


Isorhapontigenin
Based on 4 publication(s) in Google Scholar
Isorhapontigenin is an orally active dietary polyphenol. Isorhapontigenin acts as a potent antioxidant that reduces the production of reactive oxygen species (ROS). Isorhapontigenin promotes the binding of JUN to the AP-1 site on the SESN2 promoter, induces SESN2 transcription, triggers MAPK8-dependent JUN activation, and upregulates the expression of PPAR-α, PGC-1α and CPT-1A to facilitate fatty acid oxidation. Isorhapontigenin induces autophagy, apoptosis and preadipocyte differentiation; it inhibits tumor growth, cell invasion, NF-κB transcriptional activity, the PI3K/Akt signaling pathway, STAT1 phosphorylation and MMP-2 expression. Isorhapontigenin alleviates oxidative stress, inflammatory cytokine release and triglyceride accumulation; it increases intracellular ATP levels and promotes Nrf2 nuclear translocation. Isorhapontigenin improves insulin sensitivity in adipose tissue and glucose tolerance, and reduces postprandial blood glucose, insulin and free fatty acid levels. Isorhapontigenin is applicable to research on bladder cancer, liver injury, chronic obstructive pulmonary disease, acute lung injury and type 2 diabetes.
Nur für Forschungszwecke. Wir verkaufen nicht an Patienten.
- Reinheit: 99.82%
- CAS. Nr.: 32507-66-7
- Formel: C15H14O4
- Molecular Weight:258.27
-
Speicherung:
4°C, sealed storage, away from moisture and light
* In solvent : -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light)

To place orders, for customer services and technical support, please contact: MedChemExpress USA
Tel: 609-228-6898 E-mail: [email protected] [email protected]





-
 Biologische Aktivität
Biologische Aktivität
-
Chemical Information
-
Lösungsmittel & Löslichkeit
- Reinheit & Dokumentation
- Verweise
-
 Help & FAQs
Help & FAQs
-


Apoptosis Compound Library
HY-L003
-
Immunology/Inflammation Compound Library
HY-L007
-
Kinase Inhibitor Library
HY-L009
-
Metabolism/Protease Compound Library
HY-L012
-
NF-κB Signaling Compound Library
HY-L014
-
PI3K/Akt/mTOR Compound Library
HY-L015
-
Stem Cell Signaling Compound Library
HY-L017
-
Natural Product Library
HY-L021
-
Anti-Cancer Compound Library
HY-L025
-
Autophagy Compound Library
HY-L029
-
Anti-Aging Compound Library
HY-L034
-
Antioxidant Compound Library
HY-L037
-
Differentiation Inducing Compound Library
HY-L038
-
Anti-diabetic Compound Library
HY-L040
-
Oxygen Sensing Compound Library
HY-L045
-
Ferroptosis Compound Library
HY-L051
-
Phenols Library
HY-L057
-
Glycolysis Compound Library
HY-L058
-
Pyroptosis Compound Library
HY-L059
-
Cytoskeleton Compound Library
HY-L060
-
Orally Active Compound Library
HY-L061
-
Glutamine Metabolism Compound Library
HY-L064
-
Traditional Chinese Medicine Active Compound Library
HY-L065
-
Anti-Breast Cancer Compound Library
HY-L074
-
Anti-Lung Cancer Compound Library
HY-L075
-
Anti-Pancreatic Cancer Compound Library
HY-L077
-
Anti-Blood Cancer Compound Library
HY-L079
-
Anti-Cancer Metabolism Compound Library
HY-L083
-
Anti-Parkinson's Disease Compound Library
HY-L085
-
Neurodegenerative Disease-related Compound Library
HY-L086
-
Anti-Obesity Compound Library
HY-L087
-
Angiogenesis-Related Compound Library
HY-L088
-
Transcription Factor-Targeted Library
HY-L090
-
Lipid Metabolism Compound Library
HY-L091
-
Glucose Metabolism Compound Library
HY-L092
-
Food-Sourced Compound Library
HY-L094
-
Anti-Liver Cancer Compound Library
HY-L101
-
Anti-Colorectal Cancer Compound Library
HY-L103
-
Anti-Cancer Natural Product Library
HY-L107
-
Antidepressant Compound Library
HY-L108
-
Anti-inflammatory Traditional Chinese Medicine Active Compound Library
HY-L114
-
Plant-Sourced Natural Product Library
HY-L115
-
Anti-Prostate Cancer Compound Library
HY-L124
-
Anti-Pulmonary Fibrosis Compound Library
HY-L125
-
Osteogenesis Compound Library
HY-L131
-
Cancer Stem Cells Compound Library
HY-L135
-
Pain-Related Compound Library
HY-L139
-
Protease Inhibitor Library
HY-L147
-
Membrane Protein-targeted Compound Library
HY-L149
-
Cell Death Library
HY-L162
-
Serine/Threonine Kinase Inhibitor Library
HY-L164
-
Anti-Hematopathy Compound Library
HY-L171
-
Anti-Ovarian Cancer Compound Library
HY-L173
-
Multi-Target Compound Library
HY-L176
-
Radioprotector Library
HY-L178
-
Bioactive Compound Library Max
HY-L181
-
MCE Bioactive Compound Library
HY-L001V
-
Natural Product Library Plus
HY-L021P
-
Natural Product and Natural Product-Like Compound Library
HY-L021M
-
Bioactive Compound Library
HY-L001
-
Anti-Gastric Cancer Compound Library
HY-L184
-
Anti-Fibrosis Compound Library
HY-L185
-
Anti-Brain Cancer Compound Library
HY-L188
-
Miao Ethnicity Medicine Compound Library
HY-L190
-
Heat-Clearing and Detoxification Traditional Chinese Medicine Compound Library
HY-L194
-
Protein Kinase Compound Library
HY-L196
-
Non-Alcoholic Fatty Liver Disease (NAFLD) Compound Library
HY-L199
-
RO5 Drug-like Natural Product Library
HY-L200
-
Cell Proliferation Compound Library
HY-L201
-
High-Throughput Bioactive Compound Library
HY-L205
-
High-Throughput Natural Product Library
HY-L206
-
Ancient Chinese Classical Formulas Traditional Chinese Medicine Active Compound Library
HY-L209
-
Anti-Rheumatic Arthritis Compound Library
HY-L210
-
Diarrhea-Related Traditional Chinese Medicine Active Compound Library
HY-L224
-
Mongolian Medicine Compound Library
HY-L238
-
Lactylation Compound Library
HY-L249
-
Mass Spectrometry Natural Product Library
HY-L262


Publications Citing Use of MedChemExpress (MCE) Isorhapontigenin
More Customer Validation & Images
Customer Validation & Images

-
Cell Proliferation/Viability Assay
-
RT-PCR
-
In Vivo Efficacy Study
-
Histological Imaging/Staining
-
WB


Biologische Aktivität
|
CPT-1A |
MMP-2 |
PPARα |
PPARγ |
FOXO1 |
IL-6 |
|
Cell Line
|
Type | Value | Description | References |
|---|---|---|---|---|
| Platelet | IC50 |
1.85 μM
Compound: Isorhapontigenin
|
Antiplatelet activity in human platelet rich plasma assessed as inhibition of ADP-induced platelet aggregation preincubated for 5 mins followed by ADP addition and measured after 5 mins
Antiplatelet activity in human platelet rich plasma assessed as inhibition of ADP-induced platelet aggregation preincubated for 5 mins followed by ADP addition and measured after 5 mins
|
[PMID: 34731765] |
Isorhapontigenin (1.25-40 μM; 24 h) induces autophagy in a dose-dependent manner in UMUC3, T24T, and HeLa cells, and this autophagy contributes to its inhibition of anchorage-independent growth of UMUC3 cells[1].
Isorhapontigenin (2.5-10 μM; 12-24 h) increases SESN2 transcription via a JUN-dependent mechanism, which is required for autophagy induction and inhibition of anchorage-independent growth in UMUC3 cells[1].
Isorhapontigenin (10 μM; 12 h pre-APAP treatment, 24 h post-APAP treatment) attenuates APAP-induced FAO dysregulation in AML12 cells by upregulating PPAR-α/PGC-1α/CPT-1A signaling[2].
Isorhapontigenin (1-100 μM; 1 h pre-incubation + 24 h stimulation, 1 h pre-incubation + 10-60 min stimulation) inhibits IL-6 and CXCL8 release from primary human airway epithelial cells and A549 cells, with IC50 values for IL-6 of 17.3-19.7 μM, and suppresses NF-κB, AP-1, and PI3K/Akt/FoxO3A signaling pathways[3].
Isorhapontigenin (1-100 μM; 1 h pre-incubation + 30 min stimulation) concentration-dependently reduces intracellular ROS levels in IL-1β-stimulated A549 cells[3].
Isorhapontigenin (5-15 μM; 12 h pretreatment + 12 h LPS stimulation) exerts anti-inflammatory and antioxidant effects on LPS-challenged RAW264.7 cells by reducing pro-inflammatory mediator and ROS production[4].
Isorhapontigenin (15 μM; 0-24 h direct treatment, 12 h pretreatment + 12 h LPS stimulation) activates the Nrf2 pathway in RAW264.7 cells, and its anti-inflammatory/antioxidant effects depend on Nrf2 activation[4].
Isorhapontigenin (25 μM; 6 days) promotes adipocyte differentiation, enhances insulin sensitivity, and reduces lipolysis in 3T3-L1 preadipocytes by increasing PPARγ activity and expression[5].
Isorhapontigenin (25 μM; 0-12 h) increases PPARγ activity and stability in 3T3-L1 cells by reducing inhibitory phosphorylation and decelerating proteasomal degradation[5].
Isorhapontigenin (10-20 μM; 24 h) specifically inhibits invasion of UMUC3 and T24T human invasive bladder cancer cells, respectively, without affecting migration[6].
Isorhapontigenin (2.5-20 μM; 6-18 h) induces dose- and time-dependent upregulation of FOXO1 protein expression in UMUC3 and T24T human invasive bladder cancer cells[6].
Isorhapontigenin (2.5-20 μM; 3-9 h) upregulates foxo1 mRNA expression at the transcriptional level in UMUC3 and T24T human invasive bladder cancer cells[6].
Isorhapontigenin (10 μM; 6-18 h) enhances FOXO1 promoter activity in a time-dependent manner in UMUC3 human invasive bladder cancer cells[6].
Isorhapontigenin (2.5-10 μM; 12 h) inhibits STAT1 phosphorylation at Tyr701 in a dose-dependent manner in UMUC3 human invasive bladder cancer cells[6].
Isorhapontigenin (2.5-20 μM; 18 h) inhibits dose-dependent MMP-2 protein expression in UMUC3 and T24T human invasive bladder cancer cells[6].
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.
-
Cell Line:UMUC3, T24T, HeLa
-
Concentration:1.25 μM, 2.5 μM, 5 μM, 10 μM, 20 μM, 40 μM
-
Incubation Time:24 h
-
Result:Caused a dose-dependent increase in LC3-I to LC3-II conversion in UMUC3, T24T, and HeLa cells. Increased the percentage of GFP-LC3 puncta-positive cells and the number of puncta per cell in a dose-dependent manner in GFP-LC3-transfected UMUC3 cells.
-
Cell Line:UMUC3
-
Concentration:2.5 μM, 5 μM, 10 μM
-
Incubation Time:24 h
-
Result:Increased SESN2 and BECN1 expression at 2.5-10 μM in UMUC3 cells. Saw its induced LC3-II formation unaffected by BECN1 knockdown in UMUC3 cells. Had its LC3-II formation significantly attenuated in SESN2 knockdown UMUC3 cells. Saw its inhibitory effect on anchorage-independent growth of UMUC3 cells abolished when SESN2 was knocked down.
-
Cell Line:UMUC3
-
Concentration:5 μM, 10 μM
-
Incubation Time:6 h, 12 h, 24 h
-
Result:Increased MAPK8 phosphorylation in a dose-dependent manner. Had its induced JUN phosphorylation, SESN2 induction, LC3-II formation, SESN2 mRNA expression, and SESN2 promoter activity blocked when MAPK8 was knocked down. Saw its inhibitory effect on anchorage-independent growth of UMUC3 cells abolished when MAPK8 was knocked down.
-
Cell Line:3T3-L1 preadipocytes
-
Concentration:25 μM
-
Incubation Time:6 days (differentiation)
-
Result:Promoted lipid accumulation (quantified by Oil Red O staining optical density at 490 nm). Significantly increased mRNA and protein expression of PPARγ target genes (CEBPα, FAS, FABP4, GLUT4) and PPARγ itself. Enhanced insulin-stimulated glucose uptake (1.5-fold over basal). Reduced mRNA and protein levels of hormone-sensitive lipase (HSL).
-
Cell Line:UMUC3, T24T human invasive bladder cancer cells
-
Concentration:10 μM (UMUC3 cells); 20 μM (T24T cells)
-
Incubation Time:24 h (UMUC3, T24T cells)
-
Result:Reduced relative invasion rate of UMUC3 cells by 63.1% and T24T cells by 61.2% compared to vehicle control; did not affect cell migration under the same conditions.
-
Cell Line:UMUC3, T24T human invasive bladder cancer cells
-
Concentration:2.5-10 μM (UMUC3 cells, dose-response); 10-20 μM (T24T cells, dose-response); 10 μM (UMUC3 cells, time-course)
-
Incubation Time:12 h (UMUC3, T24T cells, dose-response); 6-18 h (UMUC3 cells, time-course)
-
Result:Induced FOXO1 protein expression in a dose-dependent manner in UMUC3 and T24T cells; caused a gradual increase in FOXO1 protein level.
-
Cell Line:UMUC3, T24T human invasive bladder cancer cells
-
Concentration:2.5-10 μM (UMUC3 cells, dose-response); 10 μM (UMUC3 cells, time-course); 10-20 μM (T24T cells, dose-response)
-
Incubation Time:6 h (UMUC3, T24T cells, dose-response); 3-9 h (UMUC3 cells, time-course)
-
Result:Upregulated endogenous foxo1 mRNA expression in a dose-dependent manner in UMUC3 and T24T cells, and in a time-dependent manner in UMUC3 cells; did not affect exogenous flag-foxo1 mRNA expression.
-
Cell Line:UMUC3 human invasive bladder cancer cells
-
Concentration:2.5-10 μM
-
Incubation Time:12 h
-
Result:Dramatically inhibited STAT1 phosphorylation at Tyr701 in a dose-dependent manner without affecting total STAT1 protein expression.
-
Cell Line:UMUC3, T24T human invasive bladder cancer cells
-
Concentration:2.5-10 μM (UMUC3 cells); 10-20 μM (T24T cells)
-
Incubation Time:18 h (UMUC3, T24T cells)
-
Result:Profoundly inhibited MMP-2 protein expression in a dose-dependent manner in both UMUC3 and T24T cells.
-
Cell Line:T24T human invasive bladder cancer cells
-
Concentration:10-20 μM
-
Incubation Time:18 h
-
Result:Attenuated MMP-2 mRNA level in a dose-dependent manner; did not affect MMP-9 mRNA level.
Isorhapontigenin (25-50 mg/kg; i.g. once of once daily for 3 consecutive days) alleviates acetaminophen-induced liver injury in mice by reducing apoptosis, inflammation, oxidative stress, and restoring fatty acid oxidation, with 25 and 50 mg/kg doses showing efficacy[2].
Isorhapontigenin (51.6 mg/kg; p.o.; daily; 7 days) effectively ameliorates LPS-induced acute lung injury in mice by reducing inflammation and oxidative stress[4].
Isorhapontigenin (25 mg/kg; i.p.; daily; 5 weeks) ameliorates type 2 diabetes in db/db mice by improving glucose and insulin homeostasis, enhancing insulin sensitivity, and modulating adipose tissue function via PPARγ regulation[5].
Isorhapontigenin (150 mg/kg/day; drinking water; daily; 20 weeks) inhibits N-Butyl-N-(4-hydroxybutyl)nitrosamine (BBN) (HY-W755252)-induced invasive bladder cancer formation in C57BL/6J mice, with 16.7% of treated mice developing high-grade muscle-invasive disease versus 100% in BBN-only controls[6].
MedChemExpress (MCE) has not independently confirmed the accuracy of these methods. They are for reference only.
-
Animal Model:C57BL/6 (male, 6-8 weeks old; Acetaminophen-induced liver injury model)[2]
-
Dosage:25 mg/kg; 50 mg/kg
-
Administration:i.g. (once, 1 hour after acetaminophen); i.g. (once daily for 3 consecutive days, acetaminophen on day 3); i.g. (once, 1 hour before acetaminophen); i.g. (once, 3 hours after acetaminophen)
-
Result:Significantly reduced serum ALT, AST, and LDH levels, centrilobular necrosis area, TUNEL-positive cells, C-PARP expression, serum TNF-α and IL-6 levels, liver MDA levels, and increased liver CAT levels at 25 and 50 mg/kg (post-treatment) compared to acetaminophen alone; reduced lipid accumulation (Oil Red O and BODIPY staining), serum and liver TG levels, increased liver ATP content and FAO activity, and upregulated PPAR-α, PGC-1α, and CPT-1A protein expression at 50 mg/kg (pre-treatment).
-
Animal Model:C57BL/6 (male, 6-8 weeks old, 20-25 g, LPS-challenged)[4]
-
Dosage:51.6 mg/kg
-
Administration:p.o.; daily; 7 days
-
Result:Reduced LPS-induced lung injury score, myeloperoxidase (MPO) activity in lung tissues, lung wet/dry weight ratio, and protein concentration in bronchoalveolar lavage fluid (BALF); suppressed LPS-induced increases in BALF concentrations of IL-1β, IL-6, and TNF-α; inhibited p-NF-κB p65 expression and IκB degradation in lung tissues; reduced malondialdehyde (MDA) formation in lung tissues; restored superoxide dismutase (SOD) and glutathione (GSH) activities in lung tissues.
-
Animal Model:db/db mice (male, 8 weeks old at treatment start, genetically diabetic model)[5]
-
Dosage:25 mg/kg
-
Administration:i.p.; daily; 5 weeks
-
Result:Reduced postprandial fasting glucose levels by 17.2% after 2 weeks and 40.0% after 5 weeks; reduced insulin levels by 23.2% after 5 weeks; reduced plasma FFA levels by 34.7% after 5 weeks compared to vehicle controls; improved glucose disposal during GTT; enhanced exogenous-insulin-stimulated glucose uptake during ITT; significantly reduced water intake; reduced white adipose tissue adipocyte diameters; significantly increased mRNA expression of PPARγ and its target genes (Fabp4, Glut4, Fas); significantly reduced mRNA and protein levels of HSL; significantly increased insulin-stimulated Akt phosphorylation in white adipose tissue.
-
Animal Model:C57BL/6J (male, 5-6 weeks old) injected with N-Butyl-N-(4-hydroxybutyl)nitrosamine[6]
-
Dosage:150 mg/kg/day
-
Administration:oral via drinking water; daily; 20 weeks
-
Result:Reduced the incidence of BBN-induced high-grade muscle-invasive bladder cancer from 100% (12/12 mice) to 16.7% (2/12 mice); caused 7 cases of papillomas and 3 cases of low-grade non-muscle-invasive bladder cancer in treated mice; up-regulated FOXO1 protein expression and down-regulated MMP-2 protein expression in mouse bladder tissues.


Chemical Information
-
CAS. Nr. 32507-66-7
-
Appearance Solid
-
Molecular Weight 258.27
-
Formel C15H14O4
-
Color White to yellow
-
SMILES
OC1=CC(O)=CC(/C=C/C2=CC(OC)=C(O)C=C2)=C1
-
Structure Classification
-
Initial Source
-
Versand
Room temperature in continental US; may vary elsewhere.
-
Speicherung
4°C, sealed storage, away from moisture and light
* In solvent : -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light)


Publications (4)
-
Journal Impact Factor
-
Most Recent
-
Int Immunopharmacol
Isorhapontigenin suppresses inflammation, proliferation and aggressiveness of rheumatoid arthritis fibroblast-like synoviocytes by targeting farnesyl diphosphate synthase. [Abstract]2025 May 23:159:114894. PMID: 40412131 -
Int Immunopharmacol
Isorhapontigenin delays senescence and matrix degradation of nucleus pulposus cells via PI3K/AKT/mTOR-mediated autophagy pathway in vitro and alleviates intervertebral disc degeneration in vivo. [Abstract]2024 Jul 26:139:112717. PMID: 39067404 -
mSphere
Exploring the potential of isorhapontigenin: attenuating Staphylococcus aureus virulence through MgrA-mediated regulation. [Abstract]2024 Jun 5:e0031724. PMID: 38837389
Isorhapontigenin purchased from MedChemExpress. Usage Cited in: mSphere. 2024 Jun 5:e0031724. [Abstract]
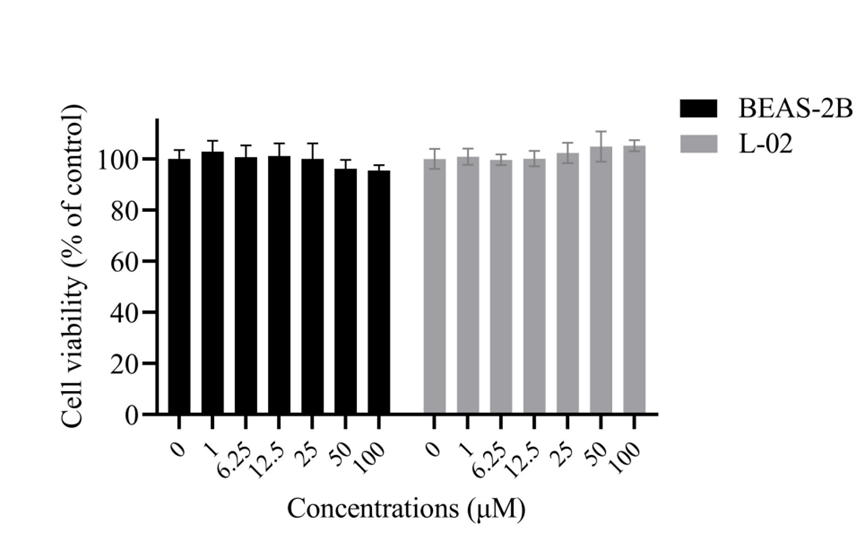
Cell viability of BEAS-2B and L-02 cell lines after 24-h exposure to various concentrations of Isorhapontigenin (0, 1, 6.25, 12.5, 25, 50, 100 μM).
Isorhapontigenin purchased from MedChemExpress. Usage Cited in: mSphere. 2024 Jun 5:e0031724. [Abstract]
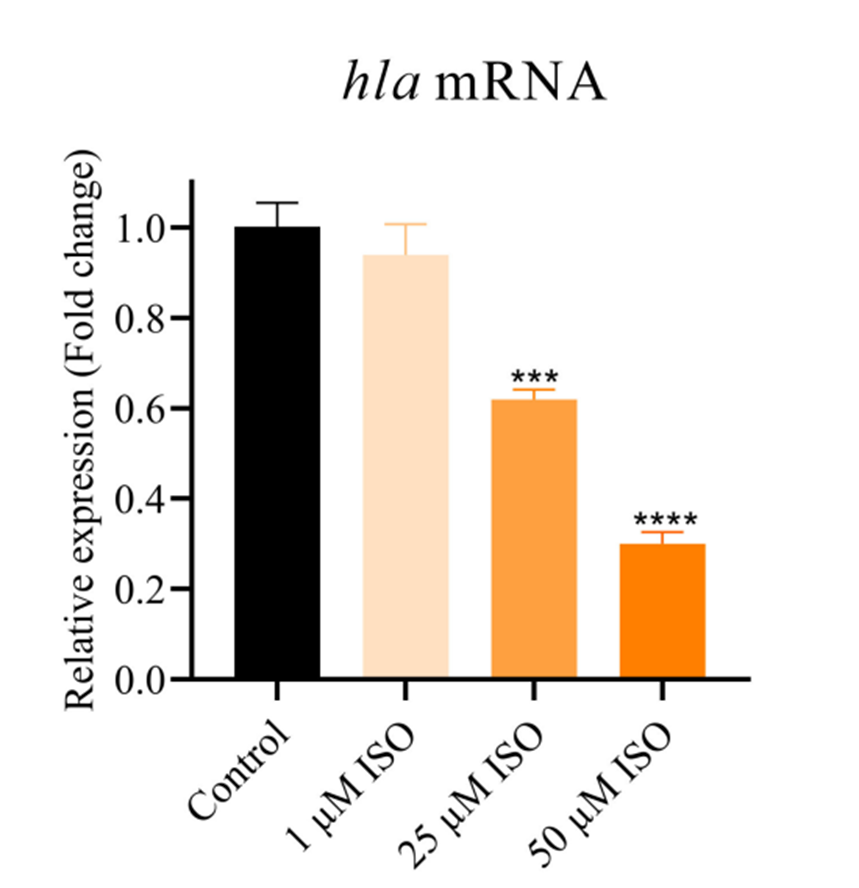
RT-qPCR analysis of the effect of Isorhapontigenin (ISO) (1, 25, 50 μM) on the expression of the hla gene.
Isorhapontigenin purchased from MedChemExpress. Usage Cited in: mSphere. 2024 Jun 5:e0031724. [Abstract]
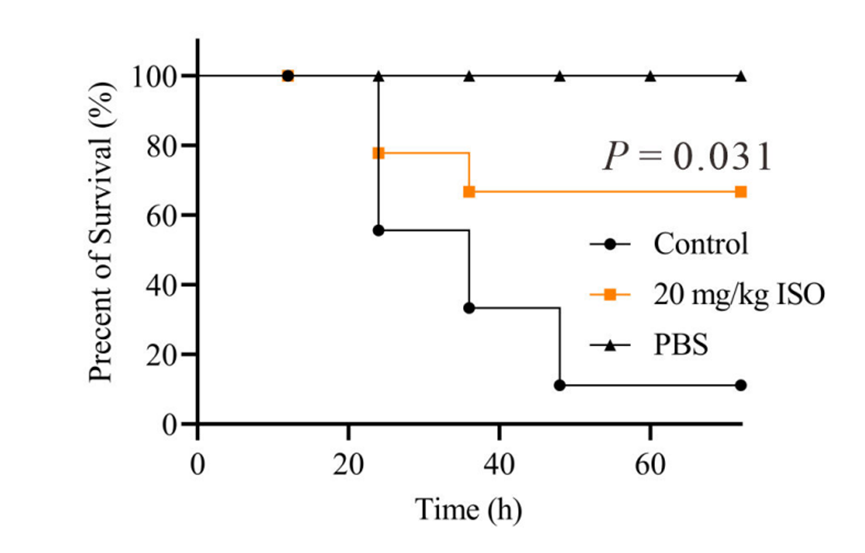
The impact of Isorhapontigenin (ISO) (20 mg/kg, s.c.) on survival rates of mice infected with a lethal dose of S. aureus Newman.
Isorhapontigenin purchased from MedChemExpress. Usage Cited in: mSphere. 2024 Jun 5:e0031724. [Abstract]
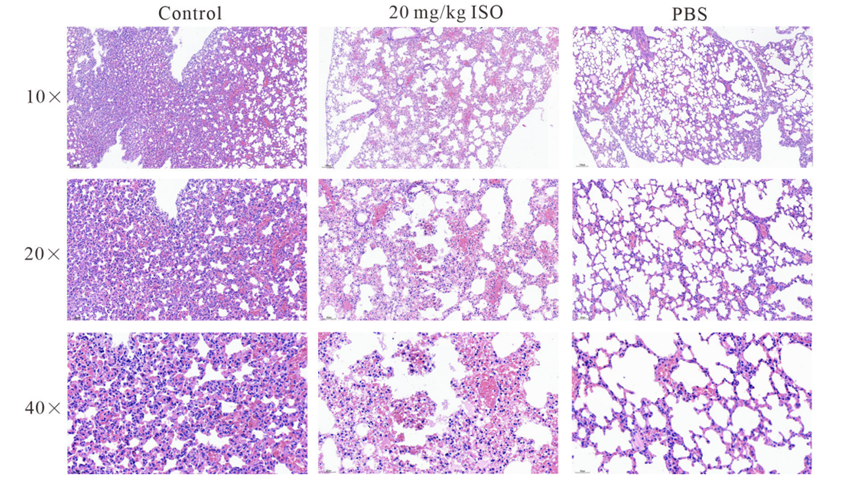
Histological analysis of the impact of Isorhapontigenin (ISO) (20 mg/kg, s.c.) on the lungs of mice, using H&E staining.
Isorhapontigenin purchased from MedChemExpress. Usage Cited in: mSphere. 2024 Jun 5:e0031724. [Abstract]
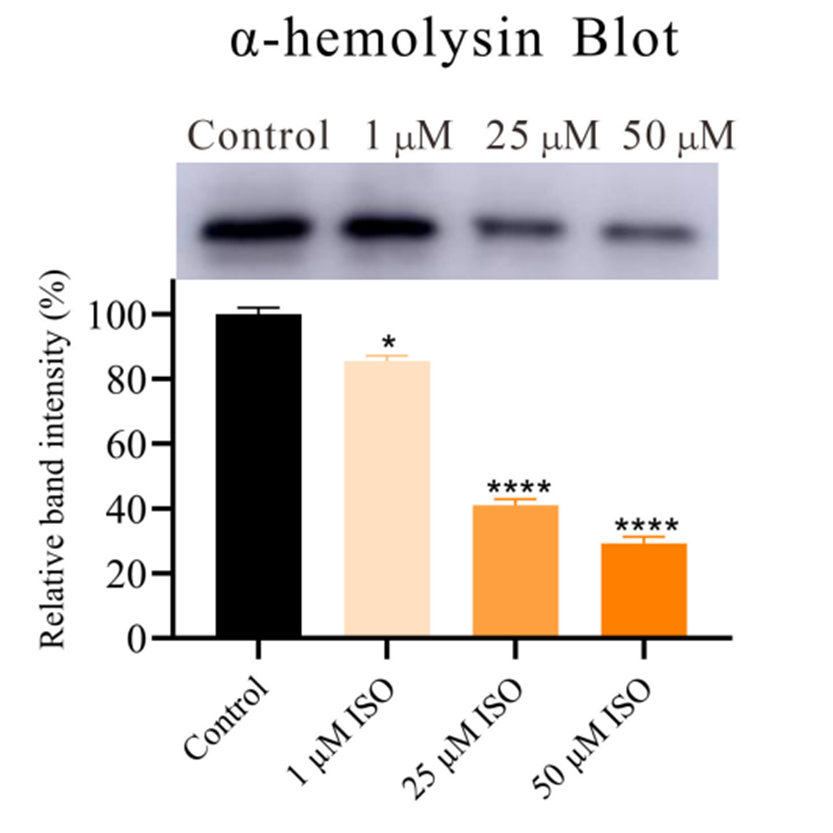
Western blot analysis of the impact of Isorhapontigenin ISO (1, 25, 50 μM) on the production of α-hemolysin.
-
Biomed Res Int
Anti-influenza A Virus Effects and Mechanisms of Emodin and Its Analogs via Regulating PPAR α/ γ-AMPK-SIRT1 Pathway and Fatty Acid Metabolism. [Abstract]2021 Sep 9;2021:9066938. PMID: 34540999


Lösungsmittel & Löslichkeit
DMSO : 50 mg/mL (193.60 mM; Need ultrasonic; Hygroscopic DMSO has a significant impact on the solubility of product, please use newly opened DMSO)
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light). When stored at -80°C, please use it within 6 months. When stored at -20°C, please use it within 1 month.
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light). When stored at -80°C, please use it within 6 months. When stored at -20°C, please use it within 1 month.
Konzentration (Stammlösung) × Volumen (Stammlösung) = Konzentration (Ziellösung) × Volumen (Ziellösung)
Select the appropriate dissolution method based on your experimental animal and administration route.
- For the following dissolution methods, please ensure to first prepare a clear stock solution using an In Vitro approach and then sequentially add co-solvents:
- To ensure reliable experimental results, the clarified stock solution can be appropriately stored based on storage conditions. As for the working solution for In Vivo experiments, it is recommended to prepare freshly and use it on the same day.
- The percentages shown for the solvents indicate their volumetric ratio in the final prepared solution. If precipitation or phase separation occurs during preparation, heat and/or sonication can be used to aid dissolution.
Add each solvent one by one: 10% DMSO 40% PEG300 5% Tween-80 45% Saline
Solubility: ≥ 2.5 mg/mL (9.68 mM); Clear solution
This protocol yields a clear solution of ≥ 2.5 mg/mL (saturation unknown).
Taking 1 mL working solution as an example, add 100 μL DMSO stock solution (25.0 mg/mL) to 400 μL PEG300, and mix evenly; then add 50 μL Tween-80 and mix evenly; then add 450 μL Saline to adjust the volume to 1 mL.
Preparation of Saline: Dissolve 0.9 g sodium chloride in ddH₂O and dilute to 100 mL to obtain a clear Saline solution.
Add each solvent one by one: 10% DMSO 90% (20% SBE-β-CD in Saline)
Solubility: 2.5 mg/mL (9.68 mM); Suspended solution; Need ultrasonic
This protocol yields a suspended solution of 2.5 mg/mL. Suspended solution can be used for oral and intraperitoneal injection.
Taking 1 mL working solution as an example, add 100 μL DMSO stock solution (25.0 mg/mL) to 900 μL 20% SBE-β-CD in Saline, and mix evenly.
Preparation of 20% SBE-β-CD in Saline (4°C, storage for one week): 2 g SBE-β-CD powder is dissolved in 10 mL Saline, completely dissolve until clear.
Please enter the basic information of animal experiments:
-
-
-
-
Recommended: Prepare an additional quantity of animals to account for potential losses during experiments.
Please enter your animal formula composition:
-
%DMSO +
Recommended: Keep the proportion of DMSO in working solution below 2% if your animal is weak.
-
%+
-
+%Tween-80 + +
-
%Saline +
The co-solvents required include: DMSO, . All of co-solvents are available by MedChemExpress (MCE). , Tween 80. All of co-solvents are available by MedChemExpress (MCE).
Working solution concentration: 0.22 mg/mL
Method for preparing stock solution: mg drug dissolved in μL DMSO. Stock solution concentration: mg/mL. * In solvent : -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light)
1. Take μL DMSO stock solution;
2. Add μL .
μL , mix evenly;
3. Then add μL Tween 80, mix evenly;
4. Then add μL
Please ensure that the stock solution in the first step is dissolved to a clear state, and add co-solvents in sequence. You can use ultrasonic heating (ultrasonic cleaner, recommended frequency 20-40 kHz), vortexing, etc. to assist dissolution.


Reinheit & Dokumentation
-
Data Sheet (295 KB)
-
SDS (476 KB)
- English - EN (476 KB)
- Français - FR (476 KB)
- Deutsch - DE (476 KB)
- Norwegian - NO (476 KB)
- Español - ES (476 KB)
- Swedish - SV (476 KB)
- Italian - IT (476 KB)
- Korean - KR (476 KB)
- Portuguese - PT (476 KB)
-
Handling Instructions (2659 KB)
Verweise
[1]. Liang Y, et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy. 2016;12(8):1229-1239. [Content Brief]
[2]. Zha H, et al. Isorhapontigenin alleviates acetaminophen-induced liver injury by promoting fatty acid oxidation. Biochim Biophys Acta Mol Basis Dis. 2025;1871(2):167575. [Content Brief]
[3]. Yeo SCM, et al. Isorhapontigenin, a bioavailable dietary polyphenol, suppresses airway epithelial cell inflammation through a corticosteroid-independent mechanism. Br J Pharmacol. 2017;174(13):2043-2059. [Content Brief]
[4]. Yao P, et al. Isorhapontigenin alleviates lipopolysaccharide-induced acute lung injury via modulating Nrf2 signaling. Respir Physiol Neurobiol. 2021;289:103667. [Content Brief]
[5]. Chu XY, et al. Isorhapontigenin Improves Diabetes in Mice via Regulating the Activity and Stability of PPARγ in Adipocytes. J Agric Food Chem. 2020;68(13):3976-3985. [Content Brief]
[6]. Jiang G, et al. Isorhapontigenin (ISO) Inhibits Invasive Bladder Cancer Formation In Vivo and Human Bladder Cancer Invasion In Vitro by Targeting STAT1/FOXO1 Axis. Cancer Prev Res (Phila). 2016 Jul;9(7):567-80. [Content Brief]



Complete Stock Solution Preparation Table
Please refer to the solubility information to select the appropriate solvent. Once prepared, please aliquot and store the solution to prevent product inactivation from repeated freeze-thaw cycles.
Storage method and period of stock solution: -80°C, 6 months; -20°C, 1 month (sealed storage, away from moisture and light). When stored at -80°C, please use it within 6 months. When stored at -20°C, please use it within 1 month.
| Optional Solvent | Concentration Solvent Mass | 1 mg | 5 mg | 10 mg | 25 mg |
|---|---|---|---|---|---|
| DMSO | 1 mM | 3.8719 mL | 19.3596 mL | 38.7192 mL | 96.7979 mL |
| 5 mM | 0.7744 mL | 3.8719 mL | 7.7438 mL | 19.3596 mL | |
| 10 mM | 0.3872 mL | 1.9360 mL | 3.8719 mL | 9.6798 mL | |
| 15 mM | 0.2581 mL | 1.2906 mL | 2.5813 mL | 6.4532 mL | |
| 20 mM | 0.1936 mL | 0.9680 mL | 1.9360 mL | 4.8399 mL | |
| 25 mM | 0.1549 mL | 0.7744 mL | 1.5488 mL | 3.8719 mL | |
| 30 mM | 0.1291 mL | 0.6453 mL | 1.2906 mL | 3.2266 mL | |
| 40 mM | 0.0968 mL | 0.4840 mL | 0.9680 mL | 2.4199 mL | |
| 50 mM | 0.0774 mL | 0.3872 mL | 0.7744 mL | 1.9360 mL | |
| 60 mM | 0.0645 mL | 0.3227 mL | 0.6453 mL | 1.6133 mL | |
| 80 mM | 0.0484 mL | 0.2420 mL | 0.4840 mL | 1.2100 mL | |
| 100 mM | 0.0387 mL | 0.1936 mL | 0.3872 mL | 0.9680 mL |


 Powered by Bioz
Powered by Bioz