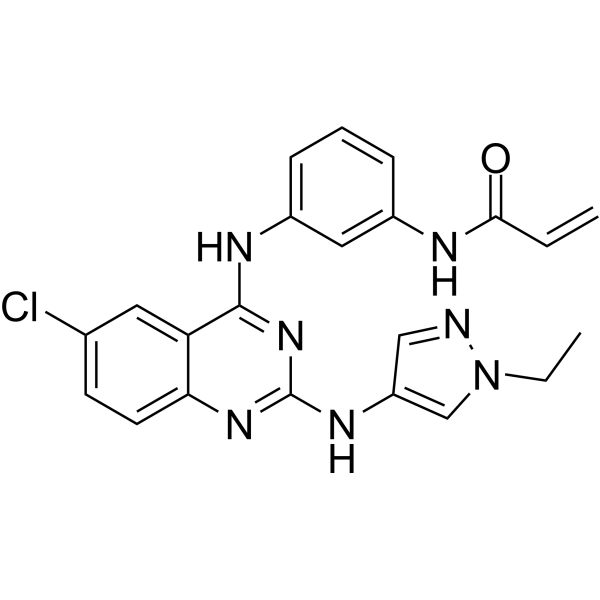
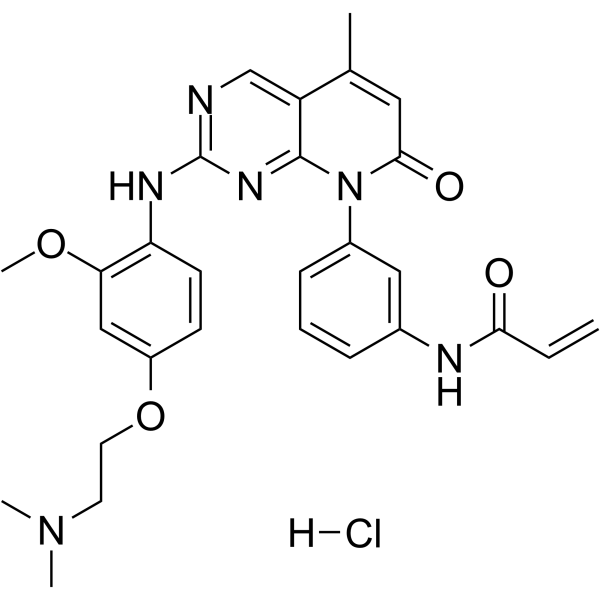
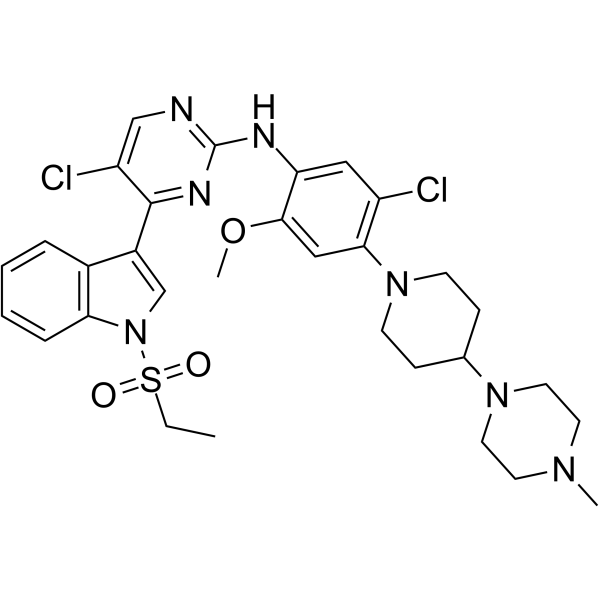
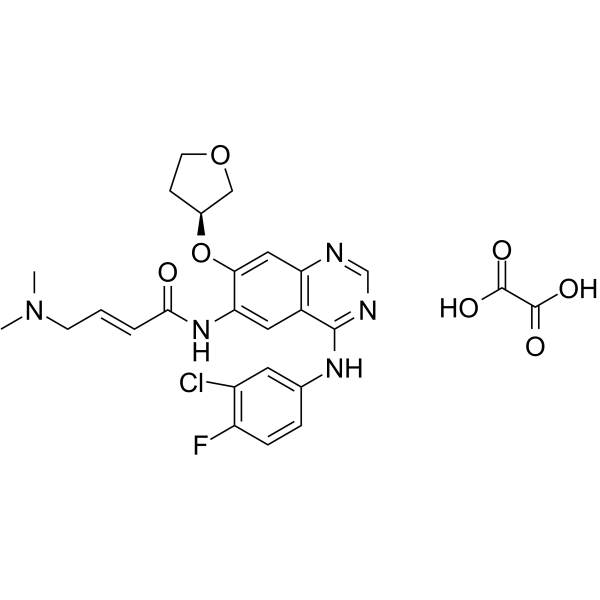
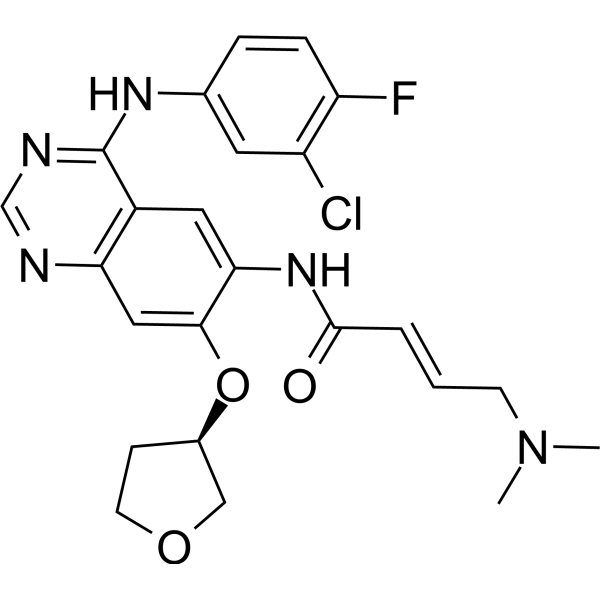
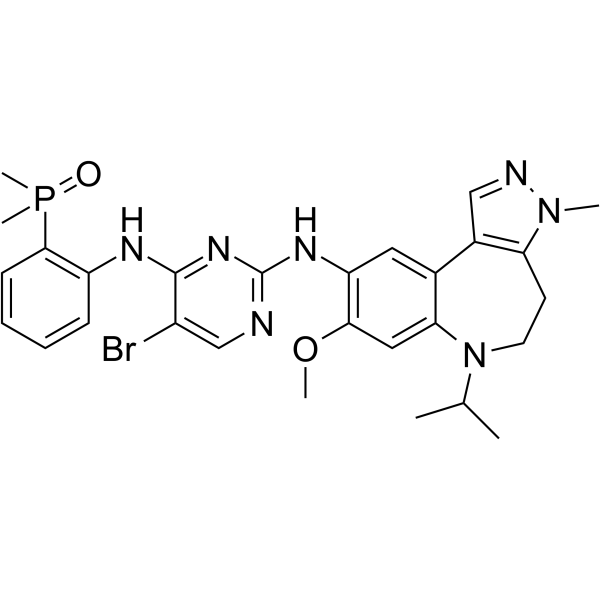
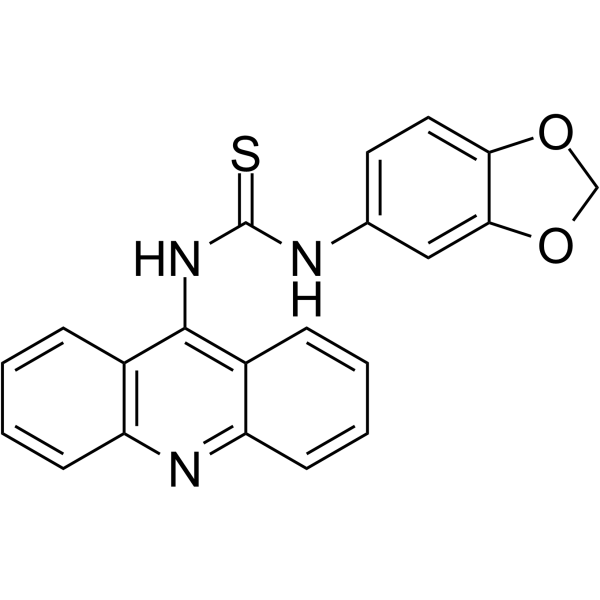
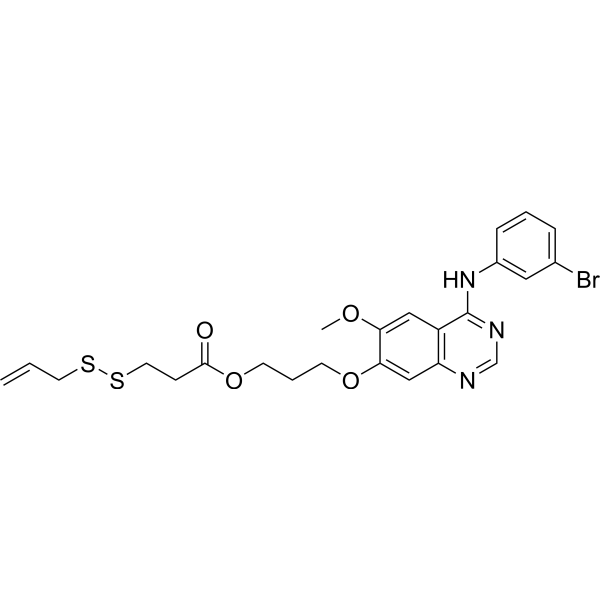
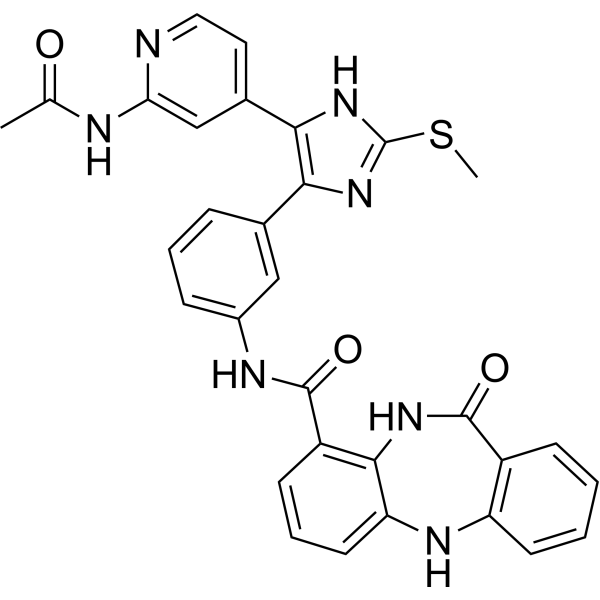
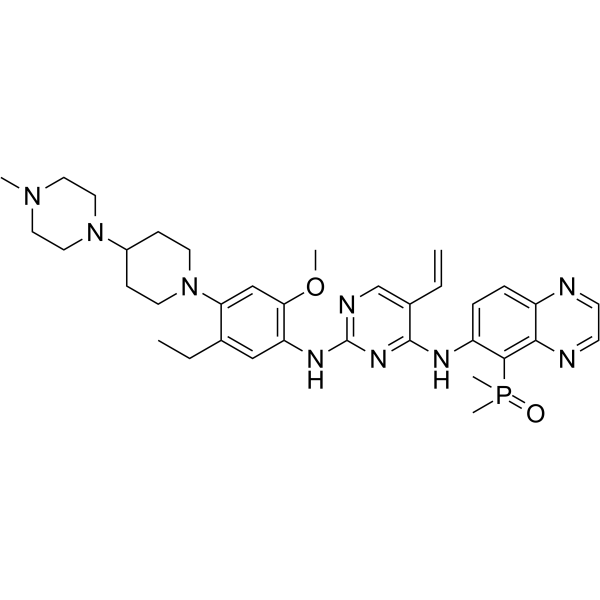
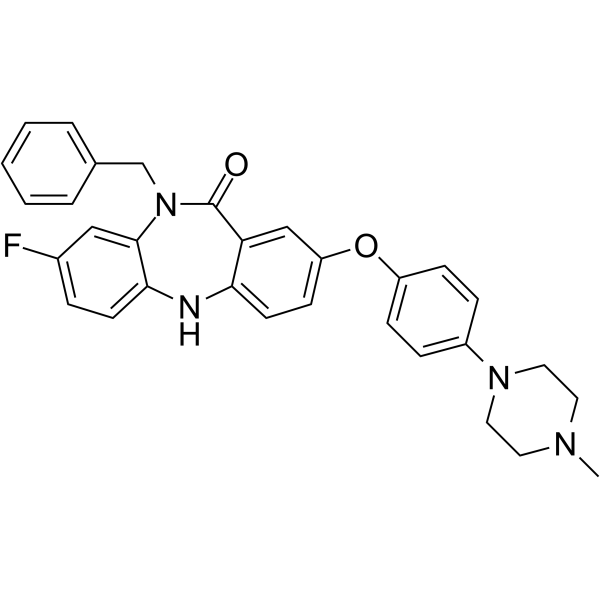
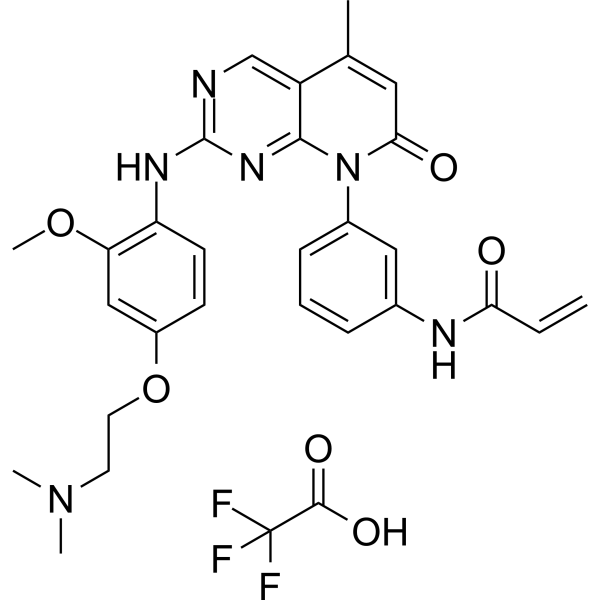
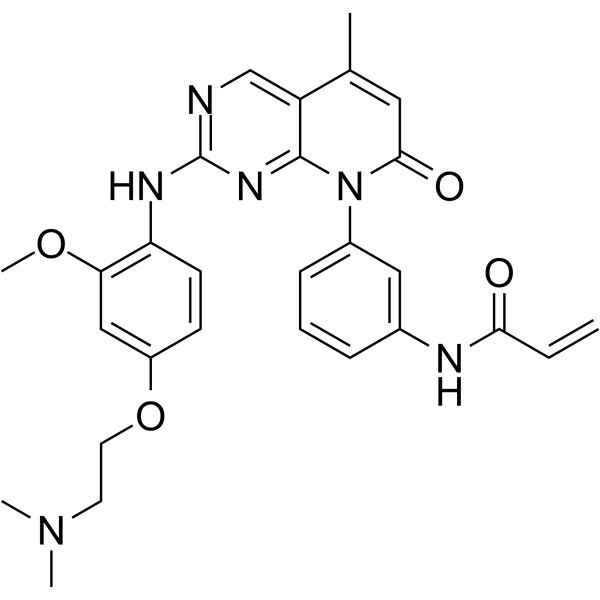
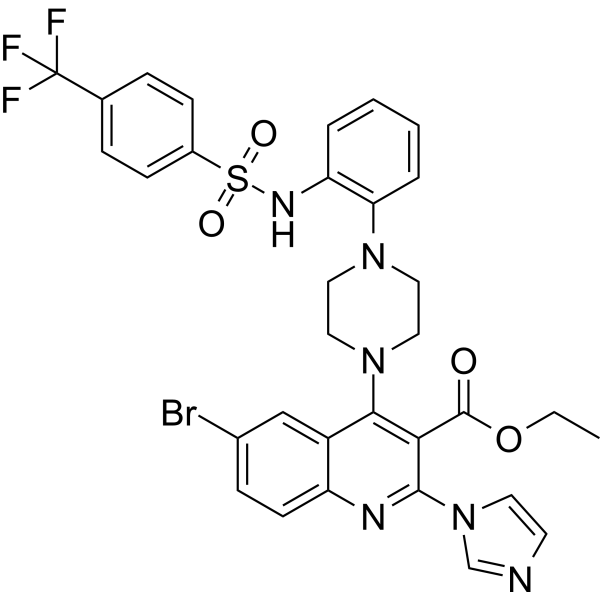
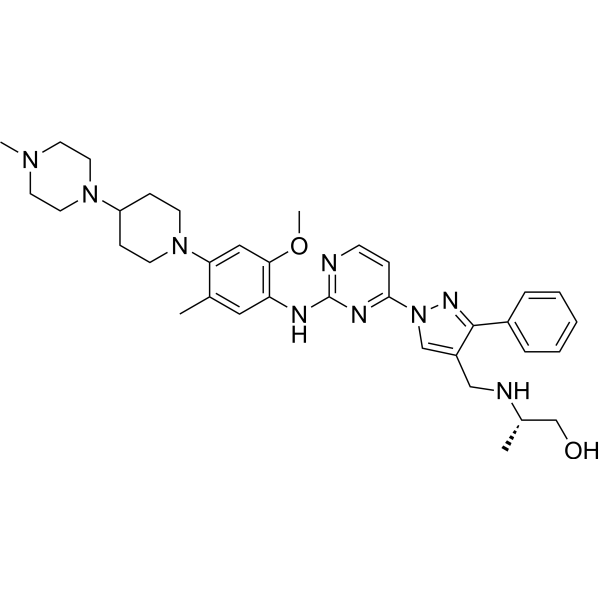
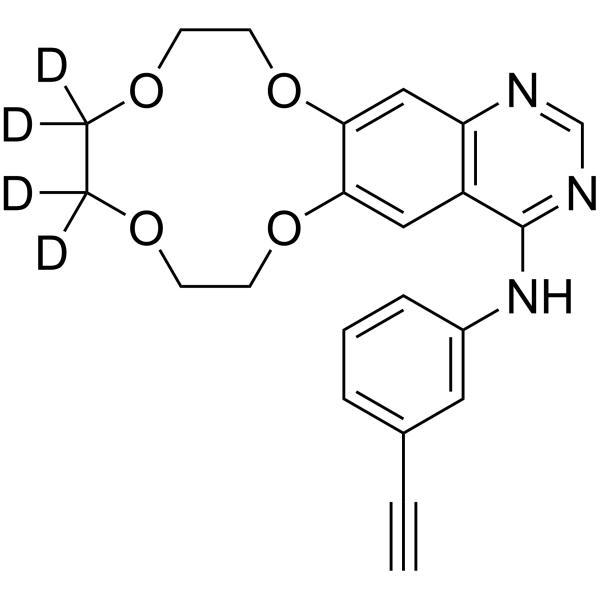
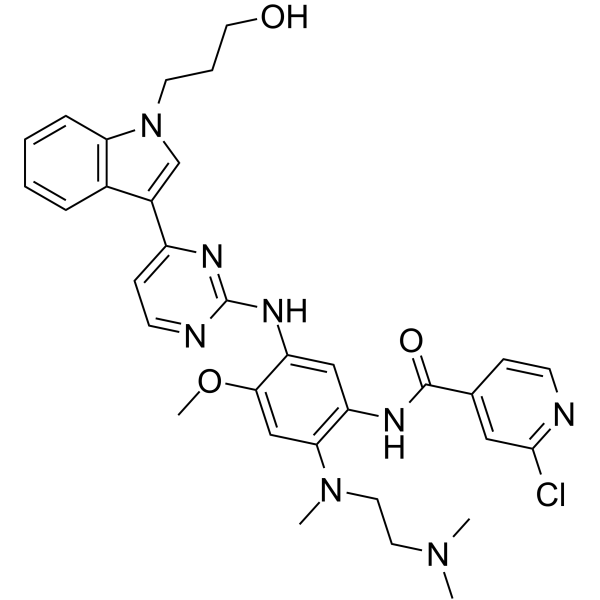
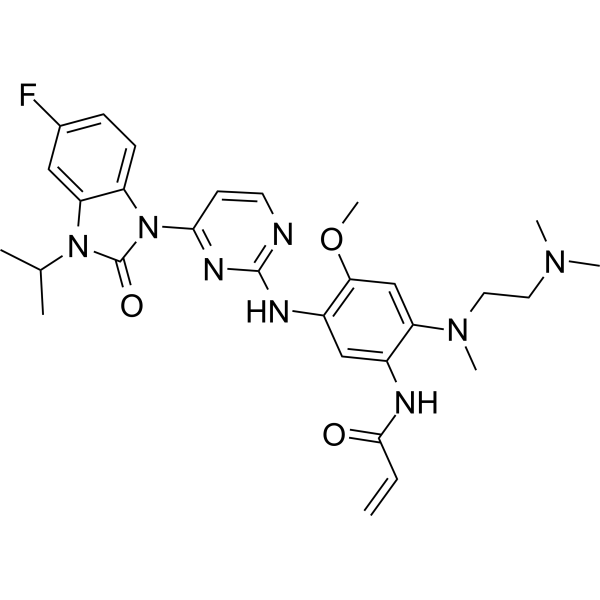
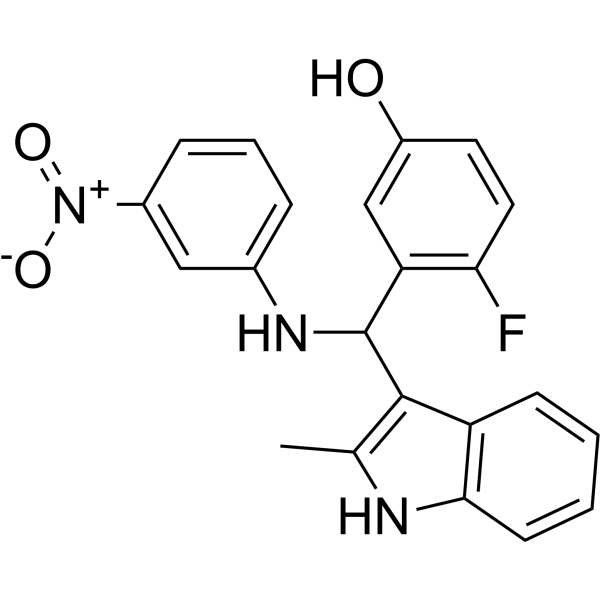
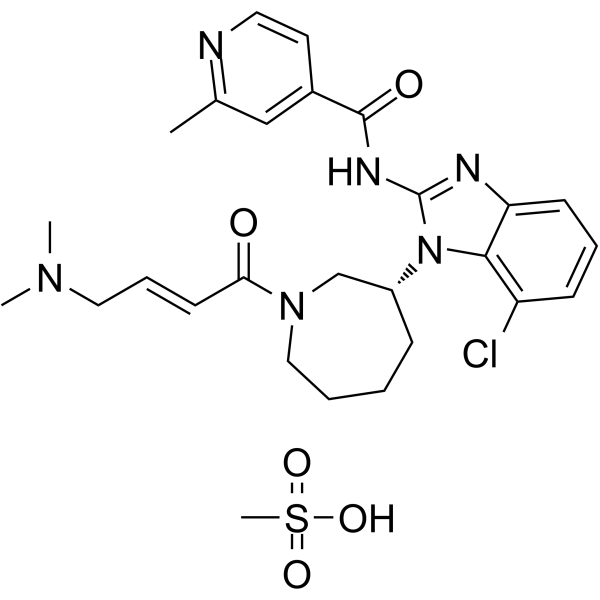
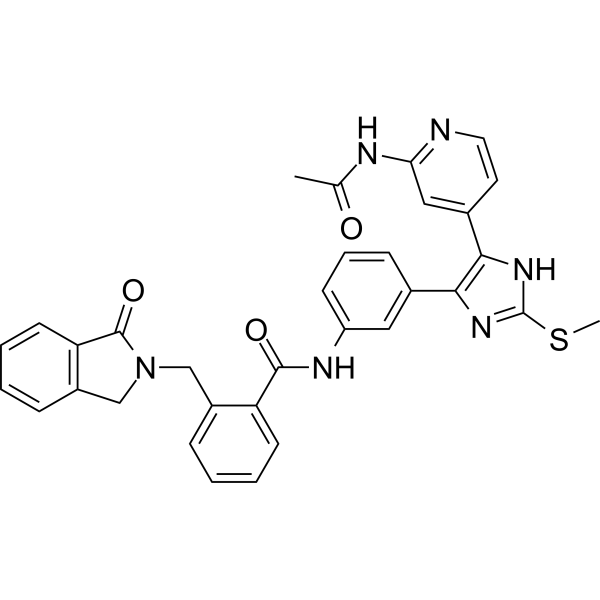
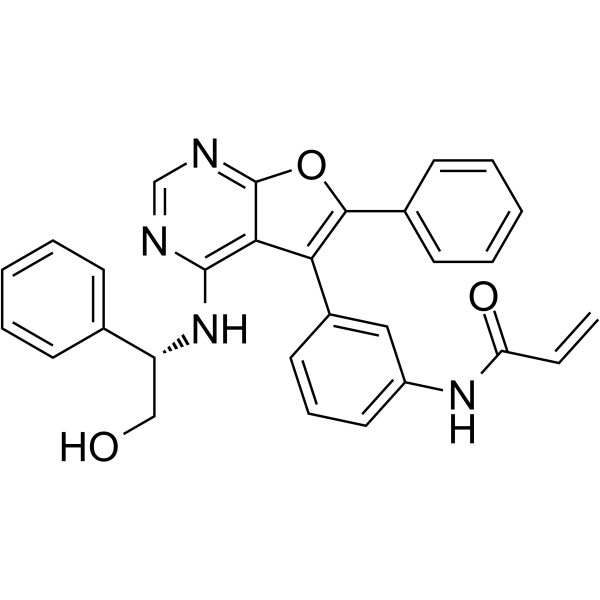
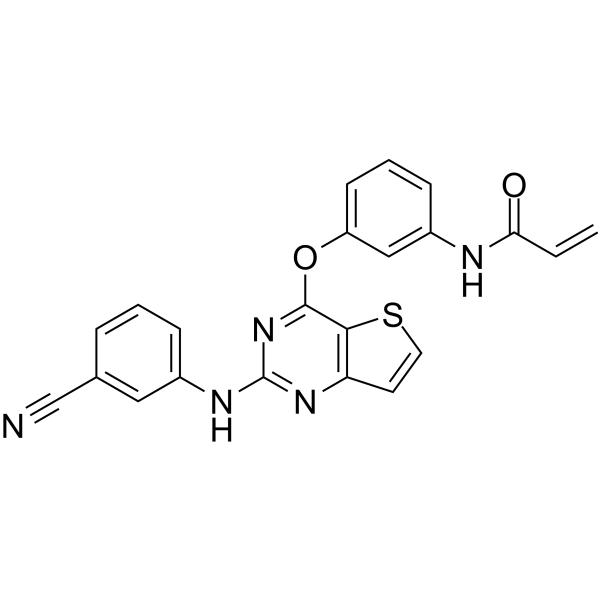
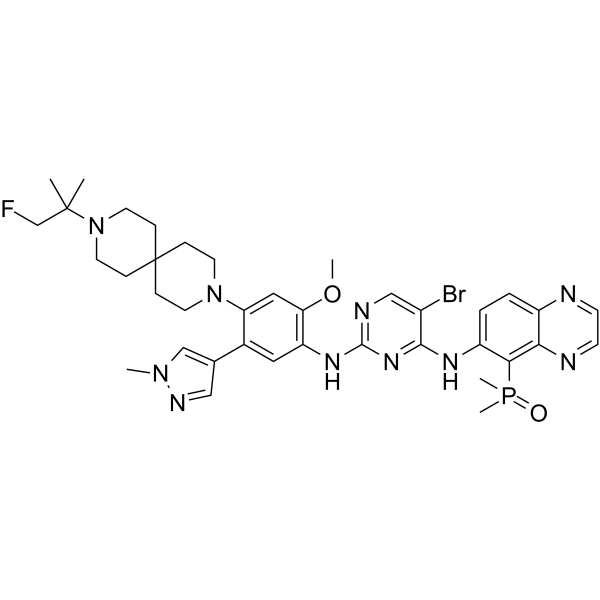
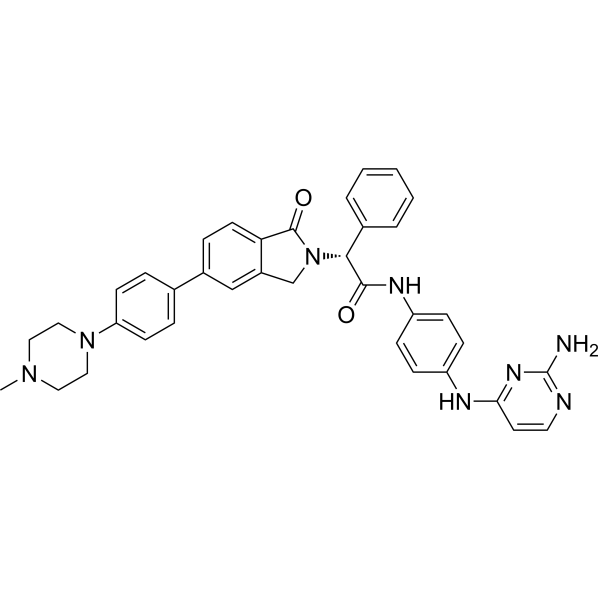
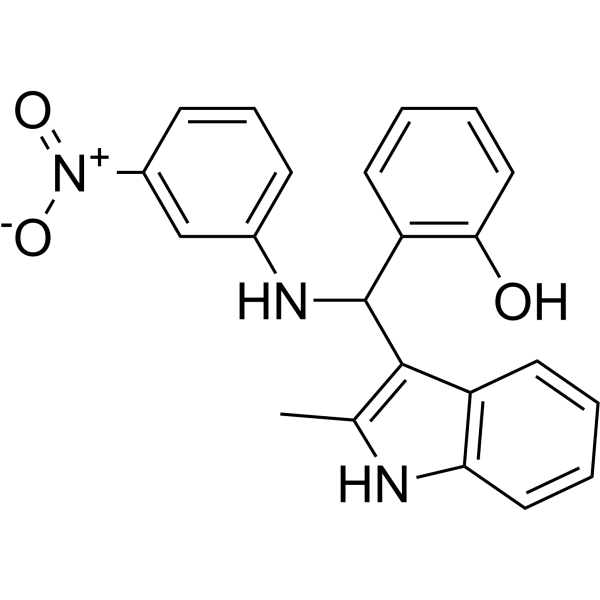
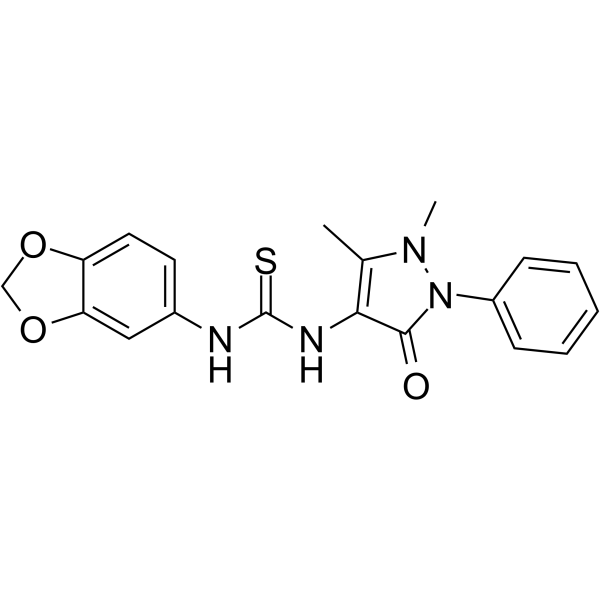
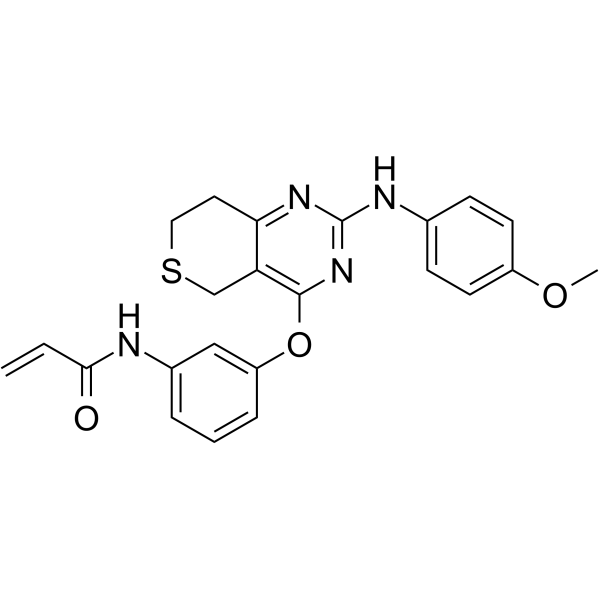
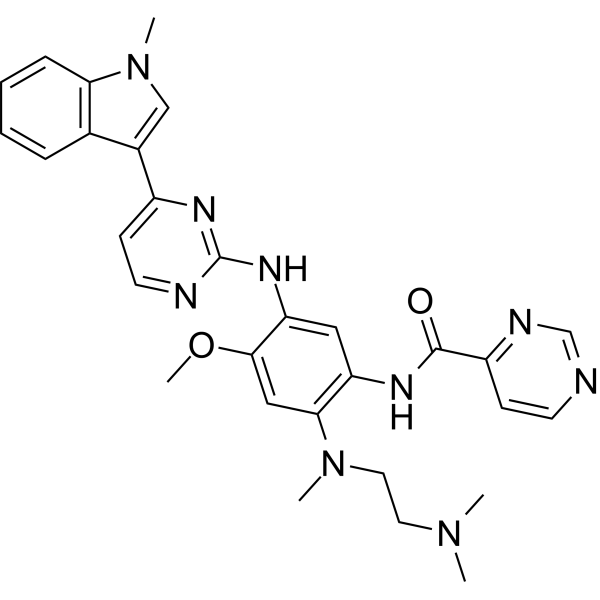
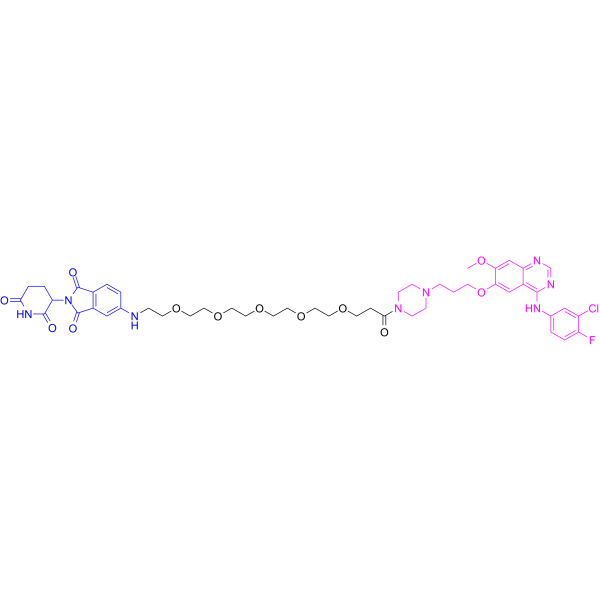
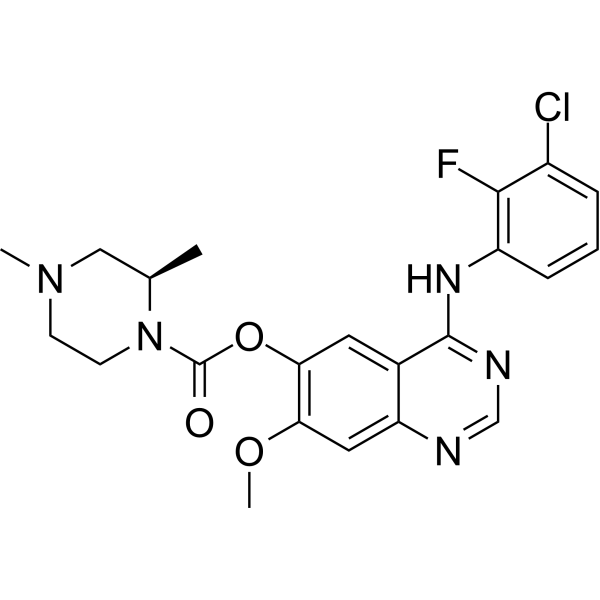
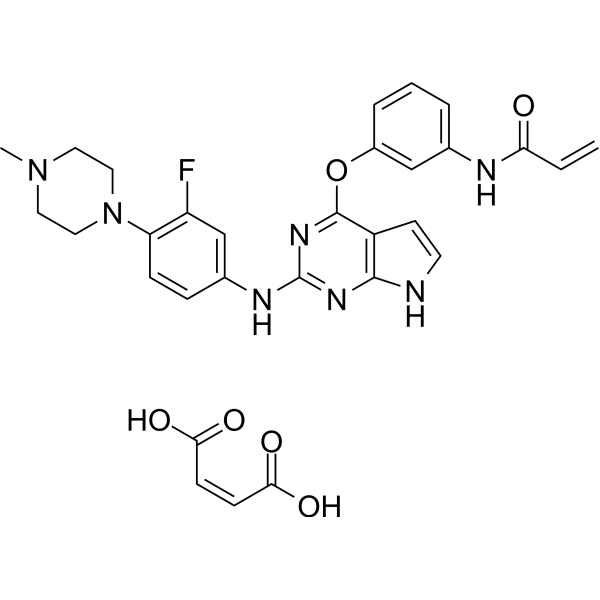
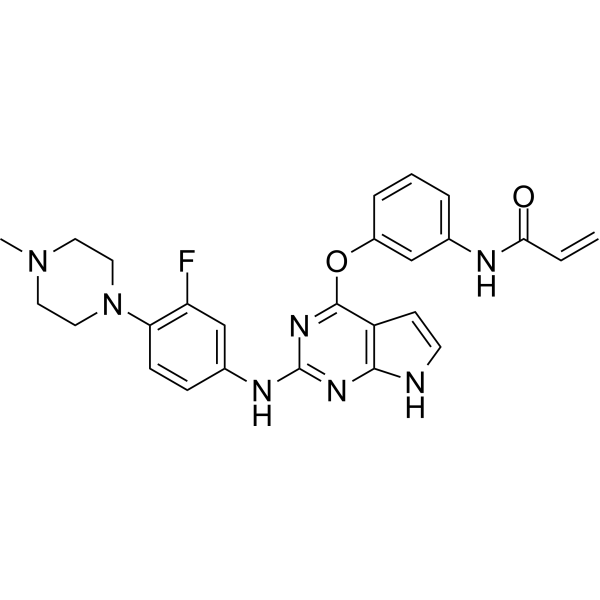
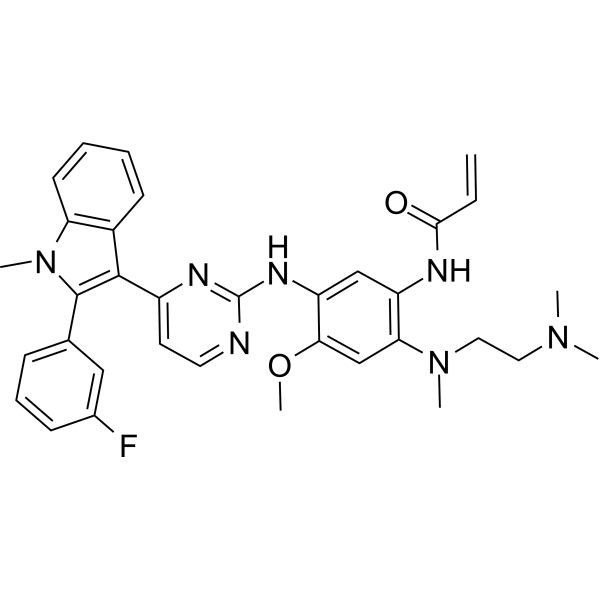
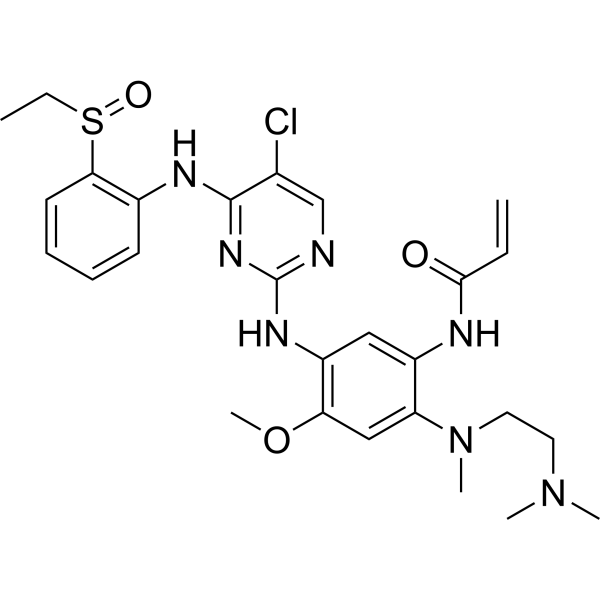
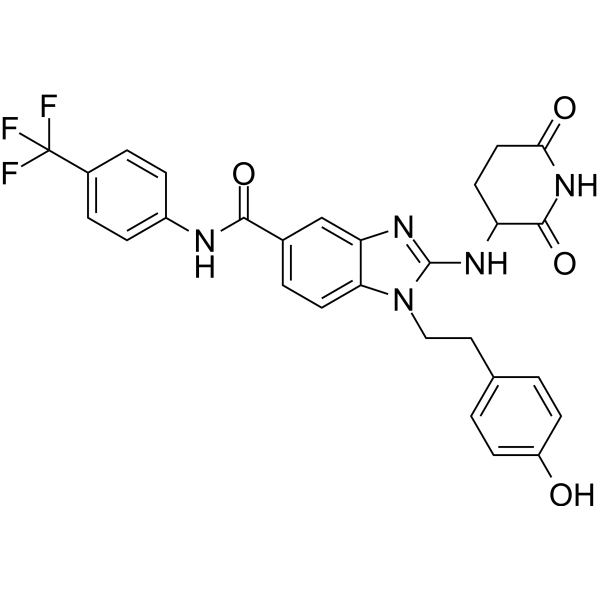
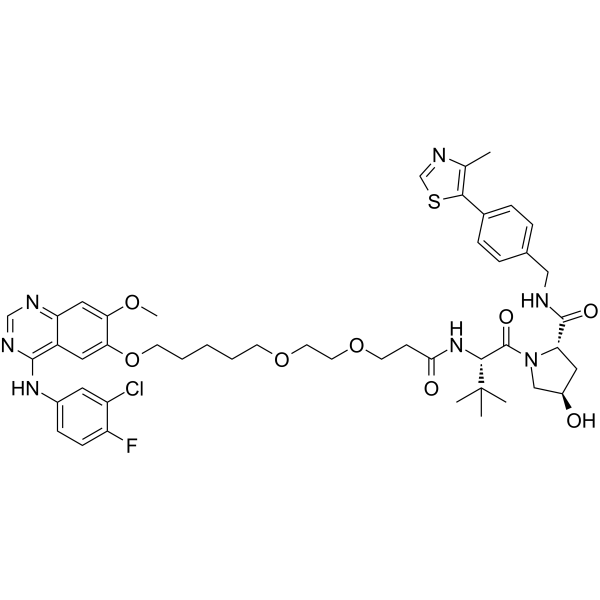
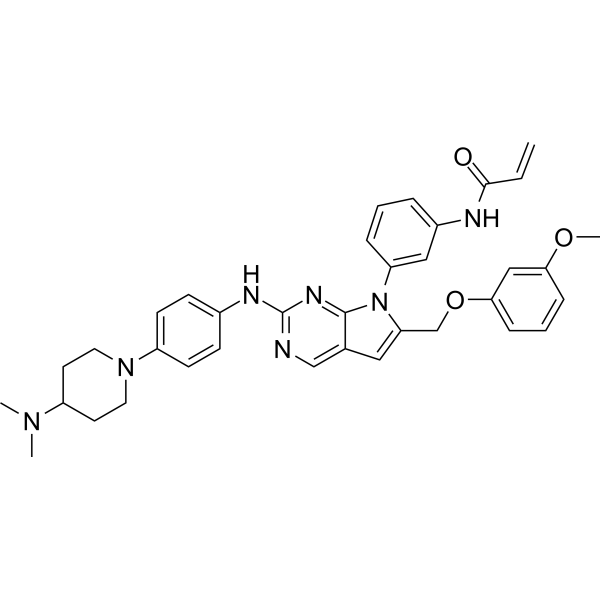
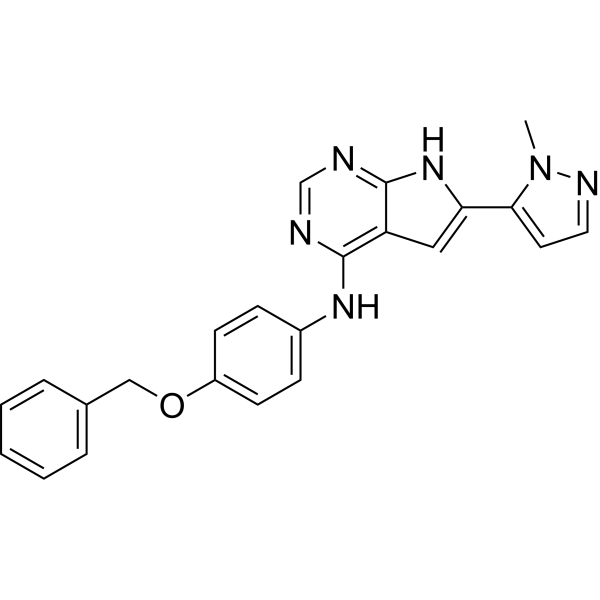
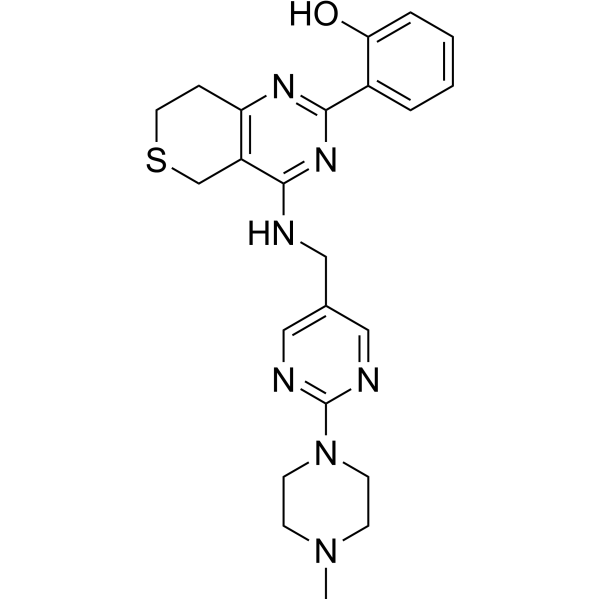
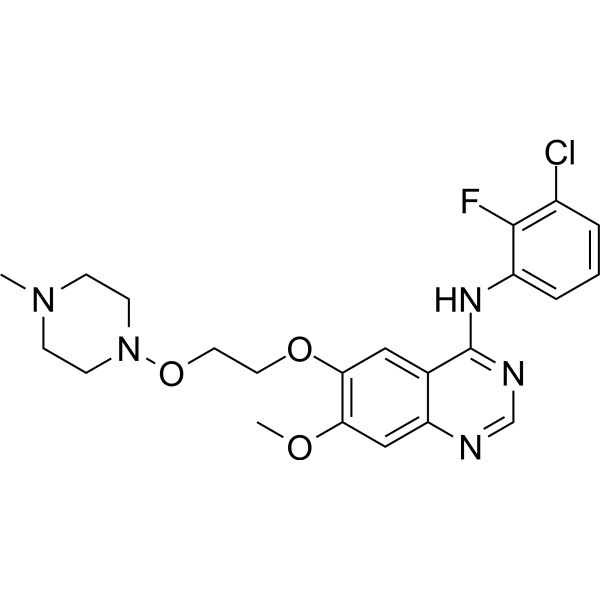
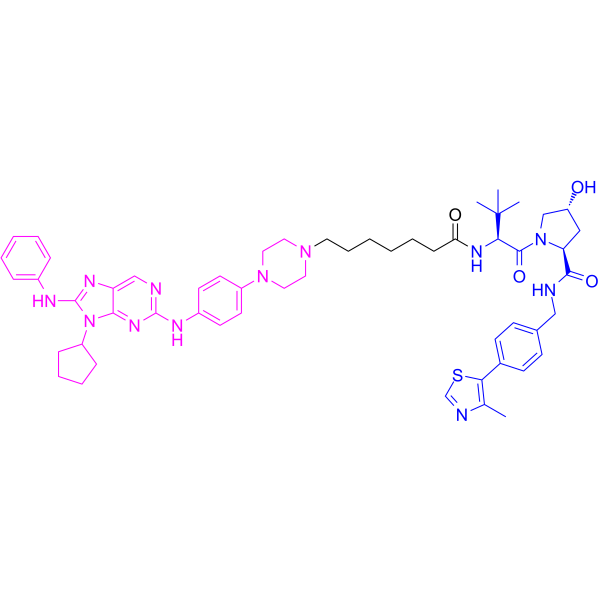
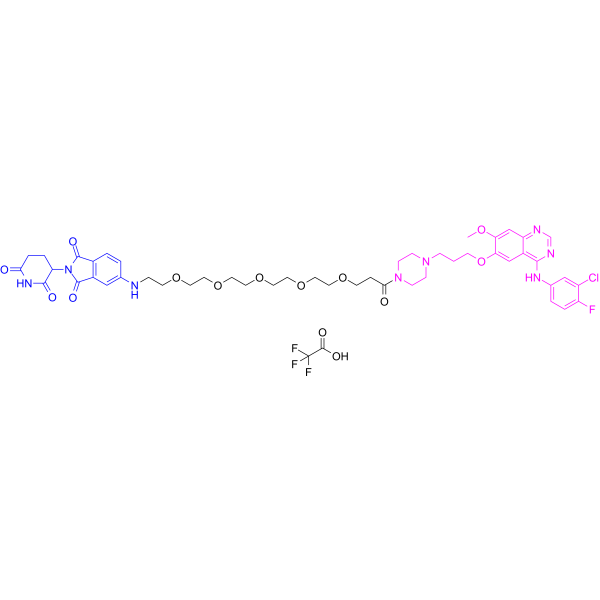
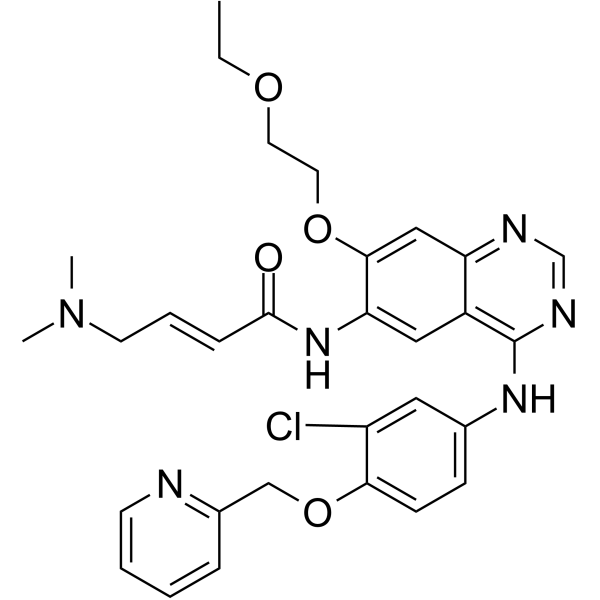
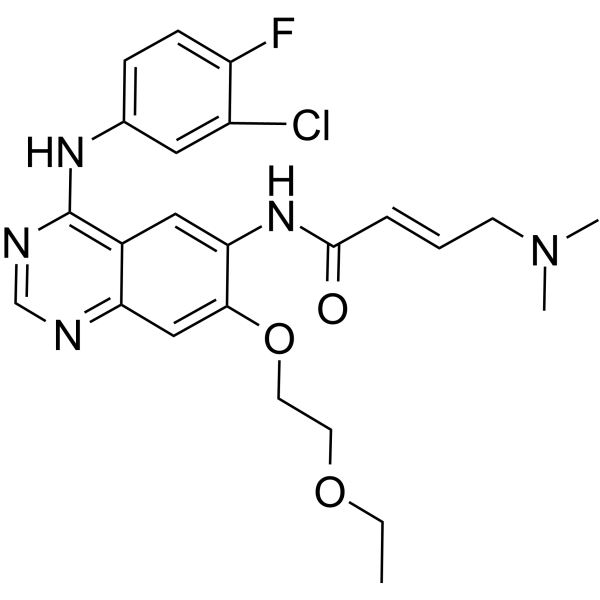
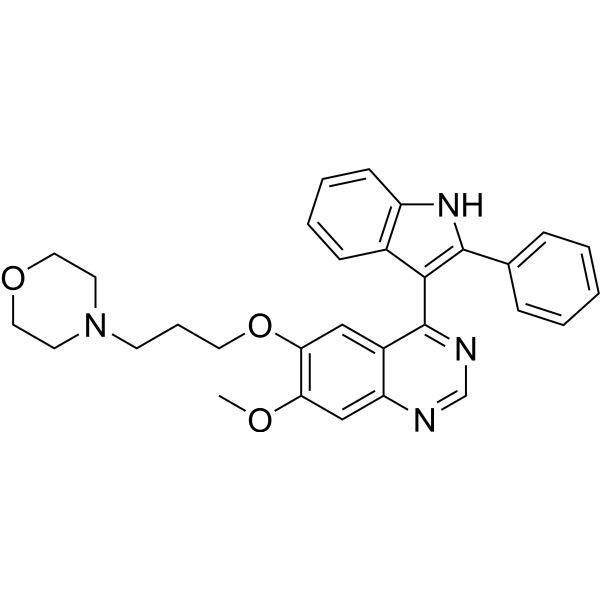
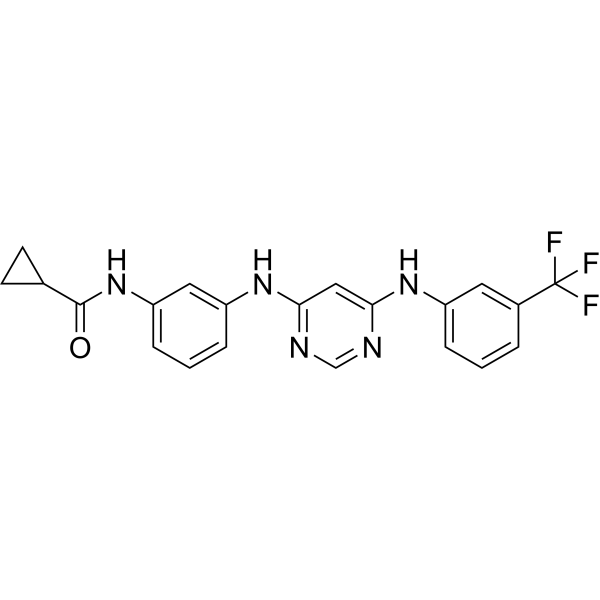
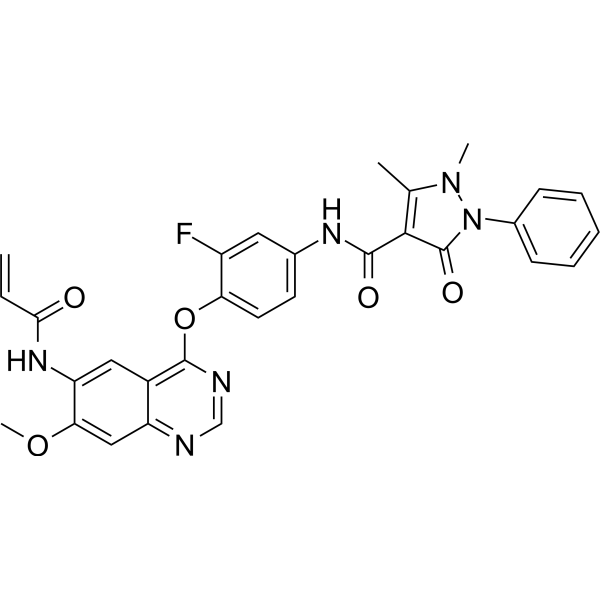
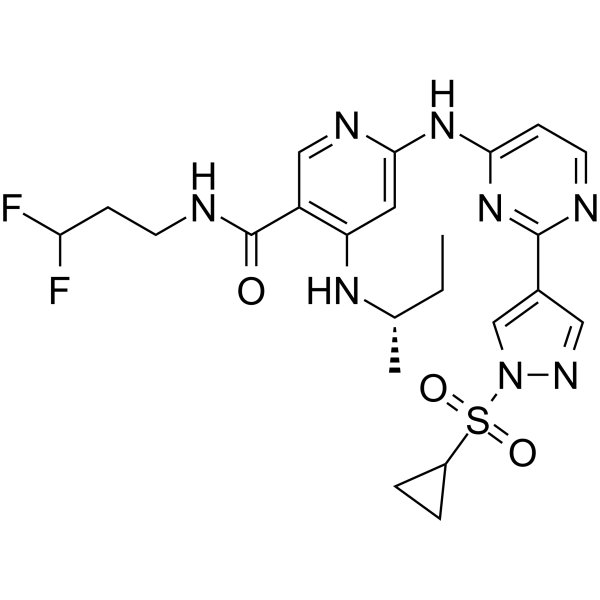
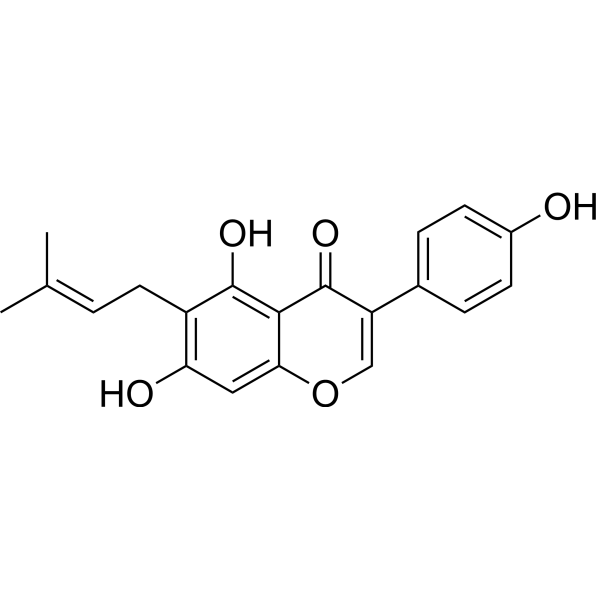
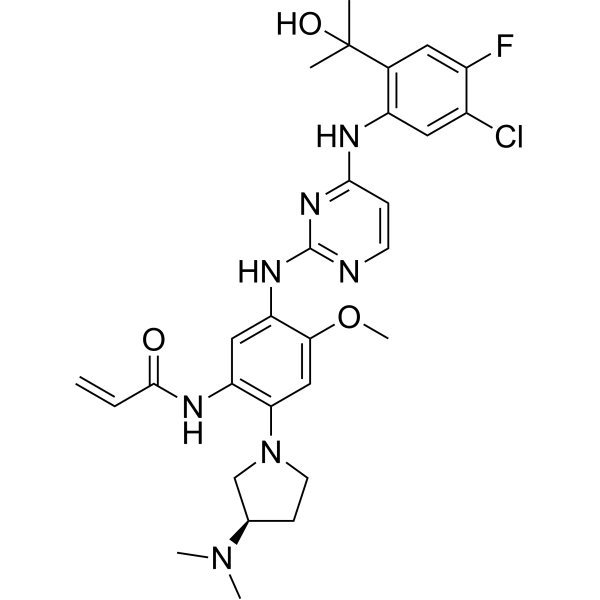
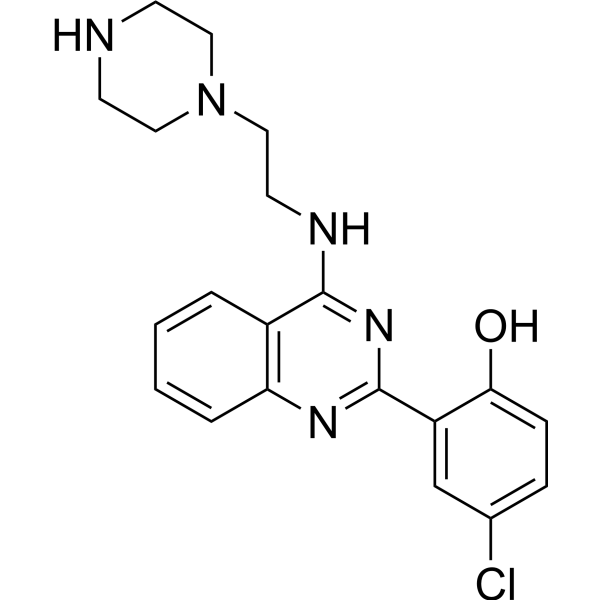
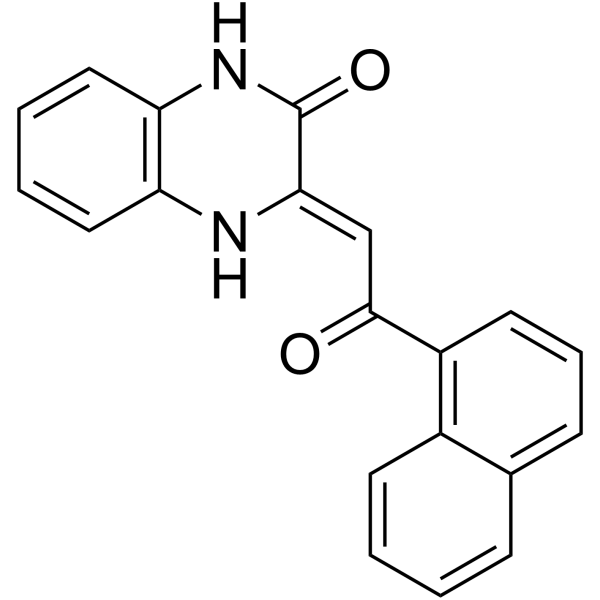
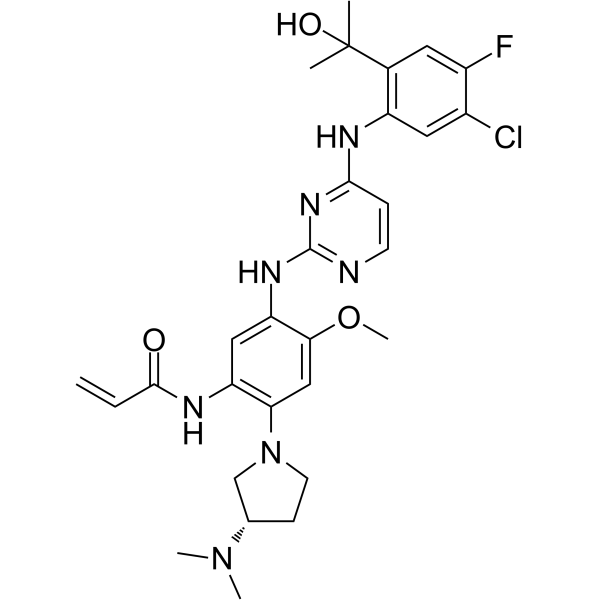
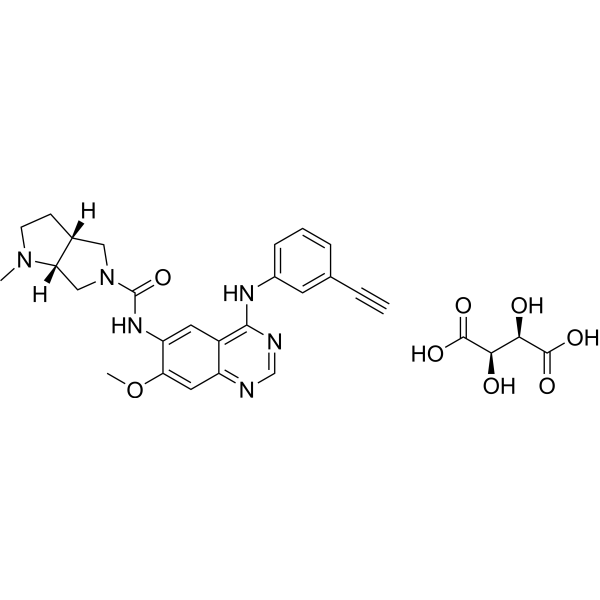
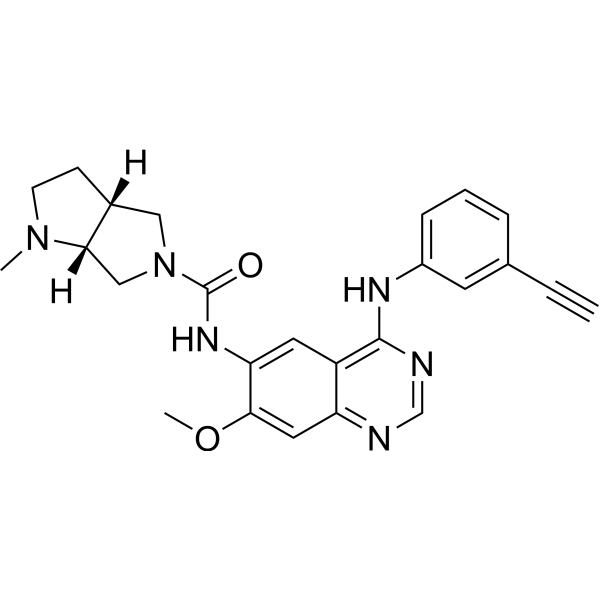
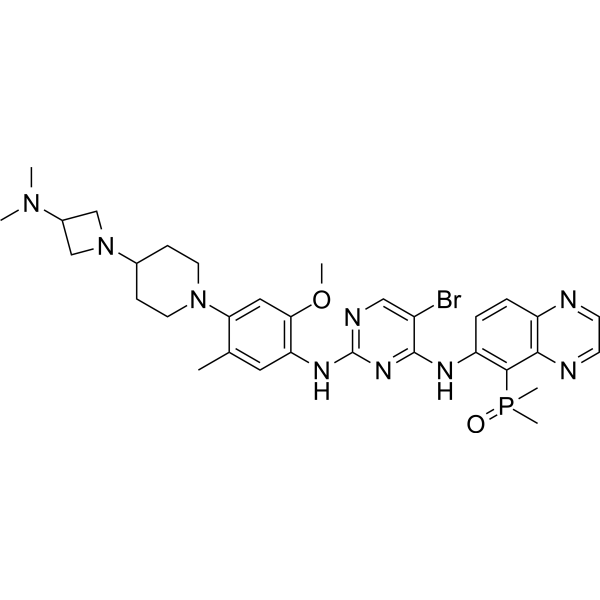
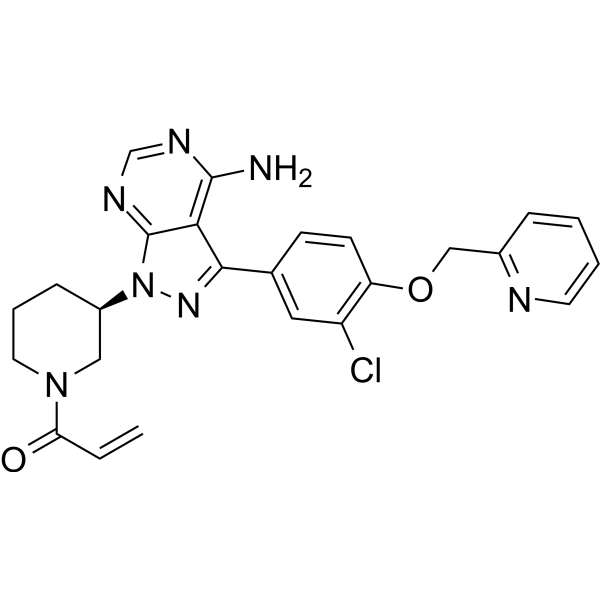
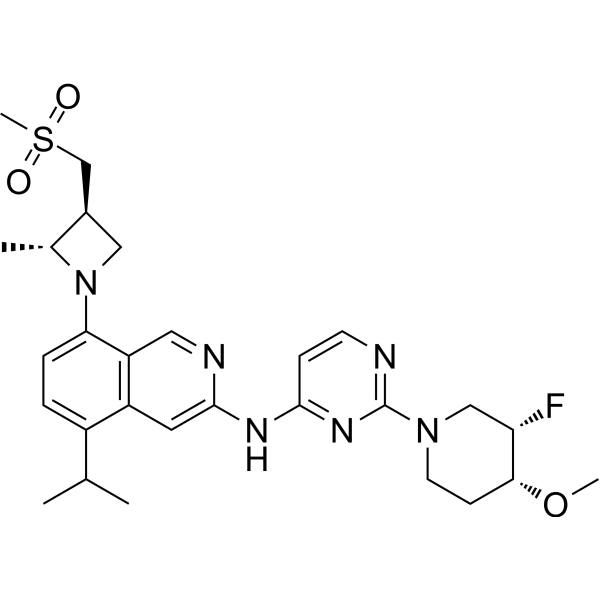
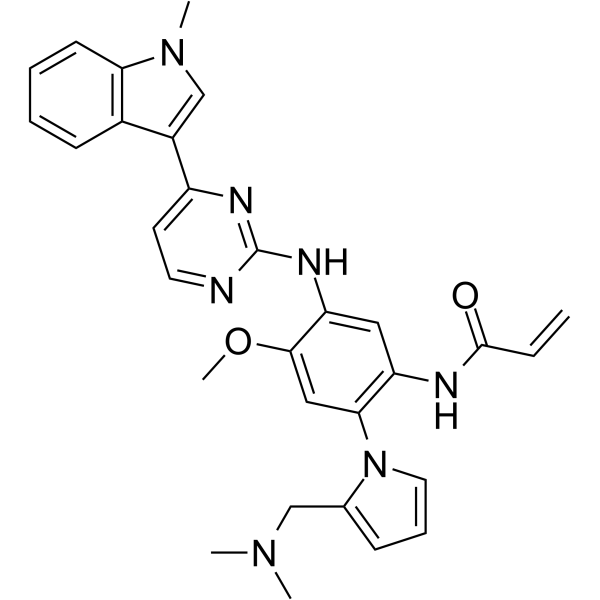
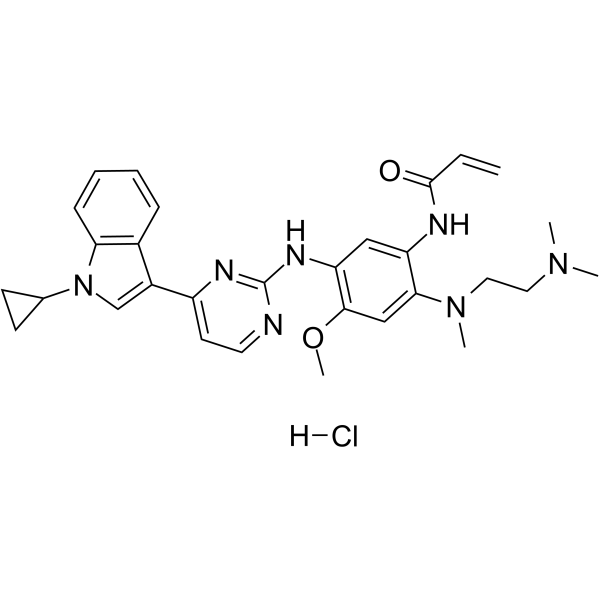
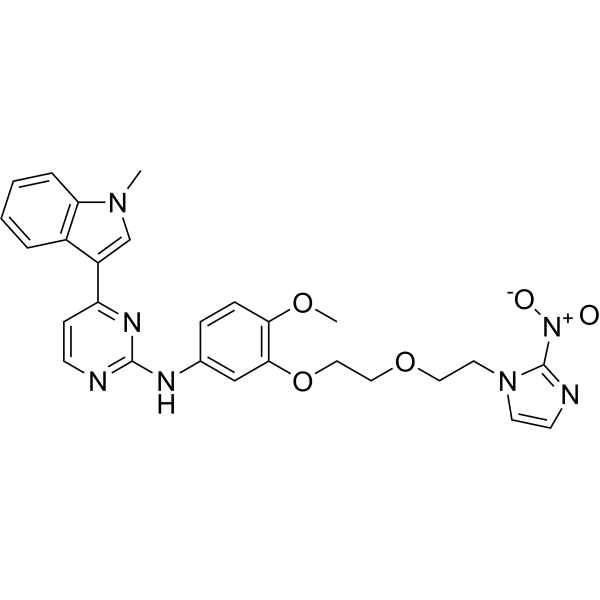
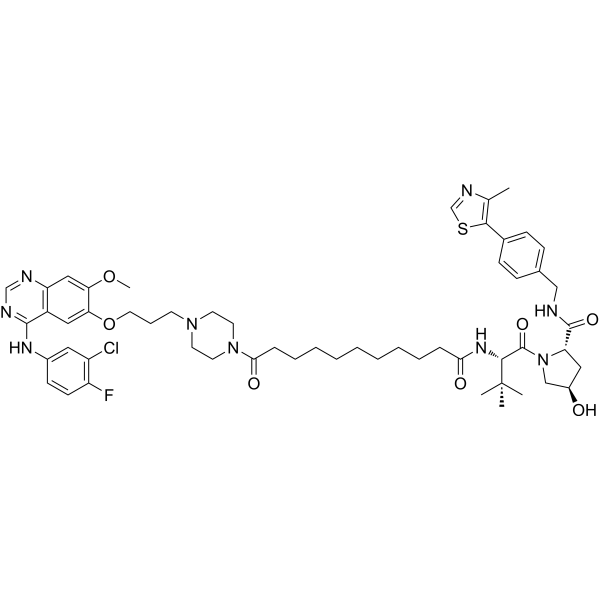
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
Afatinib (BIBW 2992) is an orally active, potent and irreversible dual specificity inhibitor of ErbB family (EGFR and HER2), with IC50 values of 0.5 nM, 0.4 nM, 10 nM and 14 nM for EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2, respectively. Afatinib can be used for the research of esophageal squamous cell carcinoma (ESCC), non-small cell lung cancer (NSCLC) and gastric cancer .
Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
Afatinib (BIBW 2992) dimaleate is an orally active, potent and irreversible dual specificity inhibitor of ErbB family (EGFR and HER2), with IC50 values of 0.5 nM, 0.4 nM, 10 nM and 14 nM for EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2, respectively. Afatinib dimaleate can be used for the research of esophageal squamous cell carcinoma (ESCC), non-small cell lung cancer (NSCLC) and gastric cancer .
AZ-5104 is an active, demethylated metabolite of AZD 9291. AZ-5104 is an EGFR inhibitor with IC50s of 1, 6, 1, 25 and 7 nM for EGFRL858R/T790M, EGFRL858R, EGFRL861Q, EGFR and ErbB4, respectively.
Osimertinib dimesylate (AZD-9291 dimesylate) is an irreversible and mutant selective EGFR inhibitor with IC50s of 12 and 1 nM against EGFRL858R and EGFRL858R/T790M, respectively.
Osimertinib (Standard) is the analytical standard of Osimertinib. This product is intended for research and analytical applications. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
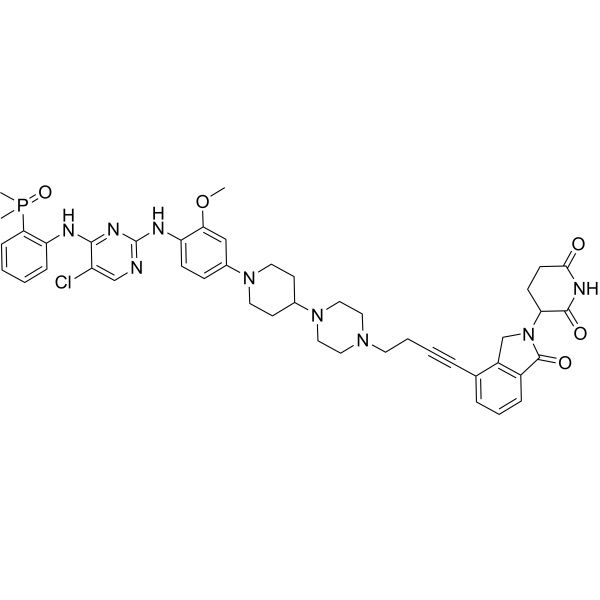
MS154 is a first-in-class E3 ligase cereblon-recruited EGFR degrader with Kd values of 1.8 nM and 3.8 nM for EGFRWT and EGFRL858R mutant, respectively. MS154 potently induces degradation of mutant, but not wild-type, EGFR in cancer cell lines in an E3 ligase-dependent manner. MS154 exhibits anticancer effects against lung cancer (blue: E3 ligase ligand (HY-103596); black: linker (HY-W096167); pink: EGFR ligand (HY-168305)) .
Icotinib (BPI-2009) is a potent, CNS-penetrant and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
EAI045 is an allosteric and the fourth-generation inhibitor of mutant EGFR with IC50s of 1.9, 0.019, 0.19 and 0.002 μM for EGFR, EGFRL858R, EGFRT790M and EGFRL858R/T790M at 10 μM ATP, respectively.
Nazartinib (EGF816) is a covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min −1 on EGFR(L858R/790M) mutant, respectively.
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
Befotertinib (D-0316) is an orally active EGFR inhibitor and ABCB1 inhibitor. Befotertinib selectively targets EGFR mutations including EGFRT790M, EGFRL858R and delE746-A750, forms covalent bonds with EGFRC797, inhibits oncogenic signaling pathways, and exerts antiproliferative effects. Befotertinib inhibits ABCB1-mediated drug efflux, activates the ATPase activity of ABCB1, acts as a chemosensitizer and apoptosis enhancer, and restores the sensitivity of multidrug-resistant cancer cells. Befotertinib can be used in research related to multidrug-resistant cancers and non-small cell lung cancer .
WZ3146 is a mutant selective EGFR inhibitor with IC50s of
2, 2, 5, 14 and 66 nM for EGFRL858R, EGFRL858R/T790M, EGFRE746_A750, EGFRE746_A750/T790M and EGFR, respectively.
WZ4002 is a mutant selective EGFR inhibitor with IC50s of 2, 8, 3 and 2 nM for EGFRL858R, EGFRL858R/T790M, EGFRE746_A750 and EGFRE746_A750/T790M, respectively.
Icotinib Hydrochloride (BPI-2009) is a potent, CNS-penetrant and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib (Hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
JBJ-04-125-02 is a potent, mutant-selective, allosteric and orally active EGFR inhibitor with an IC50 of 0.26 nM for EGFRL858R/T790M. JBJ-04-125-02 can inhibit cancer cell proliferation and EGFRL858R/T790M/C797S signaling. JBJ-04-125-02 has anti-tumor activities .
Mavelertinib is a selective, orally available and irreversible EGFR tyrosine kinase inhibitor (EGFR TKI), with IC50s of 5, 4, 12 and 3 nM for Del, L858R, and double mutants T790M/L858R and T790M/Del, respectively. Mavelertinib can be used for the research of non-small-cell lung cancer (NSCLC) .
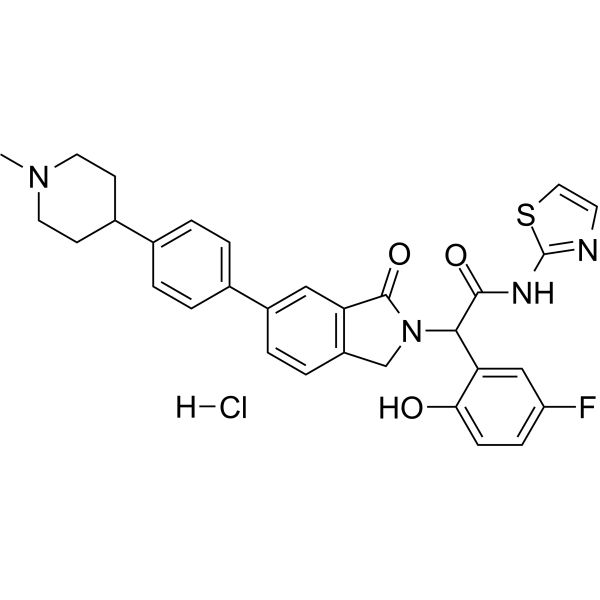
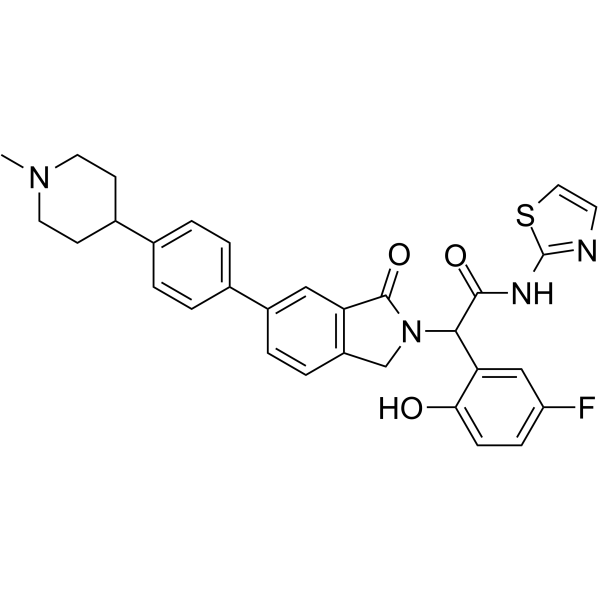
NT-1 (EGFR mutant-IN-3) is a potent mutant EGFR inhibitor and an analog of Osimertinib (HY-15772). This mutant EGFR inhibitor suppresses FGFR WT with an IC50 of 0.4 nM. NT-1 also inhibits EGFRL858R, EGFRExon 19 deletion and EGFRT790M. NT-1 exerts deeper inhibition on p-EGFR and p-ERK, and induces tumor cell apoptosis. NT-1 can be used in colorectal cancer research .
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
JND3229 is a reversible EGFRC797S inhibitor with IC50 values of 5.8, 6.8 and 30.5 nM for EGFRL858R/T790M/C797S, EGFRWT and EGFRL858R/T790M, respectively. JND3229 has good anti-proliferative activity and can effectively inhibit tumour growth in vivo. JND3229 can be used in cancer research, especially in non-small cell carcinoma .
JBJ-09-063 TFA is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFRL858R, EGFRL858R/T790M, EGFRL858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 TFA effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 TFA is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 TFA can be used for researching EGFR-mutant lung cancer .
JBJ-09-063 hydrochloride is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFRL858R, EGFRL858R/T790M, EGFRL858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 hydrochloride effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 hydrochloride is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 hydrochloride can be used for researching EGFR-mutant lung cancer .
EAI001 is a potent, selective mutant epidermal growth factor receptor (EGFR) allosteric inhibitor with an IC50 value of 24 nM for EGFRL858R/T790M. EAI001 can be used for research of cancer .
PF-6274484 is a potent EGFR inhibitor with Kis of 0.14 nM and 0.18 nM for EGFR-L858R/T790M and WT EGFR, respectively. PF-6274484 inhibits EGFR-L858R/T790M autophosphorylation in H1975 tumor cells and EGFR WT in A549 tumor cells with IC50s of 6.6 and 5.8 nM, respectively .
JBJ-09-063 is a mutant-selective allosteric EGFR inhibitor with IC50s of 0.147 nM, 0.063 nM, 0.083 nM and 0.396 nM for EGFRL858R, EGFRL858R/T790M, EGFRL858R/T790M/C797S and EGFRLT/L747S. JBJ-09-063 effectively reduces EGFR, Akt and ERK1/2 phosphorylation. JBJ-09-063 is effective across EGFR tyrosine kinase inhibitor (TKI)-sensitive and resistant models. JBJ-09-063 can be used for researching EGFR-mutant lung cancer .
Osimertinib (mesylate) (Standard) is the analytical standard of Osimertinib (mesylate). This product is intended for research and analytical applications. Osimertinib mesylate (AZD9291 mesylate) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFRL858R Recombinant Human Active Protein Kinase is a recombinant EGFRL858R protein that can be used to study EGFRL858R-related functions .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR C797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR C797S/L858R protein that can be used to study EGFR C797S/L858R-related functions .
PROTAC EGFR degrader 12 (example 1) is a PROTAC degrader targeting EGFR that can effectively degrade EGFR mutants, but has little effect on EGFR WT. PROTAC EGFR degrader 12 shows IC50s against EGFRL858R-T790M (NCI-H1975 cells), EGFRL858R (NCI-H3255 cells), and EGFRL858R-T790M-L797S (NCI-H1975+CS cells) of all <50 nM .
AV-412 free base (MP-412 free base) is an EGFR inhibitor with IC50s of 0.75, 0.5, 0.79, 2.3, 19 nM for EGFR, EGFRL858R, EGFRT790M, EGFRL858R/T790M and ErbB2, respectively.
PROTAC EGFR degrader 16 (Compound 98) a selective EGFR PROTAC degrader with DC50 values of < 50 nM in NCI-H1975 (EGFRL858R-T970M), NCI-H3225 (EGFRL858R) and NCI-H1976 + CS (EGFRL858R-T970M-L797S). PROTAC EGFR degrader 16 can be used for the study of EGFR-driven cancerssuch as non-small cell lung cancer (Pink: EGFR ligand (HY-178313); Blue: CRBN ligand (HY-W1128702); Black: Linker; EGFR ligand + Linker (HY-178311)) .
Gozanertinib is an orally active furanopyrimidine-based EGFR inhibitor with IC50s of 15 nM and 48 nM for EGFRWT and EGFRL858R/T790M, respectively. Gozanertinib can occupy the ATP-binding site. Gozanertinib has significant antitumor efficacy .
EGFR-IN-126 (compound 9d) is a potent inhibitor of EGFRL858R/T790M/C797S, with the IC50 value of 0.005 μM. EGFR-IN-126 shows antitumor effects in vivo and in vitro .
EGFR T790M/L858R-IN-2 is a potent and selective EGFRT790M/L858R inhibitor with IC50 values of 3.5, 1290 nM for EGFRT790M/L858R, EGFR WT, respectively. EGFR T790M/L858R-IN-2 decreases the expression of p-EGFR, P-AKT, P-ERK1/2. EGFR T790M/L858R-IN-2 induces Apoptosis and cell cycle arrest in the G1 phase. EGFR T790M/L858R-IN-2 shows anti-cancer activity .
EGFR T790M/L858R-IN-7 (Compound 72) is a novel pyrimidine compound that inhibits the EGFR T790M and L858R mutation with a high efficacy (93% inhibition rate at 0.05 μM). EGFR T790M/L858R-IN-7 functions by specifically binding to the kinase domain of EGFR, thereby inhibiting its phosphorylation activity .
EGFR WT/T790M/L858R-IN-1 (compound 10d) is a potent EGFR inhibitor, with IC50s of 0.097, 0.280, and 0.051?μM for EGFRWT, EGFRT790M, and EGFRL858R, respectively. EGFR WT/T790M/L858R-IN-1 can be used for the research of cancer .
EGFR T790M/L858R-IN-8 (Compound 9) is a selective EGFRT790M/L858R mutant kinase inhibitor with a IC50 of 56.8 μM, and it shows no inhibitory activity against wild-type EGFR. EGFR T790M/L858R-IN-8 can be used for the research of non-small cell lung cancer .
EGFR T790M/L858R-IN-3 (compound B1) is a EGFRL858R/T790M inhibitor with IC50 value of 13?nM. EGFR T790M/L858R-IN-3 shows anti-tumour activity in H1975 cells with an IC50 value of 0.087 μΜ. EGFR T790M/L858R-IN-3 inhibits cell migration in A549 cells and induces apoptosis in H1975 cells .
EGFR T790M/L858R-IN-9 (Compound 8) is an EGFR-L858R/T790M inhibitor that demonstrates potent inhibitory phosphorylation effects against the EGFR-L858R/T790M mutant kinase, with an IC50 value of 0.0064µM. EGFR T790M/L858R-IN-9 also inhibits the proliferation of non-small cell lung cancer (NSCLC) cells and can be utilized in cancer research .
EGFR T790M/L858R-IN-5 (example 52) is a potent inhibitor of EGFR T790M/L858R with the inhibition percentage of 92.9% in the concerntration of 0.05 μM .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR T790M/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR T790M/L858R protein that can be used to study EGFR T790M/L858R-related functions .
EGFR ligand-2 (compound C4), a covalent EGFR ligand, is a EGFR mutant inhibitor with IC50s of 21 nM and 48 nM for EGFRL858R and EGFRL858R/T790M, respectively. EGFR ligand-2 can be used to synthesize PROTAC .
Afatinib impurity 11 is an impurity of Afatinib. Afatinib is an irreversible EGFR family inhibitor with IC50s of 0.5 nM, 0.4 nM, 10 nM and 14 nM for EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2, respectively .
EGFR T790M/L858R/ACK1-IN-1 is a dual inhibitor of EGFRT790M/L858R and ACK1. IC50 values are 23 and 263 nM, respectively. EGFR T790M/L858R/ACK1-IN-1 can inhibit cell proliferation and has antitumor activity .
EGFR-IN-177 is a potent EGFR inhibitor with IC50s of 0.32, 1.04, 0.65, 0.67, 0.48, 0.55 and 0.38 nM against EGFRL858R, EGFRL858R/T790M, EGFRL858R/T790M/C797S, EGFR D746-750, EGFR D746-750/C797S, EGFR D770_N77linsNPG, and EGFR WT. EGFR-IN-177 inhibits lung cancer proliferation and EGFR phosphorylation. EGFR-IN-177 can be used for the study of cancers such as non-small cell lung cancer .
EGFR-IN-1 hydrochloride is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 hydrochloride potently inhibits Gefitinib-resistant EGFRL858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 hydrochloride displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
EGFR-IN-69 (compound 17g) is a potent EGFR inhibitor, with IC50 values of 4.3, 6.6 and 25.6 nM against EGFRL858R/T790M/C797S, EGFRL858R/T790M, and EGFR19del/T790M/C797S, respectively. EGFR-IN-69 can be used for non-small-cell-lung-cancer (NSCLC) research .
Afatinib (BIBW 2992) oxalate is an orally active, potent and irreversible dual specificity inhibitor of ErbB family (EGFR and HER2), with IC50 values of 0.5 nM, 0.4 nM, 10 nM and 14 nM for EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2, respectively. Afatinib oxalate can be used for the research of esophageal squamous cell carcinoma (ESCC), non-small cell lung cancer (NSCLC) and gastric cancer .
Osimertinib dimer is a dimer of Osimertinib (HY-15772). Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
(R)-Afatinib ((R)-BIBW 2992) is the Afatinib isomer. Afatinib (HY-10261) is an orally active, potent and irreversible dual specificity inhibitor of?ErbB?family (EGFR?and?HER2), with?IC50?values of 0.5 nM, 0.4 nM, 10 nM and 14 nM for?EGFRwt, EGFRL858R,?EGFR L858R/T790M?and HER2, respectively. Afatinib can be used for the research of esophageal squamous cell carcinoma (ESCC), non-small cell lung?cancer?(NSCLC) and gastric?cancer .
EGFR-IN-30 is a potent EGFR inhibitor with IC50s of 1-10 nM, <1 nM for EGFR (WT), EGFR (L858R/T790M/C797S), respectively. EGFR-IN-30 has potential for cell proliferative diseases, such as cancer research .
EGFR-IN-51 (Compound 6) is a potent EGFR inhibitor with IC50 values of 0.493, 102.60 and 461.63 µM against EGFR, EGFRL858R-TK and EGFR T790M-TK, respectively. EGFR-IN-51 shows cytotoxic activity against cancer cell lines and induces apoptosis .
ZSH-2117 is a covalent and selective EGFRPROTAC degrader with a DC50 of 45 nM in Ba/F3-EGFRL858R/T790M/C797S cells. ZSH-2117 significantly inhibits cell proliferation and reduces the downstream EGFR signaling proteins level of AKT and ERK. ZSH-2117 effectively inhibits tumor growth in Ba/F3-EGFRL858R/T790M/C797S xenograft mice model . Pink: EGFR ligand (HY-175162); Blue: NEDD4 ligase ligand (HY-175159); Black: linker
EGFR-IN-179 (Compound 8d) is an EGFR inhibitor (IC50: 0.068 μM for EGFRL858R/T790M/C797S; 2.56 μM for EGFR-WT-TK). EGFR-IN-179 has anticancer activity against non-small cell lung cancer .
Avitinib (Abivertinib) maleate dihydrate is a third-generation, irreversible and orally active selective EGFR inhibitor, with IC50 values of 0.18 nM, 0.18 nM, 7.68 nM and against EGFRL858R, EGFR T790M and wild-type EGFR. Avitinib maleate dihydrate is also a BTK inhibitor that induces apoptosis and inhibits phosphorylation of BTK in mantle cell lymphoma. Avitinib maleate dihydrate shows anticancer effects .
EGFR-IN-27 is a potent EGFR inhibitor with IC50s of <50 nM for EGFR Del, L858R, Del/T790M, L858R/T790M, Del/T790M/C797S, and L858R/T790M/C797S, respectively (WO2021249324A1, compound 511) .
Mutated EGFR-IN-3 (compound 3) is a potent, ATP-competitive and highly selective allosteric dibenzodiazepinone inhibitor of the EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) mutants with IC50 values of 12 nM and 13 nM, respectively .
EGFR-IN-1 TFA is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 TFA potently inhibits Gefitinib-resistant EGFRL858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 TFA displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
EGFR-IN-1 (compound 24) is an orally active and irreversible L858R/T790M mutant selective EGFR inhibitor. EGFR-IN-1 potently inhibits Gefitinib-resistant EGFRL858R, T790M with 100-fold selectivity over wild-type EGFR. EGFR-IN-1 displays strong antiproliferative activity against the H1975 cells and the first line mutant HCC827 cells. Antitumor activity .
Icotinib (Standard) is the analytical standard of Icotinib. This product is intended for research and analytical applications. Icotinib (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
EGFR-IN-101 (I-10) is a 2-phenylamino pyrimidine derivative. EGFR-IN-101 is a EGFR inhibitor. The IC50 values for EGFRL858R/T790M/C797S and Ba/F3-EGFRL858R/T790M/C797S are 33.26 and 106.4 nM, respectively. EGFR-IN-101 can be used IN the study of non-small cell lung cancer (NSCLC) .
Icotinib-d4 is the deuterium-labeled Icotinib (HY-15164A). Icotinib-d4 (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib-d4 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
LSD1/EGFR-IN-1 (compound L-1) is a potent inhibitor of LSD1, EGFRT790M/L858R and EGFRL858R/T790M/C797S, with IC50s of 6.24 and 2.06 and 5.01 μM, respectively. LSD1/EGFR-IN-1 plays an important role in cancer research .
EGFR-IN-61 (compound 22a) is a potent EGFR kinase inhibitor, with IC50 values of 42 nM (L858R/T790 M), 137 nM (L858R/T790 M/C797S), and 743 nM (WT), respectively. EGFR-IN-61 shows antiproliferative activity against A549 and H1975 cell lines, with IC50 values of 2.14 and 1.82 μM, respectively .
Mutated EGFR-IN-2 (compound 91) is a mutant-selective EGFR inhibitor extracted from patent WO2017036263A1, which potently inhibits single-mutant EGFR (T790M) and double-mutant EGFR (including L858R/T790M (IC50=<1nM) and ex19del/T790M), and can suppress activity of single gain-of-function mutant EGFR (including L858R and ex19del) as well. Mutated EGFR-IN-2 shows anti-tumor antivity .
Neptinib (NEP010) is an orally active derivative of Afatinib (HY-10261) that has stronger antitumor activity than Afatinib (HY-10261) by improving pharmacokinetics. Neptinib has a significant inhibitory effect on tumor growth in mouse non-small cell lung cancer models with different EGFR mutations. Neptinib has a certain inhibitory effect on the EGFR kinase family, with IC50 values ??of 0.24 nM, 7.25 nM, 0.46 nM and 1.79 nM for EGFRwt, EGFRL858R/T790M, EGFRL858R and EGFRT790M, respectively .
EGFR-IN-93 (compound 18) is an allosteric inhibitor of T790M/L858R double mutant EGFR. EGFR-IN-93 can be used for non-small lung cancer (NSCLC) research .
Nazartinib mesylate (EGF816 mesylate) is a novel, covalent mutant-selective EGFR inhibitor, with Ki and Kinact of 31 nM and 0.222 min −1 on EGFR(L858R/790M) mutant, respectively.
EGFR-IN-155 (compound 13a) is an EGFR inhibitor with IC50 values of 0.14 nM and 0.18 nM against EGFR TK and EGFRL858R, respectively. EGFR-IN-155 inhibits tumor growth, causes a cell cycle arrest at S phase, and and induces cell apoptosis .
EGFR-IN-9 (Compound 8) is a potent EGFR kinase inhibitor with IC50s of 7 nM, 28 nM for the wild type EGFR kinase and double mutant EGFR kinase (L858R/T790M). EGFR-IN-9 has antitumor activity .
EGFR-IN-49 is a potent and selective EGFR inhibitor with IC50s of 65.0 nM and 13.6 nM for EGFRT790M and EGFRT790M/L858R, respectively. EGFR-IN-49 induces late apoptosis in a dose-dependent manner .
EGFR-IN-120 (Compound 11eg) is an orally active EGFR inhibitor. EGFR-IN-120 inhibits EGFRL858R/T790M/C797S with an IC50 value of 0.053 μM, and has a relatively weak effect on EGFRWT (IC50: 1.05 μM). EGFR-IN-120 inhibits the phosphorylation of EGFR and main downstream effectors (STAT3, AKT, and Erk). EGFR-IN-120 induces cell cycle arrest and cell apoptosis in EGFR mutant cells. EGFR-IN-120 inhibits the proliferation of the NSCLC cells harboring EGFRL858R/T790M/C797S with an IC50 of 0.052 μM .
EGFR-IN-22 is a potent EGFR inhibitor with IC50s of 4.91 nM and 0.54 nM for wild type EGFR and EGFRL858R/T790M/C797S, respectively (CN112538072A, compound 243) .
EGFR-IN-97 (compound 6q) is a EGFR inhibitor. EGFR-IN-97 shows inhibitory activity against Ba/F3-EGFRL858R/T790M/C797S and Ba/F3-EGFRDel19/T790M/C797S cells, with IC50 values of 0.42 μM and 0.41 μM, respectively. EGFR-IN-97 can promote apoptosis of NCI-H1975-EGFRL858R/T790M/C797S cells at the concentration of 0.8 μM .
EGFR-IN-139 (compound PD 18) is an epidermal growth factor receptor (EGFR) inhibitor, with IC50s of 12.88 (wild type), 10.84 (L858R/T790M), 42.68 (L858R/T790M/C797S) nM, respectively. EGFR-IN-139 displays strong anticancer activity against A549 and H1975 cancer cell lines, which are highly expressed EGFR. EGFR-IN-139 has a strong selectivity to cancer cells. EGFR-IN-139 can be used for nonsmall cell lung cancer (NSCLC) research[1].
EGFR-IN-52 (Compound 4) is a potent EGFR inhibitor with IC50 values of 0.358, 86.02 and 432.67 µM against EGFR, EGFRL858R-TK and EGFR T790M-TK, respectively. EGFR-IN-52 shows cytotoxic activity against cancer cell lines and induces apoptosis .
EGFR-IN-133 (Compound 24) is an inhibitor for EGFR, that inhibits the EGFR wildtype, L858R/T790M, d19/T790M, L858R/T790M/C797S, and d19/T790M/C797S mutans with IC50 of 0.1, 0.044, 0.036, 0.04, and 0.054 nM. EGFR-IN-133 exhibits good pharmacokinetics characteristics with high oral exposure .
EGFR-IN-132 (Compound 23) is an inhibitor for EGFR, that inhibits the EGFR wildtype, L858R/T790M, d19/T790M, L858R/T790M/C797S, and d19/T790M/C797S mutans with IC50 of 1.6, 0.025, 0.019, 0.022, and 0.029 nM. EGFR-IN-132 exhibits good pharmacokinetics characteristics with high oral exposure .
EGFR is a driver of tumorigenesis. EGFR is mainly found in an auto-inhibited, dimerization-incompetent, state at the plasma membrane (PM). Ligand binding promotes receptor dimerization, which determines a series of structural rearrangements that are conveyed to the cytoplasmic domain allowing the formation of asymmetric dimers between the two juxtaposed catalytic domains. EGFR has multiple mutants. EGFR d746-750/T790M/C797S/L858R Recombinant Human Active Protein Kinase is a recombinant EGFR d746-750/T790M/C797S/L858R protein that can be used to study EGFR d746-750/T790M/C797S/L858R-related functions .
EGFR-IN-56 (Compound 13a) is a potent EGFR inhibitor with IC50 values of 541.7 nM and 132.1 nM against EGFRT790M and EGFRT790M/L858R, respectively. EGFR-IN-56 blocks cancer cells in G2/M phase and induce into late apoptosis .
EGFR-IN-77 (Compound 4a) is a selective EGFRT790M/L858R inhibitor, with an IC50 of 0.101 μM against EGFRT790M/L858R, 0.477 μM against EGFRL858R, and 1.771 μM against wild-type EGFR. EGFR-IN-77 exerts selective antiproliferative effects on EGFRT790M/L858R non-small cell lung cancer. EGFR-IN-77 can be used for the research of EGFRL858R/T790M double-mutant non-small cell lung cancer .
EGFR-IN-451 is an EGFR inhibitor with an IC50 value of 0.02 nM. EGFR-IN-451 also inhibits mutant EGFRL858R, EGFRT790M, and EGFRL858R/T790M with IC50 values of 0.26-34 nM. EGFR-IN-451 inhibits AKT and ERK activation and inhibits proliferation of EGFR-mutant cancer cells. EGFR-IN-451 can be used for the research of EGFR-driven cancer .
EGFR-IN-208 is an allosteric mutant EGFRL858R/T790M and EGFRL858R/T790M/C797S inhibitor, with IC50 values of 3.06 μM and 1.08 μM, respectively. EGFR-IN-208 binds to the allosteric site of EGFR and inhibits EGFR phosphorylation. EGFR-IN-208 induces apoptosis and exhibits antiproliferative effects in cancer cells. EGFR-IN-208 can be used in research related to non-small cell lung cancer .
EGFR-IN-199 is a selective and potent EGFR inhibitor with IC50 values of 1.1 nM and 1.4 nM for purified EGFR WT and EGFRT790M/L858R kinases. EGFR-IN-199 can be used for the research of cancer .
EGFR-IN-87 (Compound Example 2) is a EGFR tyrosine kinase inhibitor. EGFR-IN-87 has IC50 value of 3.1 nM, 1.3 nM and 7.1 nM for EGFR_d746-750, EGFR_L858R/T790 and EGFR_WT in A431 cells, respectively. EGFR-IN-87 can be used for cancer diseases research .
EGFR-IN-87 (Compound Example 2) hydrochloride is a EGFR tyrosine kinase inhibitor. EGFR-IN-87 hydrochloride has IC50 value of 3.1 nM, 1.3 nM and 7.1 nM for EGFR_d746-750, EGFR_L858R/T790 and EGFR_WT in A431 cells, respectively. EGFR-IN-87 hydrochloride can be used for cancer diseases research .
EGFR-IN-47 is a potent and orally active EGFRL858R/T790M/C797S inhibitor with an IC50 of 0.01 μM. EGFR-IN-47 induces cell cycle attest and cell apoptosis. EGFR-IN-47 has the potential for the research of NSCLC .
EGFR-IN-164 (Compound 4) is a selective and covalent allosteric EGFR inhibitor. EGFR-IN-164 significantly inhibits the activity of EGFRL858R/T790M/C797S kinase (IC50: 48.1 nM) and proliferation of of EGFR-mutant cells. EGFR-IN-164 can be used for drug resistance of cancer research .
EGFR-IN-7 is a potent, selective and orally active EGFR kinase inhibitor. EGFR-IN-7 has inhibitory effect for for EGFR (WT) and EGFR (mutant C797S/T790M/L858R) with IC50 values of 7.92 nM and 0.218 nM, respectively. EGFR-IN-7 can be used for the research of various cancers .
EGFR-IN-11 is a fourth-generation EGFR-tyrosine kinase inhibitor (EGFR-TKI) with an IC50 of 18 nM for triple mutant EGFRL858R/T790M/C797S. EGFR-IN-11 significantly suppresses the EGFR phosphorylation, induce the apoptosis, and arrest cell cycle at G0/G1 .
EGFR-IN-172 is a EGFR inhibitor. EGFR-IN-172 effectively inhibits the proliferation of non-small cell lung cancer (NSCLC) cells carrying the L858R, T790M and C797S drug-resistant mutations. EGFR-IN-172 inhibits EGFR phosphorylation, induces cell cycle arrest and apoptosis. EGFR-IN-172 can be used for the study of NSCLC .
EGFR-IN-55 (Compound 8a) is a potent EGFR inhibitor with IC50 values of 70 nM and 3.9 nM against EGFRWT and EGFRL858R/T790M, respectively. EGFR-IN-55 arrests NCI-H1975 cells in G0/G1 phase and shows anticancer activity .
EGFR mutant-IN-1, a 5-methylpyrimidopyridone derivative, is a potent and selective EGFRL858R/T790M/C797S mutant inhibitor with an IC50 of 27.5 nM, while being a significantly less potent for EGFRWT (IC50 >1.0 μM) .
MS9449 is a potent PROTAC EGFR degrader with Kds of 17 nM and 10 nM for EGFR WT and EGFRL858R, respectively. MS9449 effectively induces degradation of mutant EGFRs through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9449 potently inhibits the proliferation of NSCLC cells. MS9449 can be used for researching anticancer .
Zorifertinib (AZD3759) hydrochloride is a potent, orally active, BBB-penetrant, EGFR inhibitor (IC50s: 0.3, 0.2, and 0.2 nM for EGFRwt, EGFRL858R, and EGFRexon 19Del, respectively). Zorifertinib hydrochloride induces cancer cell apoptosis. Zorifertinib hydrochloride has antitumor activity, and can be used for NSCLC, HCC etc. research .
EGFR-IN-62 (compound 9h) is a potent and reversible EGFR kinase inhibitor, with IC50 values of 10 nM (L858R/T790 M), 29 nM (WT), and 242 nM (L858R/T790 M/C797S), respectively. EGFR-IN-62 shows antiproliferative activity against A549 and H1975 cell lines, with IC50 values of 2.53 and 1.56 μM, respectively. EGFR-IN-62 induces dose-dependent apoptosis process, G1/G0-phase arrestation, and the inhibition of motility on A549 and/or H1975 cell lines .
EGFR-IN-183 (Compound 5q) is an EGFR inhibitor. EGFR-IN-183 strongly binds to EGFR (T790M/L858R) mutants. EGFR-IN-183 exhibits anti-proliferative activity against MDA-MB-231, with an IC50 value of 16.4 μM. EGFR-IN-183 can be used for research on triple-negative breast cancer .
EGFR-IN-197 is an EGFR inhibitor with IC50 values of 19.5 nM and 12.0 nM against EGFRL858R/T790M and EGFRL858R/T790M/C797S, respectively. EGFR-IN-197 arrests the cell cycle of NCI-H1975 cells at the G2/M phase, while inhibiting their proliferation, colony formation and migration; it also inhibits mitochondrial translocation and upregulates mitochondrial H2S levels. EGFR-IN-197 disrupts anti-apoptotic signaling pathways by regulating apoptosis-related proteins; it induces DNA damage and activates pro-apoptotic pathways to trigger apoptosis. EGFR-IN-197 can be used in studies related to non-small cell lung cancer (NSCLC) .
MS9427 is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFRL858R, respectively. MS9427 selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 potently inhibits the proliferation of NSCLC cells. MS9427 can be used for researching anticancer .
Zorifertinib (AZD3759) is a potent, orally active, BBB-penetrant, EGFR inhibitor. At Km ATP concentrations, the IC50s are 0.3, 0.2, and 0.2 nM for EGFRwt, EGFRL858R, and EGFRexon 19Del, respectively. Zorifertinib induces cancer cell apoptosis. Zorifertinib has antitumor activity, and can be used for NSCLC, HCC etc. research .
Avitinib (Abivertinib) maleate is a third-generation, irreversible and orally active selective EGFR inhibitor, with IC50 values of 0.18 nM, 0.18 nM, 7.68 nM and against EGFRL858R, EGFR T790M and wild-type EGFR. Avitinib maleate is also a BTK inhibitor that induces apoptosis and inhibits phosphorylation of BTK in mantle cell lymphoma. Avitinib maleate shows anticancer effects .
Avitinib (Abivertinib) is a third-generation, irreversible and orally active selective EGFR inhibitor, with IC50 values of 0.18 nM, 0.18 nM, 7.68 nM and against EGFRL858R, EGFR T790M and wild-type EGFR. Avitinib is also a BTK inhibitor that induces apoptosis and inhibits phosphorylation of BTK in mantle cell lymphoma. Avitinib shows anticancer effects .
EGFR-IN-107 (compound 3r) is an orally active EGFR inhibitor with IC50 values of 0.4333 μM for EGFRWT and 0.0438 μM for EGFRL858R/T790M. EGFR-IN-107 has anti-proliferative activity and can inhibit the proliferation of H1975 cells and induce their apoptosis. EGFR-IN-107 can be used in cancer research .
ALK/EGFR-IN-1 (Compound 8l) is an ALK/EGFR dual inhibitor that blocks the phosphorylation of EGFR and ALK. ALK/EGFR-IN-1 inhibits ALK/EGFR mutants respectively, with IC50 of 4.3 nM for EGFRL858R T790M in H1975 cells and EML4-ALK in BaF3 cells, respectively. and 3.6 nM. ALK/EGFR-IN-1 may be used in NSCLC research .
EGFR-IN-154 (compound 4c) is an EGFR inhibitor with EC50 values of 0.16 μM, 21.73 μM and 41.56 μM against EGFR Del19, EGFR WT and EGFRL858R, respectively. EGFR-IN-154 shows anticancer activity on various cance cell lines. EGFR-IN-154 induces mitochondrial apoptosis, and decreases pERK1/2 and pAkt levels, but increases pJNK and pp38 levels .
EGFR-IN-81 (Compound 10i) is an EGFR inhibitor. EGFR-IN-81 inhibits EGFR WT and L858R/T790M with IC50s 4.38 nM and 5.69 nM. EGFR-IN-81 has cytotoxic activity against MCF-7 and HCT116 cells with of 2.07 μM and 6.72 μM respectively .
Gefitinib-based PROTAC 3, conjugating an EGFR binding element to a von Hippel-Lindau ligand via a linker, induces EGFR degradation with DC50s of 11.7 nM and 22.3 nM in HCC827(exon 19 del) and H3255 (L858R mutantion) cells, respectively .
EGFR-TK-IN-1 (compound 7o) is a potent mutant EGFR inhibitor with IC50 of 8.5 nM an 9.3 nM against EGFRL858R/T790M and EGFRDel19.EGFR-TK-IN-1 showes strong antiproliferative effects against EGFR mutant-driven non-small cell lung cancer (NSCLC) cells and induces cell apoptosis .
EGFR/AURKB-IN-1 (compound 7) is a dual-targeted EGFR/AURKB inhibitor, and inhibits the phpsphorylations of L858REGFR and AURKB with IC50s of 0.07 and 1.1, respectively. EGFR/AURKB-IN-1 occupies the hydrophobic region I or the αC-helix out pocket of EGFR and the back pocket of AURKB, inhibiting the growth, division and metastasis of tumor cells, thus can be used for cancer research .
EGFR-IN-104 (Compound A23) is an effective inhibitor of EGFR, with IC50 values of 0.33 μM and 0.133 μM against EGFRL858R/T790M and EGFRDel19/T790M/C797S, respectively. EGFR-IN-104 exhibits anticancer activity both in vitro and in vivo .
EGFR-IN-91 (compound 9) is an orally available EGFR inhibitor with blood-brain barrier penetrability. EGFR-IN-91 inhibits EGFRL858R/C797S and EGFRexon 19del/C797S, inducing tumor regression in xenograft (PDX) mouse models. EGFR-IN-91 has the potential to inhibit localized and metastatic non-small cell lung cancer (NSCLC) driven by EGFR mutants .
PROTAC EGFR degrader 4 is a potent PROTAC targeting mutant EGFR.PROTAC EGFR degrader 4 induces EGFRdel19 and EGFRL858R/T790M degradation with DC50s of 0.51 and 126 nM, respectively. PROTAC EGFR degrader 4 significantly inhibits growth of HCC827 and H1975 cell lines with IC50s of 0.83 and 203.1 nM, respectively. Induced EGFR degradation is related to autophagy .
MS9427 TFA is a potent PROTAC EGFR degrader with Kds of 7.1 nM and 4.3 nM for EGFR WT and EGFRL858R, respectively. MS9427 TFA selectively degrades the mutant but not the WT EGFR through both the ubiquitin/proteasome system (UPS) and autophagy/lysosome pathways. MS9427 TFA potently inhibits the proliferation of NSCLC cells. MS9427 TFA can be used for researching anticancer .
EGFR-IN-161 (Compound DD-8) is a potent and reversible inhibitor of L858R/T790M/C797S mutant EGFR kinases, with an IC50 of 0.87 nM. EGFR-IN-161 can induce apoptosis process, G1-phase arrestation, and migration inhibition in tumor cells .
EGFR/HER2-IN-5 (compound 6h) is an orally active irreversible dual inhibitor. EGFR/HER2-IN-5 inhibits EGFR with an IC50 value of 1.01 nM and demonstrates potent EGFR kinase inhibitory activities on L858R and T790M mutations. EGFR/HER2-IN-5 has potent antitumor efficacy in vivo and can be used for lung cancer research .
EGFR/HER2-IN-4(compound 6d) is an orally active irreversible dual inhibitor. EGFR/HER2-IN-4 inhibits EGFR with an IC50 value of 0.6 nM and demonstrates potent EGFR kinase inhibitory activities on L858R and T790M mutations. EGFR/HER2-IN-4 has potent antitumor efficacy in vivo and can be used for lung cancer research .
EGFR-IN-180 (Compound L15) is an EGFR inhibitor. EGFR-IN-180 shows inhibitory activity against EFGR and EGFR harboring the L858R/T790M/C797S triple drug-resistant mutation, with IC50 values of 80.96 nM and 16.43 nM, reapectively. EGFR-IN-180 can be used for the study of non-small cell lung cancer (NSCLC) .
YS-363 is a potent, selective, and orally active EGFR inhibitor, with IC50s of 0.96 nM and 0.67 nM for wild-type and L858R mutant forms of EGFR, respectively. YS-363 can induce G0/G1 cell cycle arrest and apoptosis .
EGFR-IN-12 is a 4,6-disubstituted pyrimidine and is a potent, ATP-competitive, irreversible and highly selective EGFR inhibitor with an IC50of 21 nM. EGFR-IN-12 also inhibits mutant EGFRL858R and EGFRL861Q with IC50s of 63 nM and 4 nM, respectively. EGFR-IN-12 displays strong selectivity for EGFR over HER4 (IC50 = 7640 nM) and a panel of 55 other kinases. EGFR-IN-12 induces cells apoptosis and has antitumor activity .
EGFR/c-Met-IN-1 (compound TS-41) is a dual-target inhibitor of EGFR/c-Met. The IC50 for inhibiting EGFRL858R and c-Met is 68.1 nM and 0.26 nM respectively. . EGFR/c-Met-IN-1 induces apoptosis and cell cycle arrest in A549-P cells, downregulating the phosphorylation of EGFR, c-Met, and downstream AKT. EGFR/c-Met-IN-1 inhibits tumor growth in vitro and in vivo .
EGFR-IN-175 is an orally active and selective EGFRL858R/T790M/C797S inhibitor with an IC50 of 18.94 nM. EGFR-IN-175 can induce cell apoptosis and cause G1 phase arrest. EGFR-IN-175 can downregulate p-EGFR, p-AKT, and p-ERK expression. EGFR-IN-175 can be used for the research of cancer, such as lung cancer .
EGFR-IN-140 (Compound 31) is the inhibitor for EGFR, that inhibits EGFR wildtype and EGFRL858R/T790M/C797S mutant with Ki of 0.95 nM and 2.1 nM, and inhibits EGFR del19/T790M/C797S in Ba/F3 with an IC50 of 56.9 nM. EGFR-IN-140 exhibits antitumor efficacy in mouse model .
EGFR-IN-95 (compound 5j) is an 2,4-diaminonicotinamide derivative. EGFR-IN-95 has potent inhibitory activity against EGFR del19/T790M/C797S and L858R/T790M/C797S .
EGFR-IN-186 is a potent inhibitor of EGFR with an IC50 of 0.065 µM. EGFR-IN-186 also exhibits inhibitory activity against EGFRL858R (IC50 = 0.528 µM) and EGFRT790M (IC50 = 0.465 µM). EGFR-IN-186 induces apoptosis by increasing Bax and caspase-3 levels and down-regulating Bcl-2 expression level. EGFR-IN-186 can be used for the research of non-small cell lung cancer (NSCLC) .
Wighteone (6-Isopentenylgenistein; Erythrinin B) is a prenylated isoflavone that acts as a HSP90/EGFRL858R/T790M inhibitor and antifungal agent. Wighteone reduces the expression level of HSP90, blocks EGF-induced phosphorylation of EGFR, and thereby inhibits the downstream ERK and AKT signaling pathways. Wighteone induces cell cycle redistribution, inhibits proliferation and triggers apoptosis in cancer cells. Wighteone can be isolated from Erythrina suberosa, and can also be induced to synthesize in Lotus japonicus under specific conditions. Wighteone can be used to study HER2-positive breast cancer, leukemia, non-small cell lung cancer with EGFRL858R/T790M mutation, and fungal infections .
Sunvozertinib (DZD9008) is a potent ErbBs (EGFR, Her2, especially mutant forms) and BTK inhibitor. Sunvozertinib shows IC50s of 20.4, 20.4, 1.1, 7.5, and 80.4 nM for EGFR exon 20 NPH insertion, EGFR exon 20 ASV insertion, EGFRL858R and T790M mutations, and Her2 Exon20 YVMA, and EGFR WT A431, respectively (patent WO2019149164A1, example 52) .
SH-1092 is an orally active EGFR inhibitor with an IC50 of 0.96 nM against EGFRT790M/L858R and an IC50 of 6.1 nM against wild-type EGFR. SH-1092 exhibits anticancer activity against non-small cell lung cancer. SH-1092 can be used in studies related to non-small cell lung cancer .
EGFR-IN-98 (Compound 4c) is a EGFR inhibitor. The IC50 values of L858R/T790M/C797S and Del19/T790M/C797S are 0.277 μM and 0.089 μM, respectively. EGFR-IN 98 can be used in the study of tumors .
EMI1 is an EGFR ex19del/T790M/C797S and EGFRL858R/T790M/C797S inhibitor. EMI1 can be used for the research of mutant EGFR-associated, drug-resistant non-small-cell lung cancer (NSCLC) .
EGFR-IN-1671 is a selective EGFR inhibitor with an IC50 of 0.19 nM. EGFR-IN-167 exhibits good potency against various EGFR mutants (IC50 = 0.109 nM, 0.75 nM and <0.05 nM against EGFR (L858R), EGFR (C797S) and EGFR (del19), respectively). EGFR-IN-1671 covalently engages the catalytically conserved lysine of EGFR in live mammalian cells. EGFR-IN-1671 demonstrates excellent anti-proliferative activity by inhibiting EGFR autophosphorylation. EGFR-IN-1671 can be used for the study of non-small-cell lung carcinomas (NSCLC), glioblastoma and many solid tumors .
EGFR-IN-213 is a selective inhibitor of EGFRL858R/T790M/C797S with a human IC50 of 0.48 nM. EGFR-IN-213 acts as an antiproliferative agent, inducing apoptosis and cell cycle arrest, and inhibiting colony formation, cell migration, and tube formation. EGFR-IN-213 can be used for the research of non-small cell lung cancer, chronic myeloid leukemia, gastric cancer, prostate cancer .
DDC-01-163 is an allosteric PROTAC degrader targeting EGFR. DDC-01-163 is dependent on the ubiquitin–proteasome system. DDC-01-163 can selectively inhibit the proliferation of L858R/T790M (L/T) mutant Ba/F3 cells. DDC-01-163 is effective against Osimertinib (HY-15772)-resistant cells with L/T/C797S and L/T/L718Q EGFR mutations. DDC-01-163 exhibits enhanced anti-proliferative activity against L858R/T790M EGFR-Ba/F3 cells when combined with the ATP-site EGFR inhibitor Osimertinib. DDC-01-163 can be used for the study of non-small cell lung cancer .
EGFR mutant-IN-2 (Compound D51) is an EGFR mutant inhibitor. EGFR mutant-IN-2 inhibits the EGFRL858R/T790M/C797S mutant with an IC50 value of 14 nM. EGFR mutant-IN-2 inhibits the EGFRdel19/T790M/C797S mutant with an IC50 value of 62 nM. EGFR mutant-IN-2 has favorable PK parameters, safety properties, in vivo stability, and antitumor activity .
pan-HER-IN-1 (Compound C5) is an irreversible, orally active pan-HER inhibitor with IC50 values of 0.38, 1.6, 2.2 and 3.5 nM against EGFR, HER4, EGFRT790M/L858R and HER2, respectively. pan-HER-IN-1 induces apoptosis and shows antitumor activities .
pan-HER-IN-2 (Compound C6) is a reversible, orally active pan-HER inhibitor with IC50 values of 0.72, 2.0, 8.2 and 75.1 nM against EGFR, HER4, EGFRT790M/L858R and HER2, respectively. pan-HER-IN-2 induces apoptosis and shows antitumor activities .
EGFR-IN-152 (compound D4) is a potent EGFR tyrosine kinase inhibitor, exhibiting potent EGFRL858R/T790M/C797S inhibition activity (IC50 = 40 nM). EGFR-IN-152 induces G0/G1 phase cell cycle arrest and apoptosis, thereby inhibiting colony formation and cell proliferation of non-small cell lung cancer (NSCLC) cells. EGFR-IN-152 can be used for NSCLC research .
JNK3 inhibitor-2 is a potent and selective JNK3 inhibitor with IC50 values of >100, >100, 0.25 µM for JNK1, JNK2, JNK3, respectively. JNK3 inhibitor-2 shows DDR1 and EGFR (T790M, L858R) inhibition .
(S)-Sunvozertinib ((S)-DZD9008), the S-enantiomer of Sunvozertinib, shows inhibitory activity against EGFR exon 20 NPH and ASV insertions, EGFRL858R/T790M mutation and Her2 exon20 YVMA insertion (IC50=51.2 nM, 51.9 nM, 1 nM, and 21.2 nM, respectively). (S)-Sunvozertinib also inhibits BTK .
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
PROTAC EGFR degrader 9 (Compound C6) is an orally active CRBN-based PROTAC EGFR degrader. PROTAC EGFR degrader 9 exhibits a DC50 of 10.2 nM and a Kd of 240.2 nM against EGFRL858R/T790M/C797S. PROTAC EGFR degrader 9 exhibits potent degradation activity against various EGFR mutants, while sparing the EGFRWT. (Blue: CRBN ligand (HY-A0003), Black: linker (HY-161613); Pink: EGFR inhibitor (HY-161537)) .
EGFR-IN-82 (Cmpound 8a) is a potent and orally active EGFR inhibitor with IC50 values of 0.09 and 0.06 nM for EGFRL858R/T790M/C797S and EGFRDel19/T790M/C797S, respectively. EGFR-IN-82 has no significant effect on EGFRWT. EGFR-IN-82 has anti-proliferative activity and inhibits tumor formation in nude mice. EGFR-IN-82 can be used in non-small cell lung cancer research .
CHMFL-EGFR-202 is a potent, irreversible inhibitor of epidermal growth factor receptor (EGFR) mutant kinase, with IC50s of 5.3 nM and 8.3 nM for drug-resistant mutant EGFR T790M and WT EGFR kinases, respectively. CHMFL-EGFR-202 exhibits ~10-fold selectivity for EGFRL858R/T790M against the EGFR wild-type in cells. CHMFL-EGFR-202 adopts a covalent “DFG-in-C-helix-out” inactive binding conformation with EGFR, with strong antiproliferative effects against EGFR mutant-driven nonsmall-cell lung cancer (NSCLC) cell lines .
EGFR-IN-201 is a potent EGFR inhibitor, with an IC50 of 0.091 μM against wild-type EGFR; for mutant EGFR variants, the IC50 values of EGFRT790M, EGFRL858R and EGFRC797S are 0.147 μM, 0.221 μM and 0.703 μM, respectively. EGFR-IN-201 inhibits EGFR downstream signaling proteins AKT1 (IC50 = 0.225 μg/mL) and ERK1 (IC50 = 0.705 μg/mL). EGFR-IN-201 induces G0/G1 cell cycle arrest, apoptosis and low-level necrosis in cancer cells. EGFR-IN-201 is applicable to research on cancers such as colon cancer .
EGFR-IN-165 is a potent EGFR inhibitor. EGFR-IN-165 demonstrates superior potency with IC50s of 17.18 and 64.74 nM against EGFRL858R/T790M and EGFRWT; 2.17 and 6.2 μM against NCI-H1975 cells and A431 cells. EGFR-IN-165 significantly inhibits the migration and induces G1 phase cell cycle arrest and cell apoptosis. EGFR-IN-165 can be used for the study of cancers such as non-small cell lung cancer, colorectal cancer, and head and neck squamous cell carcinoma .
EGFR-IN-160 (Compound R12) is an EGFR inhibitor (IC50 values are 1.62, 0.49 and 0.98 μM for EGFRWT, EGFRT790M and EGFRL858R/T790M/C797S, respectively). EGFR-IN-160 induces G2/M and S phase arrest and apoptosis in NCI-H522 cells, and has anticancer activity. EGFR-IN-160 has antioxidant effects against DPPH (IC50: 12.11 µM) and H2O2 (IC50: 8.89 µM) .
BLU-945 is a potent, highly selective, reversible and orally active epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKIs). BLU-945 can effectively inhibit EGFR with L858R and/or exon 19 deletion mutation, T790M mutation and C797S mutation. BLU-945 can be used for the research of lung cancer including non-small cell lung cancer (NSCLC) .
EGFR-IN-60 (Compound 7d) shows obvious inhibition of EGFRWT, EGFRT790M, EGFRL858R and JAK3 with IC50s of 83, 26, 53, and 69 nM, respectively. EGFR-IN-60 potently inhibits the growth of H1975 cells harboring EGFRT790M mutation (IC50=1.32 µM) over A431 cells overexpressing EGFRWT (IC50=4.96 µM). EGFR-IN-60 exhibits good oral absorption, potent and safe antitumor activity. EGFR-IN-60 induces cell death through apoptosis supported by increased Bax/Bcl-2 ratio .
EGFR-IN-117 (Compound 8h) exhibits inhibitory activity against EGFR mutation, targets the tumor environment, and induces apoptosis of cancer cells. EGFR-IN-117 inhibits proliferations of H1975, PC-9, and EGFR mutant cells BaF3-EGFRL858R/T790M/C797S and BaF3– C797S/Del19/T790M, with IC50 of 13 nM, 19 nM, 1.2 nM and 1.3 nM, respectively. EGFR-IN-117 exhibits antitumor efficacy in mouse models .
JS04 is a EGFRL858R/T790M kinase inhibitor. JS04 activates both endogenous and exogenous apoptosis (apoptosis) pathways and induces G2/M phase arrest of the cell cycle. JS04 is applicable to the research of drug-resistant non-small cell lung cancer .
Befotertinib (D-0316) mesylate is an orally active EGFR inhibitor and ABCB1 inhibitor. Befotertinib mesylate selectively targets EGFR mutations including EGFRT790M, EGFRL858R and delE746-A750, forms covalent bonds with EGFRC797, inhibits oncogenic signaling pathways, and exerts antiproliferative effects. Befotertinib mesylate inhibits ABCB1-mediated drug efflux, activates the ATPase activity of ABCB1, acts as a chemosensitizer and apoptosis enhancer, and restores the sensitivity of multidrug-resistant cancer cells. Befotertinib mesylate can be used in research related to multidrug-resistant cancers and non-small cell lung cancer .
PROTAC EGFR degrader 13 (compound 106) is an EGFR PROTAC degrader with a DC50 of <0.1 μM. PROTAC EGFR degrader 13 has proliferation inhibitory activity on cell Ba/F3-TEL-EGFR-T790M-L858R-C797S with an IC50 of 15.6 nM. PROTAC EGFR degrader 13 can be used for the study of EGFR-related diseases such as cancer (Pink: Target protein ligand; Blue: E3 ligand (HY-14658); Black: linker (HY-W262798); E3 ligand + linker: HY-W998306) .
Poziotinib (HM781-36B) is an orally active, irreversible pan-HER inhibitor, which effectively inhibits EGFRwt, HER-2 and HER-4 with IC50s of 3.2, 5.3 and 23.5 nM, respectively. Poziotinib (HM781-36B) also shows excellent inhibitory activities against mutated EGFRs, including EGFRT790M and EGFRL858R/T790M, with IC50s of 4.2 and 2.2 nM, respectively. Excellent antitumor activity .
Gly-PEG3-BA is an EML4-ALKPROTAC degrader. Gly-PEG3-BA effectively reduces EML4-ALK with a DC50 value of 0.50 μM in H3122 (EML4-ALK) cells. Gly-PEG3-BA effectively reduces EGFR mutant (L858R/T790M) levels with a DC50 of 20.15 μM in H1975 (EGER-L858R/T790M) cells. Gly-PEG3-BA exerts potent antiproliferation activity in H3122 (EML4-ALK) and H1975 (EGER-L858R/T790M) cells with IC50s value of 0.84 and 20.74 μM. Gly-PEG3-BA can be used for non-small lung cancer research .
Theliatinib (Standard) is the analytical standard of Theliatinib (HY-104066). This product is intended for research and analytical applications. Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
BI-4732 is an orally active, reversible, ATP-competitive EGFR inhibitor with blood-brain barrier penetration. BI-4732 inhibits the kinase activity of EGFRL858R, T790M and C797S with IC50 values of 1 nM while sparing EGFR wild-type. BI-4732 inhibits EGFR and reduces the phosphorylation of AKT, ERK, and S6K. BI-4732 demonstrates excellent intracranial anti-tumor efficacy in YU-1097 xenograft model harboring EGFR_E19del/T790M/C797S. BI-4732 can be used for the study of non-small cell lung cancer (NSCLC) .
HJM-561 is a selective and effective orally active EGFRPROTAC degrader. HJM-561 is able to overcome the triple EGFR mutations that are resistant to Osimertinib (HY-15772). HJM-561 exhibits potent degradation of EGFR Del19/T790M/C797S (DC50: 9.2 nM) and L858R/T790M/C797S (DC50: 5.8 nM), and has anti-tumor activity (pink: EGFR ligand (HY-12857); blue: CRBN ligand (HY-A0003); black: linker) .
PROTAC EGFR degrader 14 is a potent and selective EGFR PROTACdegrader with a DC50 of about 2.9 nM and a Dmax of 93.1% for EGFRL858R/T790M/C797S. PROTAC EGFR degrader 14 selectively induces EGFRC797S degradation through a VHL and proteasome-dependent manner and downregulated EGFR-associated transcriptome and exhibits good selectivity over EGFRWT. PROTAC EGFR degrader 14 induces cell cycle arrest and apoptosis and significantly inhibits tumor growth. PROTAC EGFR degrader 14 can be used for the study of nonsmall cell lung cancer (NSCLC) (Pink: Target protein ligand: (HY-143337); Blue: E3 ligand (HY-125845); Black: Linker (HY-W004688)) .
EGFR-IN-176 is an orally active and ATP-competitive EGFR mutant inhibitor (particularly C797S-mediated EGFR triple mutant). EGFR-IN-176 effectively inhibits subsequent AKT signaling and induces apoptosis in Ba/F3 and PC-9 cells expressing EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S. EGFR-IN-176 selectively inhibits EGFR signaling in cell lines harboring EGFR triple mutation and shows no inhibitory effect against A431 cells that express wild-type EGFR. EGFR-IN-176 can effectively inhibit the enzymatic activity of ALK (IC50 < 0.5 nM). EGFR-IN-176 can be used for the study of non-small cell lung cancer (NSCLC) .
PROTAC EGFR degrader 7 (compound 13b) is a potent and selective CRBN-recruiting PROTAC EGFRL858R/T790M degrader, with a DC50 of 13.2 nM. PROTAC EGFR degrader 7 inhibits NCI–H1975 cells proliferation, with an IC50 of 46.82 nM. PROTAC EGFR degrader 7 significantly induces apoptosis and G2/M phase arrest in NCI–H1975 cell. PROTAC EGFR degrader 7 shows antitumor activity, and can be used for non-small cell lung cancer (NSCLC) research . PROTAC EGFR degrader 7 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
EGFR-IN-173 is an orally active, pan-mutant EGFR tyrosine kinase inhibitor that targets EGFR 19del, L858R/T790M and C797S triple-mutations, potently inhibiting EGFR19del/T790M/C797S with an IC50 of 1.19 nM while showing over 100-fold selectivity for mutant over wild-type EGFR (IC50 = 19.362 μM against WT). EGFR-IN-173 significantly inhibits cell migration, induces apoptosis in non-small cell lung cancer (NSCLC) cells. EGFR-IN-173 inhibits EGFR phosphorylation and suppresses the downstream pathways (MAPK/ERK, AKT, STAT3). EGFR-IN-173 exhibits antitumor efficacy in NSCLC and Ba/F3 xenograft models. EGFR-IN-173 can be used for NSCLC research .
Almonertinib (HS-10296) mesylate is an orally available, irreversible, third-generation EGFR tyrosine kinase inhibitor with high selectivity for EGFR-sensitizing and T790M resistance mutations. Almonertinib mesylate shows great inhibitory activity against T790M, T790M/L858R and T790M/Del19 (IC50: 0.37, 0.29 and 0.21 nM, respectively), and is less effective against wild type (3.39 nM). Almonertinib mesylate is used for the research of the non-small cell lung cancer .
Almonertinib (HS-10296) is an orally available, irreversible, third-generation EGFR tyrosine kinase inhibitor with high selectivity for EGFR-sensitizing and T790M resistance mutations. Almonertinib shows great inhibitory activity against T790M, T790M/L858R and T790M/Del19 (IC50: 0.37, 0.29 and 0.21 nM, respectively), and is less effective against wild type (3.39 nM). Almonertinib is used for the research of the non-small cell lung cancer .
Almonertinib (HS-10296) hydrochloride is an orally available, irreversible, third-generation EGFR tyrosine kinase inhibitor with high selectivity for EGFR-sensitizing and T790M resistance mutations. Almonertinib hydrochloride shows great inhibitory activity against T790M, T790M/L858R and T790M/Del19 (IC50: 0.37, 0.29 and 0.21 nM, respectively), and is less effective against wild type (3.39 nM). Almonertinib hydrochloride is used for the research of the non-small cell lung cancer .
Lys-PEG3-BA is an EML4-ALK/EGFRPROTAC degrader with DC50 values of 1.32 and 19.66 μM for H3122 (EML4-ALK) and H1975 (EGFR-L858R/T790M) cells, respectively. Lys-PEG3-BA hinders proliferation via rewiring the ubiquitin- proteasome system in vitro. Lys-PEG3-BA can be used for non-small cell lung cancer research .
BI-8128 is a potent EGFR inhibitor, with IC50 values of 12, 6.7, 22, 10, and 3 nM against wild-type, T790M, C797S, T790M/C797S, and L858R/T790M/C797S mutant EGFR, respectively. BI-8128 significantly inhibits the proliferation of Ba/F3 and PC-9 drug-resistant mutant cells. BI-8128 is applicable for the research of non-small cell lung cancer .
EGFR-IN-178 is an orally active EGFR mutant inhibitor, exhibits highly selective inhibitory activity against mutants of the EGFR enzyme, including Del19 (IC50 = 3.4 nM), L858R/T790 M (IC50 = 2.9 nM), and Del19/T790 M (IC50 = 2.5 nM). EGFR-IN-178 has good activity against JAK2 (IC50 = 55.6 nM) and JAK3 (IC50 = 46.1 nM) kinases. EGFR-IN-178 can increase cellular lipid oxide MDA, meanwhile decrease GSH content, causing ferroptosis in cancer cells. EGFR-IN-178 promotes apoptosis by increasing cleaved caspase-3 expression. EGFR-IN-178 can inhibit the phosphorylation of EGFR protein and decrease the active form p-JAK2 for JAK2, induce an increase in intracellular ROS. EGFR-IN-178 can be used for the study of non-small cell lung cancer (NSCLC) .
Almonertinib (Standard) is the analytical standard of Almonertinib. This product is intended for research and analytical applications. Almonertinib (HS-10296) is an orally available, irreversible, third-generation EGFR tyrosine kinase inhibitor with high selectivity for EGFR-sensitizing and T790M resistance mutations. Almonertinib shows great inhibitory activity against T790M, T790M/L858R and T790M/Del19 (IC50: 0.37, 0.29 and 0.21 nM, respectively), and is less effective against wild type (3.39 nM). Almonertinib is used for the research of the non-small cell lung cancer .
Pro-PEG3-BA is an EML4-ALK/EGFRPROTAC degrader, degrading EML4 ALK and EGFR mutant (L858R/T790M) with DC 50 values of 0.42 and 13.50 μM, respectively. Pro-PEG3-BA hinders proliferation and induces cell cycle arrest and apoptosis of NSCLC cells in vitro. Pro-PEG3-BA shows safety profile and decreases EML4-ALK protein via rewiring the ubiquitin- proteasome system in vivo. Pro-PEG3-BA can be used for non-small cell lung cancer research .
Antiproliferative agent-34 (Compound A14) is a multi-target kinase inhibitor, with an IC50 of 177 nM and 1567 nM for EGFRL858R/T790M and EGFR WT. Antiproliferative agent-34 also inhibits JAK2, ROS1, FLT3, FLT4, PDGFRα with IC50 of 30.93, 106.90, 108.00, 226.60, 42.53 nM. Antiproliferative agent-34 inhibits H1975 and HCC827 cells proliferation with IC50 values below 40 nM under normoxic condition, and the anti-proliferation potency achieves 4–6-fold improvement (IC50 values < 10 nM) under hypoxic condition .
LS-106 is an orally active and potent inhibitor against epidermal growth factor receptor (EGFR) . LS-106 exhibits antitumor activities both in vitro and in vivo. LS-106 inhibits the kinase activities of EGFR19del/T790M/C797S and EGFRL858R/T790M/C797S with IC50 values of 2.4 nmol/L and 3.1 nmol/L, respectively, which is more potent than Osimertinib (HY-15772). LS-106 induces Apoptosis, suppresses cell proliferation of tumor cells harboring EGFR19del/T790M/C797S and leas to significant tumor regression in a C797S-mutant xenograft model .
AZ14289671 is an orally active, blood-brain barrier-penetrant tyrosine kinase (tyrosine kinase) inhibitor (TKI) that specifically targets non-small cell lung cancer (NSCLC) harboring EGFR exon 20 insertion mutations (EGFR Exon20Ins), while largely sparing wild-type EGFR to reduce off-target toxicities such as rash and diarrhea. AZ14289671 inhibits the downstream MAPK/ERK/AKT pathway, suppressing tumor cell proliferation, survival and migration. AZ14289671 can be used for NSCLC research .
MS39 (compound 6) is a PROTAC targeting EGFR. MS39 reduces the expression of EGFR and downstream signaling in HCC-827 and H3255 cells. MS39 inhibits the proliferation of H3255 cells .
Wighteone (6-Isopentenylgenistein; Erythrinin B) is a prenylated isoflavone that acts as a HSP90/EGFRL858R/T790M inhibitor and antifungal agent. Wighteone reduces the expression level of HSP90, blocks EGF-induced phosphorylation of EGFR, and thereby inhibits the downstream ERK and AKT signaling pathways. Wighteone induces cell cycle redistribution, inhibits proliferation and triggers apoptosis in cancer cells. Wighteone can be isolated from Erythrina suberosa, and can also be induced to synthesize in Lotus japonicus under specific conditions. Wighteone can be used to study HER2-positive breast cancer, leukemia, non-small cell lung cancer with EGFRL858R/T790M mutation, and fungal infections .
The EGFR protein is a receptor tyrosine kinase that can bind to a variety of ligands, such as EGF, TGFA, AREG, epigen, BTC, epiregulin, and HBEGF, to initiate signaling cascades that mediate cellular responses. This involves receptor dimerization, autophosphorylation and recruitment of adapter proteins such as GRB2, activating downstream pathways such as RAS-RAF-MEK-ERK, PI3-kinase-AKT, PLCgamma-PKC and STAT. EGFR Protein, Human (C797S, T790M, L858R, sf9, GST) is the recombinant human-derived EGFR, expressed by Sf9 insect cells, with N-GST labeled tag.
The EGFR protein is a receptor tyrosine kinase that can bind to a variety of ligands, such as EGF, TGFA, AREG, epigen, BTC, epiregulin, and HBEGF, to initiate signaling cascades that mediate cellular responses. This involves receptor dimerization, autophosphorylation and recruitment of adapter proteins such as GRB2, activating downstream pathways such as RAS-RAF-MEK-ERK, PI3-kinase-AKT, PLCgamma-PKC and STAT. EGFR Protein, Human (T790M, L858R, sf9, Strep II, GST, His) is the recombinant human-derived EGFR, expressed by Sf9 insect cells, with C-8*His, N-GST, N-Strep II labeled tag.
Osimertinib- 13C,d3 is the deuterium and 13C labeled Osimertinib. Osimertinib (AZD9291) is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M, respectively.
Osimertinib-d6 is a deuterium labeled osimertinib. Osimertinib is a covalent, orally active, irreversible, and mutant-selective EGFR inhibitor with an apparent IC50 of 12 nM against L858R and 1 nM against L858R/T790M. Osimertinib overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer .
Icotinib-d4 is the deuterium-labeled Icotinib (HY-15164A). Icotinib-d4 (BPI-2009) is a potent and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib-d4 is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Icotinib (BPI-2009) is a potent, CNS-penetrant and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
PROTAC EGFR degrader 7 (compound 13b) is a potent and selective CRBN-recruiting PROTAC EGFRL858R/T790M degrader, with a DC50 of 13.2 nM. PROTAC EGFR degrader 7 inhibits NCI–H1975 cells proliferation, with an IC50 of 46.82 nM. PROTAC EGFR degrader 7 significantly induces apoptosis and G2/M phase arrest in NCI–H1975 cell. PROTAC EGFR degrader 7 shows antitumor activity, and can be used for non-small cell lung cancer (NSCLC) research . PROTAC EGFR degrader 7 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Icotinib Hydrochloride (BPI-2009) is a potent, CNS-penetrant and specific EGFR inhibitor with an IC50 of 5 nM; also inhibits mutant EGFRL858R, EGFRL858R/T790M, EGFRT790M and EGFRL861Q. Icotinib (Hydrochloride) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Theliatinib (Xiliertinib) is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Theliatinib (Xiliertinib) tartrate is a potent, ATP-competitive, orally active and highly selective EGFR inhibitor with a Ki of 0.05 nM and an IC50 of 3 nM. Theliatinib has an IC50 of 22 nM for EGFR T790M/L858R mutant. Theliatinib shows >50-fold selectivity for EGFR than other kinases . Theliatinib (tartrate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
EGFR-IN-77 (Compound 4a) is a selective EGFRT790M/L858R inhibitor, with an IC50 of 0.101 μM against EGFRT790M/L858R, 0.477 μM against EGFRL858R, and 1.771 μM against wild-type EGFR. EGFR-IN-77 exerts selective antiproliferative effects on EGFRT790M/L858R non-small cell lung cancer. EGFR-IN-77 can be used for the research of EGFRL858R/T790M double-mutant non-small cell lung cancer .
Inquiry Online
Your information is safe with us. * Required Fields.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
MedchemExpress Validation 03
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
MedchemExpress Validation 04
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedchemExpress Validation
Western blot analysis of extracts from THP-1(lane 2(20μg), Jurkat (lane 3(20μg) and NIH3T3(lane 4(20μg) using FOXO1A (HY-P80132) Rabbit mAb. Proteins were transferred
to a PVDF membrane and blocked with 5% non-fat milk in TBST for 2 hour at room temperature. The primary antibody (1/1000) and Loading control antibody (Beta Actin, HY-P80438, 1/10000) was
used in 5% non-fat milk in TBST at 4°C overnight. Goat Anti-Mouse/Rabbit IgG-HRP Secondary Antibody (1/10000) was used for 1 hour at room temperature.
MedChemExpress values your privacy and your trust is important to us. We use cookies to enhance your website experience. Some cookies are necessary to run the website.
Privacy and Cookie Policy